

Abstract
The 70-amino-acid A13L protein is a component of the vaccinia virus membrane. We demonstrate here that the protein is expressed at late times of infection, undergoes phosphorylation at a serine residue(s), and becomes encapsidated in a monomeric form. Phosphorylation is dependent on Ser40, which lies within the proline-rich motif SPPP. Because phosphorylation of the A13 protein is only minimally affected by disruption of the viral F10 kinase or H1 phosphatase, a cellular kinase is likely to be involved. We generated an inducible recombinant in which A13 protein expression is dependent upon the inclusion of tetracycline in the culture medium. Repression of the A13L protein spares the biochemical progression of the viral life cycle but arrests virion morphogenesis. Virion assembly progresses through the formation of immature virions (IVs); however, these virions do not acquire nucleoids, and DNA crystalloids accumulate in the cytoplasm. Further development into intracellular mature virions is blocked, causing a 1,000-fold decrease in the infectious virus yield relative to that obtained in the presence of the inducer. We also determined that the temperature-sensitive phenotype of the viral mutant Cts40 is due to a nucleotide transition within the A13L gene that causes a Thr48→Ile substitution. This substitution disrupts the function of the A13 protein but does not cause thermolability of the protein; at the nonpermissive temperature, virion morphogenesis arrests at the stage of IV formation. The A13L protein, therefore, is part of a newly recognized group of membrane proteins that are dispensable for the early biogenesis of the virion membrane but are essential for virion maturation.
Vaccinia virus, the prototypic poxvirus, is a complex DNA virus that replicates solely within the cytoplasm of infected cells. Among the 200 gene products encoded by the 192-kb DNA genome are those involved in gene expression, genome replication and resolution, and virion assembly. Morphogenesis of the large, membrane-bound virion is a complex process that has been dissected by an effective combination of genetic, biochemical, and ultrastructural analyses. The onset of morphogenesis is dependent upon the dual-specificity protein kinase encoded by the F10 gene; none of the visually distinct intermediates associated with morphogenesis are seen when cells are infected under nonpermissive conditions with conditionally lethal mutants bearing lesions in the F10 gene (40, 43). The normal sequence of events can be broadly defined as follows: the condensation of electron-dense “virosomes” within the cytoplasm, the emergence of membrane crescents at the border of these virosomes, the enlargement of these crescents into oval, immature virions (IV), the appearance of a dense nucleoid within the immature virions (IVN), and the restructuring of these immature forms into mature, brick-shaped virions that contain a distinct internal core (IMV). The membrane of the virion is thought to be derived from tubulovesicular elements of the endoplasmic reticulum (ER) or ER Golgi intermediate compartment (ERGIC); biochemical analyses have identified the A17L, A14L, A13L, L1L, D8R, and H3L proteins as the major components of the membrane (15, 38).
Genetic analyses of inducible recombinants, in which the expression of a target protein is regulated by the elements of the tetracycline (TET) or lactose operon, have identified the A14L and A17L proteins as essential for membrane biogenesis (31, 34, 41, 45). Both of these proteins are inserted into membranes cotranslationally, appear to span the viral membrane twice, and are phosphorylated (1, 2, 18, 21, 35). The A14L protein is found primarily as a disulfide-linked dimer (14, 21, 33), and the A17L protein undergoes proteolytic processing at both its N′ and C′ termini (32, 44). The H3L protein is the major immunodominant protein within the virion membrane and appears to associate with the membrane posttranslationally (6, 48). Viruses containing H3 deletions are viable, although they show a small plaque phenotype, a 10-fold reduction in viral yield, and a corresponding accumulation of morphogenetic intermediates (7). The myristylated L1R protein is essential; when its expression is repressed, virion maturation arrests at the IV→IMV transition (30). The D8R protein is not involved in virion formation, but rather in cell attachment. It binds chondroitin sulfate, and its deletion compromises cell entry and becomes more debilitating when the heparan sulfate-binding A27L protein is also deleted (13).
No reports of the role of the A13L protein have been published, although some structural characterization has been performed (35, 38). It is only 70 amino acids long and has an N-terminal hydrophobic region that is implicated in cotranslational insertion of the protein into the membranes of the ER. It is accessible to proteolytic cleavage in intact virions, suggesting that the bulk of the protein is cytosolic during synthesis and extends outward from the virion surface. Although its predicted pI is 10, demonstrations that its apparent pI is 8 led to the suggestion that the protein might sustain an acidic modification such as phosphorylation (15, 35). Because our laboratory is interested both in the role of phosphorylation within the vaccinia life cycle and in virion morphogenesis, we utilized a genetic approach to study the role of the A13L protein in the viral life cycle. Here we describe the construction and/or characterization of TET-inducible recombinants and temperature-sensitive mutants, which together have enabled a biological analysis of the structure and function of the A13L protein. Cumulatively, these studies reveal that the A13L phosphoprotein is essential for virion morphogenesis, playing a primary role at the IV→IMV transition.
MATERIALS AND METHODS
Materials.
Restriction endonucleases, polynucleotide kinase, T4 DNA ligase, calf intestinal phosphatase, pancreatic RNase, S1 endonuclease, and Taq polymerase were purchased from Roche Molecular Biochemicals (Indianapolis, Ind.) and were used according to the manufacturer's instructions. 32P-labeled nucleoside triphosphates, [32P]orthophosphate, and [35S]methionine were purchased from Perkin-Elmer Life Sciences (Boston, Mass.). Rifampin, α-FLAG M2 monoclonal antibody, and 5-bromo-2′-deoxyuridine (BrdU) were acquired from Sigma (St. Louis, Mo.). Geneticin (G418), Lipofectamine Plus, and DNA and protein molecular weight standards were obtained from Invitrogen (Carlsbad, Calif.). Thin-layer cellulose F chromatography plates were purchased from EM Science, Inc. (Gibbstown, N.J.). Supersignal chemiluminescent substrate and constant-boiling HCl were purchased from Pierce (Rockford, Ill.). Isatin-β-thiosemicarbazone (IBT) was obtained from Pfaltz and Bauer (Waterbury, Conn.). Dulbecco's modified Eagle's medium (DMEM) lacking methionine or phosphate was purchased from ICN (Costa Mesa, Calif.).
Cells and viruses.
Monolayer cultures of BSC40 cells or human thymidine kinase-deficient 143 cells (TK−) were maintained in DMEM containing 5% fetal calf serum (Invitrogen). Wild-type (wt) vaccinia virus (WR strain), Cts40 (kindly provided by R. Condit, University of Florida), vtetR (expresses the TET repressor) (41), vROG8 (a viral recombinant in which G8 expression is IPTG dependent; kindly provided by B. Moss, National Institutes of Health [NIH]) (47), vindA13, and vΔindA13 (described herein) were amplified in monolayer cultures of BSC40 cells or suspension cultures of L cells. Viral stocks were prepared from cytoplasmic lysates of infected cells by ultracentrifugation through 36% sucrose; titrations were performed on BSC40 cells. When indicated, rifampin was added to a final concentration of 100 μg/ml, BrdU was added to a final concentration of 25 μg/ml, and TET was added to a final concentration of 1 μg/ml. For Cts40, 31.5 and 39.7°C were used as the permissive and nonpermissive temperatures, respectively.
Polyclonal antiserum preparation. (i) Construction of pATH11:A13.
The portion of the A13 open reading frame downstream of the N′ hydrophobic region (amino acids [aa] 18 to 70) was amplified by PCR, with genomic DNA as the template. The upstream primer (TrpU) (5′CGGAATTCATCCTATACTCGATGTAT3′) introduced an EcoRI site (in bold) upstream of aa 18; the downstream primer (TrpD) (5′CCGGATCCTTATACAGAAGATTTAAC3′) introduced a BamHI (in bold) site after the termination codon (underlined). The 162-bp PCR product was digested with EcoRI and BamHI and ligated to pATH11 DNA (17) that had been previously digested with EcoRI and BamHI and treated with calf intestinal alkaline phosphatase. The resultant plasmid fused most of the A13 open reading frame (ORF) downstream of, and in frame with, the Escherichia coli trpE gene (pATH11:A13).
(ii) Expression of trpE:A13 immunogen.
E. coli transformants containing the pATH11:A13 plasmid were induced to synthesize a 42-kDa fusion protein containing 6 kDa of A13 and 36 kDa of E. coli TrpE. The fusion protein was excised from sodium dodecyl sulfate (SDS)-polyacrylamide gels and used to immunize rabbits. The specificity of the resultant polyclonal antiserum was confirmed by immunoblotting and immunoprecipitation assays.
One-dimensional phosphoamino acid analysis.
Phosphoamino acid analysis was performed essentially as described by the manufacturers of the Hunter thin-layer electrophoresis system, model HTLE-7000 (3). 32P-A13 was immunoprecipitated from metabolically labeled extracts. The immunoprecipitates were fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to Immobilon P membranes, and exposed to autoradiographic film. Alignment of the film and the filter permitted excision of the radiolabeled band from the filter. The protein was hydrolyzed in situ by incubation at 110°C in 6 N constant-boiling HCl for 1 to 2 h. The liberated hydrolysate was dried, resuspended in PAA electrophoresis buffer (pyridine-glacial acetic acid-water [1:10:189]), mixed with phosphoamino acid markers (20 nmol of each), and spotted onto thin-layer cellulose F chromatography plates. Electrophoresis was performed in the same buffer at 1,800 V for 30 min. The plates were dried, sprayed with ninhydrin, developed at 65°C, and exposed for autoradiography.
Plaque assay.
Confluent monolayers of BSC40 cells were infected with serial dilutions of the appropriate virus. Infections were allowed to proceed at the indicated temperature(s) for 48 h postinfection (hpi) before being stained with crystal violet.
Determination of 24-h viral yield.
Confluent monolayers of BSC40 cells were infected with the vindA13, vΔindA13, or vtetR virus at a multiplicity of infection (MOI) of 2 and maintained at 37°C or with the wt or Cts40 virus and maintained at 31.5 and 39.7°C. At 24 hpi, the cells were harvested, resuspended in 1 mM Tris (pH 9.0), and disrupted by three cycles of freezing-thawing and two 10-s bursts of sonication. Viral yields were determined by titration on BSC40 cells at 37 or 31.5C, as indicated.
Immunoblot analysis.
Cell extracts, often containing phosphatase inhibitors, were subjected to fractionation by SDS-PAGE and then transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, N.H.) in 10 mM CAPS buffer [3-(cyclohexylamino)-1-propane-sulfonic acid (pH 11.3) with 10% methanol]. The filters were incubated with the appropriate primary sera and secondary antibodies (horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse immunoglobulin G [Bio-Rad, Richmond, Calif.]), developed with enhanced chemiluminescent reagents, and visualized on Kodak MR film.
S1 nuclease mapping.
