

Abstract
A library of dendrimers was synthesized and optimized for targeted small interfering RNA (siRNA) delivery to different cell subpopulations within the liver. Using a combinatorial approach, a library of these nanoparticle-forming materials was produced wherein the free amines on multigenerational poly(amido amine) and poly(propylenimine) dendrimers were substituted with alkyl chains of increasing length, and evaluated for their ability to deliver siRNA to liver cell subpopulations. Interestingly, two lead delivery materials could be formulated in a manner to alter their tissue tropism within the liver – with formulations from the same material capable of preferentially delivering siRNA to (i) endothelial cells, (ii) endothelial cells and hepatocytes, or (iii) endothelial cells, hepatocytes and tumor cells in vivo. The ability to broaden or narrow the cellular destination of siRNA within the liver may provide a useful tool to address a range of liver diseases.
Keywords: nanomaterial, RNA, dendrimers, amphiphiles, drug delivery
Graphical Abstract
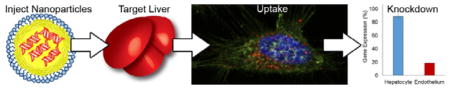
Dendrimer derivatives optimized for in vivo siRNA delivery to liver endothelial cells, hepatocellular carcinoma cells and/or hepatocytes are prepared using a combinatorial approach. The free amines on multigenerational poly(amido amine) and poly(propylenimine) dendrimers are substituted with alkyl chains of increasing length. Through formulation changes, these materials have the ability to broaden or narrow their targeted cellular subpopulation within the liver.
RNA interference (RNAi) is the process whereby a small interfering RNA (siRNA) induces the degradation of complimentary mRNA gene transcripts, thus silencing genes.[1] A key need to the broad application of RNAi is the development of safe and effective delivery systems capable of silencing genes in specific cells within the body. This type of selectivity has the potential to focus therapy, and thereby decrease side effects. Nanoformulation of siRNA is one approach towards this end, and to date the most advanced strategies are hepatocyte-specific, having both selectivity and potency in non-human primates and clinical trials.[2] There is an increasing collection of reports of siRNA delivery to tissues other than hepatocytes including tumors,[3] immune cells[4] and the endothelium.[5] However, delivery to these other tissues is often non-specific, with siRNA functionally delivered to more than just the target tissue. Here we report on the development of formulations based on dendrimeric materials where the targeting is tuned through modifying formulation parameters. Particular focus was placed on developing new delivery materials capable of silencing genes in different liver cell subpopulations, with special emphasis placed on blood vessel endothelial cells.
The chemically-modified dendrimer materials were synthesized using Michael addition chemistry by combining poly(amido amine) or poly(propylenimine) dendrimers of increasing generations with alkyl epoxides of various carbon chain lengths, as illustrated in Scheme 1. The resulting branched, amine-rich ionizable dendrimer cores that facilitate efficient complexation with negatively charged siRNA under acidic formulation conditions. Modification of the dendrimers with alkyl chains affords lipid-like properties, promoting particle formation through hydrophobic aggregation in aqueous conditions. While polycationic polymers for siRNA delivery materials are generally polydisperse and often possess random branching,[6] these modified dendrimers can be molecularly defined, with monodisperse dendrimer cores and defined branching. Poly(amido amine) and poly(propylenimine) dendrimers have been previously investigated for their utility in siRNA delivery.[7] However, the alkyl modification reported here allow for the formation of lipid-like nanoparticles with additional lipid components (excipients). These excipients can be used to tune the properties and activity of the resulting dendrimer.
Scheme 1.

Synthesis of chemically-modified dendrimer materials. Epoxide-terminated alkyl chains ranging in size from C10 to C16 were reacted with the free amines on poly(amido amine) or poly(propylenimine) dendrimers of increasing generation size. In this example, PG0, or generation 0 poly(amido amine), is reacted with an alkyl epoxide.
