

Abstract
Aberrant gene expression is a molecular hallmark of acute kidney injury (AKI). Since epigenetic processes control gene expression in a cell- and environment-defined manner, understanding the epigenetic pathways that regulate genes altered by AKI may open vital new insights into the complexities of disease pathogenesis and identify possible therapeutic targets. Here we used matrix chromatin immunoprecipitation and integrative analysis to study twenty key permissive and repressive epigenetic histone marks at transcriptionally induced Tnf, Ngal, Kim-1 and Icam-1 genes in mouse models of AKI; unilateral renal ischemia/reperfusion, lipopolysaccharide (LPS) and their synergistically injurious combination. Results revealed unexpected heterogeneity of transcriptional and epigenetic responses. Tnf and Ngal were transcriptionally upregulated in response to both treatments individually, and to combination treatment. Kim-1 was induced by ischemia/reperfusion and Icam-1 by LPS only. Epigenetic alterations at these genes exhibited distinct time-dependent changes that shared some similarities, such as reduction in repressive histone modifications, but also had major ischemia/reperfusion vs. endotoxin differences. Thus, diversity of changes at AKI genes in response to different insults indicates involvement of several epigenetic pathways. This could be exploited pharmacologically through rational-drug design to alter the course and improve clinical outcomes of this syndrome.
Keywords: acute kidney injury, gene expression, ischemia reperfusion, sepsis
Introduction
It is estimated that 2 million people worldwide die from acute kidney injury (AKI) annually, equaling the number of deaths from AIDS 1-2. Moreover, AKI is becoming an ever greater health care burden as its incidence rises at alarming rates, placing patients at risk for chronic kidney disease (CKD) and increasing the likelihood of end stage renal disease 1, 3-7. Cellular and molecular basis of AKI pathogenesis were examined in multiple studies that brought better understanding of the syndrome's pathophysiology. However treatment of AKI has remained mainly supportive and available therapeutic modalities show only minimal improvement of the high mortality associated with this disease over the last fifty years 8-9. Historically, mechanistic studies in AKI have focused on single factors or pathways. Although informative these approaches fail to capture the system-wide complexity and heterogeneity of AKI and have thus yielded limited translational and clinical breakthroughs. These considerations suggest that novel integrative approaches are needed, rather than those focused on a single gene or pathway 10, to study the temporally dynamic and functionally diverse course of AKI.
Aberrant gene expression is a molecular hallmark of AKI 11 and several gene products of renal injury, such as KIM-1 and NGAL, have been implicated in the syndrome and used as AKI biomarkers 10, 12. Still, the molecular mechanism mediating detrimental gene expression alterations are not well understood. Epigenetic processes control gene expression in a cell- and environment-defined manner. Epigenetic modifications of DNA and histones have been viewed as either transcriptionally permissive or repressive 13-14. As such, epigenetic marks can serve as sensitive indicators of transcriptional and other changes at a gene. Recent advances in transcription and epigenetic technologies allow not only measurements of rates of transcription and chromatin changes but also epigenetic modifiers bound to chromatin 15-18. An important translational application of identifying the chromatin modifiers bound to disease-causing genes is discovery of novel epigenetic targets for potential rational-design drug interventions to ameliorate renal injury.
In the clinical setting, diverse insults contribute to AKI and activate heterogeneous pathways that 10 can interact synergistically to exacerbate injury 19-20. Understanding the epigenetic landscape of genes induced by AKI may open fundamentally new insights into disease pathogenesis advancing rational-design epigenetic-based therapies. Ischemia-reperfusion (I/R) and sepsis are among the most common causes of AKI 4, but represent two clinically and pathophysiologically distinct entities 21-25. The mouse model of endotoxemia in the setting of preceding unilateral renal ischemia/reperfusion (I/R) captures the molecular events of multifactorial AKI with sepsis being a component 15, 26-27. In this model, the proximal tubular cells transcriptionally hyper-respond to endotoxin (lipopolysaccharide, LPS) leading to exaggerated renal cytokine production 15, 28.
