

Abstract
BACKGROUND. Noninvasive prenatal testing can be used to accurately detect chromosomal aneuploidies in circulating fetal DNA; however, the necessity of parental haplotype construction is a primary drawback to noninvasive prenatal diagnosis (NIPD) of monogenic disease. Family-specific haplotype assembly is essential for accurate diagnosis of minuscule amounts of circulating cell-free fetal DNA; however, current haplotyping techniques are too time-consuming and laborious to be carried out within the limited time constraints of prenatal testing, hampering practical application of NIPD in the clinic. Here, we have addressed this pitfall and devised a universal strategy for rapid NIPD of a prevalent mutation in the Ashkenazi Jewish (AJ) population.
METHODS. Pregnant AJ couples, carrying mutation(s) in GBA, which encodes acid β-glucosidase, were recruited at the SZMC Gaucher Clinic. Targeted next-generation sequencing of GBA-flanking SNPs was performed on peripheral blood samples from each couple, relevant mutation carrier family members, and unrelated individuals who are homozygotes for an AJ founder mutation. Allele-specific haplotypes were constructed based on linkage, and a consensus Gaucher disease–associated founder mutation–flanking haplotype was fine mapped. Together, these haplotypes were used for NIPD. All test results were validated by conventional prenatal or postnatal diagnostic methods.
RESULTS. Ten parental alleles in eight unrelated fetuses were diagnosed successfully based on the noninvasive method developed in this study. The consensus mutation–flanking haplotype aided diagnosis for 6 of 9 founder mutation alleles.
CONCLUSIONS. The founder NIPD method developed and described here is rapid, economical, and readily adaptable for prenatal testing of prevalent autosomal recessive disease-causing mutations in an assortment of worldwide populations.
FUNDING. SZMC, Protalix Biotherapeutics Inc., and Centogene AG.
Introduction
Noninvasive prenatal genetic testing (NIPT) of whole chromosomal aneuploidies has already altered the landscape of prenatal diagnostics in the United States (1) and increasingly worldwide. Aside from the noninvasiveness, advantages of NIPT include the rapid turnaround, relatively low cost, and simplicity of the procedure for pregnant couples. Arguably, these benefits are largely made possible because it is not necessary to construct parental haplotypes in order to accurately diagnose chromosomal copy number. For noninvasive prenatal diagnosis (NIPD) of monogenic disease, on the other hand, this is not the case. In order for NIPD to take hold in the clinical setting, it will be necessary to develop universal methodologies that apply to the diagnosis of any mutation, maternal or paternal, regardless of inheritance. Although some universal techniques for NIPD have already been described, each one requires time-consuming and sophisticated parental haplotype construction in advance of test interpretation (2–4). The classic haplotype construction methodology is simpler to implement because it involves the collection of DNA samples from several family members for linkage analysis. Nevertheless, this process is often complicated or sometimes made impossible by low compliance, couple privacy concerns, or the unavailability of living first-degree relatives. To address these issues, researchers have developed various molecular and statistical techniques for family-independent haplotyping (reviewed in ref. 5). Unfortunately, the described molecular techniques are too expensive, too time consuming, and/or too labor intensive for use in a clinical setting. Moreover, statistical approaches, which rely on high-throughput analysis of population data, are not appropriate for clinical application. Hence, the question arises as to whether a rapid, cost-effective, and routine test can be implemented for NIPD of monogenic disorders, with less reliance on blood sample collection from relatives of the pregnant couple. For mutations with increased prevalence in certain populations, also known as founder mutations, the answer to this question may be in the affirmative.
Indeed, medical centers around the world offer invasive prenatal diagnostic services for local population-specific founder mutations on a routine basis. Depending on the carrier frequency within the population, founder mutation tests often comprise a significant component of the overall molecular testing in such health care laboratories. Some examples of common founder mutations for which prenatal testing would be relevant include those implicated in long QT syndrome within the Finnish population (6); the delF508 mutation in CFTR, which causes cystic fibrosis in populations of mixed European descent (7); a mutation in the SERPINA1 gene, which causes α1-antitrypsin deficiency in Scandinavians of mixed European descent (8); a mutation in Colombians, which causes early-onset Alzheimer’s disease (9); and scores of founder mutations in the Tunisian (10) and Ashkenazi Jewish (AJ) (11) populations. The common denominator among all population-specific mutations is that they each appear with their own mutation-flanking molecular fingerprint or haplotype. Hypothetically, this fingerprint could be used as a tool for NIPD, although this has never been attempted for the aforementioned mutations. Nevertheless, if successfully implemented, this tool would alleviate the hassle of constructing family-specific haplotypes for founder mutation NIPD. Moreover, the use of mutation-specific fingerprints would also eliminate the need for sophisticated molecular haplotyping methods in NIPD, thereby effecting major savings with regard to test duration, reagent cost, and labor expenditure.