Products were purified using flash chromatography to remove any unreacted starting materials. The products contained a mixture of different substitutions as well as chiral isomers when examined using thin layer chromatography (0.4 < Rf < 0.8 for an 87.5:11:1.5 CH2Cl2:MeOH:NH4OHaq solvent system). These materials were screened for siRNA delivery using a HeLa cell line that stably expressed both firefly and Renilla luciferase.[8] Modified dendrimer nanoparticles were complexed with siRNA against firefly luciferase at a 5:1 ratio of modified dendrimer to siRNA, by mass. The Renilla luciferase was used as an internal viability control. For this initial high-throughput screen, modified dendrimers were only formulated with 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (C14PEG2000), at a 4:1 molar ratio of modified dendrimer to C14PEG2000. As shown in Figure 1a, all evaluated dendrimers demonstrated significant reduction in the expression of firefly luciferase when compared to PBS-treated controls, with differential activity dependent on the specific chemistry used. Nanoparticle uptake into HeLa cells was verified using confocal microscopy for dendrimers formulated with Cy5.5-labelled siRNA (Figure 1b & 1c).
Figure 1.

(a) A representative subset of the full in vitro screen of modified dendrimers showing HeLa luciferase luminescence after knockdown of firefly luciferase at a 25 nM siRNA dose. Renilla luminescence was used as an internal control to both gauge off-target effects and modified dendrimer-induced toxicity. Dendrimers (including a Lipofectamine positive control) are listed on the x-axis and luciferase luminescence is shown on the y-axis. For nomenclature, P = poly(amido amine), D = diaminobutane amine poly(propylenimine) tetramine, G# = generation number and C# = number of carbons in the alkyl chain. N=3 and error bars are ± S.D. (b, c) Confocal images of PG1.C12 (b) and PG1.C15 (c) nanoparticle delivering Cy5.5-labelled siRNA into HeLa cells at a 50 nM dose. Nanoparticles are red, cell membrane green and nuclei blue. Scale bar = 10 μm.
Modified dendrimers were validated in vivo for the simultaneous delivery of siRNA to both liver endothelial cells and hepatocytes. Using the same formulation conditions as were used in the initial in vitro screen, modified dendrimers were co-formulated with two siRNAs against tie2 and Factor VII (FVII). Tie2 was selected as a target for silencing because it is an endothelial cell-specific target gene.[9] FVII, meanwhile, has been previously established as a robust target for hepatocyte-specific delivery.[8, 10] After co-formulation, modified dendrimer nanoparticles were injected into the tail veins of healthy 8 week old female C57BL/6 mice. After 2 days, mice were euthanized, and tie2 gene and FVII protein levels were quantified (Figure 2a).
Figure 2.

(a) An example of an in vivo multigenerational modified dendrimer screen using C14PEG2000 as the only formulation excipient. Modified dendrimer nanoparticles were co-formulated with tie2 and FVII siRNAs. The total siRNA dose was 3.7 mg kg−1; 1.2 mg kg−1 of FVII siRNA and 2.5 mg kg−1 tie2 siRNA. The co-formulated particles simultaneously caused knockdown of both genes in the liver 2 days after tail vein injection. (b) In vivo performance of PG1.C12 and PG1.C15 modified dendrimer nanoparticles co-formulated tie2 and FVII siRNAs two days after tail vein injection. C14PEG2000 was the only excipient. For the higher dose, a total siRNA dose was 1.125 mg kg−1 (1 mg kg−1 tie2 siRNA and 0.125 mg kg−1 FVII siRNA) was used. The lower dose had a total siRNA dose of 0.51 mg kg−1 (0.5 mg kg−1 tie2 siRNA and 0.01 mg kg−1 FVII siRNA). * = p < 0.04 (t-test) for comparison between doses. (c, d) Optimized nanoparticles containing C14PEG2000 and cholesterol as excipients were co-formulated with FVII and tie2 siRNAs. The combined siRNA dose was 2.6 mg kg−1; 2.5 mg kg−1 tie2 siRNA and 0.1 mg kg−1 FVII siRNA. Expression was measured 2 days after tail vein injections. N = 3 and * = p < 0.02 for comparisons between formulations. (c) Both formulations of PG1.C15 resulted in tie2 knockdown in liver endothelial cells. However, the low excipient 98:1:1 formulation stopped delivery to hepatocytes. (d) Both PG1.C12 formulations caused knockdown in the liver endothelium, while the low excipient 97:1:2 formulation prevented hepatocyte delivery. (e, f) The reduced excipient formulations that resulted in hepatocyte exclusion also increased the mean size of the nanoparticles and reduced their polydispersity. All error bars are ± S.D.