We applied high throughput RT-qPCR and matrix chromatin immunoprecipitation (ChIP) platforms 29-30 to systematically map the heterogeneity of transcriptional and epigenetic responses in AKI caused by LPS, I/R, and their combination (LPS+I/R) in time course animal experiments.
Results
In the current study we utilized a model of endotoxin hyperresponsiveness in the setting of I/R (Fig. S1) to examine several issues associated with heterogeneity of transcriptional and epigenetic responses in AKI. Specifically, we asked: i) Which AKI-related genes exhibit endotoxin transcriptional hyperresponsiveness in the setting of I/R (i.e., a synergistic effect of I/R+LPS)? ii) Which of the established AKI biomarker mRNAs report endotoxin hyperresponse in the setting of I/R? iii) What is the time course of the endotoxin transcriptional hyperresponse for different genes? iv) What are the similarities and differences in epigenetic alterations associated with transcriptional responses in a group of representative I/R and endotoxin-induced genes?
AKI is characterized by heterogeneous mRNA responses
A high throughput RT-qPCR microplate platform was used to assess mRNA levels of a panel of large number of genes previously implicated in AKI including several biomarkers (Ngal, Kim-1, Klotho, Netrin1, IL-18, L-FABP, Timp2, angiotensinogen (Agt) and Igfbp7) 10, 31-32. Cluster analysis of gene expression profiles demonstrated highly dynamic patterns across time points and types of injury (Fig. 1). While the AKI-associated genes could be broadly divided into those that were up or down-regulated during injury, a closer inspection revealed significant diversity in transcriptional responses.
Fig.1. Cluster analysis of temporal gene expression profiles in AKI.
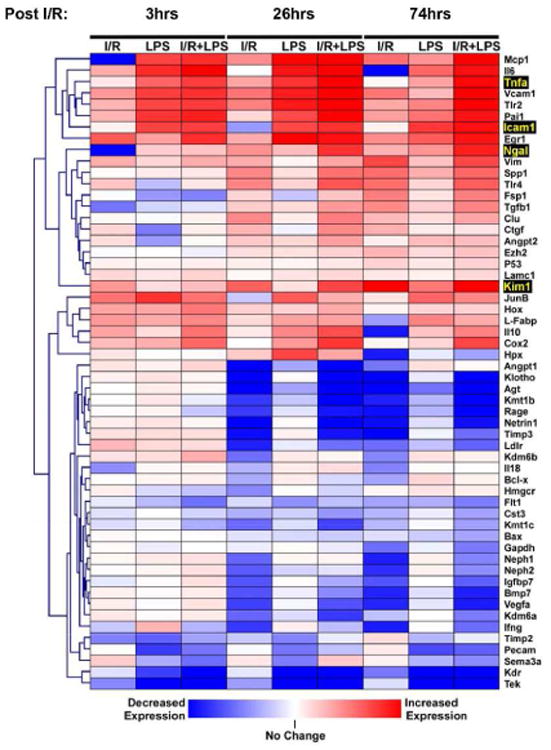
The timing of harvesting the kidneys post I/R injury was 3, 26 and 76 hrs, and 2 hrs after either LPS or saline treatment (Methods). Total RNA from mice renal cortex was used in RT reactions with oligo dT. cDNA was used in real time PCR (qPCR) with gene specific primers (Table S1, supplement). mRNA level of a given gene in each sample was normalized to β-actin transcript. The mRNA levels of 56 AKI-associated genes were measured using and clustered based on expression pattern differences across time and injury model. Note the diverse transcriptional responses to injury of co-clustered genes as highlighted in the figure. Data are represented as mean, n=6 mice from each group. Four AKI candidate genes—Tnf, Ngal, Icam-1, and Kim-1—were selected for further in-depth epigenetic analysis.