A 632-bp fragment containing the A13 ORF, the TET operator, and upstream sequences was released from pUC/NEO-op-A13 by digestion with BamHI. This fragment was treated with calf intestinal alkaline phosphatase and radiolabeled with [γ-32P]ATP in the presence of polynucleotide kinase. The fragment was then cleaved with MlyI, and the resultant 401-bp fragment containing a single 5′ radiolabel was resolved electrophoretically, purified on glass beads (42), and used as a probe for S1 nuclease analysis. Total RNA was prepared from BSC40 cells (RNeasy Midi kit; Qiagen, Valencia, Calif.) at 8 hpi with either vtetR or vindA13 (MOI = 10) in the presence or absence of TET (1 μg/ml). Purified RNA (10 μg) was mixed with 40 ng of probe (40,000 cpm/reaction) in a 30-μl reaction mixture containing 40 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 400 mM NaCl, 80% formamide, and 1 mM EDTA. The samples were denatured by incubation at 65°C for 10 min and then allowed to anneal for 3 h at 37°C. The samples were diluted 10-fold with a cocktail containing 280 mM NaCl, 30 mM sodium acetate, 4.5 mM ZnSO4, 5% glycerol, and 400 U of S1 nuclease per ml. The digestion was allowed to proceed for 30 min at 37°C. The samples were then subjected to organic extraction and ethanol precipitation; the reactions were visualized by autoradiography after resolution on a 6% urea-acrylamide gel.
Construction of vindA13.
Overlap PCR was used to generate a 632-bp DNA fragment which included 213 bp of the A13 ORF flanked by 59 bp of downstream (toward A12) and 341 bp of upstream (toward A14) sequence. This construct also inserted the 19-bp operator sequence from the TET operon (5′TCCCTATCAGTGATAGAGA3′) between the transcriptional and translational start sites of the A13 gene (9-11, 40). Viral genomic DNA was used as the template for the following primers: 4, 5′TTGGATCCGTGCTAATCGCTGGA3′; 1, 5′TTGGATCCACTGCTTCTAACGGCTA3′; B, 5′CCCTATCAGTGATAGAGAATGATTGGTATTCTTTT3′; and C, 5′CTCTATCACTGATAGGGATATTTATTGACAATAACT3′. Primers 1 and 4 insert BamHI sites (in bold) at the ends of the final PCR product; primers B and C insert the operator sequences (italicized) between the transcriptional and translational start sites (underlined) of the A13 ORF. In the first round of PCR, primers 1 plus B and C plus 4 were used to generate fragments of 292 and 357 bp, respectively, that overlapped by 17 bp. The products were purified on glass beads, and aliquots of each were used together as the template for a second round of amplification using primers 1 and 4. The final 632-bp product was digested with BamHI and ligated to pUC:NEO DNA which had been previously digested with BamHI and treated with calf intestinal alkaline phosphatase (16, 20). The resultant plasmid was designated pUC:NEO-OP-A13.
Insertion of the TET operator-regulated A13 gene into the endogenous A13 locus was achieved through transient dominant selection. Cells were infected with the vtetR virus (MOI = 0.03) and transfected at 3 hpi with pUC:NEO-OP-A13 DNA. From this point on, TET (1 μg/ml) was included in the culture medium at all times. At 19 hpi, the medium was replaced with fresh medium containing G418 (3 mg/ml). At 48 hpi, the cells were harvested and G418r viruses were isolated by two rounds of plaque purification in the presence of G418. These G418r viruses should contain a partial duplication of the A13 locus in which the endogenous and exogenous (operator-containing) copies of the gene flank the inserted pUC:NEO sequences. This genomic structure was confirmed by performing PCR with primers that flanked the transcriptional start site of the A13 gene. Reactions performed with primers U1 (5′GGAGGCGAATTTAAATTCGG3′) and D1 (5′CGTCCATATATCTCGACA3′) amplified products of 224 and 243 bp from the endogenous and operator-containing alleles, respectively. These viruses were then subjected to three rounds of plaque purification in the absence of G418 in order to allow the duplicated region to resolve by homologous recombination. The same PCR was performed to distinguish viruses that had resolved to contain only the wt allele from those retaining the operator-containing allele. Plaques of interest were then expanded for further study and designated vindA13.
Attempted construction of A13 deletion mutant.
PCR was used to generate a 351-bp fragment of the A12 gene (includes 46 bases of the A13 ORF) and a 377-bp fragment of the A14 gene. The primers used to generate the fragments were as follows: A, 5′AAGGTACCTAGGAGCACCAGAAGT3′; B, 5′GGGGATCCGGATCATATTAGTTCAT3′; C, 5′CCGGATCCCAAGCTTTTGACAATAACTTCACT3′; and D, 5′GCTCTAGATGATGCTTATGATTGG3′. Primers A and B were used to generate an A12 fragment containing KpnI and BamHI sites (in bold) at the 5′ and 3′ termini, respectively; primers C and D amplified an A14 fragment containing BamHI and HindIII/XbaI (bold) sites at the termini. The products were purified with glass beads, digested with either KpnI plus BamHI or BamHI plus XbaI (as appropriate), and ligated simultaneously to pBSIIKS vector DNA (Stratagene, La Jolla, Calif.) that had been previously digested with KpnI and XbaI and treated with calf intestinal alkaline phosphatase. The resultant plasmid, pBSIIKS-A12,A14, was then used as the backbone for the next round of cloning. A 3.2-kb fragment containing the p7.5 promoter and the LacZ gene was released from pSC65 (provided by B. Moss [4]) by using BamHI and HindIII. The purified fragment was inserted into pBSIIKS-A12,A14 DNA which had been previously digested with BamHI and HindIII and treated with calf intestinal alkaline phosphatase. The resulting plasmid was designated pA12-LacZ-A14.
BSC40 cells were infected with wt virus (MOI = 0.03) and transfected at 3 hpi with linearized pA12-LacZ-A14 DNA. Cells were harvested at 48 hpi. To screen for the generation of LacZ+ viral recombinants, we infected the cells with serial dilutions of the harvested cells and overlaid them with agarose containing X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (0.033%). Blue plaques were picked and PCR was performed to determine whether the LacZ-containing cassette had inserted via homologous recombination, with the accompanying deletion of A13, or via random integration into nonhomologous regions within the genome. Screening for the loss or presence of A13 was performed by using the following primers: A13:A, 5′GGGAATTCCATATGATTGGTATTCTTT3′; and A13:B, 5′CCGGATCCTTATACAGAAGATTTAAC3′.
Construction of vΔindA13 virus. (i) Construction of vTK:tetR↔indA13 virus. (a) Plasmid construction.
The pJS4 plasmid (kindly provided by B. Moss, NIH) contains two divergent viral promoters adjacent to independent polylinker sequences (4); integration into the viral genome is enabled by the presence of flanking TK sequences. We used this plasmid to insert (i) the tetR gene and (ii) a TET-regulated copy of the A13 gene. First, a 624-bp BamHI fragment encoding the TET repressor gene (9-11) was inserted into vector DNA which had been previously digested with BamHI and treated with calf intestinal alkaline phosphatase. The resultant plasmid was named pJS4-tetR.
Overlap PCR was then used to generate a 275-bp DNA fragment containing 213 bp of the A13 gene, the 19-bp tetO sequence, and 43 bp of the flanking upstream sequence (toward A14). The primers used to generate this fragment were as follows: A, 5′GGGATCGATTTATACAGAAGATTTAAC3′; B, 5′CCCTATCAGTGATAGAGAATGATTGGTATTCTTTT3′; C, 5′CTCTATCACTGATAGGGatatttaTTGACAATAACT3′; and D, 5′GGGGATCCAAGCTTGATATATTGCATACTT3′. The template was viral genomic DNA. Primers A and D inserted ClaI and HindIII/BamHI (in bold) sites at the termini of the final PCR product; primers B and C inserted the operator sequence (italicized) between the transcriptional (small capital letters, bold) and translational (underlined) start sites of the A13 ORF. In the first round of PCR, primers A plus B and C plus D were used to generate fragments of 230 and 62 bp, respectively, that overlapped by 17 bp. The products were used together as the template for a second round of PCR performed with primers A and D. The final 275-bp product was digested with ClaI and HindIII and ligated to pJS4-tetR DNA which had previously been digested with ClaI and HindIII and treated with calf intestinal alkaline phosphatase. The final plasmid was designated pJS4:tetR↔indA13.
(b) Virus construction.
Insertion of the tetR↔indA13 cassette into the TK locus of the viral genome was achieved through homologous recombination. Cells were infected with the wt virus (MOI = 0.03) and transfected with pJS4:tetR↔indA13 DNA at 3 hpi. At 48 hpi, the cells were harvested and TK− virus was isolated by two rounds of plaque purification on human TK− cells in the presence of BrdU (25 μg/ml). Insertion of the tetR↔indA13 cassette was confirmed by PCRs using the following primers directed to the tetR sequence: Tetr5′, 5′CAGCGCATTAGAGCTC3′; and Tetr3′, 5′TTAAGACCCACTTTCACA3′. Plaques of the correct genotype were expanded; this viral recombinant was designated vTK:tetR↔indA13.
(ii) Deletion of endogenous A13 allele within vTK:tetR↔indA13. (a) Plasmid construction for generation of a pA12-neo-A14 targeting construct.
The pBSIIKS-A12,A14 plasmid described above was digested with BamHI and HindIII, and a 1,500-bp BamHI-HindIII fragment representing p7.5-NEO (excised from the pUCNeo plasmid [16, 20]) was inserted by ligation. In this final plasmid, designated pA12-neo-A14, the NEO gene was placed between the A12 and A14 ORFs in place of the A13 gene.
(b) Virus construction for targeted disruption of endogenous A13 locus.
BSC40 cells were infected with vTK:tetR↔indA13 (MOI = 0.03) and transfected at 3 hpi with pA12-neo-A14 DNA that had been linearized with ScaI. From this time on, TET (1 μg/ml) was included in the culture medium at all times. At 19 hpi, the medium was replaced with fresh medium that also contained G418 (2.5 mg/ml). At 48 hpi, the cells were harvested and subjected to two rounds of plaque purification in the presence of TET and G418. After each round of purification, PCR was performed to confirm the presence of the NEO gene as well as the absence of A13. The following primers were used to screen for the retention or deletion of A13: A13:5′, 5′GGAGGCGAATTTAAATTCGG3′; and A14:A, 5′TTGGATCCGTGCTAATCGCTGGA3′. The final recombinant, from which the endogenous A13 locus had been deleted and in which an inducible A13 allele was present within the TK locus, was designated vΔindA13.
Metabolic labeling. (i) Metabolic labeling with 32PPi.
Monolayers of BSC40 cells were infected with wt virus (MOI = 10) and metabolically labeled with 32PPi from 3 to 17 hpi. Alternatively, cells infected with vΔindA13 and transfected with plasmids encoding 3X-FLAG-A13 variants (see below) were metabolically labeled from 6 to 24 hpi. Labeling was performed by incubating cells in the presence of phosphate-free DMEM (ICN-Flow) supplemented with l-glutamine, 5% phosphate-free fetal calf serum (prepared by dialysis against Tris-buffered saline [25 mM Tris-HCl, pH 7.4, 136 mM NaCl, 2.7 mM KCl]), and 100 μCi of 32PPi per ml. The cells were harvested and lysates were analyzed by immunoprecipitation as described below.
