

Abstract
Ependymal tumors across age groups are currently classified and graded solely by histopathology. It is, however, commonly accepted that this classification scheme has limited clinical utility based on its lack of reproducibility in predicting patients’ outcome. We aimed at establishing a uniform molecular classification using DNA methylation profiling. Nine molecular subgroups were identified in a large cohort of 500 tumors, 3 in each anatomical compartment of the CNS, spine, posterior fossa, supratentorial. Two supratentorial subgroups are characterized by prototypic fusion genes involving RELA and YAP1, respectively. Regarding clinical associations, the molecular classification proposed herein outperforms the current histopathological classification and thus might serve as a basis for the next World Health Organization classification of CNS tumors.
Graphical Abstract
INTRODUCTION
Ependymal tumors are neuroepithelial malignancies of the CNS that occur in both children and adults (Korshunov et al., 2010). These tumors can arise along the entire neuroaxis comprising the hemispheres, the hindbrain, and the spinal cord (Louis et al., 2007). In children, 90% of ependymomas (EPNs) occur intracranially, with two-thirds being located in the posterior fossa (PF) and one-third within the supratentorial (ST) compartment (Kilday et al., 2009). Across all age groups, more than 20% of primary spinal cord tumors are of ependymal lineage (Ostrom et al., 2014). The clinical behavior of ependymal tumors is highly variable, and approximately 40% of patients are incurable because of the paucity of effective treatment options available (Gajjar et al., 2014; Korshunov et al., 2010; Merchant et al., 2009; Ostrom et al., 2014). The 10-year overall survival (OS) is about 64% in pediatric patients and ranges from 70% to 89% in adult patients (Ostrom et al., 2014). Tumors in infancy are associated with a particularly poor survival rate of only 42%– 55% at 5 years after diagnosis (Gatta et al., 2014). The extent of surgical resection has for a very long time been the only clinical prognostic marker in EPN known to be associated with survival (Merchant et al., 2009). The current standard of care includes maximal safe surgical resection, followed by focal radiotherapy (Merchant et al., 2009). Nevertheless, a number of reports indicate that a subset of patients with radically resected ST tumors will not recur even without adjuvant therapy (Venkatramani et al., 2012), exemplifying the need for better patient stratification. Furthermore, although adjuvant chemotherapy is still part of many trial protocols, especially in young children in an attempt to avoid or delay radiation therapy, multiple clinical trials have failed to show a survival benefit from adding chemotherapy either at the time of primary diagnosis or at recurrence (Bouffet and Foreman, 1999; Bouffet et al., 2009).
Accurate histopathological diagnosis according to the World Health Organization (WHO) classification for CNS tumors (Louis et al., 2007) is challenging for ependymal tumors. In particular, distinction between grade II EPNs and grade III anaplastic EPNs is often difficult, with poor interobserver reproducibility, even if performed by the most experienced neuropathologists (Ellison et al., 2011; Tihan et al., 2008). Grade I EPNs, i.e., myxopapillary EPNs (MPEs) (occurring in the spine) and subependymomas (SEs) (occurring across all compartments), generally have more readily distinguishable histopathological characteristics. However, complicating the grading of EPNs is the fact that many tumors show isolated areas each representing distinct grades, resulting in the challenge of predicting which component of the tumor will influence the overall biologic behavior.
Despite histopathological similarities among variants of EPN at different anatomical sites, its molecular biology is heterogeneous, with distinct genetic and epigenetic alterations as well as diverse transcriptional programs (Carter et al., 2002; Dyer et al., 2002; Korshunov et al., 2010; Mack et al., 2014; Mendrzyk et al., 2006; Parker et al., 2014; Wani et al., 2012; Witt et al., 2011). Functional cross-species studies provide evidence that these molecular differences reflect regionally discrete cells of origin (Johnson et al., 2010; Parker et al., 2014; Taylor et al., 2005). An association between neurofibromatosis type 2 (i.e., hereditary germline mutations of the NF2 gene), as well as sporadic mutations in NF2, has long been known as a hallmark genetic aberration of spinal EPN (Ebert et al., 1999; Rubio et al., 1994). Other single markers, including immunohistochemistry-based markers, have thus far failed to adequately reflect this biological heterogeneity and cannot reliably distinguish between histological grades and subgroups of EPNs. The single molecular marker that has repeatedly shown an association with unfavorable outcome is gain of chromosome arm 1q (Godfraind et al., 2012; Kilday et al., 2012; Korshunov et al., 2010; Mendrzyk et al., 2006; Modena et al., 2012), particularly in PF EPNs of childhood. Homozygous deletion of the CDKN2A/B locus is associated with inferior prognosis, mainly in ST tumors (Korshunov et al., 2010).
Only recently, large-scale genomic and epigenomic studies have revealed the first driver genes in ST EPNs. Fusions between RELA, which encodes an NF-kB component, and the poorlycharacterized gene C11orf95 caused by a local chromosome shattering event (chromothripsis) on chromosome 11 were seen in >70% of ST EPNs (Parker et al., 2014). Strikingly, this fusion alone is sufficient to drive tumorigenesis when aberrantly expressed in neural stem cells (Parker et al., 2014). For PF EPNs, two distinct molecular subgroups were consistently identified in two independent studies using different methods and non-over-lapping patient cohorts (Wani et al., 2012; Witt et al., 2011). These subgroups (provisionally termed PF Group A and Group B, or PFA and PFB) are associated with distinct transcriptomic, genetic, epigenetic, and clinical features and as such are much more informative than WHO grading alone (Archer and Pomeroy, 2011).
The current study aimed to establish a uniform molecular classification of all ependymal tumors that adequately reflects the full biological, clinical, and histopathological heterogeneity across the major anatomical compartments, age groups, and grades.
RESULTS
DNA Methylation Profiling of Ependymal Tumors Identifies Nine Distinct Molecular Subgroups
There is a growing body of evidence that DNA methylation patterns of tumor cells, specifically of promotor regions, represent a very stable molecular memory of the respective cell of origin throughout the disease course (Hoadley et al., 2014; Hovestadt et al., 2014), thus making this assessment particularly suitable for tumor classification. To attempt such a classification for ependymal tumors across all age groups and anatomical compartments, we generated genome-wide DNA methylation profiles for 500 ependymal tumors using the Illumina 450k methylation array. Clinical parameters of the entire cohort are presented in Figure S1 and Table 1.
Table 1.