Based on their ability to simultaneously silence genes in both liver endothelial cells and hepatocytes, PG1.C12 and PG1.C15 were selected as the top performing materials. These two materials had an identical core consisting of the first generation poly(amido amine) dendrimer (i.e. PG1), but varied in the length of the substituted alkyl chains (i.e. C12 vs. C15; refer to Scheme S1, and Figures S1 & S2 in Supporting Information). PG1.C15 nanoparticles were larger in diameter and were more resistant to degradation in blood serum (Figures S3 & S4, respectively, in Supporting Information). The apparent nanoparticle pKa values of the two lead materials were ≥ 5.5 (Figure S5 & Table S1 in Supporting Information). Previous studies have shown that apparent nanoparticle pKa values ≥ 5.5 often correlate to hepatocyte delivery,[11] which corresponds to the findings reported here. The lead materials’ affinity for the liver was further confirmed and visualized with biodistribution studies (Figure S6 in Supporting Information). Tie2 knockdown was also quantified in the endothelium of other organs, but the effect was most potent in the liver (Figure S7 in Supporting Information). Moreover, no significant silencing was observed in immune cell populations when these materials were complexed with siRNA against the pan-leukocyte marker CD45 (Figure S8 in Supporting Information).
To establish the relative potency of siRNA delivery to endothelial cells and hepatocytes, a dose response was generated with both PG1.C12 and PG1.C15 nanoparticles. In these experiments, particles were co-formulated with FVII and tie2 siRNA (Figure 2b). As was done in the initial in vivo screens, these co-formulated particles used C14PEG2000 as the only excipient. As shown in Figure 2b, significant knockdown of tie2 in the liver endothelium was achieved with an siRNA dose of 1 mg kg−1, while a similar degree of FVII knockdown was observed using only an siRNA dose of 0.125 mg kg−1. Because these nanoparticles were formulated to contain both siRNAs, the difference in siRNA efficiency for each gene indicates that both nanoparticle formulations had a higher silencing potency in hepatocytes, and a reduced potency in liver endothelial cells. Thus, in order to compensate for reduced potency in the liver endothelium, a higher loading of the desired endothelial-specific siRNA would be required for a strategy designed to silence both endothelial and hepatic gene expression to the same extent.
After establishing which siRNA doses were necessary for gene knockdown in endothelial cells and hepatocytes, we next sought to boost nanoparticle performance through changes in formulation. Cholesterol is an important component in the lipid envelope of viruses[12] and has been used in many potent nanoparticle formulations.[2a–c, 2e] Thus, we sought to evaluate the effects of altering the amount of cholesterol in these formulations. For these studies, the total dose of siRNA as well as the ratio of tie2:FVII siRNA were kept constant so only the cholesterol composition was varied. Moreover, the siRNA dose used in these studies was informed by previous experiments (Figure 2b) in order to control for differences in siRNA efficiency between the two cell types. Figure 2c shows that PG1.C15 nanoparticles formulated at a 90:2:8 mass ratio of modified dendrimer:cholesterol:C14PEG2000 targeted both hepatocytes and liver endothelial cells, as was seen previously in cholesterol-free formulations. However, when the amount of cholesterol and C14PEG2000 was reduced to achieve a 98:1:1 formulation ratio, the same nanomaterial demonstrated increased potency and specificity in liver endothelial cells and significantly reduced potency in hepatocytes. Similarly, endothelial-specific targeting with reduced hepatocyte potency was seen for the PG1.C12 when the formulation was changed from 89:2:9 to 97:1:2, though the efficiency of PG1.C12 as a delivery materials is not as potent as PG1.C15 (Figure 2d). Negative controls for nanoparticles formulated with a non-functional scrambled siRNA showed no knockdown for any of the tested formulations in either cell type (Figure S9 in Supporting Information). Interestingly, nanoparticles in the endothelial-specific formulations were larger in diameter than those that did not demonstrate similar selectivity (Figure 2e & 2f). While all of the modified dendrimer nanoparticles evaluated here resulted in no dramatic increase in blood plasma cytokine levels at 48 hours following administration, formulations with higher cholesterol content resulted in fewer fluctuations in cytokine levels relative to baseline (Figure S10 in Supporting Information).
Therefore, though the PG1.C12 and PG1.C15 modified dendrimers both can enable simultaneous delivery of siRNAs to multiple liver cell types, they can be formulated such that the potency of delivery to hepatocytes is reduced while delivery to endothelial cells is maintained or enhanced. This was achieved by co-formulating with cholesterol as an additional excipient in the nanoparticles, while maintaining a constant 5:1 mass ratio for modified dendrimer:siRNA. Depending on the ratio of cholesterol and C14PEG2000 in the formulations, the targeted liver cell subpopulation can be varied.
