

Abstract
Objectives: Recent studies using next-generation sequencing (NGS) analysis disclosed the importance of the intrinsic activation of the B-cell receptor (BCR) pathway in the pathogenesis of sporadic Burkitt lymphoma (sBL) due to mutations of TCF3/ID3 genes. Since no definitive data are available on the genetic landscape of endemic Burkitt (eBL), we first assessed the mutation frequency of TCF3/ID3 in eBL compared with sBL and subsequently the somatic hypermutation status of the BCR to answer whether an extrinsic activation of BCR signaling could also be demonstrated in Burkitt lymphoma.
Methods: We assessed the mutations of TCF3/ID3 by RNAseq and the BCR status by NGS analysis of the immunoglobulin genes (IGs).
Results: We detected mutations of TCF3/ID3 in about 30% of the eBL cases. This rate is significantly lower than that detected in sBL (64%). The NGS analysis of IGs revealed intraclonal diversity, suggesting an active targeted somatic hypermutation process in eBL compared with sBL.
Conclusions: These findings support the view that the antigenic pressure plays a key role in the pathogenetic pathways of eBL, which may be partially distinct from those driving sBL development.
Keywords: BCR, Burkitt lymphoma, NGS, Clonality analysis
The B-cell receptor (BCR) is essential for normal B-cell development and maturation. There is increasing evidence that BCR signaling is implicated in B-cell malignancies. A role for BCR signaling in lymphoma has been inferred by studying immunoglobulin genes in human lymphomas and engineering mouse models.1 Among the different types of B-cell malignancies, several different tactics are applied for activation of the BCR signaling.2 Although they convey on the same pathway, understanding where the signaling is modulated may help unravel which B-cell malignancies would likely respond to which kind of targeted therapies.
Roles for antigen-dependent and antigen-independent BCR signaling have now been described for several different lymphoma types.2 Evidence for a causal relationship between hepatitis C virus infection and lymphomagenesis has been found in a subset of splenic marginal zone lymphomas.3 Chronic exposure to microbial antigens has been implicated in the pathogenesis of other types of marginal zone lymphomas, such as Helicobacter pylori in mucosa-associated lymphoid tissue lymphoma and Chlamydia psittaci in ocular adnexa lymphoma.4-6 An increasing number of antigens bound to the BCR on chronic lymphocytic leukemia cells have been identified, including autoantigens expressed on dying cells, as well as viral, bacterial, and fungal antigens.7
On the other hand, in diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL), next-generation sequencing (NGS) studies have revealed recurrent mutations of genes belonging to the BCR pathway, converging in the two different types of intrinsically deregulated downstream BCR signals—namely, chronic active signaling in DLBCL and tonic signaling in BL.8,9
In DLBCL, mutations affecting the BCR pathway are particularly enriched in cases showing the activated B-cell–like (ABC) molecular phenotype. In particular, mutations of CD79B and CARD11 have been identified in about 10% of ABC DLBCL cases. These mutations are sufficient to intrinsically activate survival signaling in the malignant B cells and obviate the need for upstream BCR signaling.8 Most of sporadic Burkitt lymphomas (sBLs) (∼70%) harbor gain-of-function mutations affecting the TCF3 gene or mutations disrupting the TCF3-negative regulator ID3.9-11 The overall consequence of ID3 and TCF3 mutants in BL is an impairment of TCF3/ID3 inhibitory heterodimerization that results in an increasing expression of BCR genes and activation of a tonic form of BCR signaling, which is antigen independent.9-11
Taking into consideration that no definitive data are available on the genetic landscape of endemic BL (eBL),10,12 as well as the different environment in which eBL emerges, characterized by an uninterrupted presence of malaria parasites, a never-ending chronic antigenic stimulation, splenomegaly, and concurrent infections by viruses, bacteria, and parasites,12 in the present study we aim to better understand the BCR activation pathway in eBL compared with sBL. To this end, we first analyzed the frequency of the TCF3 or its negative regulator ID3 gene mutations by RNAseq and Sanger sequencing and subsequently looked for a possible association with somatic hypermutation status (SHM) of BCR by NGS analysis and Sanger sequencing of immunoglobulin genes as indicative of an active ongoing interaction with antigens.
Materials and Methods
Patients
A total of 37 BL samples preserved in RNA later (RNA stabilization Reagent; Qiagen, Valencia, CA) were collected from the Department of Human Pathology of the Lacor Hospital, Uganda; from the Department of Medical Biotechnologies, University of Siena, Italy; and from “L.A Seragnoli” Haematopathology Unit, Policlinico S. Orsola, Bologna, Italy. Formalin-fixed, paraffin-embedded samples were available for all. Overall, 26 BL samples were collected from endemic areas in Uganda, Africa (eBL), while the other 11 BL cases were collected in Italy (sBL). Clinical epidemiologic details of the 37 BL cases are summarized in Table 1.
Table 1.
Clinical-Epidemiologic Characteristics of Burkitt Lymphoma Cases
| Case No. | Site | Age, y | Sex | EBV |
|---|---|---|---|---|
| eBL78 | Abdomen | 6 | M | + |
| eBL48 | Jaw | 10 | M | + |
| eBL43 | Gum | 5 | M | + |
| eBL15 | Lymph node | 9 | F | + |
| eBL32 | Jaw | 7 months | M | + |
| eBL35 | Jaw | 7 | M | + |
| eBL36 | Abdomen | 4 | M | + |
| eBL84 | Jaw | 9 | F | + |
| eBL52 | Jaw | 7 | M | + |
| eBL22 | Jaw | 6 | M | + |
| eBL49 | Jaw | 7 | M | + |
| eBL34 | Jaw | 10 | F | + |
| eBL51 | Jaw | 9 | M | + |
| eBL23 | Jaw | 7 | M | + |
| eBL19 | Abdomen | 4 | M | + |
| eBL40 | Jaw | 7 | M | + |
| eBL63 | Abdomen | 7 | F | + |
| eBL80 | Gum | 7 | M | + |
| eBL74 | Abdomen | 5 | M | + |
| eBL28 | Gum | 10 | M | + |
| eBL50 | Abdomen | 8 | F | + |
| eBL69 | Jaw | 8 | F | + |
| eBL31 | Palate | 7 | M | + |
| eBL20 | Jaw | 7 | M | + |
| eBL57 | Palate | 10 | F | + |
| eBL62 | Jaw | 9 | M | + |
| sBL4169 | Lymph node | 39 | M | – |
| sBLE9 | Abdomen | 21 | M | – |
| sBL4452 | Lymph node | 46 | M | – |
| sBLE4 | Abdomen | 53 | M | – |
| sBL4324 | Abdomen | 52 | F | – |
| sBLE2 | Lymph node | 31 | F | – |
| sBLE3 | Lymph node | 43 | F | – |
| sBLE5 | Abdomen | 40 | M | – |
| sBL2552 | Abdomen | 28 | M | + |
| sBL8274 | Abdomen | 23 | F | – |
| sBLE8 | Abdomen | 33 | M | – |
eBL, endemic Burkitt lymphoma; sBL, sporadic Burkitt lymphoma; +, positive; –, negative.
