

Abstract
PURPOSE
To determine the frequency and prognostic association of molecular markers by anatomic tumor site in patients with stage III colon carcinomas.
Experimental Design
In a randomized trial of adjuvant FOLFOX + cetuximab, BRAFV600E and KRAS (exon 2) mutations and DNA mismatch repair (MMR) proteins were analyzed in tumors (N=3,018) in relationship to tumor location including subsite. Cox models were used to assess clinical outcome including overall survival (OS).
RESULTS
KRAS codon 12 mutations were most frequent at the splenic flexure and cecum; codon 13 mutations were evenly distributed. BRAF mutation frequency sharply increased from transverse colon to cecum in parallel with deficient (d) MMR. Non-mutated BRAF or KRAS tumors progressively decreased from sigmoid to transverse (all p<0.0001). Significantly poorer OS was found for mutant KRAS in distal [HR, 1.98 (1.49–2.63); p<.0001] vs proximal [1.25 (0.97–1.60), p=.079] cancers. BRAF status and outcome were not significantly associated with tumor site. Proximal vs distal dMMR tumors had significantly better outcome. An interaction test was significant for tumor site by KRAS (padjusted=.043) and MMR (padjusted=.010) for OS. Significant prognostic differences for biomarkers by tumor site were maintained in the FOLFOX arm. Tumor site was independently prognostic with a stepwise improvement from cecum to sigmoid (OS: padjusted=0.001).
CONCLUSIONS
Mutations in BRAF or KRAS codon 12 were enriched in proximal cancers whereas non-mutated BRAF/KRAS were increased in distal tumors. Significant differences in outcome for KRAS mutations and dMMR were found by tumor site, indicating that their interpretation should occur in the context of tumor location.
Keywords: colon cancer, adjuvant, tumor site, BRAF, KRAS, MSI, prognosis
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide and is a leading cause of cancer-related death (1). The majority of colon cancers, especially distal cancers, display chromosomal instability (CIN) in association with frequent loss of heterozygosity and aneuploidy (2). CIN tumors are enriched in KRAS mutations that occur as early events during tumorigenesis (3). Alternatively, a subset of CRCs have microsatellite instability (MSI) and hypermutation due to deficient DNA mismatch repair (dMMR) (4). These tumors most commonly develop dMMR due to epigenetic inactivation of the MLH1 MMR gene and the CpG island methylator phenotype (CIMP), along with frequent oncogenic BRAF (V600E) mutations (5, 6). Colon cancers with BRAFV600E, including those with dMMR, are believed to arise via a serrated neoplasia pathway named for the pattern of mucosal crypts in precursor polyps, and have a strong predilection for the proximal colon (7). Mutations in BRAFV600E are mutually exclusive with KRAS, and both lead to the activation of MEK/ERK signaling pathway (3, 8) that promotes tumor progression. While BRAF mutation is associated with adverse outcome in the metastatic CRC (9, 10), less is known about its association or that of KRAS with patient outcome in earlier stage disease.
The primary site of colon cancer is typically categorized as located proximal to the splenic flexure, at or distal to this flexure, or in the rectum based upon their embryological origin. Mutations in BRAFV600E and dMMR are known to be enriched in cancers of the proximal colon (9, 10), although their subsite distribution and that of KRAS remain unknown. While the mechanisms underlying tumor site/subsite-related biological differences are poorly understood, distal colon cancers show more frequent chromosomal aberrations whereas proximal cancers have higher mutation rates even after excluding the subset of hypermutated dMMR tumors (11). There is a need to examine the subsite distribution of relevant molecular alterations and to examine their prognostic association by anatomic site to gain biologic insights and further guide prognostication. Mortality rates have been reported to be higher in proximal compared to distal colon cancers, including lymph node-positive, i.e., stage III, tumors (12–14). However, studies examining tumor site and prognosis generally lack data on key molecular markers that precludes their adjustment in multivariable models, and cohorts have not received standard adjuvant chemotherapy with the FOLFOX regimen.
In this report, we examined the frequency of molecular alterations by primary tumor subsite, and tested the hypothesis that the association of BRAFV600E and KRAS mutations with clinical outcome depends on their anatomic site within the colon. We also determined whether tumor site-related differences in clinical outcome persist after adjustment for BRAFV600E and KRAS mutations as well as by MMR status. Our study population consists of stage III colon cancers from an adjuvant chemotherapy trial that found no survival benefit for the addition of cetuximab to standard FOLFOX chemotherapy (15). In contrast to prior reports in the N0147 cohort that were limited to tumors lacking KRAS mutations based upon the primary endpoint analysis (15, 16), the current study includes patients with KRAS mutated tumors and reports mature overall survival (OS) data for the trial.
METHODS
Study Population
Patients with resected, stage III (any T, N1 or N2, M0) colon adenocarcinomas participated in a phase III study (15) of which 2,686 were concurrently randomized to mFOLFOX6 or mFOLFOX6 + cetuximab [NCCTG N0147]. Study participants in the clinical trial were from multiple institutions across North America. The trial was amended post-initiation to restrict randomization to patient tumors lacking KRAS exon 2 mutations given demonstration of predictive utility for anti-EGFR antibody therapy in metastatic disease (17). Post-amendment, patients with mutated (n=326) or uninterpretable (n=6) KRAS results were treated at investigator discretion [97% with treatment information received FOLFOX] and followed for recurrence/survival (total N= 3018). Central pathology review was performed that included confirmation of the diagnosis and evaluation of T and N stage. Stratification factors included: histologic grade [high: poor/undifferentiated) vs low: well/moderately differentiated)], T stage (T1,2 vs T3 vs T4) and N stage (N1: 1–3 vs N2: ≥4 metastatic regional lymph nodes. Tumor site was categorized as proximal or at or distal to the splenic flexure. The study was approved by Mayo Clinic Institutional Review Board and NCCTG (now part of Alliance for Clinical Trials in Oncology). Each participant signed an IRB-approved, protocol-specific informed consent in accordance with current guidelines. Data quality was ensured by review of data by the Alliance Statistics and Data Center and by the study chairperson per Alliance policies.