The monogenic disorder type I Gaucher disease is especially prevalent in Israel due to increased frequency of acid β-glucosidase (GBA) founder mutations, N370S (c.1226A>G or p.N409S, GenBank NM_001005741.2) and 84GG (c.84dupG, GenBank NM_001005741.2), in the AJ population (12) (the AJ population comprises 36% of the overall population in Israel; ref. 13). Unlike 84GG, which is deleterious in homoallelic form and much less prevalent in the AJ population (14), the N370S mutation appears in 1 of every 17 AJ individuals (15). In addition, most patients with type I Gaucher disease treated at our Gaucher Clinic (SZMC) are N370S homozygotes. Taken together, these research amenable circumstances led us to develop a cost-effective, expeditious test for NIPD of GBA N370S as a proof-of-principle for NIPD of autosomal recessive founder mutations in general.
As type I Gaucher disease is a relatively mild genetic disorder with safe and effective treatments (16), N370S testing was deemed an ideal candidate for NIPD assay calibration, given that (a) most mutation carrier couples do not opt to terminate homozygous N370S fetuses regardless of circumstance (17), therefore there was no clinical or ethical indication to provide test results to couples during pregnancy, and, (b) because the disease is treatable, children of at-risk couples are commonly tested in infancy to determine the need for surveillance and treatment. Therefore, the information gained by performing NIPD for type I Gaucher disease and confirmation of test results in the children born corresponded to information that would have been clinically indicated irrespective of this study.
Thus, by means of highly targeted next-generation sequencing (NGS), we show that fine mapping of a founder mutation fingerprint is a potentially valuable asset for NIPD of an autosomal recessive disease in the AJ population, and this strategy may prove beneficial for NIPD of many other worldwide autosomal recessive founder mutation conditions as well.
Results
Study description.
Eight pregnant AJ couples, of which one or both partners were heteroallelic carriers of GBA N370S, were enrolled in the study (Figure 1). Although families 1 and 4 were at risk of giving birth to a homozygote N370S child (unlike families 2, 3, 5, 6, 7, and 8 in which one parent of the fetus did not carry any mutation in GBA), we tested all couples strictly for proof-of-principle purposes. Plasma samples were collected from female participants at the time points indicated in Figure 1 for DNA extraction and targeted high-throughput sequencing of GBA-flanking SNPs. To enhance diagnostic accuracy, we elected to defer direct mutation sequencing in favor of a more specific and sensitive linkage-based analytical regimen. This methodology strengthens diagnostic confidence with increasing fetal haplotype size (measured by the number of SNPs in the inferred fetal haplotype) (Supplemental Tables 1–3; supplemental material available online with this article; doi:10.1172/JCI79322DS1). To accomplish this goal, we first sequenced GBA-flanking SNPs (up to ±250 kb distance from GBA) of the parents and their first-degree relatives in families 1 and 2, so as to construct parental haplotypes. However, these family-based haplotypes were of limited size (Supplemental Table 4). Therefore, we sought a larger haplotype sequence to aid fetal diagnosis by mapping a consensus N370S founder region surrounding the GBA gene.
Figure 1. Pedigrees of GBA mutation carrier families in this study.

Mutations in GBA are indicated in red. Individuals with unknown genotypes at sample collection are shaded in gray. WT denotes a WT GBA allele; wk, denotes the week of gestation at which maternal plasma was collected.
Fine mapping of the consensus AJ N370S founder haplotype region.
To fine map the N370S founder region, we sequenced 7 unrelated homoallelic AJ mutation carriers on our targeted GBA-flanking SNP panel. Six of these homoallelic patients with type I Gaucher disease were homozygotic for all 490 SNPs on our initial sequencing panel. The seventh sample shared the same haplotype within and 3′ to GBA, but a heterozygous region was clearly identified 144,388 nucleotides 5′ to the gene and beyond (at rs2306124, dbSNP 138). Hence, this sample was used to demarcate a preliminary consensus founder haplotype (Figure 2A). To further clarify the N370S sequence, we then crossed the preliminary version with linkage-based N370S haplotypes from the families under investigation in this study in addition to 3 other unrelated heteroallelic AJ N370S mutation carrier duos (2 first-degree relatives, each of whom carries the same mutation). This analysis identified a recombined region only 17,858 nucleotides upstream of GBA (at rs148168407, dbSNP 138) (Figure 2B), but remarkably, not a single recombination event was identified in the entire 219-kb region downstream of GBA in any of the 20 N370S chromosomes analyzed. Not coincidentally, this 3′ conserved region has been previously characterized as a nonrecombination hot spot by the HapMap Consortium (18). Moreover, previous studies have identified a conserved AJ N370S founder haplotype that extends even further downstream of GBA (12, 19). Nevertheless, although it is likely that the founder haplotype is longer than we initially mapped, our SNP-sequencing panel still successfully linked a sizable amount of GBA-flanking SNPs (153 altogether) to a consensus AJ haplotype sequence (Figure 2C and Supplemental Table 5). The next question was to determine whether this population-based haplotype could be used as a diagnostic tool for NIPD.
Figure 2. Fine mapping of the consensus AJ N370S founder haplotype region.