All cases were previously analyzed by gene expression profile (GEP) analysis and showed a signature consistent with molecular BL.13,14
Total RNA was extracted from BL samples with Trizol, according to manufacturer’s procedure (Invitrogen, Life Technologies, Carlsbad, CA). The study was approved by each institutional ethical committee, and written permission and informed consent have been obtained before sample collection in accordance with the Declaration of Helsinki.
TCF3 and ID3 Mutation Assessment by RNAseq
The paired-end libraries (2 × 75 base pairs [bp]) were prepared according to the TruSeq RNA sample preparation v2 protocol (Illumina, San Diego, CA). The resulting libraries were sequenced on a HiScan SQ (Illumina) following the manufacturer's instructions. Briefly, 2 μg Poly(A)+ RNA was purified from total RNA using poly-T oligo–attached magnetic beads and then used for fragmentation into 130- to 290-bp fragments. The first strand of complementary DNA (cDNA) synthesis was performed using reverse transcriptase enzyme (SuperScript II; Invitrogen, Life Technologies) and random hexamer primer, followed by generation of double-stranded cDNA. AmpureXP beads (Beckman Coulter, Brea, CA) were used to purify the cDNA, and an end-repair step was performed to convert the overhangs, resulting from fragmentation, into blunt ends by 3′ to 5′ exonuclease activity. A single “A” nucleotide was added to the 3′ ends of the blunt fragments to prevent them from ligating to one another during the adapter ligation reaction. This approach was adopted to ensure a low rate of chimera (concatenated template) formation. Subsequently, sequencing adapters were added to the ends of the double-stranded (ds)–cDNA fragment, and a polymerase chain reaction (PCR) reaction was used to selectively enrich those ds-cDNA fragments that had adapter molecules on both ends, amplifying the amount of ds-cDNA in the final libraries. Last, PCR libraries products were purified by AmpureXP beads, and quality control analysis was assessed using a DNA-1000 (Agilent, Waldbronn, Germany). The quantification was performed by the Quant-it PicoGreen dsDNA Assay Kit according to the manufacturer’s protocol (Invitrogen, Life Technologies). Sequence variants were obtained using the SAVI (Statistical Algorithm for Variant Identification) algorithm independently for each sample. Candidate somatic mutations were obtained by eliminating common germline variants and recurrence. Sanger sequencing was used for validation and confirmation of TCF3 and ID3 single-nucleotide variants in sBL.9-11,15,16
PCR Amplification and High-Throughput Sequencing by Roche 454 GS Junior Instrument
In total, 1 µg total extracted RNA per case was transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. Resulting cDNA was subjected to PCR amplification of the IGVH locus by using modified BIOMED-2 framework region 1 (FR1) consensus primers in conjunction with a consensus J segment primer.17
In this assay, each of the seven BIOMED-2 primers at the 5′ end contained an additional sequence, Multiplex Identifiers, that barcodes the sample Table 2. Thermal cycling was performed on an Applied Biosystems 2720 Thermal Cycler using the BIOMED-2 protocols. Small DNA fragments were removed using the AMPure PCR purification system (Agencourt, Beverly, MA) following the manufacturer's protocol (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/454SeqSys_AmpliconLibraryPrepMethodManual_Jun2013.pdf). Amplicons were subsequently quantified using the Quant-iT PicoGreen dsDNA reagent (Invitrogen Corporation, Life Technologies). All amplicons were then pooled at an equimolar ratio. Subsequently, the sample pool was diluted to a final concentration of 1 × 107 PCR fragment molecules/μL.
Table 2.
Modified FR1 BIOMED-2 Primersa
 |
NGS, next-generation sequencing.
Forward and reverse primers are in blue, key sequences are in orange and yellow, and Multiplex Identifier (MID) sequences in green.
The amplicon-PCR–derived fragments were annealed to carrier beads and clonally amplified by emulsion PCR (emPCR). The emPCR was performed according to the manufacturer's protocol (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunioremPCRAmplificationMethodManualLib-A_March2012.pdf). The beads were isolated and compartmentalized into droplets of an aqueous PCR buffer in oil emulsion. Subsequently, the emulsions were broken by isopropanol to facilitate collection of the amplified fragments bound to their specific beads. The beads carrying single-stranded DNA templates were enriched, counted, and deposited into the picotiter plate for sequencing (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJuniorSequencingManual_Jan2013.pdf).
Data analysis was performed using the Roche (Basel, Switzerland) proprietary software package for the GS Junior system. Image acquisition, image processing, and signal processing were performed during the run.
An unmutated chronic lymphocytic leukemia (CLL) case, BL cell lines, and reactive lymph node sequencing were applied as control samples for monoclonality and polyclonality.
Bioinformatical Analysis
By using the GS Junior system, we obtained, for each sample, an average depth of 3,700 reads with a minimum depth of 772 reads and a maximum depth of 7,869 reads.
The pooled raw sequences obtained by GS Junior sequencing were split by using the 454 seqTool script and custom script in Perl language according to different sample-specific tag sequences.