Overall Comparison of Clinical Data Associated with the Nine Molecular Subgroups of Ependymal Tumors
| SP- |
PF- |
ST- |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular Subgroup | SE | MPE | EPN | SE | EPN-PFA | EPN-PFB | SE | EPN-YAP | EPN-RELA | |||||||||
| Number of patients | 7 | 26 | 21 | 33 | 240 | 51 | 21 | 13 | 88 | |||||||||
|
| ||||||||||||||||||
| Age (Years) | ||||||||||||||||||
|
| ||||||||||||||||||
| Range | 22–68 | 9–66 | 11–59 | 39–76 | 0–51 | 10–65 | 25–70 | 0–51 | 0–69 | |||||||||
|
| ||||||||||||||||||
| Median | 49 | 32 | 41 | 59 | 3 | 30 | 40 | 1.4 | 8 | |||||||||
|
| ||||||||||||||||||
| >18 | 7 | 100% | 19 | 95% | 20 | 95% | 29 | 100% | 2 | 1% | 38 | 81% | 13 | 100% | 1 | 8% | 20 | 24% |
|
| ||||||||||||||||||
| 4—8 | 0 | 0% | 1 | 5% | 1 | 5% | 0 | 0% | 93 | 41% | 9 | 19% | 0 | 0% | 4 | 30% | 44 | 52% |
|
| ||||||||||||||||||
| <4 | 0 | 0% | 0 | 0% | 0 | 0% | 0 | 0% | 131 | 58% | 0 | 0% | 0 | 0% | 8 | 62% | 20 | 24% |
|
| ||||||||||||||||||
| NA | 0 | 6 | 0 | 4 | 14 | 4 | 8 | 0 | 4 | |||||||||
|
| ||||||||||||||||||
| Gender | ||||||||||||||||||
|
| ||||||||||||||||||
| Male | 3 | 43% | 14 | 54% | 12 | 63% | 25 | 76% | 154 | 65% | 21 | 41% | 15 | 71% | 3 | 25% | 57 | 65% |
|
| ||||||||||||||||||
| Female | 4 | 57% | 12 | 46% | 7 | 37% | 8 | 24% | 84 | 35% | 30 | 59% | 6 | 29% | 9 | 75% | 31 | 35% |
|
| ||||||||||||||||||
| NA | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 1 | 0 | |||||||||
|
| ||||||||||||||||||
| Ratio (male:female) | 1:1 | 1:1 | 1.7:1 | 3:1 | 1.8:1 | 0.7:1 | 2.5:1 | 0.3:1 | 1.8:1 | |||||||||
|
| ||||||||||||||||||
| Localization | ||||||||||||||||||
|
| ||||||||||||||||||
| PF | 0 | 0% | 0 | 0% | 0 | 0% | 29 | 100% | 231 | 99% | 49 | 98% | 0 | 0% | 1 | 8% | 4 | 5% |
|
| ||||||||||||||||||
| ST | 0 | 0% | 1 | 6% | 1 | 5% | 0 | 0% | 2 | 1% | 1 | 2% | 12 | 100% | 12 | 92% | 82 | 95% |
|
| ||||||||||||||||||
| SP | 7 | 100% | 17 | 94% | 19 | 95% | 0 | 0% | 0 | 0% | 0 | 0% | 0 | 0% | 0 | 0% | 0 | 0% |
|
| ||||||||||||||||||
| NA | 0 | 8 | 1 | 4 | 7 | 1 | 9 | 0 | 2 | |||||||||
|
| ||||||||||||||||||
| Histologic Grade | ||||||||||||||||||
|
| ||||||||||||||||||
| I | 5 | 71% | 20 | 83% | 2 | 10% | 21 | 66% | 0 | 0% | 0 | 0% | 14 | 67% | 0 | 0% | 0 | 0% |
|
| ||||||||||||||||||
| II | 2 | 29% | 4 | 17% | 17 | 85% | 10 | 31% | 69 | 30% | 29 | 59% | 7 | 33% | 5 | 42% | 20 | 23% |
|
| ||||||||||||||||||
| III | 0 | 0% | 0 | 0% | 1 | 5% | 1 | 3% | 160 | 70% | 20 | 41% | 0 | 0% | 7 | 58% | 66 | 77% |
|
| ||||||||||||||||||
| NA | 0 | 2 | 1 | 1 | 11 | 2 | 0 | 1 | 2 | |||||||||
|
| ||||||||||||||||||
| Level of Resection | ||||||||||||||||||
|
| ||||||||||||||||||
| Complete | 3 | 60% | 1 | 100% | 5 | 71% | 5 | 71% | 116 | 53% | 22 | 50% | 3 | 60% | 8 | 73% | 41 | 65% |
|
| ||||||||||||||||||
| Incomplete | 2 | 40% | 0 | 0% | 2 | 29% | 2 | 29% | 102 | 47% | 22 | 50% | 2 | 40% | 3 | 27% | 22 | 35% |
|
| ||||||||||||||||||
| NA | 2 | 25 | 14 | 26 | 22 | 7 | 16 | 2 | 25 | |||||||||
|
| ||||||||||||||||||
| Radiotherapy | ||||||||||||||||||
|
| ||||||||||||||||||
| Yes | 0 | 0% | 0 | 0% | 0 | 0% | 2 | 14% | 158 | 73% | 40 | 85% | 1 | 11% | 5 | 50% | 54 | 74% |
|
| ||||||||||||||||||
| No | 0 | 0% | 2 | 100% | 8 | 100% | 12 | 86% | 59 | 27% | 7 | 15% | 8 | 89% | 5 | 50% | 19 | 26% |
|
| ||||||||||||||||||
| NA | 7 | 24 | 13 | 19 | 23 | 4 | 12 | 3 | 15 | |||||||||
|
| ||||||||||||||||||
| Chemotherapy | ||||||||||||||||||
|
| ||||||||||||||||||
| Yes | 0 | 0% | 0 | 0% | 0 | 0% | 0 | 0% | 163 | 75% | 3 | 6% | 0 | 0% | 7 | 70% | 26 | 36% |
|
| ||||||||||||||||||
| No | 0 | 0% | 1 | 100% | 8 | 100% | 7 | 100% | 54 | 25% | 44 | 94% | 6 | 100% | 3 | 30% | 46 | 64% |
|
| ||||||||||||||||||
| NA | 7 | 25 | 13 | 26 | 23 | 4 | 15 | 3 | 16 | |||||||||
|
| ||||||||||||||||||
| Progression | ||||||||||||||||||
|
| ||||||||||||||||||
| Yes | 0 | 0% | 1 | 50% | 2 | 22% | 3 | 33% | 135 | 61% | 14 | 30% | 0 | 0% | 2 | 20% | 45 | 57% |
|
| ||||||||||||||||||
| No | 5 | 100% | 1 | 50% | 7 | 78% | 6 | 67% | 87 | 39% | 33 | 70% | 6 | 100% | 8 | 80% | 34 | 43% |
|
| ||||||||||||||||||
| NA | 2 | 24 | 12 | 24 | 18 | 4 | 15 | 3 | 9 | |||||||||
|
| ||||||||||||||||||
| Death | ||||||||||||||||||
|
| ||||||||||||||||||
| Yes | 0 | 0% | 0 | 0% | 0 | 0% | 0 | 0% | 60 | 27% | 2 | 4% | 0 | 0% | 0 | 0% | 19 | 25% |
|
| ||||||||||||||||||
| No | 5 | 100% | 1 | 100% | 9 | 100% | 7 | 100% | 159 | 73% | 44 | 96% | 6 | 100% | 10 | 100% | 57 | 75% |
|
| ||||||||||||||||||
| NA | 2 | 25 | 12 | 26 | 21 | 5 | 15 | 3 | 12 | |||||||||
|
| ||||||||||||||||||
| Survival probability (%) | ||||||||||||||||||
|
| ||||||||||||||||||
| 5-year PFS | 100 | 50 | 88 | 83 | 33 | 73 | 100 | 66 | 29 | |||||||||
|
| ||||||||||||||||||
| 5-year OS | 100 | 100 | 100 | 100 | 68 | 100 | 100 | 100 | 75 | |||||||||
|
| ||||||||||||||||||
| 10-year PFS | 100 | NA | NA | NA | 24 | 56 | 100 | NA | 19 | |||||||||
|
| ||||||||||||||||||
| 10-year OS | 100 | NA | NA | NA | 56 | 88 | 100 | NA | 49 | |||||||||
See also Figure S1.