After investigating gene silencing in the endothelial cells and hepatocytes of normal, non-diseased livers, silencing was examined in liver tumor cells. Liver tumors contain a heterogeneous mixture of cells, including cancerous cells, normal hepatic cell types, and endothelial cells.[13] Using the minimal excipient formulation (4:1 modified dendrimer:C14PEG2000 molar ratio and 5:1 modified dendrimer:siRNA mass ratio), PG1.C12 and PG1.C15 were evaluated for delivery to hepatocellular carcinoma cells in Met-driven tumors.[14] Unlike normal adult hepatocytes, tumor cells in these hepatocellular carcinomas specifically express and secrete Alpha-fetoprotein (AFP) into the blood.[15] Thus, as a model system for delivery to hepatocellular carcinoma, modified dendrimer nanoparticles formulated with siRNA for AFP were evaluated in this Met-driven AFP-expressing tumor model. As shown in Figure 3, PG1.C12 and PG1.C15 showed a 51% and 92% knockdown (respectively) of AFP at a 1 mg kg−1 dose. Thus, in the context of these two lead materials, changing the length of the substituted alkyl chains adds an additional formulation guideline for delivery to various liver cell types. At the siRNA doses and ratios reported here, cholesterol-free formulations of PG1.C12 preferentially silenced genes in endothelial cells and healthy hepatocytes, and reduced silencing in the tumors. In contrast, cholesterol-free formulations of PG1.C15 increased silencing in tumor cells but had similar efficacy in endothelial cells and healthy hepatocytes at the same dose (Figures 2b & 3b).
Figure 3.

(a) Serum protein Western blot showing in vivo alpha-fetoprotein (AFP) knockdown in hepatocellular carcinoma cells using PG1.C12 and PG1.C15 modified dendrimers. Nanoparticles were formulated at a 4:1 modified dendrimer:C14PEG2000 molar ratio and a 1 mg kg−1 siRNA dose was used. (b) Densitometry analysis of the blot showed that both modified dendrimers caused a significant decrease in the amount of AFP in the blood. PG1.C15 was the more potent of the two formulations. N = 3, error bars are 1 S.D. and connecting lines indicate a significant difference (p < 0.05, t-test).
This work describes the potential to bias silencing to multiple tissues, including the liver endothelium, hepatocellular carcinoma cells, and hepatocytes, in a controlled and tunable way, via formulation. With the two lead modified dendrimers presented here, preferential silencing can be influenced by varying the excipient ratio and changing the length of substituted alkyl chains. Nanoparticle formulations with higher endothelial cell selectivity were larger in diameter (Figures 2e & 2f). These larger sizes may have played a role in selective delivery by affecting transport of nanoparticles through the fenestrated endothelium of the liver, thus reducing hepatocyte access and uptake.[16] Furthermore, the changes in excipient content may have altered blood serum protein adsorption to the nanoparticles, which may have in turn altered liver cell subpopulation uptake. Lipid-based nanoparticles are known to exchange components with the serum and adsorb proteins.[17] Apolipoprotein E is a serum protein that can adsorb to nanoparticles and enhance uptake into hepatocytes.[18] Thus, one hypothesis is that the endothelial-specific formulations reduce their affinity for apolipoprotein E, which may subsequently diminish hepatocyte uptake and gene knockdown.
In regards to hepatocellular carcinoma gene silencing efficiency, a number of factors, including particle size, charge, stability and retention time, may have contributed to the observed difference in potency. While these modified dendrimers contained the same core, the lengths of their substituted alkyl chains differed; PG1.C12 had C12 alkyl chains while PG1.C15 had longer C15 chains. Once formulated, PG1.C15 nanoparticles were larger and more stable in blood serum (Figures S3 & S4 in Supporting Information). The increased serum stability may have increased the circulation time of the intact nanoparticles, allowing them more opportunity to reach their target. Moreover, the larger size of the PG1.C15 nanoparticles may have improved their retention time within the tumor.[19] Additionally, as indicated by zeta potential data (Table S1 in Supporting Information), PG1.C15 nanoparticles were more prone to aggregation. Perhaps, after nanoparticle entry into the tumor, their effective size increased through accretion, which possibly enhanced nanoparticle retention even further.