To reduce the single-nucleotide variant complexity, each single subset of sequences was clustered by cap3 software (http://bioweb.pasteur.fr/seqanal/interfaces/cap3.html) with more than 98% overlap identity by obtaining a list of consensus sequencing, and therefore the sequencing reads that differed by up to two single-nucleotide substitutions or gaps were aggregated.18 By using these stringent criteria, artifactual clonotypes were minimized. For each consensus sequence corresponding to different intrasample clones, we calculated the number of raw sequences used to indicate the abundance of each different rearrangement present in the sample. Finally, the consensus sequencing was multialigned by the Clustalw (http://www.ebi.ac.uk/tools/clustalw2) program with default parameters.19
Characteristic 454 sequencing errors, such as insertions/deletions near homopolymer tracts, were also identified, and sequences containing such errors were also discarded. Low-frequency clonotypes that differed by up to 1% from another clonotype with significantly higher frequency were deemed to be the result of technical artifact (PCR or sequencing artifacts), and the sequence differences were corrected for clonotype aggregation.
Validation of NGS Data by Sanger Sequencing and Subcloning
To validate the reliability of NGS data, 26 eBL and 11 sBL samples were amplified in two separated reactions. Both reactions were carried using the FR1 VH-JH BIOMED-2 primers as previously described.15
Purified PCR amplicons were subjected to direct sequencing and cloning into the pCRII-TOPO vector (TOPO TA Cloning Kit; Invitrogen, Paisley, UK) and were used to transform TOP 10 cells (Invitrogen) according to the manufacturer’s instructions. At least 20 clones per case were sequenced in both directions with an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Weiterstadt, Germany) using Sp6 reverse and T7 forward primers, respectively.
The sequences obtained from direct sequencing and cloning were analyzed with the IMGT/V-QUEST (http://www.imgt.org/IMGT_vquest/vquest?livret=0&Option=humanIg; Marie-Paule Lefranc [Marie-Paule.Lefranc@igh.cnrs.fr], Université de Montpellier, CNRS, LIGM, IGH, SFR, Montpellier, France).20,21
Sequence Data Analysis
All the obtained sequences were aligned with IMGT tools for nucleotide analysis of immunoglobulin (IG) V-(D)-J repertoires, polymorphisms, and IG mutations.21
In NGS data, the following two parameters have been analyzed for each V-D-J combination: (1) the number of B-cell clusters, defined as sets of sequences having the same unique V-D-J and heavy complementarity-determining region 3 (VH CDR3) rearrangement (number of clusters), and (2) the total number of sequences obtained in this V-D-J combination (number of reads). Clusters composed of more than 50 reads and showing the same VDJ and CDR3 but variation in the VH domain were considered subclusters, indicating intraclonal diversity.
The following additional information was also extracted: (1) IGHV gene usage and percentage of homology to germline considering the 2% cutoff to discriminate between mutated and unmutated immunoglobulin genes; (2) the number of nucleotide mutations associated with amino acid changes (replacement mutation) and the number of nucleotide mutations not associated with amino acid changes (silent mutation) of the rearranged immunoglobulin genes22-24; and (3) the amino acid changes, classified as very similar (+ + +), similar (+ + –, + – +), dissimilar (– +, – + –, + –), and very dissimilar (– – –) according to hydropathy, volume, and IMGT physicochemical classes (http:/www.imgt.org/IMGT_vquest/vquest?livret=0&Option=humanIg).21,25 These amino acid changes are useful to understand affinity maturation mechanisms, which result in extensive alteration of the structural protein conformation that is important in determining the binding to the antigens.
All “nonubiquitous” sequence changes from the germline were evaluated and further characterized as follows: (1) unconfirmed mutation (a mutation observed in only one subclusterd sequence from the same specimen [“unique”]) and (2) confirmed mutation (a mutation observed more than once among subcluster sequences from the same specimen [“partially shared”]). Where a clonal expansion was identified, the sequences that were more mutated were chosen as representative of the cluster to examine the characteristics of the mutations.
Statistical Analysis
Descriptive statistics for discrete parameters included counts and frequency distributions. The significance of the bivariate relationship was assessed by the Fisher exact test. For all comparisons, a significance level of P at .05 was set, and all statistical analyses were performed using GraphPad InStat version 3.05 (GraphPad Software, San Diego, CA).
Results
TCF3 and ID3 Are Mutated in eBL
ID3 and TCF3 single-nucleotide variants (SNVs) were found in few eBL cases. Specifically, ID3 SNVs were found in six (23%) of 26 eBL cases, while TCF3 was found in three (11.5%) of 26 eBL cases. Since one sample (BL23) was found to carry SNVs in both genes, eight (30.7%) of 26 eBL cases had ID3 and/or TCF3 variants Table 3. Sanger resequencing of genomic DNA confirmed ID3 and TCF3 mutations in 30% and 64% of eBL and sBL samples, respectively. In three samples, matched DNA obtained from peripheral blood was available; therefore, we could assess the absence of the SNVs, indicating that the recognized variants corresponded to somatic mutations Figure 1.
Table 3.
ID3 and TCF3 Single-Nucleotide Variants in eBL
| Sample ID | Position | Reference/Variant | Chromosome | Symbol | Amino Acid Change | Type of Mutation |
|---|---|---|---|---|---|---|
| eBL23 | NM_002167:702 | C/G | chr1:23885621 | ID3 | I99M | Missense |
| NM_002167:650 | T/C | chr1:23885673 | ID3 | V82A | Missense | |
| NM_002167:649 | G/A | chr1:23885674 | ID3 | V82I | Missense | |
| NM_002167:700 | A/C | chr1:23885623 | ID3 | I99L | Missense | |
| eBL49 | NM_002167:595 | C/T | chr1:23885728 | ID3 | L64F | Missense |
| NM_002167:571 | C/T | chr1:23885752 | ID3 | P56S | Missense | |
| eBL50 | NM_002167:616 | C/T | chr1:23885707 | ID3 | Q71 | Nonsense |
| NM_002167:592 | C/T | chr1:23885731 | ID3 | Q63 | Nonsense | |
| eBL22 | NM_002167:595 | C/T | chr1:23885728 | ID3 | L64F | Missense |
| eBL69 | NM_002167:430 | G/A | chr1:23885893 | ID3 | G9S | Missense |
| eBL80 | NM_002167:515 | C/T | chr1:23885808 | ID3 | P37L | Missense |
| eBL19 | NM_001136139:1692 | T/A | chr19:1612366 | TCF3 | N551K | Missense |
| eBL20 | NM_001136139:1692 | T/G | chr19:1612366 | TCF3 | N551K | Missense |
| eBL23 | NM_001136139:1709 | T/A | chr19:1612349 | TCF3 | V557E | Missense |
eBL, endemic Burkitt lymphoma.