Hundreds of GBA-flanking SNPs (±250 kb from GBA) were sequenced in order to identify a conserved N370S founder haplotype. (A) NGS-based homozygosity mapping with 7 unrelated homozygote N370S Gaucher patients (denoted as H1–H7) (14 N370S chromosomes) was used to identify a preliminary founder haplotype. (B) A representative linkage-based inference of a familial N370S haplotype (hapN370S). This linkage analysis was performed for 6 different heteroallelic GBA N370S mutation carrier duos (6 N370S chromosomes from 6 sets of 2 first-degree family members carrying the N370S mutation). The resultant alleles were each compared separately to the haplotype from A until a consensus N370S haplotype was demarcated with a 5′ cutoff. (C) Ultimately, the consensus AJ N370S founder haplotype (composed of 153 SNPs) used for NIPD was constructed from 20 different AJ N370S chromosome sequences. Note that this analysis set a 5′ cutoff for the conserved N370S haplotype, but a 3′ cutoff could not be established. WT denotes a WT allele.
Preliminary NIPD of an autosomal recessive founder mutation.
For pilot testing, families 1 and 2 offered 3 different avenues with which to assess the utility of the consensus N370S haplotype. This was because both parents (of the fetus) in family 1 were N370S carriers in addition to the mother in family 2 (Figure 1). For family 1, the N370S carrier father was completely homozygous for the entire consensus N370S sequence. This precluded the use of the N370S haplotype for NIPD of his allele. Nonetheless, the familial mutation-linked haplotype of the father in family 1 did facilitate the identification of his WT allele in the fetus (Figure 3A and Supplemental Tables 6 and 7). Regarding the maternal alleles in families 1 and 2, the consensus N370S haplotype proved to be quite valuable. The family-based maternal N370S-linked haplotype was clearly identified in family 1 plasma DNA. This fetal haplotype was completely concordant with the consensus N370S haplotype (Figure 3B and Supplemental Tables 7 and 8). For family 2, the family-based maternal N370S haplotype could not reliably discern which allele was transmitted to the fetus because the fetal haplotype was determined on differing SNP positions. On the other hand, the longer consensus N370S haplotype clearly matched the inferred maternal haplotype in the fetus, indicating inheritance of the N370S allele (Figure 3C and Supplemental Tables 7 and 9). Thus, in this case, the consensus N370S sequence was crucial to the diagnosis of the maternal allele in the family 2 fetus.
Figure 3. Noninvasive fetal allele identification based on familial and/or the N370S founder haplotype.

Illustrations depict the immediate GBA-proximal locus and SNPs that were deep sequenced for the construction and typing of fetal alleles (as indicated in the “Haplotype legend”). (A) In family 1, the paternal WT allele was diagnosed by inference from the family-based N370S-linked haplotype (red squares). The consensus N370S haplotype could not be used to phase the paternal allele in the fetus due to paternal homozygosity in the founder haplotype region. (B) On the other hand, the maternal N370S allele in family 1 was readily identified (in multiple sites) via the consensus haplotype, and this result was corroborated by equivalent matches to the family-based maternal N370S haplotype. (C) For the family 2 maternal allele, the fetal N370S haplotype could only be matched to a single polymorphic site in the family-based haplotype (red square). This site and 2 other fetal SNPs were definitively matched to the N370S mutation by comparison to the founder N370S haplotype. Therefore, in this case, it would not have been possible to reliably diagnose the maternal allele in the fetus without the N370S founder sequence.
Extended fine mapping of the consensus AJ N370S founder haplotype region.
Although initial testing of families 1 and 2 showed promising results regarding the utility of the consensus N370S haplotype for incorporation into NIPD, it was clear that for expanded N370S testing in a clinical setting a more sophisticated sequencing panel would be required to facilitate setup of a universal assay for noninvasive prenatal Gaucher disease testing. Our concerns with the initial 490-SNP sequencing panel were 4-fold. As evidenced by HapMap (18) and deCode (20) data, meiotic recombination is quite infrequent in the immediate human GBA-flanking locus (±250 kb), which was the small target of our pilot sequencing panel. In this genomic context, homozygosity of an N370S mutation carrier parent, such as in the family 1 father, would be expected to occur commonly because DNA is rearranged at a reduced rate in the peri-GBA locus. Along these lines, low recombination rates translate into low genotypic complexity, which, in turn, leads to limited availability of linkage-informative SNPs, which are crucial to fetal haplotyping. Thus, small family-based haplotypes, which generally handicap fetal haplotyping, such as that of the family 1 father (3 SNPs) and that of the family 2 mother (11 SNPs; Supplemental Table 4), would be predicted to represent the majority as opposed to the minority of cases. Another reason to consider looking beyond a distance of 250 kb from GBA would be to complete fine mapping of the 3′ boundary of the consensus N370S sequence, which proved so beneficial for fetal typing of family 1 and 2 maternal N370S-paired alleles. Finally, N370S aside, the implementation of a larger targeted sequencing panel should hypothetically be used to diagnose any mutation in GBA via familial linkage analysis, regardless of whether the mutation is a founder allele or not. For all these aforementioned reasons, we designed a newer and much improved targeted deep-sequencing panel to sequence 10 times the amount of GBA-flanking SNPs (~5,000 SNPs) across an 8-fold-sized genomic region (GBA ± 2 Mb) before moving forward with NIPD for other families in the study.