BRAFV600E and KRAS Gene Mutations
Mutation status was determined using genomic DNA extracted from macrodissected tumor tissue collected prospectively. Testing for the BRAF c.1799T>A (V600E) mutation in exon 15 was performed using a multiplex allele-specific, real-time polymerase chain reaction (RT-PCR)-based assay and an automated sequencing technique (6). Primer sequences included: (wild-type forward [NED-TGATTTTGGTCATGCTACAGT]; mutant forward [6-Fam-CAGTGATTTTGCTCTAGCTTCAGA]; and reverse [GTTTCTTTCTAGTAACTCAGCAGC]. KRAS status was analyzed in extracted DNA using the DxS mutation test kit KR-03/04 (DxS, Manchester, UK), assessing for seven different mutations in codons 12 and 13 (18). For both genes, analysis was performed in a Clinical Laboratory Improvement Amendments (CLIA)-compliant laboratory at Mayo Clinic.
DNA Mismatch Repair Protein Expression
MMR protein (MLH1, MSH2 and MSH6) expression was analyzed in formalin-fixed, paraffin-embedded tumor sections as previously described (19). Monoclonal antibodies included mouse anti-human MLH1 (clone G168-15, Biocare Medical, Concord, CA), anti-human MSH2 (clone FE11, Biocare), and anti-human MSH6 (clone BC/44; Biocare). MMR protein loss was defined as the absence of nuclear staining in tumor cells in the presence of nuclear staining in normal colonic epithelium and lymphocytes. Tumors were categorized as deficient (d) vs proficient (p) MMR if loss of at least one MMR protein was detected.
STATISTICAL ANALYSIS
We examined the relationship between the anatomic location of the primary tumor and biomarkers with time-to-recurrence (TTR), disease-free survival (DFS), and OS. All biomarker assays were interpreted with investigators blinded to patient outcomes. Treatment arms were pooled for all biomarker analyses given the lack of statistically significant differences in patient outcome or interactions between any of the biomarkers and treatment. Analysis within the FOLFOX alone treatment arm was also performed based upon clinical relevance. The Cochran-Armitage test for trend was used to assess the relationship between tumor site and biomarker status. Kaplan-Meier methods were used to describe the distributions of outcome (20). Univariate Cox proportional hazard models (21) were used to explore the associations of patient/tumor characteristics and biomarkers with outcome. Multivariate models were adjusted for stratification factors (see Study Population), tumor site, age, sex, treatment, and status of MMR, BRAF, or KRAS. TTR and DFS were calculated as the time from randomization until disease recurrence, or were censored at the date of last disease assessment if no recurrence occurred. For DFS, patients having died without recurrence were considered an event using date of death. OS was calculated as the time from randomization until date of death; surviving patients were censored for OS at the date of last contact/follow-up. All patients were censored at 5 years (DFS, TTR) and 8 years post-randomization (OS). Patients were analyzed according to treatment arm assignment including those who withdrew or were deemed ineligible. Median follow-up for 2,394 (79%) surviving patients was 4.9 years (range 0.0–8). Two-sided p-values are reported; values <0.05 were considered statistically significant and were not adjusted for multiple comparisons. Analyses were performed using SAS version 9.3 (SAS Institute Inc, Cary NC) and R version 3.02 (22).
RESULTS
Patient, Tumor, and Clinicopathologic Characteristics
Characteristics of the study population of stage III colon cancer patients (N=3,018) are shown stratified by primary tumor site or subsite and by molecular markers (Table 1). The overall frequency of KRAS exon 2 and BRAFV600E mutations was 36% (1042/2905) and 12.2% (346/2831), respectively. Among KRAS mutant colon cancers, 811 (77.8%) had codon 12 mutations and 231 (22.2%) had codon 13 mutations. BRAFV600E and KRAS mutations were mutually exclusive, and mutant BRAFV600E was associated with older age, female sex, high grade histology, and N2 stage (Table 1). Deficient MMR (dMMR) was detected in 11% (329/2904) of cancers and among them, 149 (47%) carried BRAFV600E mutations; 49 (15%) had KRAS mutations (Table 1). Tumors with KRAS mutations were significantly less likely to have dMMR (Table 1). There was a nearly equal distribution of proximal and distal cancers. Patients with proximal tumors were significantly older (median age of 60 vs 56 years; p<0.001), more often female (50 vs 45%, p=0.015), and their tumors were more likely to have high grade histology (32% vs 17%, p< 0.001) and higher T stage (p <0.001) [T1, 2 (12 vs 19%), T3 (74 vs 71%), T4 (14 vs 11%) ] (Table 1). Proximal vs distal tumors were significantly more likely to carry mutations in KRAS [41% vs 30%; p<0.001] or BRAFV600E [20% vs 4%; p<0.001], and to have dMMR [20% vs 3%, p<0.001] (Table 1).
Table 1.