Figure 1.
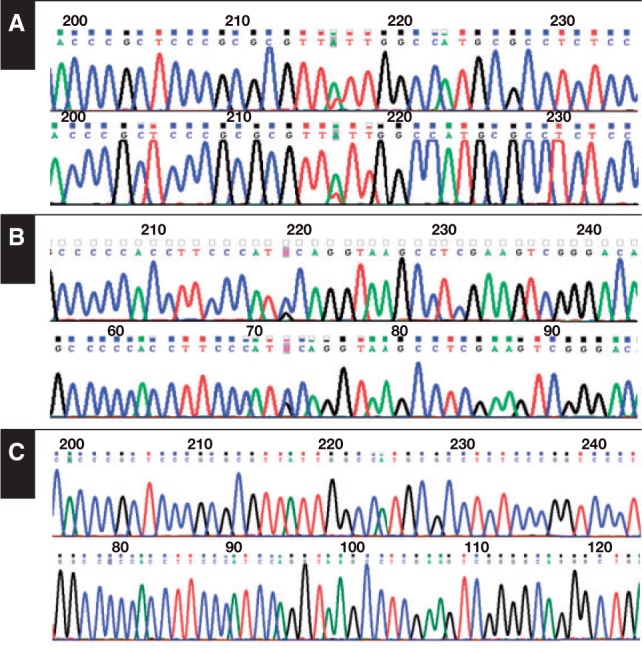
ID3 and/or TCF3 mutations in endemic Burkitt lymphoma (eBL) and sporadic Burkitt lymphoma (sBL). The number and percentage of eBL and sBL cases with ID3 and/or TCF3 mutations are as follows: eight (30%) of 18 for eBL and eight (64%) of 11 for sBL. Example of biallelic mutation in the TCF3 (A) and ID3 (B) genes identified in sBL by Sanger sequencing. C, TCF3 and ID3 wild-type sequences (top row and bottom row, respectively).
Comparison of Sequencing Data Derived From the NGS Analysis With Sanger Sequencing
Multiplex FR1 immunoglobulin H PCR products were generated and subjected to clonality analysis via 454 NGS, followed by data processing, and compared with capillary electrophoresis.
A total of 130,000 sequences/reads were generated with an average depth of 3,700 reads, with a minimum depth of 772 reads and a maximum depth of 7,869 reads for each sample. All 37 cases were clonal on NGS using the criterion that a clonal cluster(s) must be at least fourfold more abundant than the next largest clonotype of the background.26 In particular, the presumed monoclonal clusters ranged from 554 to 6,780 reads, representing from 61.8% to 99.7% of the total reads. On the other hand, the polyclonal background was composed by high percentages of single reads, ranging from 0.01% to 2.04% of the total reads.
When all the sequences were aligned with IMGT tools for nucleotide analysis of immunoglobulin (IG), polymorphisms, and IG mutations,21,24 clusters showing identical IGHV, IGHD, and IGHJ usage and CDR3 regions as the presumed monoclonal clusters were detected, ranging from 63 to 470 reads, with a range from 8.6% to 35.5% of the total reads.
Distinct clusters showed specific mutational profiles. They exhibited shared as well unique somatic mutations and sequences with different lengths for the presence of nucleotide deletion. This finding is indicative of intraclonal diversity and points to the emergence from a common ancestor. This phenomenon was particularly evident in all eBL cases, while only one case of sBL showed different subclusters without differences in length but only differences in point mutations Table 4 and Figure 2.
Table 4.
NGS and Sanger Sequencing Analysis of IgV Genes in Burkitt Lymphoma
| Case No. | VH Usage | No. of Clusters | Range, Mean (%) | R/S | VD-D/VS-S | In/Out Frame | Intraclonal Heterogeneity | |
|---|---|---|---|---|---|---|---|---|
| NGS | Sanger | |||||||
| eBL48 | 1-2*04 | 6 | 2 | 17-20 (90.58-92.24) | 12/7 | 9/3 | 6/0 | Yes |
| eBL43 | 1-3*01 | 4 | 1 | 28-35 (87.85-90.27) | 27/8 | 23/4 | 4/0 | Yes |
| eBL78 | 1-69*06 | 4 | 2 | 9-12 (95.83-96-87) | 6/6 | 5/1 | 4/0 | Yes |
| eBL15 | 1-69*05 | 6 | 3 | 15-19 (91.67-92.87) | 7/12 | 1/6 | 4/0 | Yes |
| eBL32 | 1-69*04 | 5 | 2 | 18-19 (93.15-93.61) | 13/6 | 10/3 | 5/0 | Yes |
| eBL35 | 1-69*10 | 4 | 1 | 23-28 (85.21-87.95) | 21/7 | 11/10 | 4/0 | Yes |
| eBL36 | 1-69*06 | 6 | 2 | 5-6 (97.26-97.76) | 4/2 | 4/0 | 6/0 | Yes |
| eBL84 | 1-69*04 | 5 | 3 | 39-42 (79.81-81.28) | 30/10 | 21/9 | 5/0 | Yes |
| eBL52 | 1-69*06 | 7 | 3 | 20-27 (90.62-93.05) | 21/6 | 14/7 | 3/0 | Yes |
| eBL22 | 1-8*02 | 4 | 1 | 20-24 (91.67-92.53) | 14/10 | 12/2 | 4/0 | Yes |
| eBL49 | 3-7*01 | 5 | 2 | 15-21 (92.71-94.79) | 14/7 | 12/2 | 5/0 | Yes |
| eBL34 | 3-9*01 | 3 | 1 | 24-29 (87.34-89.24) | 19/10 | 9/9 | 5/0 | Yes |
| eBL51 | 3-11*01 | 3 | 1 | 12-15 (93.27-94.61) | 13/2 | 8/4 | 3/0 | Yes |
| eBL23 | 3-15*01 | 8 | 5 | 22-33 (86.66-93.33) | 17/6 | 9/5 | 5/0 | Yes |
| eBL19 | 3-23*01 | 10 | 4 | 5-13 (95.20-95.65) | 11/2 | 10/1 | 3/0 | Yes |
| eBL40 | 3-23*01 | 5 | 2 | 32-39 (86.48-88.88) | 26/13 | 18/8 | 5/0 | Yes |
| eBL63 | 3-23*01 | 3 | 2 | 14-16 (93.57-94.37) | 14/1 | 12/2 | 3/0 | Yes |
| eBL80 | 3-23*01 | 4 | 1 | 11-13 (93.98-95.58) | 9/4 | 8/1 | 4/0 | Yes |
