

Abstract
In Alzheimer’s disease, microglia cluster around β-amyloid deposits, suggesting that these cells are important for amyloid plaque formation, maintenance and/or clearance. We crossed two distinct APP transgenic mouse strains with CD11b-HSVTK mice, in which nearly complete ablation of microglia was achieved for up to 4 weeks after ganciclovir application. Neither amyloid plaque formation and maintenance nor amyloid-associated neuritic dystrophy depended on the presence of microglia.
Introduction
CD11b-HSVTK (TK) mice express the thymidine kinase of herpes simplex virus under the CD11b (also known as ITGAM) promoter. HSVTK is a suicide gene that converts antiviral nucleotides analog prodrugs such as ganciclovir (GCV) into a monophosphorylated form, which is then transformed into a toxic triphosphate by cellular kinases. Thus, GCV treatment leads to selective ablation of proliferating myeloid cells1. TK mice were bred with APPPS1 transgenic mice, which develop robust amyloid pathology that is accompanied by a strong microglia activation as early as 2 months of age2. To restrict HSVTK expression in APPPS1; TK mice to resident microglia and to overcome the reported GCV-mediated myelotoxicity1, we generated chimeric mice harboring congenic GFP-labeled wild-type bone marrow (Supplementary Methods). At 5 months, we treated bone marrow chimeric APPPS1; TK and APPPS1 (negative littermates without the CD11b-HSVTK gene) mice with oral GCV for 4 weeks. This treatment caused a ~30% decrease in microglia number in the neocortex (and most other brain regions) in APPPS1; TK mice compared with APPPS1 control mice (Supplementary Fig. 1). Despite the decrease in microglia number we found no change in the morphology and number of congophilic or Aβ-immunoreactive amyloid deposits (Supplementary Fig. 1). Consequently, no change in transgenic human Aβ40 or Aβ42 was detected using western blotting and ELISA (Supplementary Fig. 1).
To test whether a more complete microglial ablation induces a change in cerebral amyloidosis in APPPS1 mice, we used several alternative GCV administration protocols in TK mice. The most successful approach was to deliver GCV directly into the ventricle (intracerebroventricular, icv) via an osmotic pump (50 mg ml−1, 0.25 μl h−1 flow rate). In contrast with systemic GCV administration, this icv application benefited from the fact that adoptive bone-marrow transfer appeared to be dispensable. We found a 40% reduction in the number of Iba1-positive microglia in the neocortex and in other brain regions after 1 week of icv GCV administration and a 90% reduction after 2 weeks (Supplementary Fig. 2). Concomitant with the ablation of microglia, we observed a temporary activation of astrocytes (Supplementary Fig. 2). We found no apparent change in the number or morphology of neurons and no alteration in endogenous murine Aβ40 and Aβ42 levels in TK mice lacking microglia (Supplementary Fig. 2).
To investigate the effect of a complete microglial ablation on cerebral amyloidosis, we icv GCV-treated 3-month-old amyloid-bearing APPPS1; TK and APPPS1 mice. GCV administration for 2 weeks resulted in a 95% reduction of the number of Iba-1 positive microglia (Fig. 1a–c). GVC treatment for 4 weeks, which allowed us to study plaque homeostasis in the absence of microglia, caused a 97% reduction in the number of microglia in GCV-treated APPPS1; TK mice compared with APPPS1 mice. Qualitatively similar ablation was observed using CD11b, F4/80 or CD68 as microglial markers (data not shown). The efficacy of microglia ablation was confirmed by conventional ultrastructural analysis (detection of microglia was independent of the expression of activation markers) and by in vivo multiphoton imaging. Both techniques showed at least a 90% reduction in the microglial cell number (Supplementary Fig. 3).
Figure 1.