Patient Tumor and Clinicopathologic Characteristics
| MMR | BRAF | KRAS | Tumor Site | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dMMR N=329 |
pMMR N=2575 |
P-value | Mutated N=346 |
Non- mutated N=2485 |
P-value | Mutated N=1042 |
Non- mutated N=1863 |
P-value | Proximal N=1511 |
Distal N=1463 |
P-value | |
| Age | 0.0011 | <0.0011 | 0.7061 | <0.0011 | ||||||||
| Median | 61.0 | 58.0 | 65.0 | 57.0 | 58.0 | 58.0 | 60.0 | 56.0 | ||||
| Range | (22.0–86.0) | (19.0–85.0) | (31.0–86.0) | (19.0–85.0) | (22.0–85.0) | (19.0–86.0) | (19.0–86.0) | (19.0–85.0) | ||||
| Treatment | 0.0042 | <0.0012 | <0.0012 | 0.0442 | ||||||||
| mFOLFOX6 | 158 (48%) | 1453 (56%) | 155 (45%) | 1409 (57%) | 700 (67%) | 909 (49%) | 808 (53%) | 836 (57%) | ||||
| mFOLFOX6+cetuximab | 171 (52%) | 1122 (44%) | 191 (55%) | 1076 (43%) | 342 (33%) | 954 (51%) | 703 (47%) | 627 (43%) | ||||
| Sex | <0.0012 | <0.0012 | 0.3112 | 0.0152 | ||||||||
| Female | 189 (57%) | 1189 (46%) | 224 (65%) | 1119 (45%) | 507 (49%) | 870 (47%) | 754 (50%) | 665 (45%) | ||||
| Male | 140 (43%) | 1386 (54%) | 122 (35%) | 1366 (55%) | 535 (51%) | 993 (53%) | 757 (50%) | 798 (55%) | ||||
| Histologic Grade | <0.0012 | <0.0012 | <0.0012 | <0.0012 | ||||||||
| High | 172 (52%) | 546 (21%) | 167 (48%) | 543 (22%) | 212 (20%) | 508 (27%) | 482 (32%) | 253 (17%) | ||||
| Low | 157 (48%) | 2029 (79%) | 179 (52%) | 1942 (78%) | 830 (80%) | 1355 (73%) | 1029 (68%) | 1210 (83%) | ||||
| Lymph Node Involvement | 0.5292 | <0.0012 | 0.0112 | 0.2652 | ||||||||
| 1–3 | 198 (60%) | 1503 (58%) | 170 (49%) | 1486 (60%) | 642 (62%) | 1058 (57%) | 866 (57%) | 868 (59%) | ||||
| >=4 | 131 (40%) | 1072 (42%) | 176 (51%) | 999 (40%) | 400 (38%) | 805 (43%) | 645 (43%) | 595 (41%) | ||||
| Stage | 0.0122 | 0.0072 | 0.0792 | <0.0012 | ||||||||
| Missing | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | ||||
| T1 or T2 | 32 (10%) | 410 (16%) | 37 (11%) | 389 (16%) | 159 (15%) | 278 (15%) | 185 (12%) | 272 (19%) | ||||
| T3 | 252 (77%) | 1855 (72%) | 253 (73%) | 1803 (73%) | 736 (71%) | 1375 (74%) | 1119 (74%) | 1033 (71%) | ||||
| T4 | 45 (14%) | 309 (12%) | 56 (16%) | 292 (12%) | 146 (14%) | 210 (11%) | 207 (14%) | 157 (11%) | ||||
| Tumor Subsite | <0.0012 | <0.0012 | <0.0012 | |||||||||
| Missing | 30 | 134 | 24 | 139 | 55 | 109 | 99 | 26 | ||||
| Cecum | 90 (30%) | 474 (19%) | 91 (28%) | 461 (20%) | 277 (28%) | 289 (16%) | 587 (42%) | 0 (0%) | ||||
| Ascending colon | 88 (29%) | 362 (15%) | 91 (28%) | 337 (14%) | 177 (18%) | 271 (15%) | 471 (33%) | 0 (0%) | ||||
| Hepatic flexure | 43 (14%) | 79 (3%) | 39 (12%) | 83 (4%) | 35 (4%) | 89 (5%) | 127 (9%) | 0 (0%) | ||||
| Transverse colon | 41 (14%) | 178 (7%) | 49 (15%) | 168 (7%) | 82 (8%) | 137 (8%) | 227 (16%) | 0 (0%) | ||||
| Splenic flexure | 8 (3%) | 102 (4%) | 7 (2%) | 99 (4%) | 49 (5%) | 61 (3%) | 0 (0%) | 116 (8%) | ||||
| Descending colon | 6 (2%) | 144 (6%) | 9 (3%) | 136 (6%) | 54 (5%) | 96 (5%) | 0 (0%) | 156 (11%) | ||||
| Sigmoid colon | 23 (8%) | 1102 (45%) | 36 (11%) | 1062 (45%) | 313 (32%) | 811 (46%) | 0 (0%) | 1165 (81%) | ||||
| Tumor Site | <0.0012 | <0.0012 | <0.0012 | |||||||||
| Missing | 8 | 34 | 5 | 38 | 19 | 25 | ||||||
| Proximal | 283 (88%) | 1168 (46%) | 287 (84%) | 1126 (46%) | 599 (59%) | 852 (46%) | ||||||
| Distal | 38 (12%) | 1373 (54%) | 54 (16%) | 1321 (54%) | 424 (41%) | 986 (54%) | ||||||
| KRAS | <0.0012 | <0.0012 | <0.0012 | |||||||||
| Missing | 5 | 40 | 2 | 6 | 60 | 53 | ||||||
| Mutated | 49 (15%) | 981 (39%) | 0 (0%) | 1000 (40%) | 599 (41%) | 424 (30%) | ||||||
| Non-mutated | 275 (85%) | 1554 (61%) | 344 (100%) | 1479 (60%) | 852 (59%) | 986 (70%) | ||||||
| KRAS Submutation | <0.0012 | <0.0012 | ||||||||||
| Missing | 5 | 40 | 2 | 6 | 0 | 0 | 60 | 53 | ||||
| Non-mutated | 275 (85%) | 1554 (61%) | 344 (100%) | 1479 (60%) | 0 (0%) | 1863 (100%) | 852 (59%) | 986 (70%) | ||||
| Codon 12 Mutant | 28 (9%) | 773 (31%) | 0 (0%) | 780 (31%) | 811 (78%) | 0 (0%) | 458 (32%) | 338 (24%) | ||||
| Codon 13 Mutant | 21 (6%) | 207 (8%) | 0 (0%) | 220 (9%) | 231 (22%) | 0 (0%) | 141 (10%) | 86 (6%) | ||||
