

Abstract
Purpose
Sonic hedgehog (SHH), an activating ligand of smoothened (SMO), is overexpressed in > 70% of pancreatic cancers (PCs). We investigated the impact of vismodegib, an SHH antagonist, plus gemcitabine (GV) or gemcitabine plus placebo (GP) in a multicenter phase Ib/randomized phase II trial and preclinical PC models.
Patients and Methods
Patients with PC not amenable to curative therapy who had received no prior therapy for metastatic disease and had Karnofsky performance score ≥ 80 were enrolled. Patients were randomly assigned in a one-to-one ratio to GV or GP. The primary end point was progression-free-survival (PFS). Exploratory correlative studies included serial SHH serum levels and contrast perfusion computed tomography imaging. To further investigate putative biologic mechanisms of SMO inhibition, two autochthonous pancreatic cancer models (KrasG12D; p16/p19fl/fl; Pdx1-Cre and KrasG12D; p53R270H/wt; Pdx1-Cre) were studied.
Results
No safety issues were identified in the phase Ib portion (n = 7), and the phase II study enrolled 106 evaluable patients (n = 53 in each arm). Median PFS was 4.0 and 2.5 months for GV and GP arms, respectively (95% CI, 2.5 to 5.3 and 1.9 to 3.8, respectively; adjusted hazard ratio, 0.81; 95% CI, 0.54 to 1.21; P = .30). Median overall survival (OS) was 6.9 and 6.1 months for GV and GP arms, respectively (95% CI, 5.8 to 8.0 and 5.0 to 8.0, respectively; adjusted hazard ratio, 1.04; 95% CI, 0.69 to 1.58; P = .84). Response rates were not significantly different. There were no significant associations between correlative markers and overall response rate, PFS, or OS. Preclinical trials revealed no significant differences with vismodegib in drug delivery, tumor growth rate, or OS in either model.
Conclusion
The addition of vismodegib to gemcitabine in an unselected cohort did not improve overall response rate, PFS, or OS in patients with metastatic PC. Our preclinical and clinical results revealed no statistically significant differences with respect to drug delivery or treatment efficacy using vismodegib.
INTRODUCTION
Pancreatic cancer (PC) is the fourth leading cause of cancer mortality in the United States, with 38,460 deaths annually.1 Five-year survival for all stages combined is only 6%. Gemcitabine had beenthe backbone treatment for years in advanced disease,2 until the introduction of FOLFIRINOX (fluorouracil, leucovorin, irinotecan, and oxaliplatin)3 and gemcitabine plus albumin-bound nab-paclitaxel4 regimens, both reported after initiation of our trial. Despite numerous attempts, most gemcitabine combinations with molecularly targeted therapies have failed to demonstrate a significant improvement in overall survival (OS),5–8 with the exception of gemcitabine plus erlotinib, which has demonstrated a statistically significant but clinically modest benefit.9
Vismodegib (Erivedge; Genentech, South San Francisco, CA), a synthetic small-molecule inhibitor of smoothened (SMO) in the hedgehog (Hh) pathway,10,11 has demonstrated clinical benefit in basal cell carcinoma and medulloblastoma—both harboring recurrent Hh pathway mutations in SMO or protein patched homolog 1 (PTCH1).12,13 Vismodegib is US Food and Drug Administration approved for the treatment of patients with advanced basal cell carcinoma. Trials applying various SMO inhibitors to other tumors harboring genomic activation of the Hh signaling pathway are under way, some within novel clinical trial designs.14 However, no single-agent activity was observed in early phase I studies within molecularly unselected patients with advanced and pretreated PC.15,16
Nevertheless, the Hh pathway has been reported to be critical for tumor progression in preclinical PC models17,18 and has been considered a potential therapeutic target.19–21 Sonic Hh (SHH) is overexpressed in approximately 70% of PCs and has been shown to be an early and late mediator of PC tumorigenesis.22–24 Tumor-derived SHH has influenced and promoted tumor growth in preclinical PC systems by activating Hh signaling in stroma.25–28 This paracrine Hh signaling may establish and maintain the desmoplastic stroma observed in PC, creating a barrier for proper drug penetration.29 Hh pathway inhibition with IPI-269609, an SMO inhibitior,30–32 reportedly led to increased tumor perfusion, enhanced tumor delivery of gemcitabine when coadministered, and improvement in survival in a genetically engineered murine PC model,29 forming the rationale for human clinical trials in PC.
Given this background, we hypothesized that inhibition of the Hh pathway would be synergistic with gemcitabine and would lead to improved progression-free survival (PFS) compared with gemcitabine alone for metastatic PC.33 This article reports the final results of a phase Ib (n = 7) and multicenter randomized phase II trial (n = 106) comparing gemcitabine plus vismodegib (GV) with gemcitabine plus placebo (GP) in patients with advanced PC; there were no significant differences in overall response rate (ORR), PFS, or OS between these two groups. To further investigate the putative biologic mechanisms of SMO inhibition, we used two autochthonous PC models (KrasG12D; p16/p19fl/fl; Pdx1-Cre [KPP] and KrasG12D; p53R270H/wt; Pdx1-Cre [KR]) to recapitulate the phase II clinical trial. In contrast to recent reports with IPI-926 (saridegib),29,34–36 our preclinical and clinical results with vismodegib are concordant and demonstrate no statistically significant differences with respect to tumor growth, drug delivery, or treatment efficacy.