The first priority, in terms of test implementation, was to use the new expanded sequencing panel to complete fine mapping of the founder N370S haplotype. As mentioned above, the original sequencing panel successfully demarcated a 5′ boundary for the consensus sequence that was approximately 17 kb upstream of GBA and at least 219 kb downstream. When repeating the same exercise (as that described in Figure 2) using the large sequencing panel, the 5′ boundary for the consensus haplotype mapped approximately 28 kb upstream of GBA (at SNP rs914615, dbSNP141). This 11 kb discrepancy between fine-map boundaries is quite remarkable, given that, due to technical reasons, the newer panel did not incorporate many of the SNPs sequenced previously with the older panel. Nevertheless, the preliminary 5′ cutoff, based only on N370S homozygotes, strikingly mapped to the exact same SNP position (SNP rs2306124) 144,388 nucleotides 5′ to GBA in both panels. Thus, given this concordance between old and new sequencing panels regarding the 5′ N370S consensus sequence, it was especially edifying that the 3′ boundary of the haplotype fine mapped to a position that is roughly 650 kb downstream of GBA (at SNP rs1055184, dbSNP 141) by the new and improved panel. This expanded N370S-linked 670-kb-sized sequence (composed of 301 SNPs; Supplemental Table 10) would already seem quite large for what is considered to be an ancient founder allele. Yet, remarkably, previous studies have shown that the N370S haplotype should, in fact, extend much further downstream from GBA, up to a full Mb from the gene (12, 19). Indeed, after careful scrutiny of the new sequencing data, we found that, among 16 sequenced N370S chromosomes from 8 N370S homozygotes, 15 chromosomes shared a near-consensus haplotype that extended 1.1 Mb 3′ to GBA (Figure 4A). Therefore, we postulated that, if a 250-kb consensus sequence could be used to haplotype fetal alleles in families 1 and 2 (as in Figure 3), then a 1.1-Mb sequence might prove even more useful for typing of N370S chromosomes in most mutation carrier families in general.
Figure 4. Extended fine mapping of the consensus and near-consensus AJ N370S founder haplotype region as a tool for phasing fetal haplotypes.

Illustration depicting the GBA locus (±2 Mb) and thousands of SNPs that were deep sequenced for the construction and typing of fetal alleles according to the analytical pipeline (as indicated in the key). (A) An extended deep-sequencing panel was used to better fine map the conserved N370S founder haplotype, as in Figure 2. Accordingly, a 301-SNP haplotype (termed “full-consensus N370S haplotype”) was identified in all N370S chromosomes in this study (28 chromosomes altogether). In addition, the consensus haplotype was found to extend 500 kb further downstream of GBA (620 additional SNPs) in 15 of 16 chromosomes from 8 N370S homozygotes. Furthermore, in all N370S homozygotes (but not all N370S carriers), the consensus haplotype was found to extend another 120 kb upstream of GBA (100 additional SNPs). Altogether, these extended haplotypes were termed “near-consensus N370S haplotypes.” (B) The N370S haplotype from each N370S carrier parent in the study was carefully mapped according to homozygous regions and family-based linkage analysis. After comparison to the near-consensus haplotype in A, new parent-specific 5′ and/or 3′ demarcations of the N370S near-consensus haplotype were set (this haplotype was termed the “parent-specific consensus N370S haplotype”). (C) In this example, deep sequencing of the GBA-flanking region in a fetus identified stretches of a linkage-based parental N370S haplotype that resided outside of the consensus N370S region. In addition, some stretches of fetal sequence could not be phased according to family-based linkage. (D) When unphased fetal sequence, such as in C, fell within the parent-specific consensus N370S haplotype (as determined in B), the consensus information was used to phase the fetus (here, with the N370S-linked haplotype), thereby increasing confidence in the diagnostic test result.
To make effective use of the expanded near-consensus N370S haplotype without allowing haplotype errors to corrupt downstream fetal analysis, we carefully inspected each N370S chromosome in all mutation carrier parents in the study (families 1 through 8) using the large sequencing panel. We found that, in some cases, we could detect recombination in the true parent-specific N370S-linked sequence with respect to the founder mutation near-consensus haplotype (Figure 4B). These discrepancies were applied on an allele-specific basis toward refinement of the consensus sequence, so that it could be used for the analysis of fetal haplotypes, which were not phased by conventional family-based linkage analysis (Figure 4C). In such scenarios, the parent-specific near-consensus N370S haplotype was appropriated for the resolution of unphased fetal haplotypes to increase confidence in the final NIPD test result (Figure 4D). For example, if an N370S carrier mother and her immediate family members (whose samples were used for standard linkage analysis) were all heterozygous for the same SNP loci, the mother’s genotypes were considered informative, even though linkage could not set phase on her N370S-linked haplotype. In this case, it was possible that the mother’s unphased SNPs were located within the fine-mapped mother-specific consensus N370S haplotype (as in Figure 4B) and genotyped in her fetus (as in Figure 4C). When this occurs, the correct haplotype can be identified in the fetus, even though conventional family-based linkage analysis fails. More examples of this new approach to NIPD will be illustrated below.