| BRAF | <0.0012 | <0.0012 | <0.0012 | |||||||||
| Missing | 11 | 104 | 42 | 40 | 98 | 88 | ||||||
| Mutated | 149 (47%) | 190 (8%) | 0 (0%) | 344 (19%) | 287 (20%) | 54 (4%) | ||||||
| Non-mutated | 169 (53%) | 2281 (92%) | 1000 (100%) | 1479 (81%) | 1126 (80%) | 1321 (96%) | ||||||
| MMR | <0.0012 | <0.0012 | <0.0012 | |||||||||
| Missing | 7 | 35 | 12 | 34 | 60 | 52 | ||||||
| Proficient (p) MMR | 190 (56%) | 2281 (93%) | 981 (95%) | 1554 (85%) | 1168 (80%) | 1373 (97%) | ||||||
| Deficient (d) MMR | 149 (44%) | 169 (7%) | 49 (5%) | 275 (15%) | 283 (20%) | 38 (3%) | ||||||
Kruskal Wallis
Chi-Square
We examined the association of mutations in BRAFV600E and KRAS in stage III colon carcinomas with primary tumor subsite. The distribution of molecular markers was significantly associated with anatomic tumor subsite within the colon (all p<0.0001) (Table 1, Fig. 1). KRAS mutations were more evenly distributed across the subsites compared to BRAFV600E mutations or dMMR. The highest rates of KRAS codon 12 mutations were found in cancers at the cecum and splenic flexure, whereas the rate of KRAS codon 13 mutations was similar across tumor subsites (Fig. 1). Cancers lacking mutations in KRAS and BRAFV600E were significantly more likely to be located in the distal colon with a progressive decrease from sigmoid to transverse colon. An abrupt increase in BRAFV600E mutant cancers was seen beginning in the transverse colon and extending to the cecum with a peak in frequency at the hepatic flexure (Fig. 1). The distribution of BRAFV600E mutations in tumors directly paralleled that of dMMR with both being highest in tumors at the hepatic flexure followed by the ascending colon and cecum (Table 1). Interestingly, the distribution of mutant BRAFV600E tumors by subsite was similar in dMMR and pMMR tumors. Tumors with dMMR and a non-mutated BRAF gene were also predominantly located in the proximal colon (Fig. 1). In contrast, pMMR tumors with non-mutated BRAF were more commonly distal with the highest rate detected in the sigmoid colon.
Figure 1.
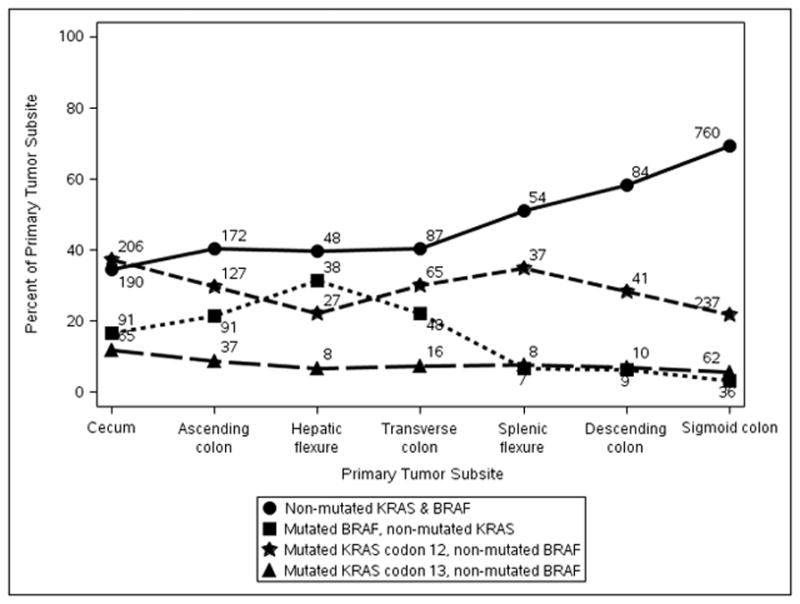
Frequency of Molecular Alterations by Primary Tumor Subsite in Stage III Colon Carcinomas. Data are shown for KRAS and BRAFV600E mutations as a combined variable. Separation of KRAS mutations into those located in codon 12 vs codon 13 of exon 2 are shown by tumor subsite.
Association of Molecular Markers with Prognosis
Mutations in KRAS or BRAFV600E were each associated with significantly poorer time-to-recurrence (TTR), disease-free survival (DFS) and overall survival (OS) in univariate models (all p<.0094) [Suppl Table e-1]. Adverse outcome was found for patients whose tumors carried KRAS codon 12 or codon 13 mutations compared to those whose tumors were non-mutated for BRAFV600E (Fig. 2). Univariately, dMMR was not significantly associated with the time-to-event variables (Suppl Table e-1). No statistically significant interactions were observed for any of the biomarkers and adjuvant treatment arm.
Figure 2.

Time-to-recurrence (% Recurrence-free; top] and overall survival (% Alive; bottom] for the combined variable of KRAS (codons 12, 13) and BRAF mutation status among the overall cohort of stage III colon cancer patients treated with adjuvant FOLFOX-based chemotherapy.
Clinical variables that were significantly associated with patient outcome in a multivariable analysis included patient sex, T and N stage and histologic grade (Suppl Table e-2). After adjustment for covariates, mutations in KRAS or BRAFV600E were each significantly associated with worse TTR, DFS and OS in the overall cohort (Suppl Table e-2; Fig. 2). Patients whose tumors had non-mutated KRAS and BRAF had significantly better outcome compared to those with a mutation in either gene (Fig. 2). In the overall cohort, dMMR was associated with a statistically significantly delay in TTR [HR 0.71, 95% CI (0.55–0.93; p=.0144], but not better DFS (23) or OS compared to pMMR after adjustment for covariates (Suppl Table e-2).