The ability to bias delivery to liver endothelial cells may prove useful when the target genes are expressed in both hepatocytes and endothelial cells, but therapy requires only silencing in the endothelium. A potential example of this could include the treatment of inflamed liver endothelium, which often occurs in liver disease. In contrast, it is possible that delivery to both hepatocytes and endothelium could be useful when diseases affect both tissues, such as in the case of ischemia/reperfusion injuries caused by liver surgery and transplantation.[20]
Alkyl substitution of regularly branched dendrimer structures to produce amphiphiles rich with primary, secondary and tertiary amines is a facile strategy to prepare nanoparticles that can condense siRNA for targeted delivery. Moreover, the use of molecularly defined, regularly-branched, ionizable dendrimers as the core structure for these materials is advantageous with respect to clinical translation. Future formulations of these materials could be considered for the delivery of other therapeutic nucleic acids, such as mRNA, microRNA, and DNA.
Experimental Section
Refer to the Supporting Information section online via the link at the end of the document.
Supplementary Material
Acknowledgments
The authors thank Dr. Carmen Barnes, Dr. Yizhou Dong and J. Robert Dorkin for useful advice. Special thanks to MIT’s Division of Comparative Medicine and Swanson Biotechnology Center. Funding was provided by the National Institutes of Health Centers of Cancer and Nanotechnology Excellence (U54 CA151884), the United States Army Medical Research and Materiel Command’s Armed Forces Institute of Regenerative Medicine (W81XWH-08-2-0034) and Alnylam Pharmaceuticals. R.L. is a shareholder and member of the Scientific Advisory Board of Alnylam. R.L and D.G.A have sponsored research grants from Alnylam. O.F.K. designed experiments and performed all work with the assistance of other authors.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]; b) Elbashir SM, Lendeckel W, Tuschl T. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Dong Y, et al. Proc Natl Acad Sci USA. 2014;111:3955–3960. doi: 10.1073/pnas.1322937111. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Novobrantseva TI, et al. Mol Ther Nucleic Acids. 2012;1:e4. doi: 10.1038/mtna.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Love KT, et al. Proc Natl Acad Sci USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Maier MA, et al. Mol Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jayaraman M, et al. Angewandte Chemie. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Coelho T, et al. N Engl J Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 3.a) Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ. Proc Natl Acad Sci USA. 2010;107:981–986. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xue W, et al. Proc Natl Acad Sci USA. 2014 [Google Scholar]
- 4.Novobrantseva TI, et al. Mol Ther Nucleic Acids. 2012;1 [Google Scholar]
- 5.a) Dahlman JE, et al. Nature nanotechnology. 2014;9:648–655. doi: 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Santel A, et al. Gene Therapy. 2006;13:1360–1370. doi: 10.1038/sj.gt.3302778. [DOI] [PubMed] [Google Scholar]
- 6.a) Grayson ACR, Doody AM, Putnam D. Pharm Res. 2006;23:1868–1876. doi: 10.1007/s11095-006-9009-2. [DOI] [PubMed] [Google Scholar]; b) Zintchenko A, Philipp A, Dehshahri A, Wagner E. Bioconjugate Chem. 2008;19:1448–1455. doi: 10.1021/bc800065f. [DOI] [PubMed] [Google Scholar]; c) Katas H, Alpar HO. J Control Release. 2006;115:216–225. doi: 10.1016/j.jconrel.2006.07.021. [DOI] [PubMed] [Google Scholar]; d) Lee DW, Yun KS, Ban HS, Choe W, Lee SK, Lee KY. J Control Release. 2009;139:146–152. doi: 10.1016/j.jconrel.2009.06.018. [DOI] [PubMed] [Google Scholar]; e) Jere D, Jiang HL, Kim YK, Arote R, Choi YJ, Yun CH, Cho MH, Cho CS. Int J Pharm. 