PATIENTS AND METHODS
The Data Supplement provides detailed information on methods.
RESULTS
Clinical Trial
Patient characteristics.
Seven patients were enrolled onto the phase Ib open-label GV portion, and no safety issues were identified. For the randomized phase II part of the trial, 111 patients were enrolled at 13 sites between February 2010 and June 2012, stratified by Karnofsky performance score (80 v 90 or 100) and disease status (newly diagnosed v recurrent; Fig 1). Of these, four patients withdrew consent before starting treatment (two from each arm), and one patient (randomly assigned to GV) was subsequently found to have been ineligible and never started therapy. Analyses were based on the remaining 106 patients. Patient characteristics were similar between treatment arms, except for the incidence of peritoneal metastases, which were higher in the GP arm (9% v 23%; Table 1).
Fig 1.
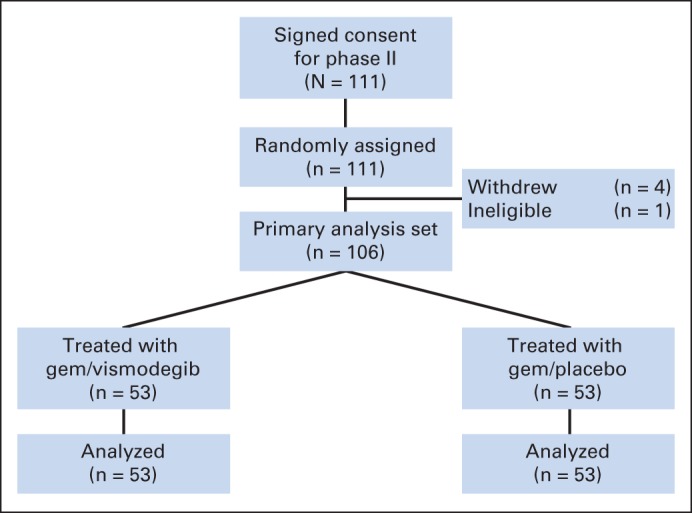
CONSORT diagram of clinical trial enrollment and treatment in phase II trial. gem, gemcitabine.
Table 1.
Baseline Patient Demographic and Clinical Characteristics
| Characteristic | No. (%) |
||
|---|---|---|---|
| GV (n = 53) | GP (n = 53) | Total (N = 106) | |
| Age, years | |||
| Median | 64 | 64 | 64 |
| Range | 49-82 | 39-84 | 39-84 |
| Sex | |||
| Male | 31 (58) | 27 (51) | 58 (55) |
| Female | 22 (42) | 26 (49) | 48 (45) |
| Race | |||
| White | 40 (77) | 45 (88) | 85 (83) |
| African American | 10 (19) | 6 (12) | 16 (16) |
| Asian | 1 (2) | 0 (0) | 1 (1) |
| Other | 1 (2) | 0 (0) | 1 (1) |
| Missing | 1 | 2 | 3 |
| Karnofsky performance score | |||
| 100 | 19 (36) | 17 (32) | 36 (34) |
| 90 | 14 (26) | 20 (38) | 34 (32) |
| 80 | 20 (38) | 16 (30) | 36 (34) |
| Disease status at enrollment | |||
| Newly diagnosed | 48 (91) | 48 (91) | 96 (91) |
| Recurrent metastatic | 5 (9) | 5 (9) | 10 (9) |
| Primary tumor location | |||
| Head | 23 (43) | 24 (46) | 47 (45) |
| Neck/uncinate | 1 (2) | 2 (4) | 3 (3) |
| Body | 16 (30) | 11 (21) | 26 (25) |
| Tail | 13 (25) | 13 (25) | 26 (25) |
| Site of metastasis* | |||
| Liver | 41 (77) | 44 (83) | 85 (80) |
| Lung | 12 (23) | 14 (26) | 26 (25) |
| Peritoneum | 5 (9) | 12 (23) | 17 (16) |
| Other | 8 (15) | 10 (19) | 18 (17) |
| Crossover to GV | 22 (42) | ||
Abbreviations: GP, gemcitabine plus placebo; GV, gemcitabine plus vismodegib.
Patients may have > one primary or metastatic site.
Safety.
Median number of cycles was four (range, one to 12 cycles) in the GV arm and three (range, zero to 14 cycles) in the GP arm. Combination therapy with GV was generally well tolerated and did not result in unexpected toxicities (Table 2). There were no statistically significant differences in the rate of adverse events (AEs) between the arms. Four patients in the GV arm and two in the GP arm withdrew from treatment as a result of AEs. Sixteen patients (GV, n = 10; GP, n = 6) died while receiving treatment.
Table 2.