Association of Molecular Markers with Outcome by Primary Tumor Site
Distal vs proximal cancers with mutated vs non-mutated KRAS had poorer outcome that achieved statistical significance for OS [distal HR, 1.98 (1.49–2.63); p<.0001 vs proximal 1.25 (0.97–1.60), p=.079], and showed a trend for stronger impact for TTR and DFS (Table 2). The interaction between mutations in KRAS and primary tumor site for OS was statistically significant (pinteraction= .043) in the overall cohort [Table 2]. The association of BRAFV600E mutations with poor outcome did not differ by primary tumor site based upon interaction tests that were not statistically significant. However, a statistically significant interaction was found between MMR status and primary tumor site for TTR, DFS, and OS (all pinteraction ≤ 0.01) [Table 2]. In this regard, significantly better outcome was observed for dMMR cancers of the proximal [TTR: HR=0.57, 95% CI (0.42–0.78), p≤0.001; OS: HR=0.71, 95% CI (0.53–0.97), p=0.03] but not the distal colon compared to pMMR tumors (Table 2). In the distal colon, dMMR vs pMMR cancers were associated with poor outcomes [TTR: HR=1.86, 95% CI (1.11–3.12), p=0.019]; OS: HR=1.85, 95% CI(0.99–3.46), p=0.054](Table 2), as we previously reported for DFS (16).
Table 2.
Multivariate Models within Proximal * and Distal Tumor Site
| Biomarker | DFS: N(Events): 1389 (477) HR (95% CI), p value |
OS: N(Events): 1389 (365) HR (95% CI), p value |
TTR: N(Events): 1389 (417) HR (95% CI), p value |
|---|---|---|---|
| MMR: dMMR vs pMMR | |||
| Overall | 0.82 (0.64–1.04), 0.106 | 0.83 (0.63–1.09), 0.182 | 0.71 (0.55–0.93), 0.014 |
| Proximal | 0.69 (0.53–0.91), 0.009 | 0.71 (0.53–0.97), 0.029 | 0.57 (0.42–0.78), 0.0004 |
| Distal | 1.75 (1.05–2.93), 0.032 | 1.85 (0.99–3.46), 0.054 | 1.86 (1.11–3.12), 0.019 |
| MMR* Site Interaction P-value | 0.004 | 0.010 | 0.0003 |
| BRAF: Mutated vs Non-mutated | |||
| Overall | 1.36 (1.08–1.71), 0.009 | 1.70 (1.31–2.20), <.0001 | 1.42 (1.11–1.81), 0.005 |
| Proximal | 1.35 (1.03–1.76), 0.028 | 1.58 (1.17–2.12), 0.003 | 1.40 (1.05–1.86), 0.024 |
| Distal | 1.25 (0.75–2.11), 0.394 | 1.79 (0.99–3.24), 0.053 | 1.42 (0.84–2.39), 0.190 |
| BRAF* Site Interaction P-value | 0.707 | 0.903 | 0.994 |
| KRAS: Mutated vs Non-mutated | |||
| Overall | 1.45 (1.24–1.69), <.0001 | 1.52 (1.26–1.84), <.0001 | 1.49 (1.27–1.76), <.0001 |
| Proximal | 1.27 (1.03–1.57), 0.027 | 1.25 (0.97–1.60), 0.079 | 1.29 (1.03–1.61), 0.028 |
| Distal | 1.66 (1.34–2.07), <.0001 | 1.98 (1.49–2.63), <.0001 | 1.72 (1.37–2.17), <.0001 |
| KRAS* Site Interaction P-value | 0.151 | 0.043 | 0.162 |
Adjusted for age, treatment, sex, t-stage, histologic grade, and nodal status in addition to the variables shown. The results of these variables within site are similar to the overall model.
Since both the N0147 (15) and PETACC-8 (24) clinical trials in stage III cancers did not find a benefit for the addition of cetuximab to standard adjuvant FOLFOX, only biomarker data from the FOLFOX treatment arm are directly relevant to clinical practice. Accordingly, analysis of biomarkers by tumor site within the FOLFOX treatment arm was performed with adjustment for covariates. The association of mutations in KRAS and BRAF, as well as MMR status, with clinical outcome by tumor site was found to be consistent with the overall cohort (Fig. 3). Specifically, patients with distal vs proximal cancers with mutated vs non-mutated KRAS had poorer outcome (Fig. 3A). The association of mutated vs non-mutated BRAF with adverse outcome was more evident in proximal cancers yet the interaction test did not reach statistical significance as previously indicated (Fig. 3B). In the FOLFOX alone arm, the association of dMMR vs pMMR with better outcome variables was restricted to proximal cancers as was found in the overall cohort (Fig. 3C). In distal tumors, the association of dMMR vs pMMR with significantly worse outcome did not achieve statistical significance in the FOLFOX alone arm.
Figure 3.



Association of KRAS (A) or BRAF (B) mutations and DNA mismatch repair status (C) with time-to-recurrence (% Recurrence-free; top) or overall survival (% Alive; bottom) stratified by primary tumor site [proximal (left); distal (right)]. Data are shown in stage III colon cancer patients from the FOLFOX alone treatment arm.
Prognostic Impact of Primary Tumor Site
Tumors of the proximal (vs distal) colon were significantly associated with poorer outcome for TTR, DFS, and OS in the overall cohort after adjustment for covariates (all p<0.05; Suppl Table e-2). Multivariable analysis by tumor subsite demonstrated that OS improved significantly, and a similar trend was observed for TTR, with distal migration of the primary tumor following the anatomic order from cecum to sigmoid colon (i.e., cecum, ascending, hepatic flexure, transverse, splenic flexure, descending, and sigmoid) [TTR, p=0.0646; OS, p=0.0011](Fig. 4). Patients with cecal cancers had the worst outcome whereas those with sigmoid tumors had the best outcome as illustrated by Forest plots for TTR and OS (Fig. 4). Specifically, patients with cecal and ascending colon cancers had 5 year OS rates of 70% (66–74) and 74% (69–78) respectively, whereas OS rates for patients with descending or sigmoid cancers were 89% (83–94) or 85% (83–87) (Fig. 4). Similar results were found for 5-year TTR by tumor site (cecum 67%, ascending 67%; descending 71%, sigmoid 73%).