Unsupervised hierarchical clustering of the DNA methylation data using 10,000 probes with highest SD across the entire dataset identified nine distinct subgroups of ependymal tumors, three within each major compartment of the CNS: spine (SP), PF, or ST (Figure 1A). Cluster analyses using different numbers of probes (5,000, 20,000, or 50,000) gave the same results (data not shown). Subgroup stability was further confirmed by bootstrap analysis (bootstrap probability 0.999; p = 0.001). Based on associations with anatomical location, histology, and genetic alterations, as outlined below, we annotated these nine subgroups as SP-SE (n = 7 samples), SP-MPE (n = 26), SP-EPN (n = 21), PF-SE (n = 33), PF-EPN-A (n = 240), PF-EPN-B (n = 51), ST-SE (n = 21), ST-EPN-YAP1 (n = 13), and ST-EPN-RELA (n = 88). One of the subgroups within each compartment was enriched with grade I SEs, and we therefore labeled them as SP-SE, PF-SE, and ST-SE. Interestingly, some grade II EPNs, confirmed after pathology re-review, also clustered with these SE subgroups (Figures 1A and S2A). We then turned our attention to the non-SE-like subgroups (a total of six) within each of the three major anatomical compartments. Within the spinal compartment, SP-MPE and SP-EPN showed a relatively good concordance with the histopathological subtypes MPE (grade I) and EPN (grade II) (Figure 1A). However, for the two remaining PF and ST subgroups, we found no concordance with histological grading. Within the PF, the two non-SE-like subgroups represented previously described Group A and Group B EPNs of the PF (Mack et al., 2014; Wani et al., 2012; Witt et al., 2011), which for standardization purposes we termed PF-EPN-A and PF-EPN-B. In the ST compartment, we suspected that the largest subgroup probably represented EPNs with recurrent C11orf95-RELA fusions, as described by Parker et al. (2014). Indeed, when we performed RNA sequencing and/or targeted analysis for the two major types of C11orf95-RELA fusion transcripts (Figure S2B; Table S1), we found these in 22 of 25 (88%) ST-EPN-RELA EPNs tested for RELA fusion types 1 and 2, suggesting that the majority of cases in this subgroup harbor these specific types of fusions. Interestingly, in one case of the ST-EPN-RELA subgroup, negative for RELA fusion types 1 and 2, we detected a PTEN-TAS2R1 fusion leading to a frame shift and subsequent disruption of PTEN (Table S1). Loss of PTEN expression/activity has previously been demonstrated to induce NF-kB activity through activation of Akt/ mTOR (Dan et al., 2008), suggesting that alternative mechanisms may exist that lead to activation of this key pathway in the ST-EPN-RELA subgroup. We did not systematically test for other less frequent fusions involving RELA as described by Parker et al. (2014) (RELA fusion types 3-7), which likely account for the majority of RELA fusion type 1 and 2 negative cases within the ST-EPN-RELA subgroup.
Figure 1. Methylation Profiling Identifies the Existence of Nine Distinct Epigenetic Subgroups of Ependymal Tumors.

(A) Heat-map representation of an unsupervised clustering of DNA methylation profiles of 500 ependymal tumors. Each row represents a probe; each column represents a sample. The level of DNA methylation (beta value) is represented with a color scale as depicted. For each sample, subgroup association, anatomical location, histopathological diagnosis, and patient age are indicated.
(B) Heat map of methylation levels in primary ependymal tumors and corresponding recurrent diseases. Molecular subgroup, anatomical location, histopathological diagnosis, and tumor type are indicated. Equal numbers at top of dendrogram and arrows at the bottom indicate that samples derive from the same individual.
Molecular Subgroups Remain Stable at Relapse
To evaluate further whether molecular subgroup classification remains stable at disease recurrence, we performed hierarchical clustering of DNA methylation profiles of 45 primary ependymal tumors and their corresponding recurrent tumor specimens. In addition to primary tumors representing SP-MPE, PF-SE, PF-EPN-A, PF-EPN-B, or ST-EPN-RELA subgroups, the cohort comprised of 48 tumors from the time of first or second recurrence. Notably, inspection of cluster groups relative to patient identification number (1–45) showed that all recurrent ependymal tumors clustered without exception directly next to their corresponding primaries or very close to them, in all cases within the same subgroup that was attributed to the primary tumor (Figure 1B). These results strongly suggest that DNA methylation profiling remains a reliable tool for subgroup identification not only at initial diagnosis, but also at the time of recurrence.
YAP1 Gene Fusions Define a Distinct Subgroup of ST EPNs
As RELA fusions were identified exclusively in the ST-RELA-EPN subgroup, we next asked which genetic alterations might underlie the other ST EPN subgroup in children, especially as we identified several cases in this subgroup (9 of 13) with copy number aberrations on chromosome 11 around the YAP1 locus (Figure 2A). Parker et al. (2014) had already identified YAP1 fusions in two ST EPN samples that were negative for a RELA fusion. Therefore, it was tempting to speculate that these cases might represent a separate molecular subgroup of ST EPN. To test this hypothesis, we performed RNA sequencing for 7 of the 13 cases that clustered with this subgroup and for which sufficient high-quality RNA was available. Consistent with our findings on the DNA copy-number level, we detected the previously identified YAP1-MAMLD1 fusion (Parker et al., 2014) in six of seven tumors and another gene fusion containing YAP1 and an uncharacterized gene, FAM118B, in the remaining tumor (Figures 2B and 2C). All YAP1 fusions identified by RNA sequencing could be verified by Sanger sequencing (Figure 2B). In the YAP1-MAMLD1 fusions, exons 1–5 or 1–6 (out of 9) of YAP1 (according to reference sequence GenBank: NM_001130145) are fused in frame to exons 2–7 or 3–7 of MAMLD1 (according to reference sequence GenBank: NM_005491). In the other fusion, YAP1 exons 1–7 are fused in frame to the entire coding region encoded by exon 3–9 of FAM118B (according to reference sequence GenBank: NM_024556). In both fusion proteins, most of the N-terminal domains of the YAP1 protein are retained, and for MAMLD1, most of the C-terminal part is retained (Figure 2C). Importantly, no single YAP1 fusion was identified by RNA sequencing in any of 48 additional ependymal tumors of the other molecular subgroups of the PF or ST region. This demonstrates that YAP1 fusion-positive cases characterize another molecular subgroup of ST EPNs, annotated as ST-EPN-YAP1, that are distinct from RELA fusion-positive ST EPNs.
Figure 2. Recurrent YAP1 Fusion Transcripts in a Distinct ST Subgroup.

(A) Copy-number variation plots of four ST tumors classified as ST-EPN-YAP1 based on DNA methylation. Red rectangular marks chromosomal arm 11q highlighting existence of global and/or focal aberrations. Sample identifications are indicated.
(B) Electropherograms of the fusion transcripts detected in the tumor samples depicted in (A). Numbers flanking the indicated breakpoint (dashed line) represent upstream and downstream fusion sites.
(C) Predicted YAP1 fusion products at protein level. Red dashed lines indicate fusion sites. Proteins are drawn to scale, and protein domains are indicated at the bottom.
Molecular Subgroups Show Distinct Copy Number Profiles
Genome-wide DNA copy number profiles, generated by using the combined intensity values of the methylation probes (Hovestadt et al., 2013), showed strong differences in numbers and patterns of DNA copy number alterations (CNAs) between the molecular subgroups (Figure 3). Strikingly, gene amplifications, frequently seen in other brain tumors like medulloblastoma and glioblastoma, were almost totally absent in ependymal tumors. The same is true for homozygous gene deletions with the exception of CDKN2A deletions, which were commonly found in the ST-EPN-RELA subgroup (14 of 88; 16%), but not in other subgroups (p < 0.001). This subgroup also shows frequent loss of the entire chromosome 9 or chromosomal arm 9p (46 of 88; 52%), which is not seen in any other subgroup (p < 0.001). Most CNAs across all subgroups involved gains or losses of whole chromosomes indicating aneuploidy. Gains and losses of chromosomal arms were much less frequent. Interestingly, ST-EPN-RELA tumors, but not tumors from the other eight subgroups, frequently displayed dramatic copy number changes, reminiscent of chromothripsis (38 of 88; 43%), i.e., rearrangements of entire chromosomes or chromosomal arms with alternating copy number states (Korbel and Campbell, 2013; Parker et al., 2014; Rausch et al., 2012). Chromosome 11, where the C11orf95 and RELA genes are located, was always involved in these events, but sometimes other chromosomes were affected as well, yet to a much lesser extent (Figures 3 and S3; Table S2). Since Parker et al. (2014) extensively demonstrated based on whole-genome sequencing data that C11orf95-RELA fusions resulted from chromothripsis of chromosome 11q, we inferred that the observed massive fragmentations of chromosome 11q in ST-EPN-RELA tumors are based on this phenomenon. No chromothripsis was detected in tumors classified as ST-EPN-YAP1, even though YAP1 is also located on chromosome 11. Instead, these cases frequently displayed focal CNAs around the YAP1 locus (Figure 2A). Surprisingly, besides the CNAs on chromosome 11, the remainder of the genome is relatively stable in most ST-EPN-YAP1 tumors, which is in contrast to the ST-EPN-RELA EPNs that typically show an abundance of CNAs. Predominantly stable genomes were also seen in SEs of all three anatomical compartments (SP-SE, PF-SE, ST-SE). As expected, this was also true for the PF-EPN-A subgroup. The only CNA that was frequently observed in both SP-SE and PF-SE, but not ST-SE, was a complete or partial loss of chromosome 6, in line with previous findings (Kurian et al., 2008). Loss of chromosome 6 is also frequently seen in PF-EPN-B tumors, where it is the most frequent CNA (42 of 51 tumors, 82%), and to a lesser extent in SP-EPN, PF-EPN-A, and ST-EPN-RELA tumors. For PF-EPN-A tumors, the most frequent CNA observed was gain of 1q (60 of 240; 25%), which was also seen in PF-EPN-B (9 of 51; 18%), and ST-EPN-RELA tumors (21 of 88; 24%). Many more CNAs are seen in both SP-MPE and SP-EPN, with the most frequent event being loss of 22q in SP-EPN tumors (19 of 21; 90% show loss of 22q). This frequent involvement of the 22q locus is not surprising as it includes NF2, which is known to be frequently mutated in spinal EPNs (Ebert et al., 1999; Slavc et al., 1995). However, loss of 22q is not entirely restricted to SP-EPN, as it is also seen at lower frequencies in other subgroups (Figure 3). It remains to be seen whether NF2 also plays a role in these subgroups or whether other genes on 22q are involved. Finally, aside from ST-EPN-RELA tumors displaying chromothripsis, tumors in the PF-EPN-B subgroup show by far the highest degree of genomic instability, with many gains and losses of entire chromosomes or (less frequent) chromosomal arms in each tumor (Figure 3). Altogether, these data show that ependymal tumors of the nine molecular subgroups indeed represent genetically highly distinct entities.