Grade 3 to 5 Toxicities at Least Possibly Related to Treatment
| Toxicity | No. (%) |
P | |
|---|---|---|---|
| GV (n = 53) | GP (n = 53) | ||
| Neutropenia | 15 (28) | 12 (23) | .66 |
| Fatigue | 7 (13) | 4 (8) | .53 |
| Anorexia | 5 (9) | 2 (4) | .44 |
| Vomiting | 5 (9) | 2 (4) | .44 |
| WBC decreased | 5 (9) | 7 (13) | .76 |
| Platelet count decreased | 6 (11) | 5 (9) | 1.0 |
| Anemia | 4 (8) | 7 (13) | .53 |
| Nausea | 4 (8) | 3 (6) | 1.0 |
| Elevated AST | 5 (9) | 3 (6) | .72 |
| Elevated ALT | 3 (6) | 2 (4) | 1.0 |
| Blood bilirubin increased | 3 (6) | 1 (2) | .62 |
| Hyperglycemia | 3 (6) | 3 (6) | 1.0 |
| Hypokalemia | 4 (8) | 2 (4) | .68 |
| Alkaline phosphatase increased | 2 (4) | 2 (4) | 1.0 |
| Lymphocyte count decreased | 2 (4) | 5 (9) | .44 |
| Hyponatremia | 1 (2) | 5 (9) | .20 |
Abbreviations: GP, gemcitabine plus placebo; GV, gemcitabine plus vismodegib.
Efficacy.
ORRs were similar in the two arms (GV: complete response [CR], 0 [0%]; partial response [PR], 4 [8%]; stable disease [SD], 27 [51%]; disease control [ie, CR + PR + SD], 31 [58%]; GP: CR, 1 [2%]; PR, 6 [11%]; SD, 20 [38%]; disease control, 27 [51%]). The difference in response rates (8% v 13%) was not significant (P = .53; Data Supplement).
The primary end point of the study was PFS. At the final analysis, events (progression or death) occurred in 48 patients (91%) receiving GV and 51 (95%) receiving GP. Median PFS was 4.0 months for GV and 2.5 months for GP (adjusted hazard ratio [HR], 0.83; 95% CI, 0.55 to 1.23; Fig 2A; Data Supplement).
Fig 2.

(A) Progression-free and (B) overall survival by treatment arm. Blue, gemcitabine plus vismodegib; gold, gemcitabine plus placebo. Hazard ratio (HR) after adjusting (adj) for Karnofsky performance score and disease status (newly diagnosed v recurrent).
Median OS was 6.9 months for GV and 6.1 month for GP (adjusted HR, 0.96; 95% CI, 0.64 to 1.44; Fig 2B; Data Supplement). No survival differences were noted in a preplanned secondary analysis of OS that censored patients receiving GP at first progression, before crossover to GV (P = .69). Note that patient crossover did not affect the primary end point (ie, PFS), because crossover happened after the event occurred. For patients receiving GP who crossed over at progression (n = 22 [42%]), median PFS was 1.8 months, and median OS was 2.9 months (Data Supplement). One-year survival rates in the GV and GP arms were 15% and 25%, respectively (P = .3). OS and PFS did not differ significantly by Karnofsky performance score (P = .66 and .42, respectively; Data Supplement). Mortality and disease progression rates were consistent and uniformly high across all centers.
Clinical Trial Correlative Results
SHH serum levels.
Median pretreatment plasma SHH level pooled for both treatment arms was 1.01 ng/mL (GV arm, 1.01 ng/mL; GP arm, 1.06 ng/mL). SHH levels did not change significantly with subsequent cycles (P = .087), nor was there a difference between treatment groups (P = .85) or patients with cancer (n = 89) and normal controls (n = 40; P = .4) (Figs 3A and 3B). SHH serum levels did not correlate with age in either patients with cancer (r = 0.17; P = .13) or controls (r = 0.03; P = .87; Figs 3C and 3D).
Fig 3.

Clinical trial translational correlatives. Serum SHH levels (A) comparing controls (n = 40) with patients with pancreatic cancer enrolled onto trial (n = 89), (B) by treatment group (gemcitabine plus vismodegib [GV] or gemcitabine plus placebo [GP]) with increasing treatment (TX) cycle, and association with age in (C) patients with cancer and (D) controls. (E) Radiologic correlatives evaluating association of baseline tumor perfusion with tumor response to therapy.
Radiologic tumor perfusion.
By univariable analysis, baseline computed tomography (CT) perfusion of the primary tumor was not associated with response to therapy, expressed as percent change in tumor size (r = −0.09; P = .77; Fig 3E; Data Supplement).
Murine Translational Correlatives
SMO inhibition does not reveal quantifiable changes to tumor stroma in vivo.
Previous preclinical studies have suggested that SHH is abnormally expressed in genetically engineered models of PC.29,37 SHH expression and pathway activation were confirmed in the KPP genetically engineered mouse model of PC by transcriptional gene analysis and immunohistochemistry. SHH immunoreactivity exhibited focal staining throughout tumors, predominantly in mucinous and well-defined ductal epithelial cells (data not shown), consistent with previous reports.26 Comparative transcriptional analysis of normal pancreas and tumor revealed significantly elevated expression of Hh ligands SHH and Indian Hh (IHH), as well as SMO, glioma-associated oncogene family zinc finger 1 (GLI1), GLI2, Hh Interacting Protein (HHIP), and PTCH1 (Data Supplement).