(ii) Metabolic labeling with [35S]methionine.
Infected monolayers were rinsed with methionine-free DMEM (ICN-Flow) supplemented with l-glutamine and then radiolabeled in the same medium supplemented with 100 μCi of [35S]methionine (EXPRESS label; Perkin-Elmer Life Sciences) per ml. The time and duration of the labeling reactions are indicated in the figure legends.
Immunoprecipitation.
For immunoprecipitation analysis, cell extracts were prepared in 1× phospholysis buffer (10 mM NaPO4 [pH 7.4], 100 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) and clarified by centrifugation; after incubation with the indicated antisera, antigen-antibody complexes were retrieved on protein A-Sepharose (Sigma, St. Louis, Mo.) and analyzed by SDS-PAGE and autoradiography. Where appropriate, phosphatase inhibitors (1 mM sodium orthovanadate, 1 mM sodium fluoride, and 40 mM β-glycerolphosphate) were included in the lysis and wash buffers.
Genome resolution assay.
Confluent 35-mm-diameter dishes of BSC40 cells were infected with wt or vΔind A13 virus (with or without TET) at an MOI of 2 or with vROG8 (without IPTG) (47) at an MOI of 15. An additional culture was infected with wt virus in the presence of 60 μm IBT. At 18 hpi, the cells were harvested, washed with ice-cold phosphate-buffered saline (PBS), resuspended in 50 mM NaCl-10 mM EDTA-20 mM Tris (pH 8.0), and incubated for 10 min on ice. The samples were adjusted to 0.6% SDS and 0.7 μg of proteinase K per ml and incubated at 37°C for 6 h. The DNA was purified by organic extraction and ethanol precipitation, digested with BstEII, fractionated by electrophoresis through a 1% agarose gel, run in 1× TAE (40 mM Tris acetate, 1 mM EDTA), and then transferred to a ZetaProbe membrane in 0.4 M NaOH. The membrane was probed with a radiolabeled PvuII-EcoRI fragment excised from pSV9 (a gift from M. Merchlinsky) to visualize the DNA fragments corresponding to the telomeres of the monomeric viral genome and the concatemer junction of replication intermediates (22, 23). The blot was washed, dried, and exposed for autoradiography.
Electron microscopy.
Confluent 60-mm-diameter dishes of BSC40 cells were infected with vΔindA13 in the presence or absence of TET (1 μg/ml) at an MOI of 2 and incubated at 37°C or with wt or Cts40 virus and incubated at 31.5 and 39.7°C. At 17 hpi, the cells were rinsed with ice-cold PBS and then fixed in situ with 1% glutaraldehyde in PBS (on ice for 60 min). The cells were then removed from the dish by gentle scraping, collected by centrifugation, and processed for conventional transmission electron microscopy (40). Samples were examined on a Hitachi H-600 electron microscope.
Electron microscopy of rifampin-synchronized cultures.
Dishes (60-mm diameter) of BSC40 cells were infected with either wt virus or vΔindA13 (MOI = 2). Rifampin (100 μg/ml) was added 10 min before infection and was present continuously until 12 hpi. At 12 hpi, the cells were either harvested immediately (0 min) or refed with warm medium and incubated at 37°C for the indicated times (5 to 180 min) prior to being harvested. Upon harvesting, the cells were washed with ice-cold PBS and fixed with 1% glutaraldehyde in PBS for 10 min on ice, followed by incubation for 20 min at 37°C. The cells were processed for conventional electron microscopy as described above.
Construction of A13 expression constructs containing Ser→Ala substitutions and a 5′ 3X-FLAG epitope tag.
Mutant alleles of A13 encoding serine-to-alanine substitutions were generated by PCR or overlap PCR, as necessary, according to the strategy shown in Table 1. The following primers were used: U-flag, 5′GCGGATCCATATGATTGGTATTCTTTTG3′; D, 5′CCGGATCCTTATACAGAAGATTTAAC3′; 68,69B, 5′CCGGATCCTTATACAGCAGCTTTAAC3′; 60,61,68,69B, 5′CCGGATCCTTATACAGCAGCTTTAACTAGATTATACAATGCAGCAATATGATCCTTTTCCAG3′; 35,40T, 5′GCTCCGAATTTAAATGCGCCTCCTCC3′; 35,40B, 5′CATTTAAATTCGGAGCTGGATTCGGA3′; 21,29T, 5′GTATAATAAGATCAAGAACGCACAAAATCCG ′; 21,29B, 5′CTTGATCTTATTATACATCGCGTATAGGATGG3′; 35T, 5′CGAATCCAGCTCCGAATT3′; 35B, 5′ATTTAAATTCGGAGCTGGATTCGG3′; 40T, 5′GTCCGAATTTAAATGCGCCTCC3′; and 40B, 5′GTTCTGGAGGAGGCGCATTTAAAT3′. The U-flag primer inserts an NdeI site at the 5′ terminus, overlapping the translational start site of A13 (underlined); primers D and 60,61,68,69B insert a BamHI site (bold) at the 3′ terminus downstream of the translational stop site (underlined) of the A13 ORF.
TABLE 1.
Construction of A13 expression constructs
| Mutant | Ser residues changed (positions) | PCR 1 primers | PCR 2 primers | PCR 3 primers (products 1 + 2 used as template) |
|---|---|---|---|---|
| 1 | 21, 29 | U-flag + 21,29B | 21,29T + D | U-flag + D |
| 2 | 60, 61, 68, 69 | U-flag + 60,61,68,69B | ||
| 3 | 21, 29, 60, 61, 68, 69 | U-flag + 21,29B | 21,29T + 60,61,68,69B | U-flag + 60,61,68,69B |
| 4 | 35, 40, 60, 61, 68, 69 | U-flag + 35,40B | 3540T + 60,61,68,69B | U-flag + 60,61,68,69B |
| 5 | 21, 29, 35, 40, 60, 61, 68, 69 | U-flag + 35,40B (3 as template) | 3540T + 69,61,68,69B | U-flag + 60,61,68,69B |
| 6 | 35 | U-flag + 35B | 35T + D | U-flag + D |
| 7 | 40 | U-flag + 40B | 40T + D | U-flag + D |
A wt copy of the A13 ORF served as the template for mutants 2 to 4, 6, and 7; mutant 3 served as the template for PCRs 1 and 2 of mutant 5. The mutant alleles were ligated into pJS4-3X-FLAG (a derivative of pJS4 that accepts NdeI-BamHI inserts and leads to the addition of an in-frame N′ 3X-FLAG epitope) (A. Punjabi and P. Traktman, unpublished data) DNA that had already been digested with NdeI and BamHI and treated with calf intestinal phosphatase. Plasmids were prepared from E. coli transformants by the use of Qiagen Endofree Maxi kits and subjected to automated DNA sequencing.
Infection-transfection assay.
Dishes (35-mm diameter) of confluent BSC40 cells were infected with vΔindA13 at an MOI of 2 in the presence or absence of TET (1 μg/ml). At 3 hpi, 5 μg of supercoiled DNA (pJS4-3XFLAG or pJS4-3XFLAG:A13 constructs) was transfected into infected cells by the use of Lipofectamine Plus reagent (Invitrogen). Cells were harvested at 24 hpi. The lysates were subjected to fractionation on a 17% polyacrylamide gel for subsequent immunoblot analysis.
Marker rescue.
Confluent 35-mm-diameter dishes of BSC40 cells were infected with Cts40 virus (MOI = 0.03) at 31.5°C. At 3 hpi, the cells were transfected with calcium phosphate precipitates containing 3.5 μg of pUC1246, pUC1246:A17, pUC1246:A14, or pUC1246:A13 DNA that had been previously linearized by digestion with ScaI. The cells were shifted to 39.7°C at the time of transfection. At ∼48 hpi, the cells were harvested, and the yield of total and temperature-insensitive viruses was determined by titration at 31.5 and 39.7°C.
DNA sequence analysis of the A13 allele within Cts40.
The A13 allele was amplified from the Cts40 stock by using primers A (5′GCGGATCCATATGATTGGTATTCTTTTG3′) and B (5′CCGGATCCTTATACAGAAGATTTAAC3′). Two independent PCRs were performed, and the resulting PCR products were digested with NdeI and BamHI (underlined) before being ligated to pET16B which had previously been digested with the corresponding enzymes and treated with calf intestinal alkaline phosphatase. Two plasmid clones derived from each of the independent PCRs were subjected to automated DNA sequencing.
Antibody neutralization assay.
The wt virus was incubated for 30 min on ice with various concentrations (0, 1:20, 1:100, and 1:500) of preimmune serum or anti-A13 serum. After the incubation period, the samples were titrated on BSC40 monolayers, which were stained at 48 hpi.
Preparation of digital figures.
Sequences were aligned by using the Clustal V method and Lasergene software (DNASTAR Inc., Madison, Wis.). Original data were scanned on a SAPHIR scanner (Linotype-Hell Co., Hauppauge, N.Y.) and were adjusted with Adobe Photoshop software (Adobe Systems Inc., San Jose, Calif.). Graphs were plotted with SigmaPlot (SPSS Science, Chicago, Ill). Final figures were assembled and labeled with Canvas software (Deneba Systems, Miami, Fla.).
RESULTS
Our goal for this work was to perform a structural and functional analysis of the A13 protein. Figure 1A depicts the amino acid sequence of this protein along with several noteworthy features. The N terminus (residues 1 to 20) is extremely hydrophobic and is likely to represent a transmembrane domain; N-terminal sequence analysis of virion proteins indicated that this region is retained in the mature protein rather than being removed by proteolytic processing. The blue line indicates the portion of the protein that was used for preparation of the anti-A13 antiserum. The black circles indicate the eight serine residues that are available for phosphorylation. The amino acid composition of the nonhydrophobic portion (residues 21 to 70) of the protein is quite unusual. There are 10 asparagine residues (N) and a central cluster of 7 proline residues (P). Of the charged residues, six are basic (K), whereas only three are acidic (E and D), accounting for the predicted pI of 10. A comparative sequence alignment of the A13 homologs encoded by a variety of poxviruses is shown in Fig. 1B. The hydrophobic nature of the N terminus is retained, although the exact sequence is only conserved in the top few sequences included in the alignment. The rest of the protein is quite divergent. The proteins from vaccinia virus, cowpox virus, camelpox virus, monkeypox virus, and variola virus share almost complete identity. However, in the more divergent sequences, even such striking motifs as the proline-rich region are lost. Several hydrophobic or aromatic residues are conserved in the final 20 amino acids, however.
FIG. 1.