Figure 3. Molecular Subgroups of Ependymal Tumors Show Distinct Copy-Number Profiles.

Overview of chromosomal aberrations in the nine molecular subgroups of ependymal tumors. DNA methylation array-based copy-number variation plots were scored for loss (red), gain (green), no change (= balanced; gray), or chromothripsis (purple) for all chromosomal arms. Additional focal aberrations were scored for chromosome 11q only (blue). Results were plotted as frequencies at which these aberrations occurred within each molecular subgroup; p values on the right indicate whether there was a significant difference in the distribution of these frequencies across the nine subgroups (chi-square test).
Transcription Profiles Reveal Distinct Druggable Pathways in Molecular Subgroups
To investigate whether (epi)genetically defined molecular subgroups of ependymal tumors also show subgroup-specific transcriptional differences, we generated gene expression profiles for all tumor samples, for which sufficient high-quality RNA was available. This resulted in 209 gene expression profiles generated on the Affymetrix U133 Plus2.0 array. Unfortunately, ependymal tumors of the SP-SE subgroup were not included because no RNAs were available. Unsupervised hierarchical cluster analysis of the gene expression data almost perfectly recapitulated the molecular subgroups derived by DNA methylation profiling (Figure S4). To identify genes that are specifically (over)expressed in only one of the subgroups, we performed supervised analyses by comparing gene expression patterns of each subgroup against all other subgroups. This resulted in the identification of multiple genes that are either exclusively expressed or at least highly overex pressed specifically in one of the subgroups. A heat map showing the expression of these signature genes across all molecular subgroups is displayed in Figure 4A. In addition, gene expression patterns of two representative genes per subgroup are displayed (Figure 4B). The full list of signature genes (Table S3) provides a first hint of the underlying biology, pathway activation, and potential drug targets in the distinct molecular subgroups. For instance, KIT, targetable by several multikinase inhibitors, is only expressed at high levels in PF-SE tumors and not in other subgroups (Figure 4B). Another example is RELA, the transcription factor involved in activating the NF-kB pathway, which is only expressed in ST-EPN-RELA tumors (Parker et al., 2014). In order to get an even better insight in the biological processes and pathways that play a role in each of these subgroups, we performed gene-ranked pathway enrichment analysis with g:Profiler (Reimand et al., 2011) and used Cytoscape Enrichment Map to visualize processes and pathways that distinguished the different molecular subgroups or anatomical compartments (Figure 4C). Gene sets were compiled from the Gene Ontology (GO) biological processes, as well as Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, and CORUM databases of pathways and protein complexes. Significant gene sets (false discovery rate [FDR] < 0.05, p < 0.01) (Table S3) were visualized as interaction networks (Figure 4C). Interestingly, while several gene sets involved in brain development clearly play a role in all subgroups, as expected, other gene sets were specific to certain subgroups. For instance, cell cycle genes, cell migration genes or genes involved in MAPK signaling were most active in PF-EPN-A tumors and ST-EPN-RELA tumors, while cAMP/carbohydrate metabolism or dopamine signaling genes were more active in ST-EPN-YAP1 tumors. Ciliogenesis genes were exclusively found in PF-EPN-B tumors. The clear differences in active biological processes and pathways identified between the various molecular subgroups of ependymal tumors, in line with previous data for the PF subgroups (Witt et al., 2011), further support their distinct origins and may suggest possible avenues for future subgroup-specific targeted therapies.
Figure 4. Transcription Profiles of Ependymal Tumors Reveal Subgroup-Specific Gene Signatures and Pathways.

(A) Heat-map representation of signature genes across the molecular subgroups of ependymal tumors generated from supervised gene expression analyses. Subgroup affiliation of samples (column) and signatures (row) is indicated by color codes. SP-SE subgroup cases were not included due to unavailability of RNA.
(B) Relative expression levels of two representative signature genes per subgroup as compared with the other subgroups are shown as box plots. Box plots represent the interquartile range (IQR), with the median represented by a solid line.
(C) Pathway enrichment analysis comparing each of the molecular subgroups SP-MPE (dark green), SP-EPN (yellow), PF-SE (deep pink), PF-EPN-A (orange), PF-EPN-B (blue), ST-SE (dark violet), ST-EPN-YAP1 (cyan), and ST-EPN-RELA (red) against all other subgroups and a collection of normal brain control samples. Distinct pathways and biological processes between the molecular subgroups of ependymal tumors are illustrated (FDR corrected p < 0.01). Nodes represent enriched gene sets, which are grouped and annotated by their similarity according to related gene sets. Node size is proportional to the total number of genes within each gene set. The illustrated network map was simplified by manual curation to remove general and uninformative sub-networks. SP-SE samples were not included due to lack of RNA.
Molecular Subgroups Correlate with Clinicopathological Variables
The nine molecular subgroups of ependymal tumors described herein were closely associated with specific age groups (Figures 1 and S1; Table 1). All three SE-like subgroups were exclusively found in adults (18 years and older, range 22–76), with median ages of 49 years (SP-SE), 59 years (PF-SE), and 40 years (ST-SE), respectively. The other two SP subgroups, SP-MPE and SP-EPN, were also mainly found in adults, but some occurred in children (SP-MPE median age 32 years, range 9–66; SP-EPN median age 41 years, range 11–59). Patients in the PF-EPN-B subgroup were found mostly in the adolescent and young adult populations (median age 30 years, range 10–65). In contrast, tumors in the other PF subgroup, PF-EPN-A, were almost exclusively found in young children (median age 3 years, range 0–51). Only two patients in this subgroup were older than 18. The remaining two ST subgroups, ST-EPN-YAP1 and ST-EPN-RELA, were much more common in children (ST-EPN-YAP1 median age 1.4 years, range 0–51; ST-EPN-RELA median age 8 years, range 0–69), but a significant portion (23%) of the ST-EPN-RELA tumors was also found in adults. Gender distribution in our cohort revealed a preponderance of males over females (1.6:1). The gender distribution of all subtypes is presented in Table 1, with some significant differences between subgroups (p = 0.004). For example, among PF tumors, there was a preponderance of males represented in the PF-EPN-A (male:female = 1.8:1) and PF-SE (male:female = 3:1) subgroups, whereas in the PF-EPN-B subgroup, males were in the minority (male:female = 0.7:1).