To determine whether inhibition of SMO leads to changes in tumor stroma, we used a small molecule, HhAntag,38 a potent orally available preclinical surrogate of the US Food and Drug Administration–approved vismodegib (GDC-0449).10 Previous reports have shown that antagonism of Hh signaling resulted in increased mean microvessel density (MVD).29 HhAntag treatment on tumor-bearing KPP animals revealed no significant change in MVD (Fig 4A; P = .54; Data Supplement), despite observing pathway inhibition (Data Supplement). Moreover, HhAntag treatment did not affect intratumoral extracellular matrix deposition assessed through trichrome staining (Fig 4B; P = .29).
Fig 4.

Effects of hedgehog pathway antagonism (HhAntag) on vasculature, stromal content, and intratumoral gemcitabine metabolites in KrasG12D; p16/p19fl/fl; Pdx1-Cre (KPP) tumors. (A) Quantitation of immunohistochemical staining for meca-32 expression in pancreatic tumors of KPP mice treated with vehicle (circles; n = 13) or smoothened (SMO) inhibitor (squares; n = 13) for 10 days. Data presented as percentages of meca-32–positive areas over analyzed tumor areas for each tumor (Mann-Whitney P = .54; scale bar, 200 um). (B) Quantitative analysis of stromal content by trichromatic stain in pancreatic tumors of KPP mice treated with vehicle or SMO inhibitor for 10 days. Data presented as percentages of positive stain areas over analyzed tissue areas (Mann-Whitney P = .29). (C) Mass spectromic quantitation of intratumoral concentration of 2′,2′-difluorodeoxycytidine triphosphate (dFdCTP; active form of gemcitabine) from each tumor after treatment for 10 consecutive days with vehicle or SMO inhibitor and gemcitabine 50 mg/kg 30 minutes before tumor collection (Mann-Whitney P = 1.000). (D) Ratios of 2′,2′-difluoro 2′-deoxycytidine (dFdC) to diflurodeoxyuridine (dFdU) in pancreatic tumors from each tumor (Mann-Whitney P = .48). NS, not significant.
SMO inhibition does not affect gemcitabine drug delivery in vivo.
The modest response rates observed in patients with PC treated with gemcitabine has, in part, been attributed to poor drug delivery associated with a prohibitive tumor microenvironment. Gemcitabine undergoes intracellular conversion, from 2′,2′-difluoro 2′-deoxycytidine (dFdC) to the active triphosphate form 2′,2′-difluorodeoxycytidine triphosphate (dFdCTP), responsible for inhibition of DNA synthesis and repair. To directly measure drug delivery, dFdCTP levels were analyzed in PC tumors of KPP mice after continuous 10-day HhAntag treatment and a single dose of gemcitabine. Liquid chromatography–tandem mass spectrometry analysis revealed that dFdCTP concentrations were similar in both vehicle- and HhAntag-treated KPP mice (Fig 4C; Mann-Whitney P = 1.00). Intracellular gemcitabine is also converted to an inactivated form, diflurodeoxyuridine (dFdU). The ratio of the unprocessed form (dFdC) to inactive form (dFdU) estimates intratumoral gemcitabine exposure. The average ratio of dFdC to dFdU in KPP tumors of HhAntag-treated mice was not significantly different between vehicle and SMO inhibitor groups (Fig 4D; P = .48). Together, these results indicated that HhAntag did not increase gemcitabine delivery in this genetically engineered PC mouse model.
Pancreatic tumor progression and OS remain unaltered by Hh pathway inhibition.
Next, we investigated the therapeutic impact of HhAntag in KPP mice. Growth rates of tumors, as determined by serial ultrasound imaging, were not significantly different when comparing HhAntag and vehicle arms, indicating no single-agent effect of Hh pathway inhibition (Figs 5A and 5B). Although the combination of gemcitabine plus HhAntag significantly decreased tumor growth relative to control treatment (log-rank P = .0052), the growth rate was not different from that of gemcitabine alone, known to affect tumor growth in this model.39 Consistent with the lack of tumor growth effect, the HhAntag plus gemcitabine combination did not provide significant improvement in OS when compared with gemcitabine alone (Fig 5C). In summary, the addition of HhAntag did not affect tumor growth or animal longevity in the KPP model.
Fig 5.

Smoothened (SMO) inhibitor does not affect tumor progression or overall survival in Kras LSL-G12D; p16/p19 fl/fl; Pdx1-Cre (KPP) mice. (A) Individual tumor growth rates plotted by from serial ultrasound images as volumes depicted longitudinally by animal within each regimen. (B) Antilogged values of slopes in each longitudinal plot are graphed, and average tumor burden fold changes per day in each study group of KPP mice are shown, with approximate 95% CIs (vehicle v SMO inhibitor, P = .86; vehicle v combination, P = .0156; gemcitabine v combination, P = .18) (C) Kaplan-Meier plots of KPP mice treated with vehicle (blue, n = 15; median, 1.9 weeks), SMO inhibitor (gold, n = 16; median, 1.2 weeks), gemcitabine (gray, n = 14; median, 3.8 weeks), and gemcitabine plus SMO inhibitor combination (red, n = 12; median, 3.4 weeks; gemcitabine v vehicle, P = .0059; combination v vehicle, P = .0179; gemcitabine v combination, P = .10 [all P values from log-rank test]). HhAntag, hedgehog pathway antagonism.