Structural organization and viral conservation of A13 protein. (A) Structural features of A13 protein. A hydrophobicity plot is shown above the A13 sequence; the N terminus (aa 1 to 20) contains a predicted transmembrane domain. The region marked with the blue line was used to generate the anti-A13 serum. Residues of interest are shown in color as follows: red, Lys; purple, Glu; blue, numerous Asn residues; green, numerous, clustered Pro residues. The eight Ser residues are marked with circles; the red diamond at Ser40 marks the site of A13 phosphorylation (Fig. 8); the red arrowhead marks the Ile residue that is altered in Cts40 (Table 2 and Fig. 9). (B) Sequence alignment of poxviral A13 homologs. A sequence alignment of the A13 orthologs from vaccinia virus (VV; GenBank ID 29692238), cowpox virus (CPV; GenBank ID 20178507), camelpox virus (CMPV; GenBank ID 18640364), monkeypox virus (MPX; GenBank ID 17975037), variola virus (VAR; GenBank ID 439035), ectromelia virus (EV; GenBank ID 22164721), lumpy skin disease virus (LSDV; GenBank ID 15150543), sheeppox virus (ShPV; GenBank ID 21492557), swinepox virus (SPV; GenBank ID 18640187), Yaba-like disease virus (YLDV; GenBank ID 12085087), and molluscum contagiosum virus (MCV; GenBank ID 1492060) (sequences obtained from www.poxvirus.org by using the POCs program) is shown. Residues identical to A13 are boxed and shaded in yellow.
Temporal profile of A13 synthesis and encapsidation.
Using the anti-A13 antiserum, we were able to verify the temporal profile of A13 synthesis. Cells were metabolically labeled with [35S]Met at various times postinfection and subjected to immunoprecipitation analysis. The results are shown in Fig. 2A. No immunoreactive bands were seen in uninfected cells (lane 1) or cells harvested at 1.25 or 2.75 hpi. Likewise, no immunoreactive species were seen in extracts harvested at 4 hpi from a culture maintained in the presence of AraC, an inhibitor of DNA replication and therefore of intermediate and late gene expression. A clear band at approximately 12 kDa was seen for cells harvested at 4, 5.25, and 6.5 hpi, consistent with the expression of A13 as a late protein. The unusual electrophoretic migration of A13, indicative of an observed molecular weight of 12K rather than the predicted one of 8K, has been seen by other investigators (35, 38). We also examined purified virions by immunoblot analysis after electrophoretic resolution in the absence or presence of β-mercaptoethanol. As shown in Fig. 2B, A13 is clearly present within purified virions, as has been reported previously, and the comparable mobilities observed in the presence or absence of reducing agents indicate that the single cysteine residue does not participate in the formation of any disulfide-linked dimers.
FIG. 2.

Expression, encapsidation, and posttranslational modification of A13. (A) Temporal expression of A13. BSC40 cells were left uninfected (lane 1) or infected with wt virus (MOI = 15) in the presence or absence of AraC and were metabolically labeled with [35S]methionine for 30 min prior to being harvested. Protein lysates were prepared and subjected to immunoprecipitation with anti-A13 serum. The lanes correspond to immunoprecipitates retrieved from the following extracts: mock-infected cells (lane 1); cells infected with wt virus and harvested at 1.5, 2.75, 4, 5.25, and 6.5 hpi (lanes 2 to 6); and cells infected with wt virus in the presence of AraC and harvested at 4 hpi (lane 7). The arrow indicates A13. The electrophoretic migration of 18.4- and 14.3-kDa protein standards is shown at the left. (B) Encapsidation of A13 protein. Purified wt virions were resuspended in protein sample buffer in the presence (+) or absence (−) of β-mercaptoethanol (β-ME), boiled, resolved by SDS-17% PAGE, and subjected to immunoblot analysis with anti-A13 serum. The position of the monomeric form of A13 is indicated with an arrow. Protein standards of 18.4, 14.3, and 6.0 kDa are indicated on the left. (C) Phosphorylation of A13 protein in vivo. Cells were infected with wt virus at 37°C (lanes 1), 31.5°C (wt32; lanes 3), or 39.7°C (wt40; lanes 4), with vindH1 in the absence of IPTG (H1−; lanes 2), or with ts28 at 31.5°C (tsF1032; lanes 5) or 39.7°C (tsF1040; lanes 6). All cultures were infected at an MOI of 2 and were radiolabeled with 32PPi from 3 to 18 hpi. The lysates were subjected to immunoprecipitation analysis with anti-A13 serum (left panel), anti-A17 serum (center panel), or anti-I3 serum (right panel). The positions of A13, A17, and I3 are indicated by arrows; coprecipitated 32P-A14 (center panel, lane 2) is denoted by a diamond. Immunoprecipitates were resolved by SDS-PAGE and visualized by autoradiography. Lane M contains protein standards, with their molecular masses shown at the right, in kilodaltons.
A13 is a phosphoprotein and is phosphorylated only at a serine residue(s).
The discrepancy between the predicted and observed pIs of the encapsidated A13 protein (10 versus 8) suggested that A13 might undergo an acidifying modification such as phosphorylation (15). To test this possibility directly, we performed an immunoprecipitation analysis of extracts prepared from infected cells that had been metabolically labeled with 32PPi (Fig. 2C). As shown in the left panel, lanes 1, 3, and 4, a radiolabeled band of approximately 12 kDa was readily observed in the immunoprecipitates of extracts prepared from wt virus-infected cells, indicating that A13 is indeed a phosphoprotein. As controls, the same extracts were subjected to immunoprecipitation with sera directed against the A17 and I3 phosphoproteins (center and right panels, respectively). We were also interested in determining the role of the H1 phosphatase and the F10 kinase in modulating the phosphorylation status of A13. Therefore, we analyzed extracts prepared from cells infected with vindH1, an inducible recombinant in which the expression of H1 is repressed in the absence of IPTG (H1−, lanes 2) (20), or with ts28, a temperature-sensitive virus bearing a lesion in the F10 kinase (tsF1032, lanes 5; tsF1040, lanes 6) (40, 43). The radiolabeling of A13 did not increase when H1 phosphatase was repressed (compare lanes 1 and 2 in the left panel), suggesting that A13 is not a substrate for this enzyme in vivo. Confirmation that H1 had been repressed was seen by examining the anti-A17 immunoprecipitate performed in parallel (center panel, lane 2): the expected twofold increase in A17 labeling, as well as a dramatic increase in radiolabeling of the coprecipitated A14 protein (diamond), was seen (8, 20, 41). In contrast, an approximately twofold decrease in the radiolabeling of A13 was seen during nonpermissive infections performed with the tsF10 virus (left panel, compare lanes 1 and 6). These data suggest that F10 kinase is one of the enzymes that can phosphorylate A13; the impact of the ts lesion on A13 radiolabeling was certainly not as complete as that observed for the A17 protein (center panel, lane 6). Consistent with previous observations from our laboratory, radiolabeling of the I3 protein remained unchanged when either H1 or F10 was repressed or impaired (right panel). The 32P-labeled A13 immunoprecipitated from wt-infected cells was subjected to phosphoamino acid analysis, which indicated that A13 was phosphorylated solely at serine residues (not shown).
Generation of vindA13, an inducible recombinant in which the endogenous A13 allele is regulated by the TET operator and repressor.
To analyze the role of A13 in the viral life cycle, we attempted to generate an inducible recombinant in which the expression of A13 would be dependent upon the inclusion of TET in the culture medium. Insertion of the TET operator between the transcriptional and translational start sites of the A13 gene was accomplished by overlap PCR, and replacement of the endogenous allele with the inducible allele was accomplished by transient dominant selection, as explained in Materials and Methods. This allele replacement was undertaken in the genome of the vtetR virus, in which the TET repressor gene has been inserted into the nonessential TK locus (41). Our preliminary analysis of vindA13 is shown in Fig. 3. As shown in Fig. 3A, vindA13 was capable of forming plaques in the absence or presence of TET. A more quantitative assessment of the infectious yield after infection with vindA13 (MOI of 2) is shown in Fig. 3B; 24-h infections performed in the absence of TET yielded 10-fold less cell-associated virus than infections performed in the presence of TET.
FIG. 3.

Phenotypic analysis of vindA13. (A) Plaque assay. Confluent BSC40 cells were infected with two different dilutions of vindA13 (∼500 and 50 PFU/dish) and maintained in the presence (+) or absence (−) of TET (1 μg/ml). At 48 hpi, the medium was removed and the cells were stained with crystal violet. (B) Quantitation of 24-h viral yield. Cells were infected with vindA13 (MOI = 2) in the presence (+) or absence (−) of TET and harvested at 24 hpi. The yield of total cell-associated virus was determined by titration on BSC40 cells in the presence of TET. (C) Quantitation of A13 accumulation. Cells were infected with vindA13 (MOI = 2) in the presence (+ [lower panel]) or absence (− [upper panel]) of TET (1 μg/ml) and harvested at 24 hpi. Serial dilutions of total cell extracts were resolved by SDS-17% PAGE and subjected to immunoblot analysis with anti-A13 serum. The position of A13 is indicated with an arrow. (D) S1 nuclease analysis of A13-specific and read-through mRNA transcripts. A schematic diagram of the 371-nt radiolabeled probe used for the S1 nuclease analysis and of the transcripts expected for vtetR and vindA13 infections is shown at the top. The position of the 5′ radiolabel at the BamHI site is marked with a star; the probe contains the TET operator (triangle) inserted between the transcriptional (TAAATA) and translational (ATG) start sites of the A13 gene. The wavy lines depict the 5′ regions of the mRNAs that are predicted to be expressed during vindA13 (top) or vtetR (bottom) infections. The probe fragment protected by hybridization with the vindA13 transcript should be slightly larger than that protected by the vtetR transcript due to the presence of the 19-bp operator sequence (285 versus 263 nt) in the vindA13 allele. The autoradiograph below shows the results of the S1 nuclease protection assay. RNA was prepared from cells infected for 8 h with vtetR or vindA13 (MOI = 10) in the presence (+) or absence (−) of TET (1 μg/ml). Reactions were performed, electrophoretically resolved, and visualized by autoradiography as described in Materials and Methods. Lanes 1 and 2, control reactions in which the probe was incubated in the absence of RNA, without or with subsequent S1 nuclease digestion, respectively; lanes 3 and 4, results obtained with RNA prepared from cells infected with vindA13 in the presence or absence of TET, respectively. Protection of the full-length probe is seen with both RNA samples, but only the sample from the +TET infection contains transcripts that generate a protected fragment of 285 nt (open triangle, compare lanes 3 and 4). Lanes 5 and 6 depict the results obtained with RNA prepared from cells infected with vtetR in the presence or absence of TET, respectively. In both cases, protection of the full-length probe and the 263-nt A13-specific fragment (filled triangle) is seen.
A13 is an essential gene.
The experience of our lab and others with both IPTG- and TET-inducible recombinants is that the repression of essential genes usually engenders a more dramatic phenotype (≥2-log reduction in viral yield) than the one we observed for this sutdy. We entertained two possible explanations for the leaky phenotype of vindA13: either the repression of A13 was incomplete or A13 played a stimulatory role but was not essential for the replication of vaccinia virus in tissue culture. Therefore, we attempted to construct a deletion mutant in which the endogenous A13 gene had been replaced with a selectable marker (NEO) or reporter gene (β-galactosidase [β-Gal]) (vΔA13:neo and vΔA13:β-gal, respectively). We constructed a cassette in which the A14 and A12 genes flanked a p7.5-driven copy of either of these two reporter genes in place of the A13 gene. Linearized DNA was transfected into wt-infected cells and we rigorously attempted to isolate G418r or blue plaques (in the presence of X-Gal). No G418r plaques were obtained (not shown). Although a few blue plaques were isolated, all of these were shown to retain an intact A13 locus after repeated plaque purification and PCR analysis (not shown). Therefore, we concluded that the A13 gene was indeed essential for viral replication in tissue culture.