Risk Stratification by Molecular Subgrouping Is Superior to Histopathological Grading
Survival analyses for all patients with available outcome data (n = 388) showed remarkable differences in OS and progression-free survival (PFS) between the molecular subgroups (Figure 5). Both the PF-EPN-A and ST-EPN-RELA subgroups show a dismal outcome, with 10-year OS rates of rv50% and PFS rates of rv20%. Notably, patients comprising these subgroups are mostly pediatric. All other subgroups have a much better outcome, with 5-year OS rates around 100% and 10-year OS rates ranging from 88%–100% (Table 1). It is well known that patients with ependymal tumors within the spinal region can usually be cured by complete neurosurgical resection alone. In our study, the number of spinal tumors was not sufficient to derive clinically meaningful conclusions, but the favorable OS rates (OS 100% for all three subgroups) are consistent with published literature (McGuire et al., 2009) (Figure S5). As one example for the clinical utility of molecular subgrouping over conventional histopathology, the SE-like molecular subgroups from the PF and ST compartments all showed favorable outcomes on OS analysis. Since a substantial proportion of molecular SEs was classified as grade II and even grade III EPN (Figures 1A and S2; Table 1), molecular classification in these instances reveals tumor subgroups with favorable outcome that would not be yielded by histopathological analysis. Notably, albeit OS rates are excellent for PF-SE and ST-SE, only SE tumors from the PF show frequent progression (Figure 5). This clinical course is reminiscent of PF-EPN-B tumors, which cluster close to the PF-SE subgroup, suggesting also some biological similarities between these two subgroups. Examining other clinical variables, for either compartment-specific or subgroup-specific prognostic value, showed that extent of resection was associated with OS in PF-EPN-A (p = 0.01) and with PFS in both PF-EPN-A (p = 0.0004) and PF-EPN-B (p = 0.019) (Figure S6A). Other clinical variables that showed prognostic value in univariate analyses across all subgroups included age (outcome of children is worse than adults) and chemotherapy (patients who received chemotherapy did worse than patients who did not receive chemotherapy; Table S4). However, both these factors were strongly biased by the molecular subgrouping as the two subgroups that showed the worst outcome, PF-EPN-A and ST-EPN-RELA, were also the subgroups that were mainly pediatric and in which most patients received chemotherapy as compared with the other subgroups. Within each of these subgroups, however, patients who received chemotherapy showed no significant survival improvement compared with patients who did not receive chemotherapy (data not shown). WHO grading and radiotherapy had no prognostic value in these univariate analyses, neither within anatomical compartments nor within molecular subgroups (Figure S6B; Table S4). In line with previous reports (Godfraind et al., 2012; Korshunov et al., 2010), gain of chromosome 1q also showed strong prognostic value in the present series. However, a highly significant difference in OS between tumors with and without gain of 1q was only seen for the PF-EPN-A tumors (p = 0.00001; Figure S6C), while no significant differences in OS were seen for PF-EPN-B or ST-EPNRELA tumors, even though all three subgroups displayed similar frequencies of 1q gain (PF-EPN-A: 25%; PF-EPN-B: 18%; ST-EPN-RELA: 24%; Figure 3). Surprisingly, however, 1q gain showed a strong prognostic association regarding PFS not only in PF-EPN-A but also in PF-EPN-B tumors, but not in the ST-EPN-RELA subgroup (PF-EPN-A: p = 0.0068; PF-EPN-B: p = 0.0075; Figure S6C).
Figure 5. Molecular Subgroups of Ependymal Tumors Correlate with Distinct Clinical Outcome.

(A–D) Kaplan-Meier curves for overall (A and C) and progression-free (B and D) survival for infratentorial (A and B) and ST (C and D) molecular ependymal tumor subgroups defined by methylation profiling. The p values were computed by log rank tests between subgroups. Numbers of patients at risk are indicated. See also Figure S5.
Finally, we performed a multivariate Cox regression analysis to see whether molecular subgrouping, level of resection, and 1q gain are independent prognostic parameters for ependymal tumors. For comparison, we also included the clinical variables WHO grade and radiotherapy, even though they showed no prognostic value in the univariate analyses (Figure S6; Table S4). Results presented in Table 2 show that molecular subgrouping, level of resection, and 1q gain are all independent prognostic parameters for both OS and PFS. Additional parameters examined, including age and chemotherapy, did not have independent prognostic value in multivariate analysis. In addition, a likelihood-ratio test was performed to compare the full model including all variables with a multivariate Cox model that does not include molecular subgrouping. The resulting p values were p = 0.039 for OS and p = 0.012 for PFS, indicating that adding molecular subgrouping significantly improved the model fit. In contrast, comparing the full model with a model that omits WHO grading led to non-significant p values for OS (p = 0.79) and PFS (p = 0.56), indicating that WHO grading did not improve the model when other variables were already included (Table 2).
Table 2.
Multivariate Cox Proportional Hazard Models for Overall and PFS of Molecular Subgroups of Ependymal Tumors
| Variable | Hazard Ratioa | 95% CI | p Valueb |
|---|---|---|---|
|
| |||
| OS | |||
| 1q gain (yes versus no) | 2.44 | 1.49–4 | <0.0001 |
|
| |||
| WHO III versus WHO II | 1.06 | 0.61–1.82 | 0.827 |
|
| |||
| Age, years (<4 versus >18) | 0.46 | 0.14–1.53 | 0.21 |
|
| |||
| Age, years (4–18 versus >18) | 0.63 | 0.20–1.95 | 0.429 |
|
| |||
| Resection (STR versus GTR) | 1.79 | 1.11–2.90 | 0.017 |
|
| |||
| Chemotherapy (yes versus no) | 1.44 | 0.81–2.56 | 0.205 |
|
| |||
| Radiotherapy (yes versus no) | 0.81 | 0.43–1.52 | 0.528 |
|
| |||
| PF-SE versus PF-EPN-B | 1.45 | 0.06–33.87 | 0.814 |
|
| |||
| PF-EPN-A versus PF-EPN-B | 6.65 | 1.35–32.56 | 0.019 |
|
| |||
| ST-SE versus PF-EPN-B | 1.89 | 0.08–44.09 | 0.691 |
|
| |||
| ST-EPN-YAP versus PF-EPN-B | 1.97 | 0.08–46.31 | 0.673 |
|
| |||
| ST-EPN-RELPOS versus PF-EPN-B | 6.22 | 1.32–29.27 | 0.021 |
|
| |||
| Likelihood Ratio Test | |||
|
| |||
| Full model versus model without methylation subgroups (OS) | 0.03 | ||
|
| |||
| Full model versus model without WHO (OS) | 0.79 | ||
|
| |||
| PFS | |||
|
| |||
| 1q gain (yes versus no) | 1.79 | 1.27–2.52 | 0.001 |
|
| |||
| WHO III versus WHO II | 0.89 | 0.63–1.27 | 0.547 |
|
| |||
| Age, years (< 4 versus > 18) | 1.18 | 0.55–2.53 | 0.665 |
|
| |||
| Age, years (4-18 versus > 18) | 1.39 | 0.68–2.83 | 0.364 |
|
| |||
| Resection (STR versus GTR) | 1.79 | 1.31–2.45 | <0.0001 |
|
| |||
| Chemotherapy (yes versus no) | 0.93 | 0.64–1.34 | 0.715 |
|
| |||
| Radiotherapy (yes versus no) | 0.75 | 0.50–1.10 | 0.149 |
|
| |||
| PF-SE versus PF-EPN-B | 1.23 | 0.30–5.024 | 0.771 |
|
| |||
| PF-EPN-A versus PF-EPN-B | 2.50 | 1.13–5.56 | 0.024 |
|
| |||
| ST-SE versus PF-EPN-B | 0.29 | 0.01–5.27 | 0.407 |
|
| |||
| ST-EPN-YAP versus PF-EPN-B | 0.68 | 0.11–3.95 | 0.67 |
|
| |||
| ST-EPN-RELPOS versus PF-EPN-B | 2.66 | 1.21–5.86 | 0.015 |
|
| |||
| Likelihood Ratio Test | |||
|
| |||
| Full model versus model without methylation subgroups (PFS) | 0.01 | ||
|
| |||
| Full model versus model without WHO (PFS) | 0.56 | ||
DISCUSSION
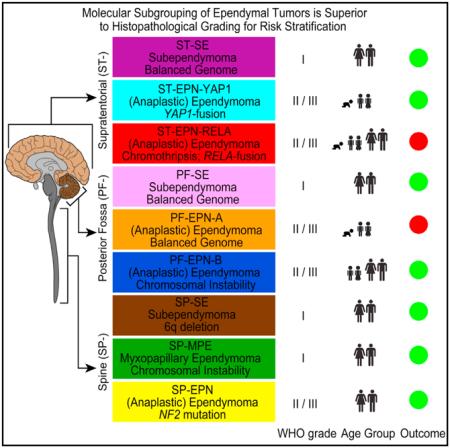
Based on genome-wide DNA methylation patterns, we identified nine distinct molecular subgroups of ependymal tumors across all age groups, three in each anatomical compartment of the CNS (SP, PF, and ST). We have shown that these molecular subgroups are genetically, epigenetically, transcriptionally, demographically, and clinically distinct. Whether they also have different cells of origin, as suggested by Johnson et al. (2010), remains to be proven and requires further functional studies, although it seems an attractive hypothesis. A robust and uniform (epi)genetic classification of ependymal tumors as presented herein may guide researchers, neuropathologists, and clinicians to a better understanding of the heterogeneity of this disease, analogous to (epi)genetic subgroups of medulloblastoma (Kool et al., 2012; Northcott et al., 2012; Taylor et al., 2012) and glioblastoma (Brennan et al., 2013; Sturm et al., 2012, 2014). Since methylation profiling can be reliably performed from very small amounts of DNA extracted from formalin-fixed and paraffin embedded tissue (Hovestadt et al., 2013), this technique lends itself to routine clinical application. Herein, we also show that molecular subgrouping remains stable throughout the course of disease, in line with previous findings for medulloblastoma (Ramaswamy et al., 2013) and anticipated from the fact that DNA methylation profiles largely reflect an epigenetic memory of the cell of origin. Molecular subgrouping may also help identifying more effective therapeutic strategies, especially for the pediatric ependymal subgroups PF-EPN-A and ST-EPN-RELA that show a dismal outcome with current treatment approaches. A graphical illustration of the key genetic and clinical features of these nine molecular subgroups of ependymal tumors is given in Figure 6.