Analysis of SMO inhibition in KR mice.
Although the KPP model represents the genetics of a subset of the PC population, previous findings were generated in a model composed of mutant KRAS and mutant p53 expression in the pancreas.40 Therefore, we reproduced all in vivo studies in the KR model. HhAntag treatment on tumor-bearing KR animals revealed a significant decrease in MVD using meca-32 (Data Supplement; P = .0418) and trend toward decrease using CD31 (Data Supplement; P = .29). Trichrome staining revealed no significant changes to the stroma of HhAntag-treated tumors (Data Supplement; P = .83). Together these findings indicated that as in the previous KPP model, the stroma of the developed KR tumors was not significantly affected by HhAntag treatment.
Finally, we observed no significant change in concentration of the gemcitabine metabolite dFdCTP in tumors after HhAntag (Data Supplement; P = .25) and no significant change in the ratio of dFdC to dFdU (Data Supplement). Furthermore, neither HhAntag nor the gemcitabine plus HhAntag combination affected tumor growth rates in KR animals compared with gemcitabine alone (Data Supplement). Consistent with the tumor growth study, HhAntag did not lead to survival benefit over vehicle or gemcitabine treatment (Data Supplement). Taken together, neither of the murine PC model genotypes (KPP or KR) demonstrated any measurable differences between gemcitabine delivery to tumors, tumor growth rate, or OS with HhAntag treatment. A significant decrease in microvessel density, consistent with previous work in transplantable models using vismodegib, was observed.41
DISCUSSION
Effective molecularly targeted treatment for advanced PC remains an unmet need. When we began this trial, extensive preclinical evidence provided rationale for the clinical evaluation of Hh pathway inhibition along with concurrent gemcitabine treatment.19,22,24,25,29,32
The addition of vismodegib to gemcitabine in this clinical study was well tolerated, no unexpected toxicities were observed, and there were no significant toxicity differences compared with the GP arm (Table 2). Unfortunately, this trial failed to meet its primary objective of a statistically significant improvement in median PFS, achieving a median PFS of 4.0 and 2.5 months in the GV and GP arms, respectively (HR, 0.83; 95% CI, 0.55 to 1.23). It should be noted, however, that the study was powered (85%) to detect an HR of 0.6, and therefore, a smaller effect, if present, may not have been detectable. Similarly, there were no differences between arms in median OS (6.9 v 6.1 months) or disease control rate (58% v 51%).
Despite a recent report suggesting that serum SHH levels were decreased in patients with PC compared with age-matched controls, we did not observe this association.42 Moreover, we hypothesized that serum SHH might change over time, potentially differentially with or without exposure to vismodegib, but as noted, this was not observed either. Consistent with these findings, Kim et al43 recently reported no change in tumor SHH protein expression in matched biopsies from patients treated for 3 weeks with vismodegib.
To investigate the discrepancy between our clinical data and previous reports,24,25,29 we sought to assess baseline primary pancreatic tumor perfusion and its association with response to therapy. Perfusion CT is a clinical technique used to provide regional maps and obtain quantitative measurements of hemodynamic parameters on the basis of the linear relationship between CT enhancement and iodinated contrast material concentration.44,45 Whole-organ perfusion of the pancreas using dynamic contrast-enhancement imaging revealed significantly lower perfusion in the tumor compared with adjacent normal pancreatic tissue.46 In addition, enhancement patterns of PCs on conventional dynamic multidetector row CT correlated with degree of angiogenesis, and these patterns were reportedly modified by degree of fibrosis.47 However, we did not observe a correlation between higher baseline tumor perfusion and improved treatment response to gemcitabine (pooled analysis, GV + GP) in this univariable analysis of a small exploratory cohort. Because there were no significant differences in clinical outcomes between the GV and GP arms, evaluation of serial changes in perfusion over time was not performed.
Much of the preclinical work supporting Hh inhibition for PC was performed with IPI-296 (saridegib).29,31 It is noteworthy that a randomized phase II trial of saridegib in PC, conducted simultaneously with our trial, was halted at interim analysis in January 2012 because of worse median PFS and median OS compared with the placebo arm.36 A recent report attempting to discern the preclinical29 to clinical36 discrepancy suggested that prolonged exposure to IPI-296 before frank tumor development (PanIN stage) ultimately led to tumors with undifferentiated histology, increased vascularity, and heightened proliferation.34 Importantly, clinical correlates to confirm the original29 or newly reported34 preclinical findings, including increased vascularity, improved drug delivery, and worsened disease resulting from IPI-296 treatment, are still lacking or unreported. Subsequently, a similar preclinical study evaluating vismodegib treatment, also at the tumor precursor stage (PanIN), reported accelerated tumor progression.35 Here, further support for the critical role of stroma early in pancreatic tumor formation was provided with elegant studies, where SHH was genetically deleted coincident with tumor suppressor loss and oncogene activation. In the absence of epithelial-derived SHH secretion, the resulting pancreatic tumors were phenotypically distinct, devoid of desmoplastic stroma, more vascularized, and more proliferative than controls.34,35 The early treatment design of these experiments is consistent with a large body of work describing the preventive nature of stroma in early tumor formation,48–50 but it does little to reconcile the preclinical and clinical discrepancy with Hh inhibition in established pancreatic tumors. The discordance between the preclinical29 (benefit) and clinical36 (detriment) results with IPI-926 in established tumor scenarios may be a result of overinterpretation of the preclinical gemcitabine model data,29 because small yet statistically significant effects do not ensure a biologically meaningful effect in patients. This lack of predictive correlation contrasts the strong predictive value of preclinical work in models harboring mutationally driven Hh pathway signaling.51 Patient-derived xenograft models may also promote better understanding in the future.