| eBL74 | 3-23*01 | 4 | 1 | 19-20 (93.06-93.40) | 18/2 | 8/10 | 4/0 | Yes |
| eBL28 | 3-30*03 | 7 | 3 | 17-26 (89.60-93.15) | 20/6 | 10/8 | 7/0 | Yes |
| eBL50 | 3-72*01 | 4 | 2 | 10-13 (91.37-92.80) | 10/3 | 9/1 | 4/0 | Yes |
| eBL69 | 3-74*01 | 4 | 2 | 18-26 (90.97-93.75) | 17/9 | 8/10 | 4/0 | Yes |
| eBL31 | 4-34*01 | 9 | 5 | 5-14 (84.15-94.79) | 9/5 | 5/3 | 5/0 | Yes |
| eBL21 | 4-34*01 | 7 | 3 | 14-15 (93.18-93.64) | 10/5 | 6/3 | 5/0 | Yes |
| eBL57 | 4-34*01 | 4 | 1 | 38-42 (80.91-82.72) | 35/7 | 28/7 | 3/0 | Yes |
| eBL62 | 4-34*01 | 5 | 2 | 26-30 (87.76-89.43) | 20/10 | 14/6 | 2/1 | Yes |
| sBL4169 | 1-3*01 | 2 | 1 | 8-9 (95.43-95.89) | 9/1 | 5/0 | 3/0 | Yes |
| sBLE9 | 3-7*01 | 1 | 1 | 8 (94.84) | 8/0 | 4/4 | 1/0 | No |
| sBL4452 | 3-13*01 | 1 | 1 | 7 (96.82) | 5/2 | 3/1 | 1/0 | No |
| sBLE4 | 3-21*01 | 1 | 1 | 8 (94.94) | 6/2 | 3/3 | 2/0 | No |
| sBL4324 | 3-23*01 | 1 | 1 | 4 (98.21) | 4/1 | 2/1 | 1/0 | No |
| sBLE2 | 3-23*01 | 1 | 1 | 11 (93.04) | 9/1 | 7/2 | 1/0 | No |
| sBLE3 | 3-23*01 | 1 | 1 | 9 (94.23) | 8/1 | 6/2 | 1/0 | No |
| sBLE5 | 3-23*01 | 1 | 1 | 3 (98.10) | 4/0 | 2/1 | 1/0 | No |
| sBL2552 | 3-74*01 | 2 | 1 | 18-19 (90.88-91.93) | 16/5 | 12/5 | 2/0 | Yes |
| sBL8274 | 4-39*01 | 2 | 2 | 13-16 (93.81-94.25) | 9/5 | 3/5 | 2/0 | Yes |
| sBLE8 | 4-39*01 | 1 | 1 | 4 (97.42) | 4/0 | 3/1 | 1/0 | No |
eBL, endemic Burkitt lymphoma; NGS, next-generation sequencing; R/S, total replacement to silent mutation ratio; sBL, sporadic Burkitt lymphoma;
VD-D, very dissimilar/dissimilar: (– – –) o (+ – –) o (– + –) o (– – +); VS-S, very similar/similar: ( ++ +) o ( ++ –) o (– ++) o (+ – +).
Figure 2.

Comparison of next-generation sequencing (NGS) analysis and GeneScan analysis in endemic Burkitt lymphoma (eBL) and sporadic Burkitt lymphoma (sBL). In NGS analysis for eBL (A) and sBL (B) and GeneScan analysis for eBL (C) and sBL (D), NGS showed the presence of multiple clusters sharing the same VDJ and CDR3 in eBL compared with sBL. E and F, sequences of clusters.
Figure 3 illustrates an example of the branching of the lymphoma clone (case eBL31) and shows that distinct subclones evolved along similar, although separate pathways, suggesting a selective pressure occurring in parallel in distinct subclones, thereby improving their BCR affinities.
Figure 3.

Evolutionary history of endemic Burkitt lymphoma (eBL) and example of clonal heterogeneity in case eBL31. In case eBL31, different clusters showing identical IGHV, IGHD, and IGHJ usage and related CDR3 regions but exhibiting different somatic mutations and sequences with different lengths for the presence of nucleotide deletion were detected by next-generation sequencing analysis. The putative progenitors are depicted with gray nuclei, and the IGHV gene rearrangement of the clonally related cells deriving from each progenitor is indicated above. Shared point mutations and acquired unique mutations are indicated above and below the line, respectively. Vertical bars depict S mutations, lollipops depict R mutations, and red squares depict nucleotide deletions.
To confirm that the presumed monoclonal clusters detected by NGS represented the major tumor cell population, we carried out Sanger sequencing of PCR products in 37 BL cases (26 eBL and 11 sBL samples) and analyzed the results. The VDJ identity of the presumed monoclonal cluster detected by NGS always corresponded to that inferred with Sanger direct sequencing.
In addition, we performed cloning of the PCR products and sequenced at least 20 clones per case. With this method, we were able to identify at least 69 different clones with the same VDJ and CDR3 region out of 150 detected by NGS (Table 4). This result can obviously be explained by the much larger amount of sequence data generated by NGS. In fact, cloning of the PCR products confirmed only the clones composed by the highest number of reads in NGS.