Figure 6. Graphical Summary of Key Molecular and Clinical Characteristics of Ependymal Tumor Subgroups.

Schematic representation of key genetic and epigenetic findings in the nine molecular subgroups of ependymal tumors as identified by methylation profiling. CIN, Chromosomal instability
The nine subgroups we identified herein showed some overlap with previously identified subgroups A to I of EPN using gene expression profiling (Johnson et al., 2010). The ST subgroups C and D in that study mainly represent our ST-EPN-RELA and ST-SE subgroups, respectively. Spinal subgroup E represents our SP-MPE subgroup, whereas the mixed spinal/PF subgroup F represents our SP-EPN and PF-EPN-B subgroups, respectively. Subgroups G, H, and I all mainly represent PF-EPN-A tumors with some PF-SE tumors. No ST-EPN-YAP1 tumors are represented in the study of Johnson et al. (2010), and subgroups A and B mainly seem to contain non-EPNs. Our data, based on a much larger cohort, are able to show that ST EPNs harboring a YAP1 fusion, as first identified by Parker et al. (2014), are molecularly and clinically distinct from ST EPNs harboring a RELA fusion. In fact, seven of seven (100%) of the ST-EPN-YAP1 cases tested harbored a YAP1 fusion, with the most common fusion being YAP1-MAMLD1 identified in six of seven (86%) and another fusion (YAP1-FAM118B), identified in one of seven (14%) of the cases. We did not find YAP1 fusions in tumors of any other subgroup for which we had RNA sequencing data available. Both YAP1-MAMLD1 and YAP1-FAM118B fusions have not been reported before in any other type of tumor and the exact function of these fusions remains to be investigated. However, it is highly likely that these YAP1 fusions comprise the oncogenic drivers in this distinct subgroup of ST EPN, also because a recent report showed that high YAP1 activity is sufficient to induce embryonal rhabdomyosarcoma (Tremblay et al., 2014). Interestingly, a similar fusion between YAP1 and MAML2 has been found in nasopharyngeal carcinomas (Valouev et al., 2014). MAMLD1 and MAML2, both members of the Mastermind gene family, are transcriptional co-activators of NOTCH signaling and probably function in the respective fusion proteins as NOTCH independent co-activators of TEAD-mediated HIPPO signaling, leading to transformation and increased proliferation (Wu et al., 2002). Whether FAM118B may have similar function is unknown, but a recent report showed that silencing FAM118B expression in HELA cells resulted in decreased proliferation (Li et al., 2014).
RELA type 1 and type 2 fusions (Parker et al., 2014) were commonly found in ST-EPN-RELA tumors, but not in any other subgroup, strongly suggesting that these C11orf95-RELA fusions are the principal drivers of ST-EPN-RELA subgroup tumors. Interestingly, while homozygous CDKN2A deletions were frequently detected in ST-EPN-RELA tumors, they were not found in ST-EPN-YAP1 tumors. Previous studies showed that homozygous CDKN2A deletions are associated with a dismal outcome (Korshunov et al., 2010; Witt et al., 2011), which fits with the present results. Moreover, in ST-EPN-RELA tumors, the recurrent RELA fusion appears to be a result of heavily rearranged genomes, in many cases in conjunction with a chromothripsis event affecting chromosome 11. In contrast, ST-EPN-YAP1 tumors never showed evidence of chromothripsis and had relatively stable genomes, with the only recurrent rearrangements affecting the locus of YAP1. What exactly is causing chromothripsis and why it is specific to ST-EPN-RELA tumors and absent in the other subgroups is unknown and requires further investigation. It is plausible that CDKN2A has a role in chromothripsis, as it affects the TP53 pathway, which has been shown to be involved in chromothripsis in other entities (Rausch et al., 2012), but we found no direct correlation between chromothripsis and focal CDKN2A deletions. In our cohort, TP53 was not sequenced, but it is known that TP53 mutations are extremely rare in EPN (Gaspar et al., 2006; Ohgaki et al., 1993).
The only other molecular subgroup for which a recurrent genetic mutation is known is the SP-EPN subgroup (Figure 6). In this group of ependymal tumors, occurring in the spinal column and affecting predominantly adults, NF2 is frequently mutated (Ebert et al., 1999; Slavc et al., 1995), and almost all tumors in this subgroup (19 of 21; 90%) showed monozygosity of 22q. Loss of 22q was also observed in several other intracranial molecular subgroups, but in these subgroups, it is unlikely that NF2 is the target, as NF2 is not frequently mutated in intracranial ependymal tumors (Singh et al., 2002). In fact, recent sequencing studies have not found a single recurrent mutation in PF EPNs or any other recurrent mutation outside the recurrent C11orf95-RELA gene fusions in ST EPNs (Mack et al., 2014; Parker et al., 2014). It therefore remains to be seen which oncogenic drivers exist in the other molecular subgroups and what role CNAs and epigenetic alterations play.
An important observation from our data is that the two ST subgroups of EPN, characterized by highly recurrent but different gene fusions, have a different clinical outcome, even though they appear in the same age groups and in the same anatomical compartment. While patients in the ST-EPN-RELA subgroup comprise high-risk patients, with a 10-year OS of around 50% and a 10-year PFS of around 20%, patients in the ST-EPN-YAP1 subgroup all survived, and only one of six recurred. While this observation is striking, definite conclusions regarding the prognosis of ST-EPN-YAP1 tumors will have to await validation in larger patient cohorts. Besides ST-EPN-RELA, the only other molecular subgroup of ependymal tumors associated with a poor outcome is the PF-EPN-A subgroup, in keeping with previously published data for this subgroup of PF EPNs (Mack et al., 2014; Wani et al., 2012; Witt et al., 2011). Patients in all other subgroups have more favorable prognosis. As we have shown in multivariate survival analyses, molecular subgrouping is superior to histopathological grading of ependymal tumors, confirming previous studies showing the inconsistent prognostic value of histopathological grading (Ellison et al., 2011; Tihan et al., 2008). In fact, the only other variables that endured as independent prognostic factors in this multivariate analysis besides molecular subgrouping were extent of surgical resection and 1q gain.