Tnf 15 and several co-clustered genes (Vcam1, Pai1, Tlr2 and Ngal) exhibited a robust endotoxin response with synergistic effects in I/R+LPS kidneys (Fig. 1). In contrast, several genes including Kim-1 and Tlr4 were up-regulated following I/R but not LPS treatment, whereas the expression of Icam1 was up-regulated by LPS alone with minimal effect of I/R. Many genes including Klotho, Netrin1, and Agt 32, were characterized by down-regulated mRNA expression following I/R (26-74hrs) but remained unresponsive to LPS. In contrast, the expression levels of several angiogeneic genes including Tek, Pecam-1 and Kdr 33, were decreased in response to LPS but they exhibited minimal response to I/R. Notably, several mRNAs whose products are being introduced as novel AKI biomarkers including IL-18, L-FABP, Timp2 and Igfbp7 changed little or not all in our AKI model. The discrepancies between the current results and the AKI biomarker literature 10, 34 may reflect species differences and/or the fact that here we examined mRNA and not protein levels. Nonetheless, our results in this mouse model suggest that no single biomarker can capture the heterogeneity of AKI transcriptional responses. For example, septic AKI signature (Icam-1 expressed in the endothelium) can be very different from I/R (Kim-1 expressed in the proximal tubules). Thus, measurements of combinatorial biomarker may be more useful to encompass responses from various injured renal tissue compartments.
Epigenetic biomarkers and therapies are emerging as important new means to diagnose and treat disease. Given their well-documented role in the pathogenesis of renal injury, and distinct temporal and injury-specific expression patterns in our animal model of AKI (Fig. 2), we chose Tnf, Ngal, Kim-1 and Icam-1 as representative AKI induced genes for in-depth epigenetic analyses.
Fig.2. Renal cortical Tnf, Ngal, Kim-1 and Icam-1 expression following unilateral kidney I/R and LPS injection.
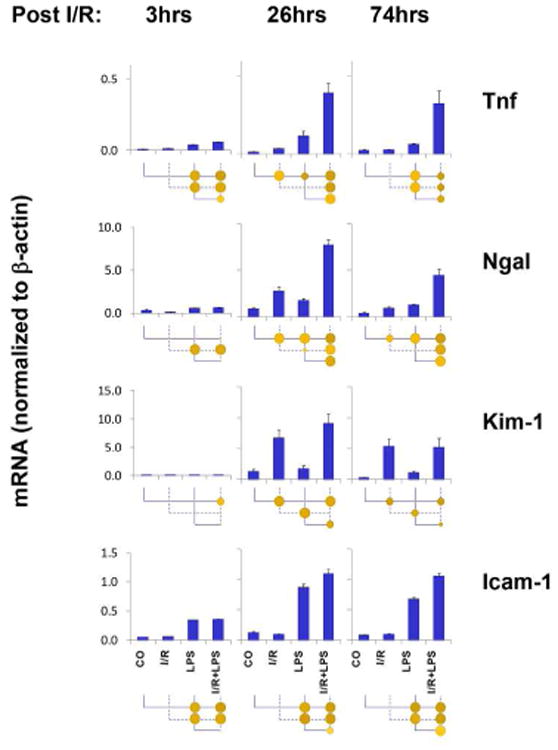
Total RNA from mice renal cortex was used in RT reactions with oligo dT. cDNA was used in real time PCR with gene specific primers (Table S1, supplement). mRNA level of a given gene in each sample was normalized to β-actin transcript. Data are represented as mean± SEM, n= 6 mice in each group. Statistical differences between two means (p value) are shown by the size of the solid circle under the x-axis: p<0.05 by small circle, p<0.01 by large circle, and no circle indicating the differences are not statistically significant (Methods and Fig.S1, supplement).