Patients with PC frequently present with advanced disease, so to model the clinical treatment scenario, we treated two independent, genetically engineered PC models when defined, measurable tumors were readily detectable. Not only did SHH pathway inhibition not improve survival, no measurable changes in gemcitabine delivery or tumor growth rate were observed in either murine model. An increase in vascular density was not observed with vismodegib treatment, in contrast to IPI-926.29 In our study, HhAntag treatment showed a significant or trending decrease in microvessel density, depending on the marker used, which is consistent with previous work in preclinical tumor models where vismodegib treatment reduced tumor growth and mean vessel density.41
It is also possible that the preclinical efficacy differences may be attributed to unique molecular properties of the agents, given that IPI-926 is a cyclopamine derivative, whereas GDC-0449 (vismodegib) is a synthetic inhibitor of SMO. For example, although both molecules occupy a similar pocket within the transmembrane domain of SMO, they may mechanistically diverge, because cyclopamine treatment leads to accumulation of SMO in the primary cilium, whereas vismodegib prevents it.52,53 Moreover, cyclopamine has been shown to have off-target effects that may lead to enhanced toxicity and/or potentially cause the stromal effects observed. Notably, a clinical trial with IPI-926 in PC was stopped because of worse survival outcome in the investigational arm.36 In our study, we did not observe increased toxicity or a detriment in survival with GV. Nevertheless, neither molecule demonstrated statistically significant improvement when combined with gemcitabine in PC.
In conclusion, we found no benefit in adding vismodegib to gemcitabine in this randomized phase II trial of molecularly unselected patients with metastatic PC; we corroborated these findings in two independent genetic murine PC models (KPP and KR). Given the discrepant results between the two tested SMO inhibitors, their selectivity may uniquely diverge, where off-target effects might explain altered outcomes. Importantly, our preclinical findings were ultimately concordant with the outcome of our clinical trial. Given that newer chemotherapy regimens have demonstrated benefit over gemcitabine, a number of ongoing trials are evaluating the role vismodegib in PC using these more active cytotoxic backbones.54
Acknowledgment
This protocol was developed at the ECCO-AACR-EORTC-ESMO Workshop on Methods in Clinical Cancer Research, Flims Switzerland. We thank Michael Vannier, MD, for his contributions to the radiologic correlative studies and Michelle Nannini, Janeko Bower, Vincent Javinal, Alfonso Arrazate, Lee Nguyen, Alfred Wong, Linda Rangell, Carmina Espiritu, and Jeff Eastham-Anderson for excellent technical assistance. We also received extensive and able technical support from the Genentech genotyping and murine reproductive technology core groups. We thank William Forrest for providing biostatistical guidance and Chunang Gu for developing the methodology to assess gemcitabine metabolites. We thank Stephen Gould and Frank Peale for engaging scientific discussions and critical input regarding this article.
Footnotes
Supported by National Cancer Institute Grant No. N01-CM-62201 under the American Recovery and Reinvestment Act of 2009.
Presented at the 48th Annual Meeting of the American Society of Clinical Oncology (ASCO), Chicago, IL, June 1-5, 2012 (interim analysis), and 49th ASCO Annual Meeting, Chicago, IL, May 31-June 4, 2013 (final analysis).