NIPD of GBA N370S using an improved targeted sequencing panel.
Having setup the framework with which to embark on streamlined NIPD for the N370S founder mutation, we returned to families 1 and 2 and retested the same samples using the expanded sequencing panel. One of the primary issues with the previous analysis involving these families was the small size of linkage-based haplotypes in the family 1 paternal N370S allele and the family 2 maternal N370S allele (Supplemental Table 4). As expected, the large sequencing panel clearly solved this issue for families 1 and 2 (and, essentially, all families in this study). Ranging from 113 to 336 phased SNPs, all parental family-based haplotypes in the current investigation were of substantial size and content to enable scoring of fetal haplotypes with generally high confidence (Supplemental Table 11). Interestingly, the family 1 father turned out to be homozygous for the entire N370S consensus and near-consensus sequence by the new panel analysis. Nonetheless, his linkage-based N370S haplotype facilitated highly unambiguous identification of his WT allele in the fetus (Figure 5A and Supplemental Table 12). This test result was clearly of much higher quality, in terms of fetal haplotype size (14 phased SNPs), in comparison with the previous test (2 phased SNPs) involving the same samples (Figure 3A). Regarding the family 1 maternal allele, there was little doubt from the previous panel whether the mother had transmitted her N370S allele to the fetus (9 phased SNPs in fetus; Figure 3B and Supplemental Table 8). In the newer panel, it was even more obvious that the mother had transmitted her N370S allele to the fetus based on clear matches between the fetal haplotype and the mother’s family-based N370S allele as well as her consensus and near-consensus N370S sequences (17 phased SNPs altogether; Figure 5B and Supplemental Table 13).
Figure 5. Noninvasive fetal allele identification based on familial, N370S consensus, and/or N370S near-consensus haplotypes.

Illustrations depict the GBA locus (±2 Mb) and SNPs that were deep sequenced for the construction and typing of fetal alleles (as indicated in the “Haplotype legend”). The numbers shown under DFM denote the distance from mutation (in Mb). The noninvasively identified fetal alleles were (A) WT paternal, (B) N370S maternal, (C) N370S maternal, (D) N370S maternal, (E) WT paternal, (F) WT maternal, (G) N370S paternal, (H) N370S paternal, (I) L444P (non-N370S) maternal, and (J) 84GG (non-N370S) maternal. Note the utility of the N370S consensus haplotype for fetal typing in B, C, and H. The near-consensus N370S haplotype also aided fetal typing in B, C, F–H, and J.
With family 2, the value and importance of the consensus N370S haplotype grew manifold after reanalysis on the larger sequencing panel. In the previous assessment, only 1 of 3 SNPs in the fetal haplotype was phased to the family-based N370S allele (Figure 3C). In the newer evaluation, the fetal haplotype was much larger, but only 5 SNPs were phased to the family-based maternal N370S haplotype, one of which was isolated on the 3′ side of GBA (1 Mb distance from the mutation). To strengthen the certainty of this test result, the fetal haplotype was compared to the parent-specific consensus and near-consensus N370S haplotype. This comparison yielded another 5 phased SNPs located 3′ to the mutation, which, together with family-based fetal alleles, led to the correct diagnosis of the N370S mutation in the family 2 fetus (based on 10 phased SNPs altogether; Figure 5C and Supplemental Table 14).
The principles set forth in these preliminary tests were subsequently put into practice for fetal allele identification involving families 3 through 8 (Figure 5, D–J, and Supplemental Tables 15–21). Of particular importance is the fact that 4 of 6 N370S-paired alleles in these families were typed noninvasively with the aid of the N370S consensus and/or near-consensus sequence (Figure 5, F–H, and J). These results thereby confirmed our assumption that the founder N370S haplotype is a valuable tool for incorporation into standard NIPD protocol. Another point to consider, which supports the use of larger sequencing panels in NIPD in general, is the fact that, even when a nonfounder GBA mutation was tested (such as that of the family 4 paternal allele; Figure 5E and Supplemental Table 16), the extended sequencing panel facilitated construction of a well-defined fetal haplotype nonetheless.