Repression of A13 synthesis is incomplete in vindA13 due to the translation of read-through transcripts.
Our inability to obtain a vΔA13 virus led us to examine the possibility that a significant level of A13 expression remained during vindA13 infections even in the absence of TET. We prepared extracts from cells infected with vindA13 in the absence or presence of TET and then performed immunoblot analyses on serial dilutions of these extracts. As shown in Fig. 3C, the omission of TET led to an ∼10-fold reduction in the level of immunoreactive A13, echoing the 10-fold reduction in viral yield (Fig. 3B) discussed above. The repression levels seen for vindA13 are not as tight as those that we have previously observed for other IPTG- and TET-inducible recombinants (16, 41).
We therefore performed S1 nuclease analyses to see whether the repression of A13-specific transcripts was also leaky (Fig. 3D). As shown schematically, hybridization of the wt A13 mRNA to the 371-nucleotide (nt) probe was predicted to generate a nuclease-resistant, radiolabeled fragment of 263 nt; hybridization of the A13 mRNA transcribed from the operator-containing allele would protect a fragment of 285 nt. The autoradiograph illustrates the electrophoretic migration of the probe (lane 1) and its sensitivity to nuclease in the absence of any hybridizing RNA sample (lane 2); lanes 3 and 5 show the results of hybridization with mRNA samples prepared from infections with vindA13 or vtetR, respectively, in the presence of TET. The predicted fragments of 285 nt (lane 3) and 263 nt (lane 5) were seen, as was protection of the full-length probe (371 nt) by read-through transcripts originating from upstream genes, as is characteristic of vaccinia virus late transcription (26). As expected, the mRNA sample prepared from vtetR infections performed in the absence of TET (lane 6) was unchanged from that prepared in the presence of TET. In contrast, when vindA13 infections were performed in the absence of TET, the 285-nt fragment generated by hybridization of the probe with the A13 transcript was no longer seen (lane 4), even when long exposures of the autoradiograph were examined (not shown). Thus, binding of the TET repressor effectively blocked transcription from the A13 promoter. However, the levels of read-through transcripts were not diminished in the absence of TET, indicating that the bound TET repressor was not effective in halting read-through transcription. Moreover, these data suggest that these read-through transcripts may be able to direct translation of the internal A13 ORF.
Generation of vΔindA13, an inducible recombinant in which a TET-regulated copy of the A13 gene has been placed within the TK locus and from which the endogenous A13 gene has been deleted: virus production and plaque formation are tightly regulated by TET.
In an attempt to generate an inducible recombinant in which the expression of A13 could be more tightly regulated, we sought to remove it from the endogenous context, in which several upstream genes are also expressed at late times and transcribed in the same direction. Therefore, we constructed a targeting vector that would lead to the integration of both a constitutively expressed tetR gene and a TET-inducible A13 allele into the nonessential TK locus via homologous recombination. Once insertion of this cassette was accomplished, the endogenous A13 allele was deleted and replaced with the NEO gene. This final recombinant was designated vΔindA13; in this recombinant, the only remaining A13 allele is under the regulation of its own promoter and the TET operator/repressor.
A preliminary characterization of vΔindA13 is shown in Fig. 4. As shown in Fig. 4A, plaque formation by the recombinant, but not the parental vtetR virus, was dependent upon the inclusion of TET in the medium. Quantitation of the viral yield from a 24-h infection (MOI = 2) with vΔindA13 indicated that the omission of TET led to a nearly 3-log decrease relative to that seen when TET was present. In contrast, the viral yield from vtetR infections was independent of TET. Because viral yield and macroscopic plaque formation showed a stringent requirement for TET, we assumed that A13 gene expression was tightly regulated by the TET repressor during vΔindA13 infections. This assumption was verified by comparing the levels of A13 protein that accumulated during repressed and induced infections with both vtetR and vΔindA13 (Fig. 4C). An immunoblot analysis of serial dilutions of vtetR-infected cell extracts showed that the inclusion or omission of TET had no effect on A13 accumulation. In contrast, the levels of A13 that accumulated during nonpermissive infections with vΔindA13 (−TET) were <3% of those seen during permissive infections (+TET). As has been seen with many inducible recombinants, the levels of A13 expressed during permissive (+TET) infections were somewhat reduced relative to those seen during wt (or vtetR) infections (approximately fourfold reduction).
FIG. 4.

Phenotypic characterization of vΔindA13. (A) Plaque assay. vtetR and vΔindA13 were titrated on BSC40 cells in the presence (+) or absence (−) of TET. At 48 hpi, the medium was removed and the cells were stained with crystal violet. (B) Quantitation of 24-h viral yield. Cells were infected with vtetR or vΔindA13 (MOI = 2) for 24 h in the presence (+) or absence (−) of TET; cell lysates were prepared and the virus was titrated onto BSC40 cells in the presence of TET. (C) Quantitation of A13 accumulation. Cells were infected with vtetR (left) or vΔindA13 (right) (MOI = 2) in the presence (+) or absence (−) of TET (1 μg/ml) and harvested at 24 hpi. Serial dilutions of total cell extracts were resolved by SDS-17% PAGE and subjected to immunoblot analysis with anti-A13 serum. The position of A13 is indicated with an arrow; protein standards of 14.8 and 6.0 kDa are shown at the left.
During nonpermissive infections with vΔindA13, gene expression, DNA replication, and genome resolution proceed normally, but proteolytic processing of virion proteins is diminished.
With the conditional lethal vΔindA13 virus in hand, we proceeded to determine which stages of the viral life cycle were compromised in the absence of the inducer. First, we examined the temporal profile of viral gene expression. Infected cells were pulse labeled with [35S]Met at sequential times after infection, and the profile of nascent proteins was visualized by autoradiography (Fig. 5A). Comparable profiles were observed for cells infected with vΔindA13 in the presence or absence of the inducer. These data imply that DNA replication, which is a prerequisite for intermediate and late gene expression, occurred independently of TET. The resolution of concatemeric replication intermediates into monomeric genomes is also required for the production of infectious progeny, and the impact of TET on this process was investigated by Southern blot analysis (Fig. 5B). During wt infections, concatemeric intermediates were efficiently processed, as evidenced by the absence of a strong band at 2.6 kb (representative of concatemer junctions) and the presence of a strong band at 1.3 kb (representative of a mature hairpin telomere) (lane 1) in BstEII-digested DNA. When the late phase of gene expression was compromised by the inclusion of the drug IBT (lane 5) or the repression of the G8 late transcription factor (lane 4), concatemer resolution was inhibited and the 2.6-kb species accumulated (29, 47). When permissive and nonpermissive infections with vΔindA13 were analyzed, concatemer resolution was found to occur with wt efficiency (lanes 2 and 3, respectively).
FIG. 5.

Impact of A13 repression on temporal profile of protein synthesis, genome resolution, and proteolytic processing. (A) Temporal profile of viral protein synthesis. BSC40 cells were infected with vΔindA13 (with or without TET) at an MOI of 5 and were metabolically labeled with [35S]methionine for 45 min before being harvested at the indicated time points (2, 4, 6, and 8 hpi). Mock-infected cells were also labeled with [35S]methionine for 45 min before being harvested (lane 1). Lysates were resolved by SDS-12% PAGE and visualized by autoradiography. The molecular masses of 14C-protein standards are indicated on the right, in kilodaltons. Representative intermediate proteins are indicated by ovals, late proteins are indicated by arrowheads, and cellular actin is indicated by a square. (B) Southern blot analysis of genome resolution. BSC40 cells were infected with wt virus or vΔindA13 (MOI = 2) in the presence (+) or absence of TET. As a control, cells were also infected with wt virus in the presence (+) of IBT or with vROG8 (MOI = 15) in the absence (−) of IPTG. At 18 hpi, the cells were harvested and viral genomic DNA was isolated, digested with BstEII, resolved electrophoretically, and subjected to Southern blot analysis by hybridization with a radiolabeled probe derived from the termini of the viral genome. The arrowheads point to the 1.3-kb fragment released from mature, monomeric genomes and the 2.6-kb junction fragment released from unresolved concatemers. The probe also hybridizes to the ∼5.5-kb BstEII fragment proximal to the telomeric regions; the slight variation in the size of this fragment in different plaque-purified isolates represents variability in the numbers of tandem repeats. The sizes of DNA standards are indicated in kilobase pairs to the left. (C) Proteolytic processing of core proteins. BSC40 cells were infected (MOI = 2) with wt virus in the presence (+) (lanes 7 and 8) or absence (lanes 1 and 2) of RIF or with vΔindA13 in the presence (+) (lanes 3 and 4) or absence (−) (lanes 5 and 6) of TET. At 8 hpi, the cells were metabolically labeled with [35S]methionine for 45 min before being harvested immediately (pulse [P]) or refed with complete medium and incubated for an additional 15 h (chase [C]). Cell lysates were resolved by SDS-12% PAGE and visualized by autoradiography. 14C-labeled protein standards are indicated at the right, with their molecular masses shown in kilodaltons. Precursor forms of the major core proteins 4a, 4b, and L4 are indicated by filled arrowheads; mature, processed forms are indicated by open arrowheads.
Because the biochemical hallmarks of the viral life cycle were unperturbed when A13 was repressed and because A13 is a known component of the virion membrane, we suspected that nonpermissive infections with vΔindA13 would arrest during the process of morphogenesis. As an initial test of this hypothesis, we investigated the extent to which the proteolytic processing of virion core precursor proteins, which occurs during the last stages of morphogenesis, was affected by A13 repression (Fig. 5C). As a positive control, a pulse-chase analysis of wt infections was examined (lanes 1 and 2); as a negative control, an infection performed with the wt virus in the presence of rifampin (RIF), an inhibitor of morphogenesis and hence of processing (27), was analyzed (lanes 7 and 8). As expected, efficient processing of the precursors (filled arrowheads) to their products (open arrowheads) was seen in the absence, but not the presence, of RIF (compare lanes 1 and 2 with lanes 7 and 8). Although a wt pattern of processing was seen during permissive infections with vΔindA13 (lanes 3 and 4), reduced levels (50%) of cleaved products were observed when A13 was repressed (lanes 5 and 6). These data suggest that virion morphogenesis is compromised when A13 levels are insufficient.
Electron microscopic analysis of the morphogenesis defect that accompanies A13 repression.