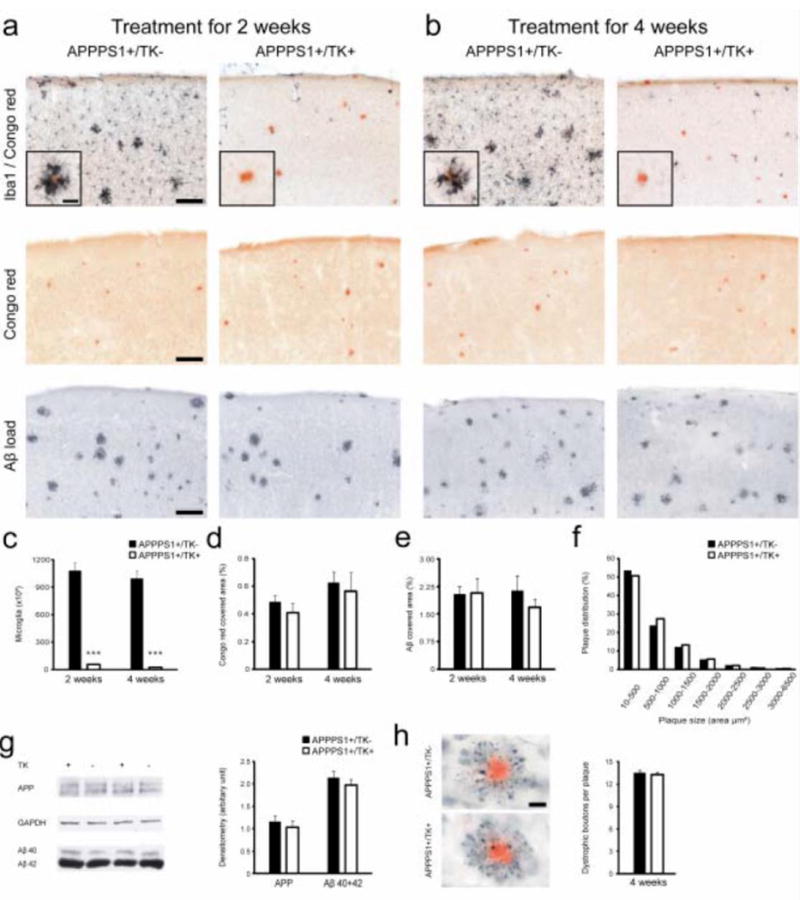
(a,b) Male 3-month-old amyloid-bearing APPPS1; TK and APPPS1 mice were treated with icv GCV for 2 or 4 weeks. Double staining for Iba1 and congophilic amyloid (top), congophilic amyloid alone (middle) and Aβ immunohistochemistry (bottom) are shown. (c) Quantitative stereological analysis of the number of Iba-1 positive microglia (n = 5 per group, *** P < 0.001). (d,e) No change in congophilic (d) or total Aβ load (e) was noted in GCV-treated APPPS1; TK mice compared with GCV-treated APPPS1 mice. (f) Plaque size distribution did not change after 4 weeks of GCV. (g) For western blotting, male APPPS1; TK mice were icv GCV-treated for 3 weeks. Two mice of each genotype are shown with GAPDH as a loading control. Densitometric analysis of all mice (n = 4–5 per group) revealed no difference in human APP and Aβ (combined Aβ40 and Aβ42) between microglia-depleted APPPS1; TK and control APPPS1 mice. Aβ ELISA of the soluble fraction revealed a 3–4-fold increase in Aβ40 (P < 0.05) and Aβ42 (P = 0.05) in microglia-deficient APPPS1; TK mice (data not shown); however, this may represent an artifact resulting from an increased release of ‘insoluble’ Aβ into the soluble fraction in the absence of microglia cells. (h) APP-immunoreactive dystrophic boutons (black) surrounding congophilic amyloid plaques (red) appeared to be unchanged in number and morphology in APPPS1; TK mice lacking microglia compared with APPPS1 mice after 4 weeks of GCV treatment (n = 5 per group, P > 0.05). Error bars represent s.e.m. Scale bars represent 100 μm (a,b), 25 μm (inserts) and 20 μm (h).
This virtually complete ablation of microglia in APPPS1; TK mice did not change congophilic amyloid load, the distribution and extent of Aβ-deposition, or plaque morphology after 2 and 4 weeks of GCV treatment (Fig. 1a–e). Plaque size distribution analysis also revealed no differences in APPPS1; TK mice harboring or lacking microglia (Fig. 1f). There was no change in human APP expression and Aβ levels in the brains of APPPS1; TK mice versus APPPS1 mice (Fig. 1g). We also found no difference between microglia-deficient APPPS1; TK mice and APPPS1 mice in terms of number and morphology of dystrophic neuritic structures near amyloid plaques (Fig. 1h), consistent with our ultrastructural observations (Supplementary Fig. 3).
APPPS1 mice start to develop cerebral amyloidosis in the neocortex at 2 months of age. Even at the earliest stages of Aβ deposition, microglia consistently surround the amyloid plaques2. To determine the extent to which microglial cells are involved with or are even the cause of Aβ deposition and plaque formation, we icv GCV-treated 6-week-old APPPS1; TK mice for 3 weeks. Again, the majority of microglia (95%) were gone. However, ablation of microglia neither prevented plaque formation nor influenced the number of plaques developing in the neocortex (Fig. 2a–e). The number and morphology of the dystrophic neuritic structures associated with congophilic amyloid deposits were also independent of microglial presence (Fig. 2f).
Figure 2.