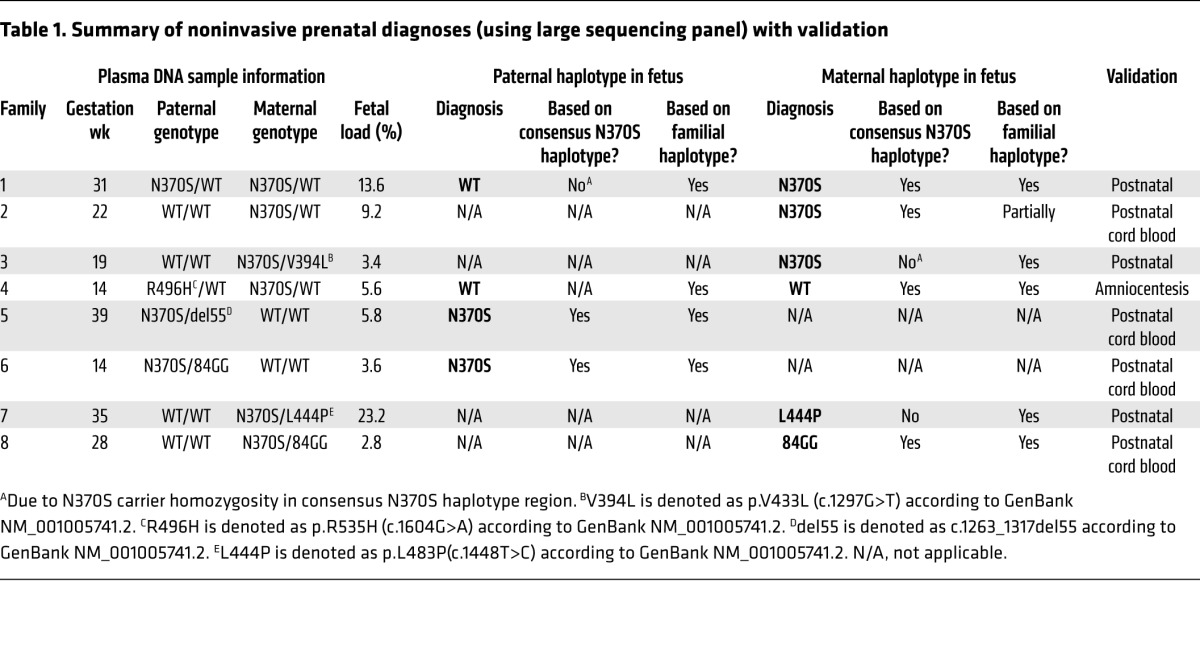
To summarize, the outcomes of this proof-of-concept study are presented in Table 1. All noninvasive test results were validated with conventional prenatal or postnatal diagnostics.
Table 1. Summary of noninvasive prenatal diagnoses (using large sequencing panel) with validation.
Discussion
Here, we describe successful NIPD of the Gaucher disease N370S founder mutation in 8 families. In addition, we have performed fine mapping of the founder AJ N370S-linked haplotype, which we assessed as a diagnostic tool for NIPD. We show that, as long as parental homozygosity does not overlap the consensus N370S sequence, the universal haplotype can serve as a valuable supplement to classic family-based haplotyping or even a highly attractive alternative in appropriate cases. Accordingly, we predict that this diagnostic strategy may be implemented for much more severe autosomal recessive founder mutation testing in relevant subpopulations worldwide.
One of the objectives of this study was to reduce monogenic disorder NIPD turnaround time for practical application in the clinic. Previously described methods require several weeks to reach a diagnosis (5), thereby increasing the risk of late-stage pregnancy termination of an affected embryo. In contrast, we performed an expeditious 5-day work flow, which can facilitate reliable diagnosis of at-risk fetuses within a short enough timeframe to enable decision making during early-stage pregnancy. In addition, previously described methods entail high-cost materials, sophisticated machinery, and/or large staff overhead, especially for haplotype construction. With our method, universal preconstructed founder haplotypes are readily accessible for free fetal DNA analysis at no extra financial or time cost.
There is also an additional benefit to utilization of universal founder haplotypes in NIPD. When performing linkage analysis to infer parental alleles ahead of fetal testing, it is not uncommon for heterozygosity to present in the mutation carrier parent and each of his/her immediate family members. In such instances, family-based linkage cannot phase parental SNPs to a specific allele (WT or mutant). However, if these unphased SNPs also lie within the conserved founder haplotype sequence, this consensus sequence can be used to phase ambiguous SNPs, which ultimately can be exploited for fetal allele identification. This boon to founder haplotype testing aided diagnosis of families 1, 2, 4, 5, 6, and 8 (Figure 5, B, C, F, G, H, and J) in the current study, and it should be expected to benefit many more founder mutation NIPD tests going forward.