Figure 4.

Clinical outcome of the overall cohort of stage III colon cancer patients by anatomic subsite of the primary tumor. Multivariate hazard ratios for TTR and OS are shown with the cecum as the reference group.
DISCUSSION
We examined the association of mutations in BRAFV600E and KRAS in stage III colon carcinomas with primary tumor subsite from patients treated in a large adjuvant chemotherapy trial. KRAS exon 2 mutations were more evenly distributed throughout the colon than were BRAFV600E mutations. KRAS codon 12 mutations had the highest rates in tumors of the cecum and splenic flexure whereas the rate of codon 13 mutations was relatively similar across tumor subsites. KRAS mutations were detected in only 15% of dMMR tumors consistent with their more frequent association with pMMR and the CIN pathway (25). The distribution of BRAFV600E mutations showed a marked increase with proximal migration and peak in frequency in tumors at the hepatic flexure. This distribution directly paralleled that of dMMR colon cancers that are known to have a strong predilection for the proximal colon (23, 26). Our findings in stage III cancers differ from those of Yamauchi et al (26) in all stages of CRC from a non clinical trial cohort where the frequencies of BRAFV600E mutations, CIMP, and MSI-H increased linearly from the rectum to the ascending colon, although both studies found that KRAS mutations were most frequent in cecal cancers. Importantly, we found that the proximal colon predominance for BRAFV600E mutations persisted when the cohort was restricted to pMMR cancers, consistent with the earlier development of BRAFV600E-mutations that appear to develop in a site-specific manner during colonic tumorigenesis. BRAF mutant colon cancers are believed develop via the serrated neoplasia pathway from precursor lesions known as sessile serrated adenomas/polyps that are characterized by frequent BRAF mutations, CIMP-high, and proximal colon predominance (7). Analysis of stage II and III colon cancers from the PETACC-3 adjuvant study (11, 27) and the TCGA series (4) also found higher rates of BRAFV600E and KRAS mutations in proximal vs distal colon cancers. While the mechanisms underlying the observed tumor site-related differences in the mutation rates of these oncogenes and dMMR remain unknown, they may reflect specific mechanisms of carcinogenesis and/or differences in embryologic origin with the proximal colon derived from the midgut and the distal colon from the hindgut. In addition, differences in colonic microbiota have been found in the proximal and distal regions of the colon that may influence molecular events and host immune responses during carcinogenesis in a site-specific manner (28, 29).
Colon cancers with BRAFV600E mutations were significantly associated with higher rates of proximal site, age, female sex, high grade histology, dMMR status and N2 stage compared to mutant KRAS tumors. These clinicopathological features were similarly associated with dMMR tumors except for higher N stage. At 5 years of median follow-up, BRAFV600E mutations were independently associated with adverse outcome, including OS, in the overall cohort (16) which is consistent with other studies (30–32). Studies also indicate that BRAFV600E mutations are associated with worse patient survival after tumor recurrence (32–34); however, such an analysis in our cohort awaits more extended patient follow-up. In the overall cohort, we found that KRAS codon 12 mutations were significantly associated with worse outcome variables compared to tumors that were non-mutated for KRAS or BRAF. Mutations at codon 13 were also significantly associated with worse DFS as we reported previously (35), and a strong trend for worse TTR and OS was also found. KRAS codon 13 mutations were of borderline significance for poorer DFS in the PETACC-8 adjuvant study that had identical treatment arms to our trial, yet had fewer codon 13 mutated tumors (136 vs 231) (36). in a database from two nationwide prospective cohort studies, the lack of association of codon 13, in contrast to codon 12, mutations may also be due to sample size since only 108 codon 13-mutated colon cancers were analyzed (37). Patients with dMMR cancers showed a statistically significant delay in TTR and showed a trend for better DFS (p=0.10) and OS (p=0.18) rates compared to patients with pMMR tumors. These data are consistent with other studies where the association of dMMR or MSI-H status with favorable prognosis is attenuated in stage III compared to stage II colon cancer patients treated with 5-FU-based adjuvant chemotherapy (38). Among stage III disease, the attenuated effect of dMMR on survival may be related to our finding of a similar proportion of dMMR and pMMR N2 tumors where both N2 subgroups showed poorer outcome in the N0147 cohort (39).
We tested the hypothesis that the association of KRAS and BRAFV600E mutations with clinical outcome depends on the anatomic site of the primary tumor. In the overall cohort, we found a statistically significant interaction between mutations in KRAS, but not BRAFV600E, and primary tumor site for OS. Specifically, KRAS mutations were associated with adverse outcome in distal vs proximal cancers that reached statistical significance for OS. Non-mutated BRAF/KRAS tumors were enriched in distal tumors, and we previously reported that this patient subgroup had a favorable clinical outcome (35). We also observed a statistically significant interaction between MMR status and tumor site for TTR, DFS and OS. Specifically, proximal but not distal dMMR cancers were associated with significantly better outcomes. In a prior study, we also reported an interaction between MMR status and tumor site for survival in patients treated with 5-FU/leucovorin alone or combined with irinotecan (CALGB 89803) (16).