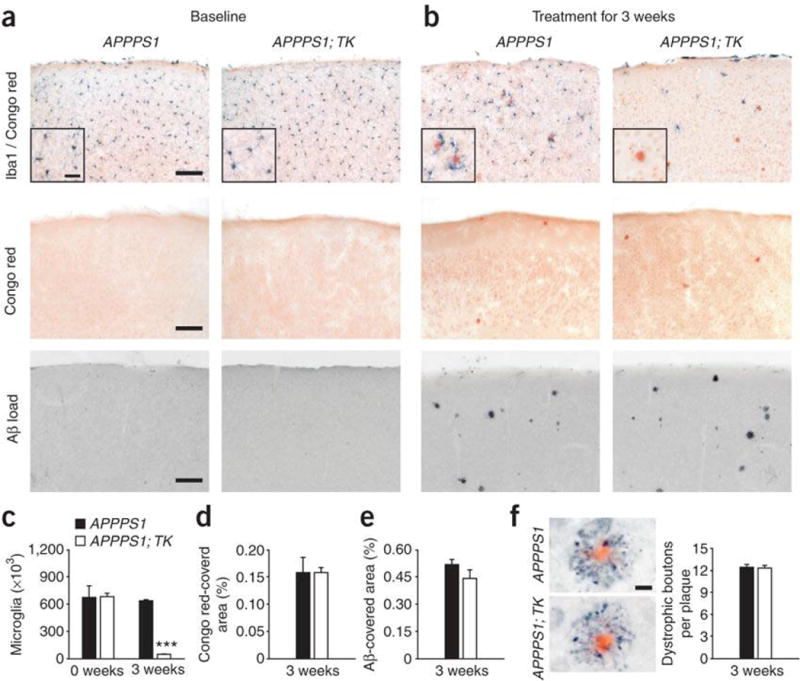
(a) Numbers of Iba1-positive microglia in 6-week-old male APPPS1; TK and APPPS1 mice did not differ. The mice did not exhibit amyloid plaque deposition. Double staining for microglia (Iba1) and compact congophilic amyloid (top panels), congophilic amyloid alone (middle panels) and Aβ immunohistochemistry (bottom panels) are shown. (b) Both APPPS1; TK and APPPS1 mice develop amyloid plaques irrespective of microglia 3 weeks after icv GCV treatment. (c) Quantitative stereological analysis of total microglia revealed identical numbers of cells at baseline (0 weeks) in both genotypes and a 95% reduction in microglia 3 weeks after GCV treatment in APPPS1; TK mice compared with APPPS1 mice (n = 5 per group, *** P < 0.001). (d,e) No group difference in congophilic amyloid and Aβ load was seen after GCV treatment. (f) APP-immunoreactive dystrophic boutons (black) surrounding congophilic amyloid plaques (red) were indistinguishable in number and morphology in APPPS1; TK mice lacking microglia and APPPS1 control mice 3 weeks after GCV application (n = 5 per group, P > 0.05). Error bars represent s.e.m. Scale bars represent 100 μm (a), 25 μm (inserts) and 20 μm (f).
The icv GCV treatment of APPPS1; TK mice for 3–4 weeks regularly resulted in microhemorrhages, which occurred predominately in the thalamic region. This microbleeding seemed to be the probable cause of the lethal phenotype of the TK mice after 4 weeks of icv GCV and may have also facilitated the influx of small conglomerates of Iba1-positive bone marrow–derived macrophages at that time (data not shown). To avoid these effects, which may be caused by myelotoxicity of GCV leaking from the CNS into the periphery1, we treated APPPS1; TK mice with a lower dose of GCV (1 mg ml−1, 0.25 μl h−1 for 4 weeks). We found no behavioral abnormalities in these GCV-treated mice. This lower dose only partially ablated microglia in the neocortex. However, because of its anatomical location close to the infusion site and the ventricles, we observed an almost complete depletion of microglia in the dorsal part of the hippocampus, without signs of hemorrhages and in the absence of invading peripheral cells (Supplementary Fig. 4). Again, no change in the Aβ load and congophilic amyloid plaques was found (Supplementary Fig. 4).
To exclude the possibility that the rather aggressive amyloidosis in APPPS1 mice is accountable for the absence of changes on microglia removal, we crossed a second APP transgenic mouse strain, APP23, with the TK mice. APP23 mice develop cerebral amyloidosis at 6–8 months of age and have both dense cored and diffuse plaques, as well as vascular amyloid3. As in APPPS1 mice, congophilic amyloid lesions in APP23 mice are tightly surrounded by activated microglia4. We gave 17-month-old APP23; TK and APP23 mice icv GCV (50 mg ml−1, 0.25 μl h−1) for 2 weeks. The efficacy of microglial ablation again reached >95% in the neocortex (Supplementary Fig. 5). Although there was no change in congophilic amyloid burden and in vascular amyloid, there was a small reduction in total Aβ load, mainly a result of the reduction of diffuse amyloid, which is known to be abundant in this Alzheimer’s disease mouse model (Supplementary Fig. 5). The number and morphology of dystrophic neuritic structures associated with the congophilic amyloid deposits were unaltered in microglia-deficient APP23; TK compared with APP23 mice (Supplementary Fig. 5). A second group of APP23; TK mice (24 months of age) received low-dose icv GCV treatment (1 mg ml−1, 0.25 μl h−1) for 4 weeks. Microglia reduction was again confined to the dorsal hippocampus, with no obvious change in congophilic amyloid load (data not shown).