Pol II density measurement at Tnf, Kim-1, Ngal and Icam-1 genes confirms that transcription plays a role in their induced expression during AKI
Measurement of Pol II levels at gene loci provides means to assess rates of transcription 35. We analyzed Pol II binding at Tnf, Kim-1, Ngal and Icam-1 using an antibody that recognizes both unphosphorylated and phosphorylated C-terminal domain CTD (4H8), and observed that changes in the levels of Pol II at genes generally matched mRNA responses (compare Fig.2 and 3). Specifically, we found that: i) there was endotoxin-induced Pol II hyper-recruitment in the setting of renal I/R at the Tnf and Ngal genes, ii) Pol II levels at Kim-1 gene were increased in response to I/R but not to LPS, and iii) at the Icam-1 locus Pol II responded to LPS but not I/R. Similar results were obtained with Pol II antibodies that recognize either phosphorylated (pSer2 and pSer5) or unphosphorylated (8WG16) CTD (Supplemental Figs. S2 and S3). Of note, Pol II induction was seen 3hrs post I/R, at Tnf, Kim-1 and Ngal, a time point when mRNA changes were not yet detectable (Fig.3). Taken together, these results indicate that upregulation of mRNA of these selected genes during I/R, LPS and I/R + LPS are, at least in part, transcriptionally mediated. Next we used a panel of antibodies to permissive and repressive histone marks to systematically compare epigenetic profiles of Tnf, Kim-1, Ngal and Icam-1 in this AKI model.
Fig.3. RNA polymerase II (Pol II) Tnf, Ngal, Kim-1 and Icam-1 genes following unilateral kidney I/R and LPS injection.
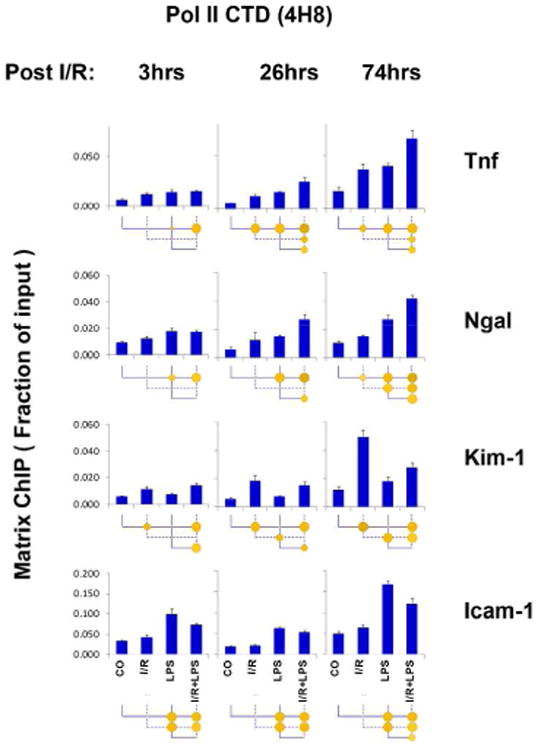
Sheared cross-linked renal cortex chromatin from mice were assayed using a monoclonal antibody to (4H8) that detects both unphosphorylated and phosphorylated C-terminal domain repeats YSPTSPS. ChIP DNA were analyzed at Tnf, Ngal, Kim-1 and Icam-1 genes in qPCR. Data represent mean ± SEM (6 animals from each group), expressed as fraction of input.