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT01064622.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Daniel V.T. Catenacci, Melissa R. Junttila, Theodore Karrison, Naoko Takebe, Ravi Salgia, Walter M. Stadler, Frederic J. de Sauvage, Hedy L. Kindler
Financial support: Daniel V.T. Catenacci, Ravi Salgia, Walter M. Stadler, Hedy L. Kindler
Administrative support: Walter M. Stadler
Provision of study materials or patients: Daniel V.T. Catenacci, Nathan Bahary, Margit N. Horiba, Sreenivasa R. Nattam, Robert Marsh, James Wallace, Mark Kozloff, Lakshmi Rajdev, Deirdre Cohen, James Wade, Bethany Sleckman, Heinz-Josef Lenz, Patrick Stiff, Pankaj Kumar, Hedy L. Kindler
Collection and assembly of data: Daniel V.T. Catenacci, Melissa R. Junttila, Nathan Bahary, Margit N. Horiba, Sreenivasa R. Nattam, Robert Marsh, James Wallace, Mark Kozloff, Lakshmi Rajdev, Deirdre Cohen, James Wade, Bethany Sleckman, Heinz-Josef Lenz, Patrick Stiff, Pankaj Kumar, Peng Xu, Les Henderson, Naoko Takebe, Xi Wang, Hedy L. Kindler
Data analysis and interpretation: Daniel V.T. Catenacci, Melissa R. Junttila, Theodore Karrison, Nathan Bahary, Les Henderson, Naoko Takebe, Ravi Salgia, Xi Wang, Walter M. Stadler, Frederic J. de Sauvage, Hedy L. Kindler
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Daniel V.T. Catenacci
Honoraria: Genentech/Roche, Eli Lilly, Amgen, Oncoplex Dx, Foundation Medicine, Taiho Pharmaceutical, Genmab
Consulting or Advisory Role: Genentech/Roche, Oncoplex Dx, Amgen
Speakers' Bureau: Foundation Medicine, Oncoplex Dx, Eli Lilly
Research Funding: Amgen, Genentech, Oncoplex Dx
Melissa R. Junttila
Employment: Genentech
Stock or Other Ownership: Genentech
Theodore Karrison
No relationship to disclose
Nathan Bahary
Consulting or Advisory Role: Bayer
Research Funding: Newlink Genetics
Margit N. Horiba
No relationship to disclose
Sreenivasa R. Nattam
No relationship to disclose
Robert Marsh
Patents, Royalties, Other Intellectual Property: Antibodies for pineal tumor determination (royalty)
James Wallace
Consulting or Advisory Role: Eli Lilly
Speakers' Bureau: Medivation/Astellas Pharma
Mark Kozloff
Honoraria: Genentech
Consulting or Advisory Role: Genentech
Speakers' Bureau: Genentech
Lakshmi Rajdev
Consulting or Advisory Role: Genentech/Roche
Deirdre Cohen
Consulting or Advisory Role: Celgene
Speakers' Bureau: Genentech
James Wade
Employment: Johnson & Johnson (I)
Stock or Other Ownership: Seattle Genetics, Celgene
Bethany Sleckman
No relationship to disclose
Heinz-Josef Lenz
Honoraria: Merck Serono, Bayer, Roche
Consulting or Advisory Role: Merck Serono, Roche, Bayer
Travel, Accommodations, Expenses: Merck Serono, Bayer, Roche
Patrick Stiff
No relationship to disclose
Pankaj Kumar
No relationship to disclose
Peng Xu
No relationship to disclose
Les Henderson
No relationship to disclose
Naoko Takebe
No relationship to disclose
Ravi Salgia
No relationship to disclose
Xi Wang
No relationship to disclose
Walter M. Stadler
Honoraria: Bayer, Caremark, MedPacto, Johnson & Johnson, Merck, Sotio, Genentech/Roche, Pfizer
Consulting or Advisory Role: Bayer, Caremark, MedPacto, Sotio, Johnson & Johnson, Merck, Genentech/Roche, Pfizer
Research Funding: Active Biotech (Inst), Bayer (Inst), Bristol-Myers Squibb (Inst), Boehringer Ingelheim (Inst), Dendreon (Inst), Exelixis (Inst), Novartis (Inst), Genentech/Roche (Inst), GlaxoSmithKline (Inst), Medivation (Inst), Pfizer (Inst), Merck (Inst), Millennium Pharmaceuticals (Inst), Johnson & Johnson (Inst)
Other Relationship: UpToDate
Frederic J. de Sauvage
Employment: Genentech
Stock or Other Ownership: Roche/Genentech
Hedy L. Kindler
Consulting or Advisory Role: Genentech/Roche
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA, 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28:3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Cutsem E, Vervenne WL, Bennouna J, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27:2231–2237. doi: 10.1200/JCO.2008.20.0238. [DOI] [PubMed] [Google Scholar]
- 7.Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group–directed intergroup trial S0205. J Clin Oncol. 2010;28:3605–3610. doi: 10.1200/JCO.2009.25.7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kindler HL, Ioka T, Richel DJ, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: A double-blind randomised phase 3 study. Lancet Oncol. 2011;12:256–262. doi: 10.1016/S1470-2045(11)70004-3. [DOI] [PubMed] [Google Scholar]
- 9.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 10.Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449: A potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett. 2009;19:5576–5581. doi: 10.1016/j.bmcl.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 11.Wong H, Chen JZ, Chou B, et al. Preclinical assessment of the absorption, distribution, metabolism and excretion of GDC-0449 (2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide), an orally bioavailable systemic hedgehog signalling pathway inhibitor. Xenobiotica. 2009;39:850–861. doi: 10.3109/00498250903180289. [DOI] [PubMed] [Google Scholar]
- 12.Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 13.Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catenacci DV. Next-generation clinical trials: Novel strategies to address the challenge of tumor molecular heterogeneity. Mol Oncol. 2015;9:967–996. doi: 10.1016/j.molonc.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clinical Cancer Res. 2011;17:2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jimeno A, Weiss GJ, Miller WH, Jr, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clinical Cancer Res. 2013;19:2766–2774. doi: 10.1158/1078-0432.CCR-12-3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hidalgo M, Maitra A. The hedgehog pathway and pancreatic cancer. N Engl J Med. 2009;361:2094–2096. doi: 10.1056/NEJMcibr0905857. [DOI] [PubMed] [Google Scholar]