In spite of its known shortcomings, histopathological grading based on WHO criteria has been used in several clinical trials to stratify EPN patients into different treatment groups, namely the Children’s Oncology Group study ACNS0121 and the German HIT 2000 Ependymoma trial. We propose a uniform and robust molecular classification system, based on nine distinct subgroups, as superior alternative to the current WHO histopathological classification. The logical next step will be to validate and further refine our findings in large, prospectively treated patient cohorts, such as cohorts of international cooperative group trial studies. We expect that a refined molecular risk stratification of patients will be of utmost value for designing prospective clinical trials that tailor therapy to the patients’ risk profile. Furthermore, the efficacy of adjuvant therapies, such as radiotherapy, chemotherapy, and/or molecular targeted therapies will need to be assessed in the context of specific molecular subgroups, as they will likely differ in their response to different therapy modalities. Thus, we believe that our proposed molecular classification of ependymal tumors will not only improve current diagnostic accuracy and prognostication, but also provide a superior platform for future clinical trial development, with the ultimate goal of improving the morbidity and mortality of children and adults with EPN.
EXPERIMENTAL PROCEDURES
Tumor Material and Patient Characteristics
Clinical samples and data were collected after receiving written informed consent according to protocols approved by the institutional review boards, including University Hospital Heidelberg, NN Burdenko Neurosurgical Institute, The Hospital for Sick Children, University of Utah, Schneider Children’s Medical Center of Israel, University of Colorado Denver, University Hospital Brno, University of California San Francisco, NYU Langone Medical Center, University of Cambridge, University of Bonn, and MD Anderson Cancer Center. At least 80% of tumor cell content was estimated in all tumor samples by staining cryosections (~5 μm thick) of the piece from which nucleic acid extraction was performed with H&E. Diagnoses were confirmed by histopathological assessment by at least two independent neuropathologists, including a central pathology review at the departments of neuropathology at the MD Anderson Cancer Center, the University Hospital Heidelberg, or the University Hospital Bonn that utilized the 2007 WHO classification for CNS tumors. The following tumor types were included: SE (WHO grade I), MPE (WHO grade I), EPN (WHO grade II), and anaplastic EPN (WHO grade III). No patient underwent chemotherapy or radiotherapy prior to the surgical removal of the primary tumor. Detailed clinical and patient characteristics are shown in Figure S1 and Table 1.
Biostatistics
Independence of chromosomal aberrations along molecular subgroups was analyzed by chi-square tests, where p values have been computed by 100,000 Monte Carlo simulations. The distribution of gender within molecular subgroups was tested against a null proportion of 0.5 using exact binomial test based on Clopper-Pearson intervals. Distribution of survival times was estimated by using Kaplan-Meier estimates and compared with the log-rank test. Prognostic impact of covariates on PFS and OS was evaluated on the basis of hazard ratios and 95% CIs from Cox’s proportional hazards regression model. Multivariate Cox’s proportional hazards regression models were used to adjust effects for additional covariates. In addition, Firth’s correction was applied to account for the monotone likelihood problem arising due to total separation of group and events (Heinze and Schemper, 2001). To compare nested Cox proportional hazards models Likelihood-ratio tests were applied.
Supplementary Material
Highlights.
DNA methylation profiling of ependymomas identifies nine molecular subgroups
YAP1 and RELA fusions characterize two distinct groups of supratentorial ependymoma
Patients with PFA or supratentorial RELA-positive ependymoma show dismal prognosis
Risk stratification by molecular subgrouping is superior to histological grading
In Brief.
Pajtler et al. classify 500 ependymal tumors using DNA methylation profiling into nine molecular subgroups. This molecular classification outperforms the current histopathological grading in the risk stratification of patients.
Significance.
DNA methylation patterns in tumors have been shown to represent a very stable molecular memory of the respective cell of origin throughout disease course, making them particularly suitable for tumor classification purposes. Methylation profiling of a large series of ependymal tumors, comprising all histological grades and anatomical locations, revealed a highly reliable classification for this clinically heterogeneous group of malignancies. Notably, the vast majority of high-risk patients (mostly children), for whom effective therapeutic concepts are desperately needed, were restricted to just two of the nine molecular subgroups identified, pointing to the clinical relevance of molecular classification. Since this analysis can be performed from minute amounts of DNA extracted from archived material, it is ideally suited for routine clinical application.
ACKNOWLEDGMENTS
This study was supported by grants from the Sander Stiftung, Stiftung Sibylle Assmus, and DKFZ/MOST/BMBF collaboration to H.W.; a grant from the German Consortium for Translational Cancer Research (DKTK) “Molecular Diagnostics of Childhood Malignancies” to P.L., S.M.P., and H.W.; a grant from the Collaborative Ependymoma Research Network (CERN) foundation to K.W., K.A., T.S.A., and S.M.P.; a grant from the German Children’s Cancer Foundation to T.P.; and a grant from The Making Headway Foundation to M.A.K., J.C.A., and D.Z. The NYU Langone Human Specimen Resource Center, Laura and Isaac Perlmutter Cancer Center, and Clinical and Translational Science Institute (CTSI) are partially supported by the Cancer Center Support Grant P30CA016087 and grant UL 1 TR000038 from the National Center for the Advancement of Translational Science (NCATS), NIH. Additional support came from the German Cancer Research Center-Heidelberg Center for Personalized Oncology (DKFZ-HIPO) and the National Center for Tumor Diseases (NCT) Precision Oncology Program. K.Z. acknowledges research support from CZ.1.05/2.1.00/03.0101 (RECAMO).
Footnotes
ACCESSION NUMBERS
The DNA methylation and gene expression data of this study have been deposited in NCBI’s Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession numbers GEO: GSE65362, GSE64415, respectively.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2015.04.002.
AUTHOR CONTRIBUTIONS
K.W.P., H.W., M.S., D.T.W.J., V.H., F.K., K.W., R.T., C.P., P.J., J.R., H.-J.W., D.C., L. Schweizer, L. Sieber, A.W., Z.H., P.v.S., R. Volckmann, J.K., R. Versteeg, T.P., and K.A. contributed to the design and conduct of experiments and to the writing. D.F., H.T., S.A., L.M.H., A.M.D., N.F., E.H., K.Z., M.G., T.S.A., N.G., J.C.A., M.A.K., D.Z., M.H., A.E.K., O.W., V.P.C., K.v.H., S.R., M.R., S.M., V.R., G.B., M.-L.Y., A.v.D., P.L., M.D.T., R.G., and D.W.E. contributed to experimental design and to the writing. A.K., M.K., and S.M.P. conceived the research and designed, directed, and wrote the study.