Cells infected with vΔindA13 in the presence or absence of TET were processed for transmission electron microscopy at 18 hpi. The full range of assembly intermediates seen in wt infections, such as virosomes (V), crescents (C), immature virions (IV), immature virions with nucleoids (IVN), and mature virions (IMV), was also seen in cells infected with vΔindA13 in the presence of TET (Fig. 6A and B). In contrast, clear defects in the progression of morphogenesis were seen when A13 was repressed (Fig. 6C to G). First and foremost, essentially no IMV were observed, and IVN were rare. In contrast, a large number of apparently normal IV accumulated within the cells (panels C and D, arrows). Electron-dense virosomes were also plentiful (panels C and D); indeed, more virosomes were seen at this time postinfection than were observed in cells infected with vΔindA13 under permissive conditions. Although normal-looking crescents were often seen at the periphery of these virosomes (panels C, E, and F), some membranous elements that had an aberrant structure were also seen. These membranes did not have the shape or rigidity of classical crescents; instead, they seemed to be more flexible and often curled off in irregular loops (panels E and F, arrowheads). Finally, we also observed electron-dense, striated structures that are thought to represent crystalloids of viral DNA (panels C and G [stars]).
FIG.6.
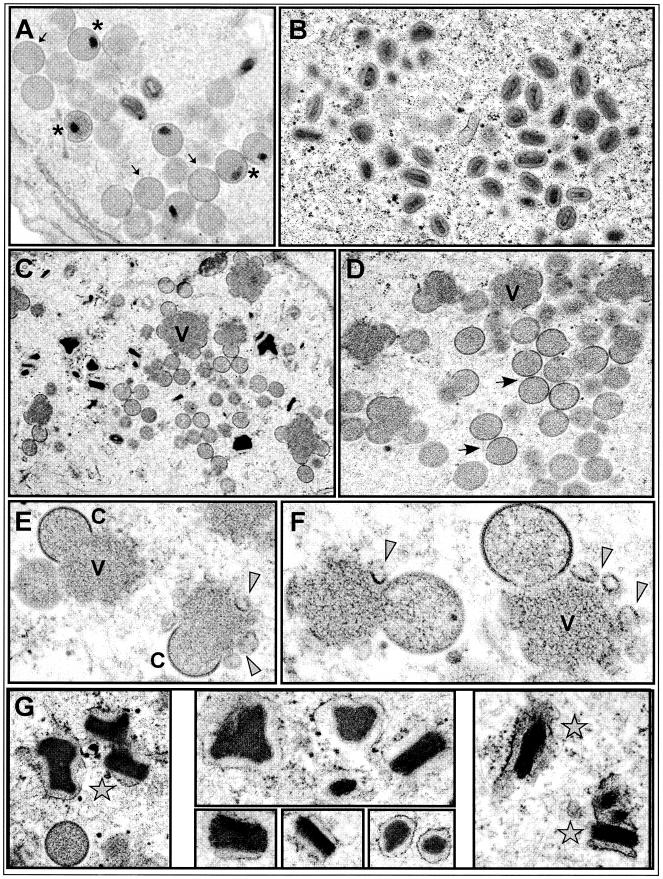
Electron microscopic analysis of vΔindA13 infections. BSC40 cells were infected with vΔindA13 (MOI = 2) in the presence (A and B) or absence (C to G) of TET, and at 18 hpi they were processed for conventional transmission electron microscopic analysis. During permissive infections, numerous groupings of IV (A, arrows), IVN (A, asterisks), and IMV (B) were seen. During nonpermissive infections, IVN and IMV were rare. Small virosomes (V) surrounded by normal crescents (C) as well as irregular fragments of membranes (arrowheads) were numerous (C to F). Immature virions were also highly abundant. Finally, these infections were characterized by the presence of many dense crystalloids (stars), which are thought to contain tightly packed viral genomes (G). Final magnifications: (A) ×25,000; (B) ×21,000; (C) ×15,000; (D) ×23,000; (E) ×44,000; (F) ×60,000; (G) left panel, ×35,000; top center panel, ×52,000; lower center panel, ×45,000, ×54,000, and ×50,000; right panel, ×43,000.
The results described above imply that both early and late defects in morphogenesis accompanied the repression of A13, including the accumulation of some malformed membranes around the virosomes, as well as a more dramatic arrest after IV formation, prior to the subsequent maturation to IVN and IMV. In order to look more closely at the timing of the morphogenesis defects, we first synchronized wt and nonpermissive vΔindA13 infections (−TET) by including RIF during the first 12 h of the infection period (27). Parallel cultures were then harvested immediately or refed with drug-free medium and allowed to incubate for a further 5, 10, 20, 45, 90, 135, or 180 min in the absence of RIF. Figure 7 shows images from representative time points; the top five panels show wt-infected cells; the bottom five panels show vΔindA13- infected cells. As seen in both 0-min panels, RIF treatment led to the characteristic arrest in morphogenesis. Numerous virosomes (V) were seen, surrounded by membranes of irregular shape (filled arrowheads) which lacked the rigidity and curvature of crescents. In wt-infected cultures released from the RIF block, a synchronous resumption of morphogenesis was seen. By 5 to 10 min postrelease, many typical crescents (C) were seen at the periphery of the virosomes. Significant numbers of IV (arrows) were first seen at 45 to 90 min postrelease, with IVN (asterisks) becoming apparent at 90 min postrelease and groupings of IMV (open arrowheads) first appearing at 135 min and becoming abundant by 180 min postrelease. A different profile was seen for the vΔindA13 infections. Again, crescents (C) appeared at 5 to 10 min postrelease, indicating that A13 is not needed for this step in membrane assembly (i.e., the insertion of D13 into the membranes [24, 25, 37]). Likewise, IV (arrows) were seen in abundance at 45 to 90 min postrelease, implying that A13 is not needed for the C→IV pathway of morphogenesis. However, even at 180 min postrelease, few IVN, and virtually no IMV, were seen. Thus, these synchronization studies indicate that the expression of A13 is needed for the IV→IVN→IMV transition.
FIG.7.
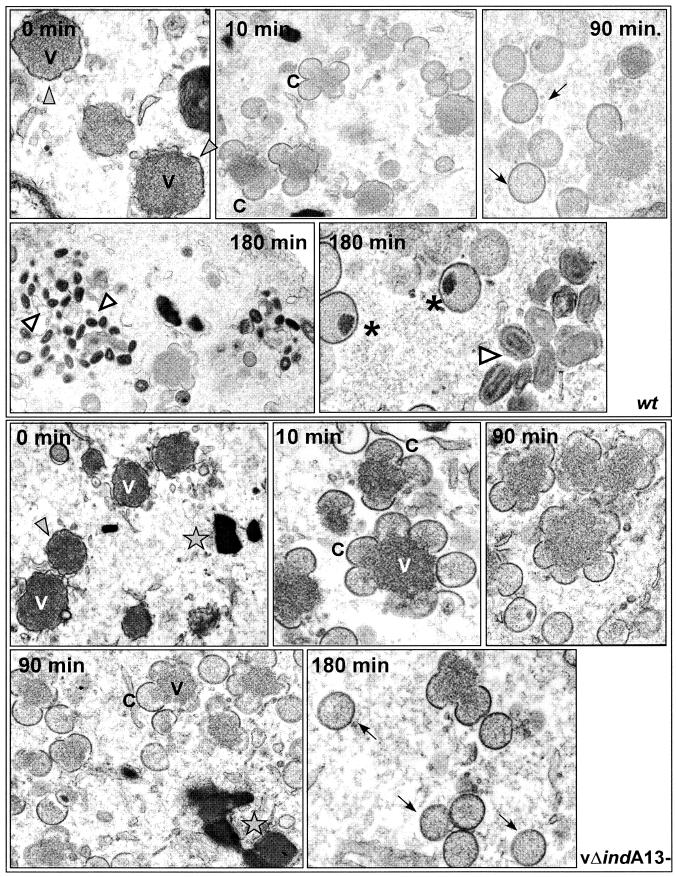
Electron microscopy of wt and vΔindA13 (−TET) infections after release from a RIF-induced morphogenesis arrest. Cells were infected with either wt virus or vΔindA13 (−TET; MOI = 2) in the presence of RIF. At 12 hpi, the cells were either harvested immediately (0 min) or the RIF was washed out and the infection was allowed to proceed for up to 180 min postrelease. Representative images of wt infections are shown in the top five panels. After RIF arrest (0 min), there were numerous small virosomes (V) surrounded by large fragments of irregular, flaccid membranes (filled arrowheads). By 10 min after release, crescents (C) had formed; by 90 min, both crescents and immature virions (arrows) were present. At 180 min postrelease, IVN (stars) and mature virions (empty arrowheads) were abundant. Representative images of vΔindA13 infections are shown in the bottom five panels. After RIF arrest, the overall profile was similar to that shown above for the wt virus; at 0 min, there were numerous small virosomes (V) surrounded by fragments of irregular, flaccid membranes (filled arrowheads). By 10 min after release, crescents (C) had formed; by 90 min, both crescents and immature virions (arrows) were seen. At 180 min, immature virions (arrows) were numerous, but those with nucleoids were extremely rare and virtually no mature virions were seen. DNA crystalloids (stars) were frequent. Final magnifications for wt panels: 0 min, ×30,000; 10 min, ×20,000; 90 min, ×30,000; 180 min, ×30,000 (left) and ×40,000 (right). Final magnifications for vΔindA13 panels: 0 min, ×15,000; 10 min, ×31,000; 90 min, ×35,000 X (upper right), ×23,000 (lower left); 180 min, ×38,000.
Phosphorylation of A13 occurs at Ser40.
The data shown in Fig. 2 confirmed our hypothesis that A13 might sustain phosphorylation in vivo; furthermore, we showed that this phosphorylation occurred exclusively on serine residues. A13 contains eight candidate serine residues. To distinguish which residues might represent phosphorylation sites in vivo, we generated constructs encoding a 3X-FLAG-wtA13 or 3X-FLAG-A13 allele, containing individual or clustered Ser→Ala substitutions. Cells were infected with vΔindA13 in the absence of TET (in duplicate), transfected with these various plasmids, and incubated in the presence of 32PPi. At 24 hpi, the cells were harvested and subjected to immunoprecipitation (Fig. 8A and B) or immunoblot analysis (Fig. 8C) with an anti-FLAG antiserum. No specific proteins were recognized by the serum in extracts prepared from mock-transfected cells (lane 1). In contrast, phosphorylated 3XFLAG-wtA13 was clearly retrieved in the immunoprecipitation analysis and appeared as a doublet in the immunoblot analysis (lane 2). Ser→Ala substitutions affecting positions 21, 29, 60, 61, 68, and 69 (lanes 3 and 4) did not block radiolabeling or the appearance of the doublet. However, the inclusion of substitutions at positions 35 and 40 (lanes 5 and 8) blocked radiolabeling and led to the disappearance of the upper band of the doublet. When only residue 35 was altered (lane 6), the results were indistinguishable from those for the wt; however, a Ser40→Ala substitution blocked radiolabeling and led to the disappearance of the upper band of the doublet. From these data, we conclude that A13 is phosphorylated at Ser40, which is found within the motif NSPPP (Fig. 1B, diamond).
FIG. 8.