Aside from founder haplotype-related advantages, a major advance in our NIPD method for Gaucher disease was evident when we transitioned from a small targeting sequencing panel (encompassing 500 kb of GBA-flanking sequence) to a larger targeted sequencing panel (encompassing 4 Mb of GBA-flanking sequence). While it is not impossible for long 4-Mb stretches of homozygosity to confound searches for perigenic informative SNP markers, this scenario is quite unlikely. Indeed, the larger sequencing panel in the current study demonstrated that at least 100 informative parental markers were identified in all cases simply by probing a larger gene-flanking region. In addition, while the larger panel added much needed complexity to the NIPD assay in terms of SNP marker availability, it also simultaneously simplified fetal typing by delineating the upper and lower boundaries of the N370S founder haplotype. As mentioned above, this was a valuable aid to downstream fetal haplotype analysis. Moreover, even in the absence of a founder allele for testing, the larger sequencing panel facilitated non-N370S GBA mutation analysis (of the paternal R496H allele, for example, in family 4; Figure 5E) because informative markers are generally easier to find in large Mb stretches of DNA. Nonetheless, when assaying large genomic regions for fetal diagnosis, one must exercise caution to avoid pitfalls resulting from the possibility that perigenic meiotic recombination might cause a WT haplotype to appear upstream of a mutation, while a mutant haplotype recombines downstream or vice versa (21). Such recombination events can lead to misdiagnosis if care is not taken to ensure that the same haplotype flanks the assayed mutation in both upstream and downstream DNA sequences. Another potential pitfall for NIPD of monogenic disorders is the phenomenon (which has already led to cases of misdiagnosis in the field of noninvasive fetal aneuploidy testing) of placental mosaicism (22–28). Given that maternal circulating fetal DNA is derived from the placenta (29), theoretically, the presence of two distinct maternal and/or paternal haplotypes in the placenta could lead to misinterpretation of cell-free fetal haplotypes. Therefore, although the outcomes in this proof-of-principle study were positive, we cannot predict how effectively two distinct fetal haplotypes will affect noninvasive fetal diagnosis of monogenic disease in the event that such a situation will arise in the future. Regardless, we do speculate that our haplotype-based testing method would provide a stronger analysis for prenatal allelic measurements than pure counting-based methods, such as those in noninvasive aneuploidy tests. At the worst, we would expect to observe an inconclusive test result as a result of placental mosaicism in our NIPD assay as opposed to outright misdiagnosis. In this case, invasive prenatal testing should still be offered to the couple in time to inform antenatal decision making.
Nevertheless, based on the exceptional preclinical results of the current study, we are encouraged by the bright prospects for noninvasive founder mutation prenatal testing in the near future. Accordingly, we predict that consensus haplotype–mediated NIPD will soon become a widespread tool for prenatal diagnosis of prevalent founder mutations in inbred subpopulations around the globe.
Methods
Further information can be found in the Supplemental Methods.
Sample collection.
Pregnant AJ couples, carrying mutation(s) in the GBA gene, were recruited at the SZMC Gaucher Clinic. Peripheral blood samples were collected from each couple, relevant mutation carrier family members, 8 unrelated AJ GBA N370S homozygotes, and 3 unrelated AJ GBA N370S heterozygote duos.
Targeted NGS.
Genomic DNA was prepared from blood samples, and plasma DNA was isolated from each pregnant female participant followed by targeted NGS of 490 SNPs and/or approximately 5,000 GBA-flanking SNPs on an Illumina MiSeq or NextSeq 500 instrument, respectively. Family-specific and N370S-specific haplotypes were constructed from NGS data and used to identify fetal alleles in each plasma-derived cell-free fetal DNA sample. All fetal diagnoses were validated with conventional prenatal or postnatal diagnostics.
Statistics.
Accuracy of identified fetal haplotypes was estimated by modeling assay-specific sensitivity/specificity as a function of inferred fetal haplotype size and genomic site–specific meiotic recombination rates (based on ref. 20 and Supplemental Tables 1–3).
Study approval.
Ethical approval for the study, including usage of materials from human subjects, was obtained from the SZMC institutional review board, and written informed consent was obtained from all study participants.
Supplementary Material
Acknowledgments
The authors thank Miriam Kott for assistance with NGS on the MiSeq instrument and Michal Bronstein for assistance with NGS on the NextSeq 500 instrument. Susanne Zielke and Sabrina Eichler supported logistics involved with the collaboration of the scientific groups. All the authors would like to thank the patients and families who participated in the study. This study was supported by the SZMC intramural Mirsky grant to D.A. Zeevi and unrestricted educational grants by Protalix Biotherapeutics Inc. and Centogene AG to the SZMC Gaucher Clinic.
Footnotes
Role of funding source: SZMC, Protalix Biotherapeutics Inc., and Centogene AG played a role in the purchase of reagents and paid for sequencing service lab fees.
Conflict of interest: David A. Zeevi and Gheona Altarescu have a patent pending on fetal haplotype identification. Arndt Rolfs serves as chief medical director and CEO of Centogene AG.
Reference information:J Clin Invest. 2015;125(10):3757–3765. doi:10.1172/JCI79322.