REFERENCES
- Archer TC, Pomeroy SL. Posterior fossa ependymomas: a tale of two subtypes. Cancer Cell. 2011;20:133–134. doi: 10.1016/j.ccr.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Bouffet E, Foreman N. Chemotherapy for intracranial ependymomas. Childs Nerv. Syst. 1999;15:563–570. doi: 10.1007/s003810050544. [DOI] [PubMed] [Google Scholar]
- Bouffet E, Tabori U, Huang A, Bartels U. Ependymoma: lessons from the past, prospects for the future. Childs Nerv. Syst. 2009;25:1383–1384. doi: 10.1007/s00381-009-0915-6. author reply 1385. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter M, Nicholson J, Ross F, Crolla J, Allibone R, Balaji V, Perry R, Walker D, Gilbertson R, Ellison DW. Genetic abnormalities detected in ependymomas by comparative genomic hybridisation. Br. J. Cancer. 2002;86:929–939. doi: 10.1038/sj.bjc.6600180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer S, Prebble E, Davison V, Davies P, Ramani P, Ellison D, Grundy R. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Am. J. Pathol. 2002;161:2133–2141. doi: 10.1016/S0002-9440(10)64491-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert C, von Haken M, Meyer-Puttlitz B, Wiestler OD, Reifenberger G, Pietsch T, von Deimling A. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am. J. Pathol. 1999;155:627–632. doi: 10.1016/S0002-9440(10)65158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison DW, Kocak M, Figarella-Branger D, Felice G, Catherine G, Pietsch T, Frappaz D, Massimino M, Grill J, Boyett JM, Grundy RG. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J. Negat. Results Biomed. 2011;10:7. doi: 10.1186/1477-5751-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin. Cancer Res. 2014;20:5630–5640. doi: 10.1158/1078-0432.CCR-14-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar N, Grill J, Geoerger B, Lellouch-Tubiana A, Michalowski MB, Vassal G. p53 Pathway dysfunction in primary childhood ependymomas. Pediatr. Blood Cancer. 2006;46:604–613. doi: 10.1002/pbc.20532. [DOI] [PubMed] [Google Scholar]
- Gatta G, Botta L, Rossi S, Aareleid T, Bielska-Lasota M, Clavel J, Dimitrova N, Jakab Z, Kaatsch P, Lacour B, et al. EUROCARE Working Group Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5—a population-based study. Lancet Oncol. 2014;15:35–47. doi: 10.1016/S1470-2045(13)70548-5. [DOI] [PubMed] [Google Scholar]
- Godfraind C, Kaczmarska JM, Kocak M, Dalton J, Wright KD, Sanford RA, Boop FA, Gajjar A, Merchant TE, Ellison DW. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. 2012;124:247–257. doi: 10.1007/s00401-012-0981-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinze G, Schemper M. A solution to the problem of monotone likelihood in Cox regression. Biometrics. 2001;57:114–119. doi: 10.1111/j.0006-341x.2001.00114.x. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al. Cancer Genome Atlas Research Network Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R, Cavalli FM, Ramaswamy V, Zapatka M, Reifenberger G, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013;125:913–916. doi: 10.1007/s00401-013-1126-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–541. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SE, White E, Eden C, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466:632–636. doi: 10.1038/nature09173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilday JP, Rahman R, Dyer S, Ridley L, Lowe J, Coyle B, Grundy R. Pediatric ependymoma: biological perspectives. Mol. Cancer Res. 2009;7:765–786. doi: 10.1158/1541-7786.MCR-08-0584. [DOI] [PubMed] [Google Scholar]
- Kilday JP, Mitra B, Domerg C, Ward J, Andreiuolo F, Osteso-Ibanez T, Mauguen A, Varlet P, Le Deley MC, Lowe J, et al. Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children’s Cancer Leukaemia Group (CCLG), Societe Francaise d’Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP) Clin. Cancer Res. 2012;18:2001–2011. doi: 10.1158/1078-0432.CCR-11-2489. [DOI] [PubMed] [Google Scholar]
- Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226–1236. doi: 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M, Milde T, Bender S, Wittmann A, Schöttler A, et al. Molecular staging of intracranial ependymoma in children and adults. J. Clin. Oncol. 2010;28:3182–3190. doi: 10.1200/JCO.2009.27.3359. [DOI] [PubMed] [Google Scholar]
- Kurian KM, Jones DT, Marsden F, Openshaw SW, Pearson DM, Ichimura K, and Collins VP. Genome-wide analysis of subependymomas shows underlying chromosomal copy number changes involving chromosomes 6, 7, 8 and 14 in a proportion of cases. Brain Pathol. 2008;18:469–473. doi: 10.1111/j.1750-3639.2008.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Fong KW, Tang M, Han X, Gong Z, Ma W, Hebert M, Songyang Z, Chen J. Fam118B, a newly identified component of Cajal bodies, is required for Cajal body formation, snRNP biogenesis and cell viability. J. Cell Sci. 2014;127:2029–2039. doi: 10.1242/jcs.143453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506:445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire CS, Sainani KL, Fisher PG. Both location and age predict survival in ependymoma: a SEER study. Pediatr. Blood Cancer. 2009;52:65–69. doi: 10.1002/pbc.21806. [DOI] [PubMed] [Google Scholar]
- Mendrzyk F, Korshunov A, Benner A, Toedt G, Pfister S, Radlwimmer B, Lichter P. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin. Cancer Res. 2006;12:2070–2079. doi: 10.1158/1078-0432.CCR-05-2363. [DOI] [PubMed] [Google Scholar]
- Merchant TE, Li C, Xiong X, Kun LE, Boop FA, Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009;10:258–266. doi: 10.1016/S1470-2045(08)70342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modena P, Buttarelli FR, Miceli R, Piccinin E, Baldi C, Antonelli M, Morra I, Lauriola L, Di Rocco C, Garrè ML, et al. Predictors of outcome in an AIEOP series of childhood ependymomas: a multifactorial analysis. Neuro-oncol. 2012;14:1346–1356. doi: 10.1093/neuonc/nos245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, Pfister SM. Medulloblastomics: the end of the beginning. Nat. Rev. Cancer. 2012;12:818–834. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H, Eibl RH, Schwab M, Reichel MB, Mariani L, Gehring M, Petersen I, Höll T, Wiestler OD, Kleihues P. Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol. Carcinog. 1993;8:74–80. doi: 10.1002/mc.2940080203. [DOI] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro-oncol. 2014;16(Suppl 4):iv1–iv63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, Lee R, Tatevossian RG, Phoenix TN, Thiruvenkatam R, et al. C11orf95-RELA fusions drive oncogenic NF-kB signalling in ependymoma. Nature. 2014;506:451–455. doi: 10.1038/nature13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14:1200–1207. doi: 10.1016/S1470-2045(13)70449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Jones DT, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Arak T, Vilo J. g:Profiler—a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MP, Correa KM, Ramesh V, MacCollin MM, Jacoby LB, von Deimling A, Gusella JF, Louis DN. Analysis of the neurofibromatosis 2 gene in human ependymomas and astrocytomas. Cancer Res. 1994;54:45–47. [PubMed] [Google Scholar]
- Singh PK, Gutmann DH, Fuller CE, Newsham IF, Perry A. Differential involvement of protein 4.1 family members DAL-1 and NF2 in intracranial and intraspinal ependymomas. Mod. Pathol. 2002;15:526–531. doi: 10.1038/modpathol.3880558. [DOI] [PubMed] [Google Scholar]
- Slavc I, MacCollin MM, Dunn M, Jones S, Sutton L, Gusella JF, Biegel JA. Exon scanning for mutations of the NF2 gene in pediatric ependymomas, rhabdoid tumors and meningiomas. Int. J. Cancer. 1995;64:243–247. doi: 10.1002/ijc.2910640406. [DOI] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat. Rev. Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tihan T, Zhou T, Holmes E, Burger PC, Ozuysal S, Rushing EJ. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s Oncology Group study and a review of prognostic factors. Mod. Pathol. 2008;21:165–177. doi: 10.1038/modpathol.3800999. [DOI] [PubMed] [Google Scholar]
- Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, Judson RN, Thway K, Nadal G, Selfe JL, et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell. 2014;26:273–287. doi: 10.1016/j.ccr.2014.05.029. [DOI] [PubMed] [Google Scholar]
- Valouev A, Weng Z, Sweeney RT, Varma S, Le QT, Kong C, Sidow A, West RB. Discovery of recurrent structural variants in nasopharyngeal carcinoma. Genome Res. 2014;24:300–309. doi: 10.1101/gr.156224.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatramani R, Dhall G, Patel M, Grimm J, Hawkins C, McComb G, Krieger M, Wong K, O’Neil S, Finlay JL. Supratentorial ependymoma in children: to observe or to treat following gross total resection? Pediatr. Blood Cancer. 2012;58:380–383. doi: 10.1002/pbc.23086. [DOI] [PubMed] [Google Scholar]
- Wani K, Armstrong TS, Vera-Bolanos E, Raghunathan A, Ellison D, Gilbertson R, Vaillant B, Goldman S, Packer RJ, Fouladi M, et al. Collaborative Ependymoma Research Network A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012;123:727–738. doi: 10.1007/s00401-012-0941-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, Benner A, Hielscher T, Milde T, Remke M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20:143–157. doi: 10.1016/j.ccr.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Sun T, Kobayashi K, Gao P, Griffin JD. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol. Cell. Biol. 2002;22:7688–7700. doi: 10.1128/MCB.22.21.7688-7700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.