Targeting of Somatic Hypermutation
All eBL cases carried IGHV genes of highly mutated status with a germline identity ranging from 79.81% to 97.26% (median, 90.54%); in sBL cases, the mutational variation compared with the germline ranged from 90.88% to 97.42% (median, 94.6%). The eBL specimens demonstrated the presence of somatic mutations in the range of five to 42 mutations. In contrast, in sBL, the range of mutations was from three to 19 (Table 4). Evaluation of the range of somatic mutations disclosed a significant difference between the two subgroups (eBL and sBL) (P< .0001).
Total mutations in eBL cases were 583 (mean, 22 mutations per case), with a significant prevalence of replacement mutations (417/583), which were more than twice that of the silent mutations (166/583) (P < .0001). The total number of mutations in sBL was 100 (mean, nine mutations/case), with a slight prevalence of replacement mutations (82/100) over silent mutations (18/100) (Table 4).
In most of the eBL cases, the majority of mutations occurred in FRs rather than CDRs, leading to a complementary-determining region (CDR)/FR ratio greater than 0.3, which would indicate antigen selection. In addition, significant accumulation of replacement as opposed to silent mutations was found in the CDRs, which form the antigen binding sites.27
Our results showed that replacement mutations most often determine substitutions of amino acids with dissimilar/very dissimilar physicochemical properties rather than similar/very similar properties, according to the standardized IMGT amino acid classes (Table 4).20,25 Based on these results, it could be suggested that somatic hypermutation in BL induces conformational changes that allow the spatial structure of the antigen binding site to better contact antigens, thus improving affinity.28
The pattern of amino acid substitutions was similar in both eBL and sBL, but in sBL, the low number of replacement mutations does not allow any significant conclusion.
Analysis of IghV Gene Usage and CDR3 Features
Clonal and in-frame VH gene sequences from BL cases derived from VH1, VH3, and VH4 families. In particular, in eBL, the comparison between the clonal VH sequences and the known germline revealed that 12 (46%) of 26 sequences were most homologous to the VH3 family (VH3-7, VH3-9, VH3-11, VH3-15, VH3-23, VH3-30, VH3-72, VH3-74) and, to a lesser extent, to the VH1 family (10/26, 38%) (VH1-2, VH1-3, VH1-8, VH1-69). The remaining four were most closely related to the VH4 family (VH4-34). In sBL, the usage of the three families was similar to that found in eBL (VH1 was found in one of 11 cases, VH3 in eight of 11 cases, and VH4 in two of 11 cases).
These results enabled us to test whether the cells from BL were derived randomly from the V gene repertoire expressed in normal peripheral blood lymphocytes or whether they exhibited a biased V gene usage. To this end, we compared the VH gene usage of this B-cell population with the rearrangements of normal peripheral B lymphocytes (PBLs).29 Our results showed that in BL cases, the VH1,VH3, and VH4 family genes were significantly overrepresented compared with PBLs (P < .05). In addition, when we looked at the individual genes, we found a preferential expression of VH3-23 (9/37, 24%), the more represented region in memory B cells, and VH1-69 (6/37, 16%) and VH4-34 (6/37, 16%). This IGHV repertoire usage is remarkably biased in comparison to the IGHV repertoire of normal B cells, where VH3-23, VH1-69, and VH4-34 are used by 12.1%, 1.4%, and 3.9%, respectively (IGHV repertoire in eBL and sBL, respectively) Figure 4.29
Figure 4.

IGHV gene usage analysis in endemic Burkitt lymphoma (eBL) (A) and sporadic Burkitt lymphoma (sBL) (B). IGHV gene usage analysis showed that eBL has a preferential usage of the VH3-23, VH4-34, and VH1-69 family gene and that sBL has a preferential usage of the VH3-23 family gene.
CDR3 from all cases was analyzed both at the nucleotide and amino acid levels. No identical or similar sequences were identified when comparing the sequences from one case with those of any other case.
The average VH CDR3 length of BL cells was similar to that of germinal center and marginal zone B cells (amino acid, 14.16 vs 14.75 and 15.01, respectively).
We determined the IGHD and IGHJ genes used by the IGHV-D-J sequences analyzed. The IGHD genes used were IGHD3 (48%) and IGHD2 (16%) and several others to a much lower extent, including IGHD1, IGHD4, IGHD5, and IGHD6. In particular, we demonstrated a bias toward IGHD3-10 (25%), which was considered statistically significant compared with the PBLs (P < .05).29,30 The IGHJ gene usage in all cases showed that IGHJ4 (56%) was used the most, followed by IGHJ3 (16%), IGHJ6 (16%), IGHJ5 (8%), and IGHJ1 and IGHJ2 (4%). In sBL, no significant difference in the distribution between the D and J segment usage has been found.
Discussion
The World Health Organization classification of BL describes three clinical variants: endemic, sporadic, and immunodeficiency related. These types are similar in morphology, immunophenotype, and genetic features. The endemic variant is associated with malaria, and Epstein-Barr virus (EBV) is found in almost all cases. The sporadic type occurs mainly throughout the rest of the world (predominantly North America and Europe) and is less frequently associated with EBV infection.13
It has been questioned whether the different BL subtypes, being characterized by peculiar epidemiologic and clinical features, may also differ in cellular biology and pathogenetic mechanisms. Interestingly, GEP studies have demonstrated that eBL and human immunodeficiency virus–BL had almost an identical molecular profile, whereas sBL cases were relatively more different.14 Differences between eBL and sBL in significant pathways such as BCR, tumor necrosis factor/nuclear factor κB, and interleukin-dependent intracellular cascades seem to reflect the different clinical contexts.12
Intriguingly, recent studies using NGS disclosed the importance of the BCR pathway in the pathogenesis of BL. In fact, mutations affecting the transcription factor TCF3 or its negative regulator ID3 were reported in around 70% of sporadic BL cases. This results in the activation of a tonic form of BCR signaling and increased expression of genes belonging to the BCR pathway.9-11
However, it should be considered that the tonic form of BCR is antigen independent and seems to be in contrast with the epidemiologic context of chronic antigen stimulation in which eBL arises.