Characterization of phosphorylation of epitope-tagged alleles of A13 containing Ser→Ala substitutions. Cells were infected with vΔindA13 (MOI = 2) in the absence of TET; at 3 hpi, cells were transfected in duplicate with empty vector (V, lane 1) or with plasmids encoding 3X-FLAG-tagged wt A13 (lane 2) or alleles containing Ser→Ala substitutions at the positions shown above the lanes (lanes 3 to 8). Cells were metabolically labeled with 32PPi from 6 to 24 hpi prior to being harvested and analyzed either by immunoprecipitation (A and B) or immunoblot analysis (C) with α-FLAG serum. Proteins were visualized by autoradiography (A and B) or chemiluminescence (C).
The temperature-sensitive phenotype of Cts40 maps to a lesion in the A13 gene.
The data described above indicate that the repression of A13 expression leads to an arrest in virion morphogenesis at the IV→IVN transition. The N-terminal 20 aa of the A13 protein are predicted to form a membrane-spanning domain, and the remaining 50 aa are predicted to extend outwards from the virion surface. These 50 aa, whose function is likely to be modulated by the phosphorylation of Ser40, are likely to mediate interactions with other molecules. As a first step towards a structure-function analysis, we scanned the collections of available ts mutants and noted that the lesion in Cts40 was predicted to map within the genomic region encoding the A8 through A17 genes (19). We obtained this mutant and performed marker rescue experiments to map the ts lesion within this mutant. As shown in Table 2, the introduction of a plasmid encoding the A13 gene, but not an empty plasmid or a plasmid encoding the A17 or A14 gene (not shown), could generate temperature-insensitive recombinants by recombination with the Cts40 genome. DNA sequence analysis of the A13 allele encoded by Cts40 indicated that the mutant contains a C→T transition at nt 143 which results in a Thr→Ile substitution at aa 48.
TABLE 2.
Marker rescue of Cts40a
| DNA | Viral yield at 39.7°C (48 hpi) (PFU/ml)
|
|
|---|---|---|
| Titrated at 31.5°C | Titrated at 39.7°C | |
| pUC1246 | 3.7 × 105 | <102 |
| pUC1246-A17 | 7.6 × 105 | <102 |
| pU1246-A13 | 2.0 × 106 | 2.8 × 104 |
BSC40 monolayer cultures were infected with Cts40 at 31.5°C; at 3 hpi, the cells were transfected with the linearized plasmid DNA indicated and shifted to 39.7°C. At 48 hpi, the cells were harvested and the viral yield was titrated at both 31.5°C (total virus) and 39.7°C (temperature- insensitive virus).
An initial characterization of the phenotype of Cts40 is shown in Fig. 9. At 31.5°C, Cts40 forms smaller plaques than the wt virus; at 39.7°C, Cts40 forms no macroscopic plaques, whereas the wt virus forms large plaques (Fig. 9A). A more quantitative measurement of the defect in virus production was obtained by quantitating the total yield of cell-associated virus from cells infected with wt virus or Cts40 at either 31.5 or 39.7°C (Fig. 9B). Whereas wt infections led to the production of nearly equivalent levels of virus at both temperatures, 600-fold less virus was produced from Cts40 infections performed at 39.7°C than at 31.5°C. An examination of Cts40-infected cells by electron microscopy (data not shown) revealed that the primary defect during nonpermissive Cts40 infections shows an overall similarity to that observed earlier for nonpermissive vΔindA13 infections (Fig. 6). Morphogenesis appears to proceed normally through the formation of IV, which accumulate to unusually high numbers; more mature forms, however, are rare. The DNA crystalloids seen frequently upon repression of A13 were less obvious than in cells infected nonpermissively with Cts40.
FIG. 9.

Phenotypic analysis of cells infected with Cts40. (A) Plaque assay. Confluent BSC40 cells were infected with either wt virus or Cts40 and incubated at 31.5 or 39.7°C for 2 days before being stained with crystal violet. (B) Quantitation of 24-h viral yield. Cells were infected with wt virus or Cts40 at either 31.5 or 39.7°C for 24 h; the total yield of cell-associated virus was determined by titration on BSC40 cells at 31.5°C. (C) Accumulation of A13 and other late proteins. BSC40 cells were infected with wt virus (lanes 1 and 4), vΔindA13 (−TET) (lanes 2 and 5), or Cts40 (lanes 3 and 6) at either 31.5°C (lanes 1 to 3) or 39.7°C (lanes 4 to 6) for 18 h. Lysates were prepared and subjected to immunoblot analysis using anti-G7, anti-L4, or anti-A13 sera.
To determine whether the temperature-sensitive phenotype of Cts40 reflected the thermolability of the A13 protein or the loss of its function at the high temperature, we prepared extracts of cells infected for 18 h (MOI = 2) at either 31.5 or 39.7°C with wt virus, vΔindA13-TET, or Cts40. These extracts were subjected to immunoblot analysis (Fig. 9C) with antisera directed against A13, L4, G7, or A17 (not shown). As expected, the repression of A13 during infections with vΔindA13-TET prevented the accumulation of detectable levels of the A13 protein; however, A13 accumulated to equivalent levels in cells infected with either wt virus or Cts40 at either temperature. Thus, the temperature sensitivity of Cts40 reflects a temperature-sensitive loss of function of the A13 protein due to the Thr→Ile substitution in the cytosolic tail of this protein. As expected, we saw no change in the accumulation of other late proteins, as shown by immunoblot analyses of L4 and G7 (and A17 [data not shown]).
DISCUSSION
The vaccinia virus A13L protein (15, 35, 38) was previously identified as a proteolytically accessible component of the external membrane of mature virions. Furthermore, cotranslational insertion of the protein into microsomal membranes had been demonstrated in vitro, and localization of the protein within the ER/ERGIC had been seen in vivo. Membrane localization is undoubtedly directed by the hydrophobic N terminus of the protein, which comprises the first 20 aa of the protein. In the work described here, we have analyzed the synthesis and posttranslational modification of A13 during infection and utilized both temperature-sensitive and TET-dependent viruses to dissect the role played by the A13 protein in the viral life cycle.
The 70-aa A13L protein is expressed at late times after infection, and unlike the membrane components A14L and A17L (1, 21, 32), does not form intra- or intermolecular disulfide bonds. A13L does undergo phosphorylation on serine residues, and this modification requires the presence of Ser40, but none of the seven other serine residues. The phosphorylation of A13L does not increase upon the repression of the virally encoded H1L dual-specificity phosphatase and diminishes only somewhat upon impairment of the virally encoded dual-specificity kinase F10L. These data suggest that A13L is likely to be phosphorylated by a cytoplasmic cellular kinase. A high-stringency search with a computer algorithm that identifies likely kinase recognition sites, found at http://scansite.mit.edu (28), predicts that Ser40 lies within an ERK1 recognition motif (NSPPP). This prediction will need to be tested empirically, but if it is accurate may point to a role for cellular kinases in the process of virion morphogenesis. Interestingly, Ser40 is conserved in the A13L homologs of vaccinia virus, cowpox virus, camelpox virus, and monkeypox virus but is absent from variola virus. In the more distantly related lumpy skin disease virus, sheeppox virus, shope fibroma virus, and Yaba-like disease virus, a Tyr residue, which might also serve as a phosphorylation site for a cellular dual-specificity kinase, occupies this position.
The proline-rich motif that surrounds Ser40 (Fig. 1A; Pro residues are in green) (aa 32 to 45) is also conserved in the same group of closely related viruses, but again is not found in the more distantly related ortho- or leporipoxviruses (Fig. 1B). This concentration of prolines is likely to have implications for the structure of the 50-aa cytoplasmic tail of the A13L protein, perhaps keeping it in an extended structure to facilitate interactions with other proteins or serving itself as a protein interaction module. Further towards the C terminus of the protein, there are several nonpolar, hydrophobic, and aromatic residues that are conserved in almost all of the A13 homologs. Thr48 is one such residue, and indeed its alteration to Ala confers a stringent temperature-sensitive phenotype (Fig. 9; also see below). Because the C-terminal tail of the A13 protein has been shown to be exposed on the virion surface, we investigated whether the treatment of virions with our polyclonal anti-A13 serum would neutralize viral infectivity. No diminution in viral titer was seen (data not shown); however, we cannot deduce whether the lack of neutralization activity merely reflects the manner in which this pool of antibodies interacts with the A13 protein or indicates that the A13 protein does not play a role in viral entry.
To approach the question of A13's function in the viral life cycle, we generated a recombinant virus in which the endogenous allele was placed under the regulation of the TET operator/repressor (41). In the absence of inducer, the virus exhibited only a mild deficit, showing a 10-fold decrease in virus production in a single infectious cycle. Our inability to isolate a virus containing a targeted deletion in A13L, however, made us believe that the A13L protein played more than a supportive role in the life cycle. We determined that the TET repressor was indeed blocking the initiation of transcription at the A13L promoter but was having no impact on the elongation of read-through transcripts initiating at upstream genes (such as A14L, A14.5L, A15L, A16L, and A17L). Moreover, the accumulation of the A13 protein was only diminished ≤10-fold, strongly suggesting that internal ORFs can be translated from these read-through transcripts. We have obtained parallel results for the F10L open reading frame (B. Unger and P. Traktman, unpublished data). Although the eukaryotic translational machinery does not, in general, allow the translation of polycistronic mRNAs, several viruses exploit mechanisms that do allow internal initiation (36). Whether viral infection stimulates these mechanisms, or induces others, has not been investigated.
As an alternative approach, we removed the inducible copy of A13 from the influence of upstream genes. We placed both the TET-regulated copy of the A13L ORF and the tetR gene within the nonessential TK locus of the viral genome and subsequently replaced the endogenous A13L gene with the NEO gene. With this recombinant (vΔindA13), <3% of the wt levels of A13L were expressed in the absence of TET, and the virus showed a conditionally lethal phenotype. In the absence of TET, plaque formation was abolished and an ∼3-log reduction was seen in the viral yield from a single, 24-h infectious cycle. Gene expression and genome replication and resolution were not affected; however, a diminution in the proteolytic processing of the major core proteins suggested that morphogenesis might have been impaired. Indeed, electron microscopic analysis confirmed that A13 expression is required for normal virion assembly. In the absence of A13, there was no block to the formation of early intermediates such as electron-dense virosomes, crescents, and immature virions. However, there was a clear block to further maturation; IV with nucleoids and IMV were rarely, if ever, seen. DNA crystalloids, which appeared to be surrounded by membranes, were numerous. Furthermore, membranous elements lacking the characteristic shape or rigidity of crescents were also found adjacent to the virosomes. These data indicated that the primary block to morphogenesis lay at the IV→IVN transition. To synchronize morphogenesis and obtain a closer look at the kinetics of crescent and IV formation, we infected cells with the wt and vΔindA13(−) viruses in the presence of RIF, which leads to the accumulation of virosomes surrounded by flaccid membranes. The cultures were then released from the RIF block and the infection was allowed to proceed for 0 to 180 min. The repression of A13L expression did not delay the appearance of crescents (5 to 10 min) or IV (90 min), but again blocked further maturation to IVN or IMV, firmly establishing that the primary role of A13L is during the IV→IMV transition. These experiments also allowed us to establish a clear timeline for wt virion maturation after release from RIF-mediated synchronization: crescents are formed after 5 to 10 min, IV are formed after 45 to 90 min, IVN are formed after 90 min, and IMV are formed after 180 min.