2009;378:194–200. doi: 10.1016/j.ijpharm.2009.05.046. [DOI] [PubMed] [Google Scholar]; f) Inoue Y, Kurihara R, Tsuchida A, Hasegawa M, Nagashima T, Mori T, Niidome T, Katayama Y, Okitsu O. J Control Release. 2008;126:59–66. doi: 10.1016/j.jconrel.2007.10.022. [DOI] [PubMed] [Google Scholar]; g) Li JG, Cheng D, Yin TH, Chen WC, Lin YJ, Chen JF, Li RT, Shuai XT. Nanoscale. 2014;6:1732–1740. doi: 10.1039/c3nr05024f. [DOI] [PubMed] [Google Scholar]; h) Watanabe K, Harada-Shiba M, Suzuki A, Gokuden R, Kurihara R, Sugao Y, Mori T, Katayama Y, Niidome T. Mol Biosyst. 2009;5:1306–1310. doi: 10.1039/b900880b. [DOI] [PubMed] [Google Scholar]; i) Nakayama Y. Acc Chem Res. 2012;45:994–1004. doi: 10.1021/ar200220t. [DOI] [PubMed] [Google Scholar]
- 7.a) Omidi Y, Hollins AJ, Drayton RM, Akhtar S. J Drug Target. 2005;13:431–443. doi: 10.1080/10611860500418881. [DOI] [PubMed] [Google Scholar]; b) Taratula O, Savla R, He HX, Minko T. Int J Nanotechnol. 2011;8:36–52. [Google Scholar]; c) Nomani A, Fouladdel S, Haririan I, Rahimnia R, Ruponen M, Gazori T, Azizi E. Afr J Pharm Pharacol. 2012;6:530–537. [Google Scholar]; d) Patil ML, Zhang M, Minko T. Acs Nano. 2011;5:1877–1887. doi: 10.1021/nn102711d. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tsai YJ, Hu CC, Chu CC, Toyoko I. Biomacromolecules. 2011;12:4283–4290. doi: 10.1021/bm201196p. [DOI] [PubMed] [Google Scholar]; f) Yu T, Liu X, Bolcato-Bellemin AL, Wang Y, Liu C, Erbacher P, Qu F, Rocchi P, Behr JP, Peng L. Angew Chem Int Ed. 2012;51:8478–8484. doi: 10.1002/anie.201203920. [DOI] [PubMed] [Google Scholar]; g) Malhotra S, et al. Biomacromolecules. 2012;13:3087–3098. doi: 10.1021/bm300892v. [DOI] [PubMed] [Google Scholar]
- 8.Akinc A, et al. Nat Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Ramsden JD, Cocks HC, Shams M, Nijjar S, Watkinson JC, Sheppard MC, Ahmed A, Eggo MC. J Clin Endocrinol Metab. 2001;86:2709–2716. doi: 10.1210/jcem.86.6.7552. [DOI] [PubMed] [Google Scholar]; b) Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]; c) Sato TN, Qin Y, Kozak CA, Audus KL. Proc Natl Acad Sci USA. 1993;90:9355–9358. doi: 10.1073/pnas.90.20.9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akinc A, et al. Mol Ther. 2009;17:872–879. doi: 10.1038/mt.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitehead KA, et al. Nat Commun. 2014;5 doi: 10.1038/ncomms5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aloia RC, Tian H, Jensen FC. Proc Natl Acad Sci USA. 1993;90:5181–5185. doi: 10.1073/pnas.90.11.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Alakhova DY, Kabanov AV. Adv Drug Deliv Rev. 2013;65:1763–1783. doi: 10.1016/j.addr.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, Kay MA, Wang R, Bishop JM. Proc Natl Acad Sci USA. 2007;104:14771–14776. doi: 10.1073/pnas.0706578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Bogorad RL, Yin H, Zeigerer A, Nonaka H, Ruda VM, Zerial M, Anderson DG, Koteliansky V. Nat Commun. 2014;5:3869. doi: 10.1038/ncomms4869. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Llovet JM, Burroughs A, Bruix J. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 16.Litzinger DC, Buiting AMJ, van Rooijen N, Huang L. BBA - Biomembranes. 1994;1190:99–107. doi: 10.1016/0005-2736(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 17.a) Cullis PR, Chonn A, Semple SC. Adv Drug Deliv Rev. 1998;32:3–17. doi: 10.1016/s0169-409x(97)00128-2. [DOI] [PubMed] [Google Scholar]; b) Chonn A, Semple SC, Cullis PR. J Biol Chem. 1992;267:18759–18765. [PubMed] [Google Scholar]
- 18.Bisgaier CL, Siebenkas MV, Williams KJ. J Biol Chem. 1989;264:862–866. [PubMed] [Google Scholar]
- 19.Albanese A, Tang PS, Chan WC. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 20.Kohli V, Selzner M, Madden JF, Bentley RC, Clavien PA. Transplantation. 1999;67:1099–1105. doi: 10.1097/00007890-199904270-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.