Since no definitive data are available on the mutations of TCF3 and ID3 for eBL, the present study was aimed to assess the frequency of these mutations in eBL and to make a comparison with sBL. Overall, eight (30.7%) of 26 eBL cases presented nonsense or missense mutations, six of 26 in ID3, three of 26 in TCF3, and one in both genes. Of note, this rate of mutation was significantly lower than that found in our sBL cases (P = .0300) and those previously reported.9-11 Therefore, our results seem to suggest that the intrinsic activation of BCR, due to mutation of TCF3/ID3, is not the more relevant pathogenetic mechanism in eBL. However, as the genetic landscape of eBL has not yet extensively studied, it is conceivable that many more genes belonging to the Wnt/β-catenin/TCF3/ID3 pathway may be also involved in eBL pathogenesis.
To answer whether an activation of BCR through extrinsic antigen-driven signaling, due to the presence of multiple pathogens,31 could be present in eBL, we analyzed SHM status of BCR by NGS of immunoglobulin genes. NGS analysis revealed intraclonal diversity, suggesting an active targeted SHM process in eBL compared with sBL. In fact, NGS analysis showed the presence of multiple clusters sharing the same VDJ and CDR3 in eBL. All these clusters carried also shared unique somatic mutations in eBL, indicating that these distinct subclones had expanded under the selective pressure of antigens, thereby fine-tuning their BCR affinities.
The probability of excess or scarcity of replacement mutations in CDR and FR regions, calculated by a multimodal distribution model, showed signs of antigen selection mostly in eBL.27 Remarkably, in all cases, we found that replacement mutations most often determine substitutions of amino acids with dissimilar/very dissimilar physicochemical properties rather than similar/very similar, suggesting an important remodeling of immunoglobulin structure that may increase affinity under antigenic pressure.28,32
In addition, most of the VH clusters did not have any mutations, deletions, or insertions that would prevent or limit translation. These findings led to the suggestion that maintenance of immunoglobulin expression and dependency on BCR may also extend to malignant cells. It is tempting to speculate that the maintenance of immunoglobulin expression and immunoglobulin-mediated clonal selection may have a similar bias since our IGHV gene usage analysis showed that BL cases have a preferential usage of the VH3-23, VH4-34, and VH1-69 genes. In this context, it is worthy of note that the VH4-34 gene encodes for the intrinsically autoreactive antibody, suggesting that autoreactive antigens could also be involved in BL pathogenesis. In addition, during acute EBV and cytomegalovirus infections, increased titers of IGHV4-34 antibodies are detected, and patients with CLL infected with EBV also have IGHV4-34 gene overrepresentation.33-35 However, important unanswered questions still remain concerning whether the VH gene repertoires of individuals living in endemic areas are skewed compared with those of individuals living in nonendemic areas and whether the African VH gene repertoires resemble non-African VH gene repertoires.
In conclusion, our data suggest a strong antigenic pressure that could potentially be related to prolonged microenvironment interactions, by which chronic stimulation of BCR with certain pathogens could be important in promoting clonal expansion of B cells expressing distinctive BCRs and growth of the neoplastic clone. For eBL, this can be related to the fact that chronic Plasmodium falciparum malaria stimulation continues to reactivate latently EBV-infected memory B cells.34-37 Yet, whether coincidental infections with additional pathogens may occur in eBL remains to be addressed. On the other hand, in sBL, the progressive acquisition of mutations in the TCF3/ID3 genes results in intrinsic tonic activation of BCR signaling.38 Therefore, neoplastic cells may grow in a way that is independent of EBV and/or other antigens.39,40 However, we cannot exclude a combining of tonic and extrinsic BCR signaling activation in a subset of BL.
Our conclusions are also supported by an elegant experimental model in which stimulation of B cells by antigen in the presence of overexpressed Myc in engineered mice gave rise to BL-like tumors that were in turn dependent on both Myc and the antigen for survival and proliferation.1 Interestingly, the growth of these murine BL could be inhibited by treatment with immunosuppressants (cyclophosphamide and cyclosporine). Intriguingly, complete remission has been achieved in patients with stage 1 to 2 BL only with cyclophosphamide treatment.41
Deciphering the pathway of BCR activation in BL may be useful to identify new therapeutic approaches since cases strongly dependent on extrinsic antigenic signaling appear to be sensitive to pathway inhibitors targeting PI3K and BTK, while cases that depend on tonic signaling are not sensitive to BTK inhibitors.42 Successful strategies to eliminate tumors will need to identify and target multiple oncogenic pathways operating in the tumor, creating a genetic bottleneck too narrow for the tumor’s evolutionary escape.
Acknowledgments
This work was supported by MIUR grant number 20104HBZ8E_004.Conflict of interest: none declared.