Since cetuximab has no role in the adjuvant therapy of colon cancer (15, 24), only data from the standard FOLFOX treatment arm are directly relevant to clinical practice. Accordingly, we performed a biomarker analysis restricted to the FOLFOX arm and found results that were highly consistent with those in the overall cohort. Specifically, the association of KRAS mutations with poorer outcomes remained more evident in distal vs proximal cancers. While the association of BRAFV600E mutations with poorer OS was again limited to proximal tumors, the interaction test was not significant. Lastly, the better prognosis of dMMR vs pMMR tumors was limited to tumors of the proximal colon in the FOLFOX alone arm. Data from the PETACC-3 adjuvant trial that evaluated 5-FU/LV + irinotecan also found significantly better OS of patients with MSI-H vs MSS/MSI-L cancers of the proximal vs distal colon (38). The association of dMMR vs pMMR with significantly poorer outcome in distal tumors in the overall cohort was less evident in the FOLFOX alone arm potentially due to the small sample size.
An unresolved issue is whether primary tumor site-related differences in outcome persist after adjustment for MMR, KRAS and BRAF status. We found that tumor site was independently associated with prognosis after adjustment for BRAFV600E and KRAS mutations as well as for MMR status in the overall cohort. Further analysis by tumor subsite revealed that anatomical migration of the primary tumor from the proximal to distal colon was significantly associated with a stepwise improvement in overall survival. Patients with cecal cancers had the worst outcome whereas those located in the sigmoid colon had the best survival rates. While the specific explanation for this finding is unknown, analysis of colon cancers from the PETACC-3 (11) and TCGA (4) studies found that proximal cancers have higher mutation rates even after excluding dMMR tumors, and potentially more deleterious mutations.
Data on molecular alterations by tumor subsite can improve our understanding of mechanisms of colon carcinogenesis with implications for clinical, epidemiological and pathological research. Our finding that primary tumor site remained prognostic after adjustment for molecular markers, suggesting its use as stratification factor for patients receiving adjuvant FOLFOX therapy. Strengths of our study include the large size of the clinical trial cohort, prospective analysis of biomarkers using uniform assay methodology in a single CLIA certified laboratory, treatment with the standard adjuvant FOLFOX regimen, and long term follow-up data. Only two other studies have examined molecular marker data from colon cancer patients treated with the adjuvant FOLFOX regimen (33, 40). Study limitations include that KRAS mutation analysis was limited to exon 2 and the inability to examine the predictive utility of the molecular markers given that all patients received adjuvant chemotherapy. We acknowledge potential sources of bias inherent in clinical trial cohorts including referral bias, the presence of strict eligibility criteria, and the requirement for tumor tissue in our study that may limit the generalizability of results.
In conclusion, mutations in BRAF or KRAS codon 12 were significantly more frequent in proximal cancers and associated with adverse outcome, whereas non-mutated BRAF/KRAS tumors were significantly enriched in distal tumors and associated with favorable outcome. The association of KRAS mutations with OS was dependent upon primary tumor site, with significantly poorer OS seen for patients with distal vs proximal cancers. The association of dMMR with significantly better outcome variables was limited to proximal tumors. These findings were maintained in the FOLFOX alone treatment arm which underscores their relevance to clinical practice. Taken together, our data demonstrate that use of KRAS and MMR status for prognostication in stage III colon cancer patients requires consideration of the location of the primary cancer within the colon.
Supplementary Material
Translational Relevance.
Among patients with CRC, there is considerable stage-independent variability in clinical outcome that is due, in part, to intratumor heterogeneity reflected in pathway-related molecular alterations. Given reported differences in the biology and prognosis of colon cancers by tumor site, we examined mutations in BRAFV600E and KRAS genes and the status of the DNA mismatch repair (MMR) system by site and their association with clinical outcome. Among patients with stage III colon carcinomas who participated in an adjuvant chemotherapy trial, we found primary tumor subsite-related differences in frequencies of BRAFV600E and KRAS mutations as well as MMR. Tumors with mutations in BRAFV600E or KRAS codon 12 were more frequent in proximal colon whereas those with non-mutated BRAF/KRAS were enriched in the distal colon. Among tumors with KRAS mutations or deficient MMR, differences in patient outcome were found by tumor site, suggesting that interpretation should occur in the context of tumor location.
Acknowledgments
Financial support: This work was supported by a National Cancer Institute Senior Scientist Award (Grant number K05CA-142885 to F.A.S). The study was also supported, in part, by grants from the National Cancer Institute to the North Central Cancer Treatment Group (NCCTG) (Grant number CA-25224); the NCCTG Biospecimen Resource (Grant number CA-114740); the Alliance for Clinical Trials in Oncology (Grant number CA31946); and the Alliance Statistics and Data Center (Grant number CA33601), and in part by unrestricted support from Sanofi U.S., Pfizer, Inc. and Imclone Systems, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
Footnotes
Disclosures: The authors report no conflicts of interest related to the manuscript content.