References
- 1.Bianchi DW, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370(9):799–808. doi: 10.1056/NEJMoa1311037. [DOI] [PubMed] [Google Scholar]
- 2.Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;487(7407):320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitzman JO, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med. 2012;4(137): doi: 10.1126/scitranslmed.3004323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo YM, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2(61): doi: 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 5.Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 2011;12(10):703–714. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marjamaa A, et al. High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann Med. 2009;41(3):234–240. doi: 10.1080/07853890802668530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morral N, et al. The origin of the major cystic fibrosis mutation (delta F508) in European populations. Nat Genet. 1994;7(2):169–175. doi: 10.1038/ng0694-169. [DOI] [PubMed] [Google Scholar]
- 8.Cox DW, Woo SL, Mansfield T. DNA restriction fragments associated with alpha 1-antitrypsin indicate a single origin for deficiency allele PI Z. Nature. 1985;316(6023):79–81. doi: 10.1038/316079a0. [DOI] [PubMed] [Google Scholar]
- 9.Lalli MA, et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer’s disease. Alzheimers Dement. 2014;10(5 suppl):S277–S283.e10. doi: 10.1016/j.jalz.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romdhane L, Kefi R, Azaiez H, Ben Halim N, Dellagi K, Abdelhak S. Founder mutations in Tunisia: implications for diagnosis in North Africa and Middle East. Orphanet J Rare Dis. 2012;7: doi: 10.1186/1750-1172-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zlotogora J. Mendelian Disorders Among Jews. [July 28, 2015];Department of Community Genetics Public Health Services Ministry of Health Israel Web site. http://www.health.gov.il/Subjects/Genetics/Documents/book_jews.pdf
- 12.Diaz GA, et al. Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid β-glucosidase mutations. Am J Hum Genet. 2000;66(6):1821–1832. doi: 10.1086/302946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demographics of Israel. [August 16, 2015];Centre for Israel and Jewish Affairs. http://www.cija.ca/resource/israel-the-basics/demographics-of-israel/
- 14.Beutler E, et al. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet. 1993;52(1):85–88. [PMC free article] [PubMed] [Google Scholar]
- 15.Horowitz M, et al. Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum Mutat. 1998;12(4):240–244. doi: 10.1002/(SICI)1098-1004(1998)12:4<240::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. 2005;129(2):178–188. doi: 10.1111/j.1365-2141.2004.05351.x. [DOI] [PubMed] [Google Scholar]
- 17.Zuckerman S, et al. Carrier screening for Gaucher disease: lessons for low-penetrance, treatable diseases. JAMA. 2007;298(11):1281–1290. doi: 10.1001/jama.298.11.1281. [DOI] [PubMed] [Google Scholar]
- 18.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz A, et al. Gaucher disease: the N370S mutation in Ashkenazi Jewish and Spanish patients has a common origin and arose several thousand years ago. Am J Hum Genet. 1999;64(4):1233–1238. doi: 10.1086/302341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong A, et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467(7319):1099–1103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- 21.Altarescu G, Eldar Geva T, Brooks B, Margalioth E, Levy-Lahad E, Renbaum P. PGD on a recombinant allele: crossover between the TSC2 gene and ‘linked’ markers impairs accurate diagnosis. Prenat Diagn. 2008;28(10):929–933. doi: 10.1002/pd.2070. [DOI] [PubMed] [Google Scholar]
- 22.Gao Y, Stejskal D, Jiang F, Wang W. False-negative trisomy 18 non-invasive prenatal test result due to 48,XXX,+18 placental mosaicism. Ultrasound Obstet Gynecol. 2014;43(4):477–478. doi: 10.1002/uog.13240. [DOI] [PubMed] [Google Scholar]
- 23.Mao J, et al. Confined placental origin of the circulating cell free fetal DNA revealed by a discordant non-invasive prenatal test result in a trisomy 18 pregnancy. Clin Chim Acta. 2014;433:190–193. doi: 10.1016/j.cca.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Hall AL, et al. Positive cell-free fetal DNA testing for trisomy 13 reveals confined placental mosaicism. Genet Med. 2013;15(9):729–732. doi: 10.1038/gim.2013.26. [DOI] [PubMed] [Google Scholar]
- 25.Pan M, et al. Discordant results between fetal karyotyping and non-invasive prenatal testing by maternal plasma sequencing in a case of uniparental disomy 21 due to trisomic rescue. Prenat Diagn. 2013;33(6):598–601. doi: 10.1002/pd.4069. [DOI] [PubMed] [Google Scholar]
- 26.Canick JA, Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat Diagn. 2013;33(7):667–674. doi: 10.1002/pd.4126. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, et al. Two cases of placental T21 mosaicism: challenging the detection limits of non-invasive prenatal testing. Prenat Diagn. 2013;33(12):1207–1210. doi: 10.1002/pd.4212. [DOI] [PubMed] [Google Scholar]
- 28.Choi H, et al. Fetal aneuploidy screening by maternal plasma DNA sequencing: ‘False positive’ due to confined placental mosaicism. Prenat Diagn. 2013;33(2):198–200. doi: 10.1002/pd.4024. [DOI] [PubMed] [Google Scholar]
- 29.Alberry M, et al. Free fetal DNA in maternal plasma in anembryonic pregnancies: confirmation that the origin is the trophoblast. Prenat Diagn. 2007;27(5):415–418. doi: 10.1002/pd.1700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.