Epigenetic patterns of permissive marks in AKI
Histone acetylation can increase in response to extracellular signals and generate a transcriptionally permissive chromatin structure 30, 36. Histones acetylated at lysine residues also serve as docking sites for recruitment of chromatin modifiers. H3K9Ac is one of the most studied permissive histone modifications. H3K9Ac levels increased at Tnf post I/R (26 and 74hrs) without endotoxin hyper-response (Fig.4). Given that Ngal Pol II profile was similar to Tnf (Fig.3), it was surprising that no increases in H3K9Ac were detected at Ngal (Fig.4). Furthermore, there was only a small increase in H3K9Ac at the I/R-responsive Kim-1 locus and no H3K9Ac changes at Icam-1 even though there was robust Pol II response to LPS (compare Figs.2 and 3). Three other acetylation marks, H3K18A, H3K27Ac and H4K5/8/12/16Ac also exhibited changes at Tnf in response to both I/R and LPS (Figs. S4-6). Given that these measurements demonstrated robust H3K9Ac, H3K18Ac, H3K27Ac and H4K5/8/12/16Ac 7 changes at Tnf but little or no changes at all other genes (Figs.4, S4-6), we concluded that up-regulation of Icam-1, Ngal, and Kim-1 occurred without changes in their histone acetylation. Nonetheless, other acetylation events that were not examined in our study may have contributed to the robust AKI-induced changes in the expression of these genes.
Fig.4. Permissive histone H3 lysine 9 acetylation (H3K9Ac) at Tnf, Ngal, Kim-1 and Icam-1 genes following unilateral kidney I/R and LPS injection.
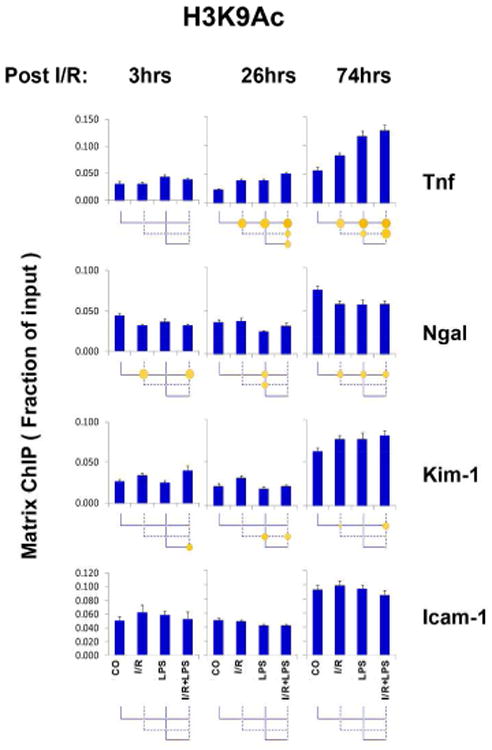
Sheared cross-linked renal cortex chromatin from mice were assayed using H3K9Ac antibody. Data represent mean ± SEM (6 animals from each group), expressed as fraction of input.
Unlike acetylation, methylation does not change lysine charge, and alters transcription by providing docking sites for chromatin modifiers. Methylation of histone H3 lysine 4 (H3K4m1, H3K4m2 and H3K4m3) marks chromatin states of active transcription14. We observed I/R-induced increases in H3K4m3 levels at Tnf at 26 and 74hrs but not at 3hrs (Fig.5). There was little or no response in H3K4m3 with LPS and there was no endotoxin hyper-response in the setting of I/R at this locus. H3K4m2 alterations at Tnf exhibited a similar pattern (Fig. S7). H3K4m3 changes at the Kim-1 and Ngal genes were similar to those seen at the Tnf locus but at a smaller magnitude. At the Icam-1 locus, we did not detect any H3K4m3 changes despite robust LPS-induced transcriptional activation of this gene (Fig.3). Permissive H3 serine 10 phosphorylation (H3pSer10) was also responsive to I/R but not LPS (Fig. S8). These results illustrate that epigenetic responses to I/R are distinct from those induced by endotoxin.
Fig.5. Permissive histone H3 lysine 4 tri-methylation (H3K4m3) at Tnf, Kim-1, Ngal, and Icam-1 genes following unilateral kidney I/R and LPS injection.