As these studies were under way, we noted that the Cts40 mutant from the Condit collection (19, 39) had been characterized as having a possible defect in morphogenesis due to a lesion within the genomic region encompassing A8 to A17. We determined that the temperature sensitivity of Cts40 is indeed due to a C→T transition within the A13 gene that leads to a Thr48→Ile substitution. The A13L protein accumulated normally during nonpermissive infections with Cts40, indicating that the amino acid substitution compromises the function, rather than the stability, of the protein. As stated above, it seems likely that the extended C-terminal tail of A13 mediates interactions with other molecules. During nonpermissive infections with Cts40, the primary defect also lay at the IV→IMV transition.
A13L joins an emerging class of membrane components that, unlike the A14L and A17L proteins, are not involved in the initial biogenesis of the virion membrane (31, 34, 41, 45) or, like the J1R protein, in the interaction of the membrane with the virosomal contents (5). In contrast, the absence or impairment of these proteins arrests the further maturation of IV. In the case of A13L, encapsidation of the viral genome is affected, leading to an accumulation of DNA crystalloids and an absence of IVN or IMV. Deletion or repression of the H3L protein, which associates with membranes posttranslationally, causes a milder phenotype, but an accumulation of crescents, IV, and DNA crystalloids can be seen (7). Indeed, the profile seen upon H3L repression, although quantitatively less significant, is qualitatively similar to that seen upon A13L repression. When the myristoylated L1R protein is repressed, the IV→IMV transition is also blocked; no data regarding the accumulation of DNA crystalloids or the relative abundance or paucity of IVN have been reported (30). When the A9L membrane protein is repressed, a mixture of normal and aberrant IV accumulate (46); some of the aberrant IV appear to be half-filled, as we have observed when the telomere-binding I6 protein is defective and DNA encapsidation fails to occur (12). Whether these membrane proteins participate directly in DNA encapsidation or in the subsequent stages of core formation and virion restructuring remains to be determined.
Future investigations of how A13L functions in virion maturation should be facilitated by the genetic tools we have established and characterized. Dissection of the interactions between the extended cytoplasmic tail of this protein and other proteins, DNA, or lipids will be key, as will be the determination of how Ser40 phosphorylation affects virion morphogenesis. The complex pathway by which the vaccinia virion assembles remains a problem of significant cell biological as well as biomedical interest.
Acknowledgments
We thank Jason Mercer for his invaluable help in determining the phosphorylation status of the mutant A13 alleles and for sharing the α-G7 antiserum prior to publication. Thanks to Kathleen Boyle as well as Jason Mercer for help during the preparation of the manuscript. We also acknowledge R. Jennifer Lopez for invaluable technical assistance and Gang Ning and Clive Wells for expert assistance with electron microscopy.
This work was supported by a grant awarded to P.T. by the NIH (2 R01 GM 53601).
REFERENCES
- 1.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Membrane topology of the vaccinia virus A17L envelope protein. Virology 261:347-356. [DOI] [PubMed] [Google Scholar]
- 2.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L kinase. J. Virol. 73:3534-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyle, W. J., G. P. van der Geer, and T. Hunter. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201:110-149. [DOI] [PubMed] [Google Scholar]
- 4.Chakrabarti, S., J. R. Sisler, and B. Moss. 1997. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques 23:1094-1097. [DOI] [PubMed] [Google Scholar]
- 5.Chiu, W. L., and W. Chang. 2002. Vaccinia virus J1R protein: a viral membrane protein that is essential for virion morphogenesis. J. Virol. 76:9575-9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.da Fonseca, F. G., E. J. Wolffe, A. Weisberg, and B. Moss. 2000. Characterization of the vaccinia virus H3L envelope protein: topology and posttranslational membrane insertion via the C-terminal hydrophobic tail. J. Virol. 74:7508-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Fonseca, F. G., E. J. Wolffe, A. Weisberg, and B. Moss. 2000. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J. Virol. 74:7518-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derrien, M., A. Punjabi, M. Khanna, O. Grubisha, and P. Traktman. 1999. Tyrosine phosphorylation of A17 during vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J. Virol. 73:7287-7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gatz, C., A. Kaiser, and R. Wendenburg. 1991. Regulation of a modified CaMV 35S promoter by the Tn10-encoded Tet repressor in transgenic tobacco. Mol. Gen. Genet. 227:229-237. [DOI] [PubMed] [Google Scholar]
- 10.Gatz, C., and P. H. Quail. 1988. Tn10-encoded tet repressor can regulate an operator-containing plant promoter. Proc. Natl. Acad. Sci. USA 85:1394-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 89:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grubisha, O., and P. Traktman. 2003. Genetic analysis of the vaccinia virus I6 telomere-binding protein uncovers a key role in genome encapsidation. J. Virol. 77:10929-10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsiao, J. C., C. S. Chung, and W. Chang. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichihashi, Y., M. Oie, and T. Tsuruhara. 1984. Location of DNA-binding proteins and disulfide-linked proteins in vaccinia virus structural elements. J. Virol. 50:929-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen, O. N., T. Houthaeve, A. Shevchenko, S. Cudmore, T. Ashford, M. Mann, G. Griffiths, and L. J. Krijnse. 1996. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J. Virol. 70:7485-7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klemperer, N., J. Ward, E. Evans, and P. Traktman. 1997. The vaccinia virus I1 protein is essential for the assembly of mature virions. J. Virol. 71:9285-9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koerner, T. J., J. E. Hill, A. M. Myers, and A. Tzagoloff. 1991. High-expression vectors with multiple cloning sites for construction of trpE fusion genes: pATH vectors. Methods Enzymol. 194:477-490. [DOI] [PubMed] [Google Scholar]
- 18.Krijnse-Locker, J., S. Schleich, D. Rodriguez, B. Goud, E. J. Snijder, and G. Griffiths. 1996. The role of a 21-kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J. Biol. Chem. 271:14950-14958. [DOI] [PubMed] [Google Scholar]
- 19.Lackner, C. A., S. M. D'Costa, C. Buck, and R. C. Condit. 2003. Complementation analysis of the Dales collection of vaccinia virus temperature-sensitive mutants. Virology 305:240-259. [DOI] [PubMed] [Google Scholar]
- 20.Liu, K., B. Lemon, and P. Traktman. 1995. The dual-specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J. Virol. 69:7823-7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercer, J., and P. Traktman. 2003. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J. Virol. 77:8857-8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merchlinsky, M., C. F. Garon, and B. Moss. 1988. Molecular cloning and sequence of the concatemer junction from vaccinia virus replicative DNA. Viral nuclease cleavage sites in cruciform structures. J. Mol. Biol. 199:399-413. [DOI] [PubMed] [Google Scholar]
- 23.Merchlinsky, M., and B. Moss. 1986. Resolution of linear minichromosomes with hairpin ends from circular plasmids containing vaccinia virus concatemer junctions. Cell 45:879-884. [DOI] [PubMed] [Google Scholar]
- 24.Miner, J. N., and D. E. Hruby. 1989. Rifampicin prevents virosome localization of L65, an essential vaccinia virus polypeptide. Virology 170:227-237. [DOI] [PubMed] [Google Scholar]
- 25.Mohandas, A. R., and S. Dales. 1995. Involvement of spicules in the formation of vaccinia virus envelopes elucidated by a conditional lethal mutant. Virology 214:494-502. [DOI] [PubMed] [Google Scholar]
- 26.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2884. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott-Raven, Philadelphia, Pa.
- 27.Moss, B., E. N. Rosenblum, E. Katz, and P. M. Grimley. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280-1284. [DOI] [PubMed] [Google Scholar]
- 28.Obenauer, J. C., L. C. Cantley, and M. B. Yaffe. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31:3635-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pacha, R. F., and R. C. Condit. 1985. Characterization of a temperature-sensitive mutant of vaccinia virus reveals a novel function that prevents virus-induced breakdown of RNA. J. Virol. 56:395-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravanello, M. P., and D. E. Hruby. 1994. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virion assembly. J. Virol. 68:6401-6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez, D., M. Esteban, and J. R. Rodriguez. 1995. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J. Virol. 69:4640-4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez, D., J. R. Rodriguez, and M. Esteban. 1993. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J. Virol. 67:3435-3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 1997. Characterization of early stages in vaccinia virus membrane biogenesis: implications of the 21-kilodalton protein and a newly identified 15- kilodalton envelope protein. J. Virol. 71:1821-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salmons, T., A. Kuhn, F. Wylie, S. Schleich, J. R. Rodriguez, D. Rodriguez, M. Esteban, G. Griffiths, and J. K. Locker. 1997. Vaccinia virus membrane proteins p8 and p16 are cotranslationally inserted into the rough endoplasmic reticulum and retained in the intermediate compartment. J. Virol. 71:7404-7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider, R. J., and I. Mohr. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130-136. [DOI] [PubMed] [Google Scholar]
- 37.Sodeik, B., G. Griffiths, M. Ericsson, B. Moss, and R. W. Doms. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi, T., M. Oie, and Y. Ichihashi. 1994. N-terminal amino acid sequences of vaccinia virus structural proteins. Virology 202:844-852. [DOI] [PubMed] [Google Scholar]
- 39.Thompson, C. L., and R. C. Condit. 1986. Marker rescue mapping of vaccinia virus temperature-sensitive mutants using overlapping cosmid clones representing the entire virus genome. Virology 150:10-20. [DOI] [PubMed] [Google Scholar]
- 40.Traktman, P., A. Caligiuri, S. A. Jesty, K. Liu, and U. Sankar. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J. Virol. 69:6581-6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Traktman, P., K. Liu, J. DeMasi, R. Rollins, S. Jesty, and B. Unger. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682-3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogelstein, B., and D. Gillespie. 1979. Preparative and analytical purification of DNA from agarose. Proc. Natl. Acad. Sci. USA 76:615-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, S., and S. Shuman. 1995. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase 2. J. Virol. 69:6376-6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitehead, S. S., and D. E. Hruby. 1994. Differential utilization of a conserved motif for the proteolytic maturation of vaccinia virus proteins. Virology 200:154-161. [DOI] [PubMed] [Google Scholar]
- 45.Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeh, W. W., B. Moss, and E. J. Wolffe. 2000. The vaccinia virus A9L gene encodes a membrane protein required for an early step in virion morphogenesis. J. Virol. 74:9701-9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Y., J. G. Keck, and B. Moss. 1992. Transcription of viral late genes is dependent on expression of the viral intermediate gene G8R in cells infected with an inducible conditional-lethal mutant vaccinia virus. J. Virol. 66:6470-6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zinoviev, V. V., N. A. Tchikaev, O. Y. Chertov, and E. G. Malygin. 1994. Identification of the gene encoding vaccinia virus immunodominant protein p35. Gene 147:209-214. [DOI] [PubMed] [Google Scholar]