Author contributions:
Conception and design: Frank A. Sinicrope, Steven R. Alberts, Daniel J. Sargent
Collection and assembly of data: Frank A. Sinicrope, Michelle R. Mahoney, Thomas C. Smyrk, Stephen N. Thibodeau, Garth D. Nelson, Richard M. Goldberg, Steven R. Alberts
Data analysis and interpretation: Frank A. Sinicrope, Steven R. Alberts, Harry Yoon, Michelle R. Mahoney, Garth D. Nelson, Daniel J. Sargent
Manuscript writing, revision and review: Frank A. Sinicrope, Michelle R. Mahoney, Gard D. Nelson, Richard M. Goldberg, Harry Yoon, Steven R. Alberts, Daniel J. Sargent
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74:3238–47. doi: 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- 3.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 4.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 6.Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41:664–8. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sweetser S, Smyrk TC, Sinicrope FA. Serrated colon polyps as precursors to colorectal cancer. Clin Gastroenterol Hepatol. 2013;11:760–7. doi: 10.1016/j.cgh.2012.12.004. quiz e54–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 9.Yaeger R, Cercek A, Chou JF, Sylvester BE, Kemeny NE, Hechtman JF, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer. Cancer. 2014;120:2316–24. doi: 10.1002/cncr.28729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–32. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Missiaglia E, Jacobs B, D’Ario G, Di Narzo AF, Soneson C, Budinska E, et al. Distal and proximal colon cancers differ in terms of molecular, pathological and clinical features. Ann Surg Oncol. 2014 doi: 10.1093/annonc/mdu275. [DOI] [PubMed] [Google Scholar]
- 12.Benedix F, Kube R, Meyer F, Schmidt U, Gastinger I, Lippert H. Comparison of 17,641 patients with right- and left-sided colon cancer: differences in epidemiology, perioperative course, histology, and survival. Dis Colon Rectum. 2010;53:57–64. doi: 10.1007/DCR.0b013e3181c703a4. [DOI] [PubMed] [Google Scholar]
- 13.Meguid RA, Slidell MB, Wolfgang CL, Chang DC, Ahuja N. Is there a difference in survival between right- versus left-sided colon cancers? Ann Surg Oncol. 2008;15:2388–94. doi: 10.1245/s10434-008-0015-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss JM, Pfau PR, O’Connor ES, King J, LoConte N, Kennedy G, et al. Mortality by stage for right- versus left-sided colon cancer: analysis of surveillance, epidemiology, and end results--Medicare data. J Clin Oncol. 2011;29:4401–9. doi: 10.1200/JCO.2011.36.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alberts SR, Sargent DJ, Nair S, Mahoney MR, Mooney M, Thibodeau SN, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–93. doi: 10.1001/jama.2012.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinicrope FA, Mahoney MR, Smyrk TC, Thibodeau SN, Warren RS, Bertagnolli MM, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31:3664–72. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. New Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 18.Angulo B, Garcia-Garcia E, Martinez R, Suarez-Gauthier A, Conde E, Hidalgo M, et al. A commercial real-time PCR kit provides greater sensitivity than direct sequencing to detect KRAS mutations: a morphology-based approach in colorectal carcinoma. J Mol Diagn. 2010;12:292–9. doi: 10.2353/jmoldx.2010.090139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinicrope FA, Rego RL, Garrity-Park MM, Foster NR, Sargent DJ, Goldberg RM, et al. Alterations in cell proliferation and apoptosis in colon cancers with microsatellite instability. Int J Cancer. 2007;120:1232–8. doi: 10.1002/ijc.22429. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Amer Statist Assn. 1958;53:457–81. [Google Scholar]
- 21.Cox DR. Regression models and life-tables. J Roy Statist Soc. 1972;34:187–220. [Google Scholar]
- 22.Team R. A language and environment for statistical computing. Vienna, Austria: R: Foundation for Statistical Computing; 2011. [Google Scholar]
- 23.Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012;18:1506–12. doi: 10.1158/1078-0432.CCR-11-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taieb J, Tabernero J, Mini E, Subtil F, Folprecht G, Van Laethem JL, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:862–73. doi: 10.1016/S1470-2045(14)70227-X. [DOI] [PubMed] [Google Scholar]
- 25.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61:847–54. doi: 10.1136/gutjnl-2011-300865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popovici V, Budinska E, Bosman FT, Tejpar S, Roth AD, Delorenzi M. Context-dependent interpretation of the prognostic value of BRAF and KRAS mutations in colorectal cancer. BMC Cancer. 2013;13:439. doi: 10.1186/1471-2407-13-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol. 2005;54:1093–101. doi: 10.1099/jmm.0.45935-0. [DOI] [PubMed] [Google Scholar]
- 29.Surana NK, Kasper DL. Deciphering the tete-a-tete between the microbiota and the immune system. J Clin Invest. 2014;124:4197–203. doi: 10.1172/JCI72332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–74. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 31.Farina-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VE, Rutten HJ, et al. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol. 2010;21:2396–402. doi: 10.1093/annonc/mdq258. [DOI] [PubMed] [Google Scholar]
- 32.Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res. 2012;18:890–900. doi: 10.1158/1078-0432.CCR-11-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gavin PG, Colangelo LH, Fumagalli D, Tanaka N, Remillard MY, Yothers G, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18:6531–41. doi: 10.1158/1078-0432.CCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popovici V, Budinska E, Tejpar S, Weinrich S, Estrella H, Hodgson G, et al. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol. 2012;30:1288–95. doi: 10.1200/JCO.2011.39.5814. [DOI] [PubMed] [Google Scholar]
- 35.Yoon HH, Tougeron D, Shi Q, Alberts SR, Mahoney MR, Nelson GD, et al. KRAS codon 12 and 13 mutations in relation to disease-free survival in BRAF-wild-type stage III colon cancers from an adjuvant chemotherapy trial (N0147 alliance) Clin Cancer Res. 2014;20:3033–43. doi: 10.1158/1078-0432.CCR-13-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blons H, Emile JF, Le Malicot K, Julie C, Zaanan A, Tabernero J, et al. Prognostic value of KRAS mutations in stage III colon cancer: post hoc analysis of the PETACC8 phase III trial dataset. Ann Oncol. 2014;25:2378–85. doi: 10.1093/annonc/mdu464. [DOI] [PubMed] [Google Scholar]
- 37.Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res. 2012;18:4753–63. doi: 10.1158/1078-0432.CCR-11-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klingbiel D, Saridaki Z, Roth AD, Bosman FT, Delorenzi M, Tejpar S. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: results of the PETACC-3 trial. Ann Oncol. 2015;26:126–32. doi: 10.1093/annonc/mdu499. [DOI] [PubMed] [Google Scholar]
- 39.Sinicrope FA, Shi Q, Smyrk TC, Thibodeau SN, Dienstmann R, Guinney J, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flejou JF, Andre T, Chibaudel B, Scriva A, Hickish T, Tabemero J, et al. Effect of adding oxaliplatin to adjuvant 5-fluorouracil/leucovorin (5FU/LV) in patients with defective mismatch repair (dMMR) colon cancer stage II and III included in the MOSIAC study. J Clin Oncol. 2013;31:Abstract 3524. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.