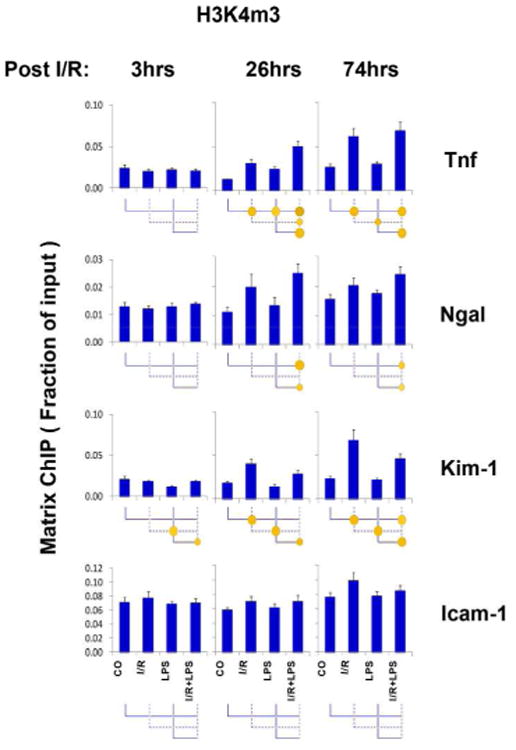
Sheared cross-linked renal cortex chromatin from mice were assayed using H3K4m3 antibody. Data represent mean ± SEM (6 animals from each group), expressed as fraction of input.
Epigenetic patterns of elongation marks in AKI
Higher levels of H3K36m3 have been correlated with enhanced transcription elongation rates 37. At the Tnf locus both I/R and LPS increased H3K36m3 levels and there was a synergistic effect for I/R+LPS at 26 and 74 hrs (Fig.6). At the Kim-1 locus, H3K36m3 increased following I/R but not LPS, and the increase was far greater at 26 and 74hrs compared to 3hrs. There was small H3K36m3 increase at 3 and 26hrs post I/R at Ngal and there was LPS hyper-response at 26hrs post I/R. Levels of H3K36m3 at Icam-1 increased in response to either I/R and LPS and the effects of I/R + LPS appeared additive. Changes in H3K36m2 were similarly responsive to either I/R or LPS (Fig.S9). H3K36m2 and H3K36m3 changes, at least in part, resembled Pol II profile at these loci (Fig.3) suggesting that they may play a role in increased transcription of these genes in response to I/R and LPS. Di-methylation of the histone H3 lysine 79 (H3K79m2), found within the nucleosome core, is also thought to be associated with transcription elongation. H3K79m2 generally increased 3 and 26hrs after I/R at all four genes. LPS alone did not have any effects on this mark (Fig.S10). Thus, overall the H3K79m2 responses were different compared to H3K36m3 and H3K36m2.
Fig.6. Transcription elongation histone H3 lysine 36 tri-methylation (H4K36m3) at Tnf, Kim-1, Ngal, and Icam-1 genes following unilateral kidney I/R and LPS injection.
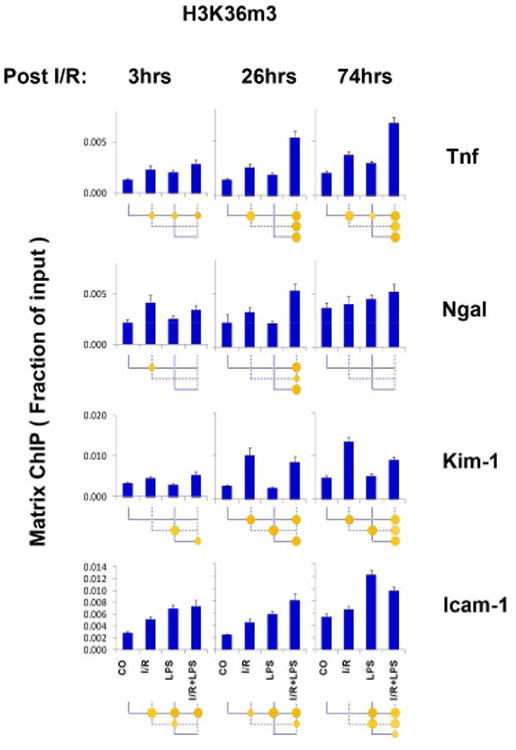
Sheared cross-linked renal cortex chromatin from mice were assayed using H3K36m3 antibody. Data represent mean ± SEM (6 animals from each group), expressed as fraction of input.