Here, we sought to clarify the controversy over the role of microglial cells in cerebral amyloidosis and Alzheimer’s disease pathogenesis5. Our results indicate that a substantial reduction or a virtually complete ablation of resident microglia (including bone marrow–derived microglia) did not alter amyloid plaque load in two distinct APP transgenic mouse models over a period of 2–4 weeks. These observations extend recent in vivo multiphoton imaging work6, suggesting that resident microglia are not important in the de novo formation of amyloid. However, in contrast with work speculating that microglia (or bone marrow–derived macrophages7, 8) may have a function in restricting plaque growth6, 9 and with compelling in vitro data on Aβ clearance and phagocytosis capability of microglia5, our results indicate that de novo formation and progression of cerebral amyloidosis in vivo in Alzheimer’s disease mouse models are controlled by nonmicroglial factors or cells other than microglia. Consistent with this, true β-amyloid phagocytotic activity of microglia and macrophages (engulfment of amyloid fibrils with the later appearance of fibrils in the lysosomal-phagosomal compartments) has not been observed in Alzheimer’s disease brains or in brains of Alzheimer’s disease mouse models10. Likewise, for peripheral amyloid, there is no undeniable evidence that macrophages phagocytose amyloid (for example, see ref. 11).
Multiple lines of evidence suggest that exogenous or genetic manipulation of factors involved in innate immune signaling or in neuroinflammatory responses, including chemokine and cytokine secretion presumably mediated by microglia, can affect amyloid deposition in APP transgenic mice5. Similarly, Aβ vaccination as targeted modulation of the immune system has been shown to prevent or to reduce cerebral amyloidosis12. As it is of therapeutic importance to understand whether and how a microglia transforms into a potentially Aβ/amyloid clearing cell, our approach of fully ablating microglia in vivo will be useful for identifying microglia-dependent or microglia-independent Aβ-clearing mechanisms.
Microglial activation adjacent to amyloid deposits has also been used to suggest that microglia-mediated neuroinflammatory responses mediate Alzheimer’s disease–associated neurodegeneration5, 13. In particular, it has been argued that cytokines, chemokines and neurotoxins generated by amyloid-activated microglia cause neuronal damage13. Even if not all aspects of Alzheimer’s disease pathology, such as severe neurodegeneration, are entirely mimicked by APP transgenic mice, our data argue against substantial microglia-driven neuritic damage occurring in Alzheimer’s disease, as neural dystrophy developed and appeared to be unaltered in the absence of microglia in the Alzheimer’s disease mouse models that we examined.
Finally, our experiments suggest that microglia can be virtually fully removed from specific brain regions of a living organism for up to 4 weeks. This is rather surprising in light of their highly dynamic nature under resting condition and their presumed role in surveying the brain, in repairing clinically silent micro-insults and in monitoring the functional state of synapses14, 15. To generalize this observation, it will be necessary to extend the time span of complete microglia ablation in the presence or absence of CNS pathology.
Supplementary Material
Acknowledgments
This work was supported by grants to M.J. (BMBF-01GI0705), F.L.H. (SFB-TR43, Exc 25 and National Institutes of Neurological Disorders and Stroke R01 NS046006) and P.M.M. (US National Institutes of Health AG017617 and NS045205).
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
Author Contributions
S.A.G., T.B., S.A.K., J.O., R.R., T.E., P.M.M. and H.W. performed the icv experiments. R.E.K., S.P. and G.W. carried out the oral treatment experiments. A.A., M.S. and S.G. provided mice and/or advice. Experimental design and manuscript preparation were mainly carried out by F.L.H. and M.J.
References
- 1.Heppner FL, et al. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 2.Radde R, et al. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sturchler-Pierrat C, et al. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stalder M, et al. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wyss-Coray T. Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 6.Meyer-Luehmann M, et al. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Town T, et al. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolmont T, et al. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jucker M, Heppner FL. Neuron. 2008;59:8–10. doi: 10.1016/j.neuron.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruys E, Timmermans HJ, van Ederen AM. Lab Anim. 1979;13:1–9. doi: 10.1258/002367779781071230. [DOI] [PubMed] [Google Scholar]
- 12.Schenk D. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- 13.El Khoury J, Luster AD. Trends Pharmacol Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Nimmerjahn A, Kirchhoff F, Helmchen F. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 15.Hanisch UK, Kettenmann H. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.