References
- 1.Refaeli Y, Young RM, Turner BC, et al. The B cell antigen receptor and overexpression of Myc can cooperate in the genesis of B cell lymphomas. PLoS Biol. 2008;6:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossi D, Ciardullo C, Gaidano G. Genetic aberrations of signaling pathway in lymphomagenesis: revelations from next generation sequencing studies. Semin Cancer Biol. 2013;23:422-430. [DOI] [PubMed] [Google Scholar]
- 3.Carbone A, Gloghini A. Relationships between lymphomas linked to hepatitis C virus infection and their microenvironment. World J Gastroenterol. 2013;19:7874-7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira MI, Medeiros JA. Role of Helicobacter pylori in gastric mucosa–associated lymphoid tissue lymphomas. World J Gastroenterol. 2014;20:684-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pannekoek Y, van der Ende A. Chlamydia psittaci infection in nongastrointestinal MALT lymphomas and their precursor lesions. Am J Clin Pathol. 2011;136:480. [DOI] [PubMed] [Google Scholar]
- 6.Carugi A, Onnis A, Antonicelli G, et al. Geographic variation and environmental conditions as cofactors in Chlamydia psittaci association with ocular adnexal lymphomas: a comparison between Italian and African samples. Hematol Oncol. 2010;28:20-26. [DOI] [PubMed] [Google Scholar]
- 7.Sutton LA, Agathangelidis A, Belessi C, et al. Antigen selection in B-cell lymphomas—tracing the evidence. Semin Cancer Biol. 2013;23P:399-409. [DOI] [PubMed] [Google Scholar]
- 8.Davis RE, Hgo VN, Lenz G, et al. Chronic active B cell receptor signaling in diffuse large B cell lymphoma. Nature. 2010;463:88-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter J, Schlesner M, Hoffmann S, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcription sequencing. Nature Gen. 2012;44:1316-1322. [DOI] [PubMed] [Google Scholar]
- 10.Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Bosch CA. Is endemic Burkitt's lymphoma an alliance between three infections and a tumour promoter? Lancet Oncol. 2004;5:738-746. [DOI] [PubMed] [Google Scholar]
- 13.Leoncini L, Raphael M, Stein H, et al. Burkitt lymphoma In: Swerdlow SH, Campo E, Harris NL, et al. , eds. WHO Classification of Tumours of Haematopietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2008:262-264. [Google Scholar]
- 14.Piccaluga PP, De Falco G, Kustagi M, et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood. 2011;117:3596-3608. [DOI] [PubMed] [Google Scholar]
- 15.Trifonov V, Pasqualucci L, Tiacci E, et al. SAVI: a statistical algorithm for variant frequency identification. BMC Syst Biol. 2013;7(suppl 2):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gebauer N, Bernard V, Feller AC, et al. ID3 mutations are recurrent events in double-hit B-cell lymphomas. Anticancer Res. 2013;33:4771-4778. [PubMed] [Google Scholar]
- 17.van Dongen JJ, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257-2317. [DOI] [PubMed] [Google Scholar]
- 18.Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson JD1, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chapter 2:Unit 2.3. [DOI] [PubMed] [Google Scholar]
- 20.Lefranc MP. IMGT databases, web resources and tools for immunoglobulin and T cell receptor sequence analysis, http://imgt.cines.fr. Leukemia. 2003;17:260-266. [DOI] [PubMed] [Google Scholar]
- 21.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT® tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569-604. [DOI] [PubMed] [Google Scholar]
- 22.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840-1847. [PubMed] [Google Scholar]
- 23.Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848-1854. [PubMed] [Google Scholar]
- 24.Zibellini S, Capello D, Forconi F, et al. Stereotyped patterns of B-cell receptor in splenic marginal zone lymphoma. Haematologica. 2010;95:1792-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pommié C, Levadoux S, Sabatier R, et al. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J Mol Recognit. 2004;17:17-32. [DOI] [PubMed] [Google Scholar]
- 26.Schumacher JA, Duncavage EJ, Mosbruger TL, et al. A comparison of deep sequencing of TCRG rearrangements vs traditional capillary electrophoresis for assessment of clonality in T-cell lymphoproliferative disorders. Am J Clin Pathol. 2014;141:348-359. [DOI] [PubMed] [Google Scholar]
- 27.Lossos IS, Tibshirani R, Narasimhan B, et al. The inference of antigen selection on Ig genes. J Immunol. 2000;165:5122-5126. [DOI] [PubMed] [Google Scholar]
- 28.Burkovitz A, Sela-Culang I, Ofran Y. Large scale analysis of somatic hypermutations in antibodies reveals which structural regions, positions and amino acids are modified to improve affinity. FEBS J. 2014;281:306-319. [DOI] [PubMed] [Google Scholar]
- 29.Brezinschek HP, Foster SJ, Brezinschek RI, et al. Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on human peripheral CD5(+)/IgM+ and CD5(–)/IgM+ B cells. J Clin Invest. 1997;99:2488-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnaout R, Lee W, Cahill P, et al. High-resolution description of antibody heavy-chain repertoires in humans. PLoS One. 2011;6:e22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt's lymphoma: a polymicrobial disease? Nat Rev Microbiol. 2005;3:182-187. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz GW, Hershberg U. Germline amino acid diversity in B cell receptors is a good predictor of somatic selection pressures. Front Immunol. 2013;4:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baptista MJ, Calpe E, Fernandez E, et al. Analysis of the IGHV region in Burkitt's lymphomas supports a germinal center origin and a role for superantigens in lymphomagenesis. Leuk Res. 2014;38:509-515. [DOI] [PubMed] [Google Scholar]
- 34.Pugh-Bernard AE, Silverman GJ, Cappione AJ, et al. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest 2001;108:1061-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kostareli E, Hadzidimitriou A, Stavroyianni N, et al. Molecular evidence for EBV and CMV persistence in a subset of patients with chronic lymphocytic leukemia expressing stereotyped IGHV4-34B-cell receptors. Leukemia. 2009;23:919-924. [DOI] [PubMed] [Google Scholar]
- 36.Chene A, Donati D, Orem J, et al. Endemic Burkitt's lymphoma as a polymicrobial disease: new insights on the interaction between Plasmodium falciparum and Epstein-Barr virus. Semin Cancer Biol. 2009;19:411-420. [DOI] [PubMed] [Google Scholar]
- 37.Moormann AM, Snider CJ, Chelimo K. The company malaria keeps: how co-infection with Epstein-Barr virus leads to endemic Burkitt lymphoma. Curr Opin Infect Dis. 2011;24:435-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolcetti R, Dal Col J, Martorelli D, et al. Interplay among viral antigens, cellular pathways and tumor microenvironment in the pathogenesis of EBV-driven lymphomas. Semin Cancer Biol. 2013;23:441-456. [DOI] [PubMed] [Google Scholar]
- 39.Dominguez-Sola D, Dalla-Favera R. Burkitt lymphoma: much more than MYC. Cancer Cell. 2012;22:141-142. [DOI] [PubMed] [Google Scholar]
- 40.Vereide D, Sugden B. Proof for EBV's sustaining role in Burkitt's lymphomas. Semin Cancer Biol. 2009;19:389-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Traoré F, Coze C, Atteby JJ, et al. Cyclophosphamide monotherapy in children with Burkitt lymphoma: a study from the French-African Pediatric Oncology Group (GFAOP). Pediatr Blood Cancer. 2011;56:70-76. [DOI] [PubMed] [Google Scholar]
- 42.Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013;13:578-591. [DOI] [PubMed] [Google Scholar]