Epigenetic patterns of repressive marks in AKI
We examined repressive modifications representing each of the core histones, H3K27m3, H3K9m2, macroH2A, H2AK119ub1, H2BK120ub1 and H4pSer1. I/R decreased H3K27m3 at Tnf within 24 and 74 hrs but not after 3hrs (Fig.S11). Similarly, late I/R H3K27m3 decreases seen at Kim-1, Ngal, and Icam-1. Late (74hrs) post I/R reductions also seen in H3K9m2 levels at Kim-1 (Fig.S12). LPS reduced H3K27m3 and H3K9m2 levels only at Icam-1 (Fig.S11-S12). Given that LPS reduced both H3K27m3 and H3K9m2 at the Icam-1 gene these modification may play a role in induction of this gene in response to endotoxin. MacroH2A, H2AK119ub1 and H2BK120ub1 were more responsive to I/R than LPS (Figs. S13-S15). Interestingly, H4pSer1 38 exhibited a bi-phasic I/R response at all four genes with increase at 3hrs and decrease at 74hrs post I/R (Fig.7).
Fig.7. Transcription repressive histone H4 serine 1 phosphorylation (H4pSer1) at Tnf, Kim-1, Ngal, and Icam-1 genes following unilateral kidney I/R and LPS injection.
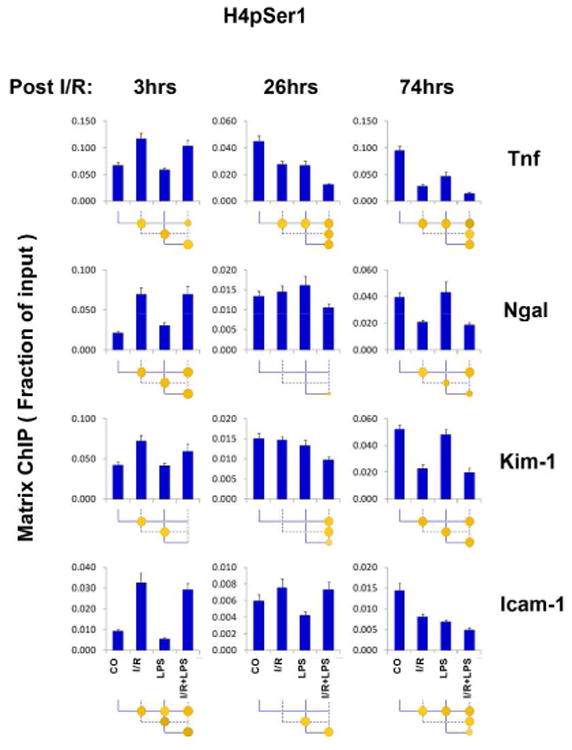
Sheared cross-linked renal cortex chromatin from mice were assayed using H4pSer1 antibody. Data represent mean ± SEM (6 animals from each group), expressed as fraction of input.
Integration analysis of mRNA, Pol II and histone modifications underscores diversity of epigenetic portraits at Tnf, Ngal, Kim-1 and Icam-1 loci in AKI
Genome-wide mapping has revealed a host of epigenetic ground-states characterized by clusters of histone modifications that coexist to control chromatin structure and function 39-40. However, little is known on how sets of functionally related epigenetic modifications respond to injury from their baseline states. To obtain an overview of the transcriptional and epigenetic signatures in AKI, we integrated mRNA, Pol II and histone modifications and summarized our findings using a heatmap (Fig. 8 and Fig.S16). Log-transformed I/R, LPS and I/R+LPS mean values normalized to the control groups of mice were used to generate gene portraits (Figs.8 and S16). The heatmap illustrates concordance between mRNA and Pol II levels indicating that, at least in part, expression of these genes in response to renal injury is transcriptionally mediated. Tnf and Ngal, two genes that exhibited synergistic interaction between I/R and endotoxin treatments had more similar profiles compared to Kim-1 and Icam-1. Still, the integrative analysis revealed a notable lack of injury-induced hyperacetylation at Ngal that was clearly observed at Tnf. Further, comparing Kim-1 and Icam-1 portraits underscored the striking differences between these two genes. Despite heterogeneity among AKI-induced genes, there was a general trend for the repressive marks to fall at 26 and 74hrs post I/R, implicating decreased silencing marks as a mechanism for increased transcription in all four genes.