- 18.Morris JP, 4th, Wang SC, Hebrok M. KRAS, hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nature Rev Cancer. 2010;10:683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu FG, Ma QY, Wang Z. Blockade of hedgehog signaling pathway as a therapeutic strategy for pancreatic cancer. Cancer Lett. 2009;283:119–124. doi: 10.1016/j.canlet.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Kelleher FC. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis. 2011;32:445–451. doi: 10.1093/carcin/bgq280. [DOI] [PubMed] [Google Scholar]
- 21.Sandhiya S, Melvin G, Kumar SS, et al. The dawn of hedgehog inhibitors: Vismodegib. J Pharmacol Pharmacother. 2013;4:4–7. doi: 10.4103/0976-500X.107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasad NB, Biankin AV, Fukushima N, et al. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619–1626. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- 24.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 25.Bailey JM, Swanson BJ, Hamada T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clinical Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian H, Callahan CA, DuPree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106:4254–4259. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theunissen JW, de Sauvage FJ. Paracrine hedgehog signaling in cancer. Cancer Res. 2009;69:6007–6010. doi: 10.1158/0008-5472.CAN-09-0756. [DOI] [PubMed] [Google Scholar]
- 28.Walter K, Omura N, Hong SM, et al. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clinical Cancer Res. 2010;16:1781–1789. doi: 10.1158/1078-0432.CCR-09-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tremblay MR, Lescarbeau A, Grogan MJ, et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926) J Med Chem. 2009;52:4400–4418. doi: 10.1021/jm900305z. [DOI] [PubMed] [Google Scholar]
- 31.Feldmann G, Fendrich V, McGovern K, et al. An orally bioavailable small-molecule inhibitor of hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725–2735. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldmann G, Habbe N, Dhara S, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008;57:1420–1430. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catenacci D, Bahary N, Horiba N, et al. Final analysis of a phase Ib/Randomized phase II study of Gemcitabine (G) plus Placebo (P) or Vismodegib (V), a hedgehog (Hh) pathway inhibitor, in patients with metastatic pancreatic cancer (PC): A University of Chicago phase II consortium study. J Clin Oncol. 2013;31(suppl 15s):245s. doi: 10.1200/JCO.2015.62.8719. abstr 4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JJ, Perera RM, Wang H, et al. Stromal response to hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A. 2014;111:E3091–E3100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madden JI. Infinity reports update from phase 2 study of saridegib plus gemcitabine in patients with metastatic pancreatic cancer. http://www.businesswire.com/news/home/20120127005146/en/Infinity-Reports-Update-Phase-2-Study-Saridegib#.Vg2sAuGJMQM.
- 37.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 38.Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 39.Singh M, Lima A, Molina R, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 40.Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: Consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 41.Chen W, Tang T, Eastham-Anderson J, et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc Natl Acad Sci U S A. 2011;108:9589–9594. doi: 10.1073/pnas.1017945108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Zaatari M, Daignault S, Tessier A, et al. Plasma Shh levels reduced in pancreatic cancer patients. Pancreas. 2012;41:1019–1028. doi: 10.1097/MPA.0b013e31824a0eeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim EJ, Sahai V, Abel EV, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clinical Cancer Res. 2014;20:5937–5945. doi: 10.1158/1078-0432.CCR-14-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miles KA. Functional computed tomography in oncology. Eur J Cancer. 2002;38:2079–2084. doi: 10.1016/s0959-8049(02)00386-6. [DOI] [PubMed] [Google Scholar]
- 45.Abe H, Murakami T, Kubota M, et al. Quantitative tissue blood flow evaluation of pancreatic tumor: Comparison between xenon CT technique and perfusion CT technique based on deconvolution analysis. Radiat Med. 2005;23:364–370. [PubMed] [Google Scholar]
- 46.Kandel S, Kloeters C, Meyer H, et al. Whole-organ perfusion of the pancreas using dynamic volume CT in patients with primary pancreas carcinoma: Acquisition technique, post-processing and initial results. Eur Radiol. 2009;19:2641–2646. doi: 10.1007/s00330-009-1453-z. [DOI] [PubMed] [Google Scholar]
- 47.Hattori Y, Gabata T, Matsui O, et al. Enhancement patterns of pancreatic adenocarcinoma on conventional dynamic multi-detector row CT: Correlation with angiogenesis and fibrosis. World J Gastroenterol. 2009;15:3114–3121. doi: 10.3748/wjg.15.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoker MG, Shearer M, O'Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci. 1966;1:297–310. doi: 10.1242/jcs.1.3.297. [DOI] [PubMed] [Google Scholar]
- 49.Dotto GP, Weinberg RA, Ariza A. Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc Natl Acad Sci U S A. 1988;85:6389–6393. doi: 10.1073/pnas.85.17.6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 51.Gould SE, Low JA, Marsters JC, Jr, et al. Discovery and preclinical development of vismodegib. Expert Opin Drug Discov. 2014;9:969–984. doi: 10.1517/17460441.2014.920816. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Zhou Z, Walsh CT, et al. Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proc Natl Acad Sci U S A. 2009;106:2623–2628. doi: 10.1073/pnas.0812110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–444. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 54.Jesus-Acosta A, O'Dwyer PJ, Ramanathan RK, et al. A phase II study of vismodegib, a hedgehog (Hh) pathway inhibitor, combined with gemcitabine and nab-paclitaxel (nab-P) in patients (pts) with untreated metastatic pancreatic ductal adenocarcinoma (PDA) J Clin Oncol. 2014;(suppl 3):32. abstr 257. [Google Scholar]