Fig.8. Integrated transcriptional and epigenetic analysis of Tnf, Kim-1, Ngal, and Icam-1 portraits following unilateral kidney injury.
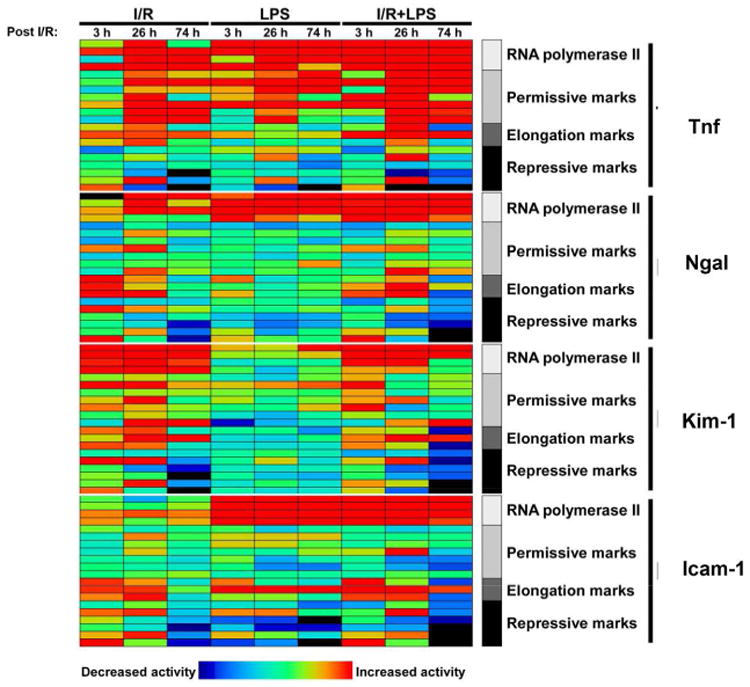
Log-transformed values for mRNA and Pol II levels, as well as several epigenetic modifications during temporal progression of AKI due to I/R, LPS and I/R+LPS are depicted as a heatmap. While there is a general increase in permissive marks and reduction of repressive marks in a time-dependent manner, note the heterogeneous response pattern of individual AKI-induced genes.
Discussion
This study represents an in-depth exploration of the epigenetic alterations at several key AKI-associated genes. We found that two commonly used models of kidney injury, I/R and LPS, have strikingly different effects on expression and epigenetic profiles of these candidate genes in a time dependent manner.
Previous experimental AKI studies have shown a host of permissive epigenetic changes at the transcriptionally induced Tnf gene 2, 15, 41. Much less is known about transcriptional and epigenetic control of Ngal, Kim-1 and Icam-1 genes in AKI. Here we found that the expression pattern of these and other AKI-related genes in response to I/R and endotoxin induced renal injury are different.
Some of the observed transcriptional heterogeneity may be attributable to cell type specific expression of AKI-associated genes. For example, Icam-1 is preferentially expressed in endothelial cells 42 thus the distinct mRNA response of this gene compared to the others (Fig.2) might represent cell type-specific mechanisms. In contrast, Kim-1, like Tnf and Ngal, is primarily expressed in the proximal tubules so the absence of Kim-1 response to LPS at the time point examined (Fig.2) suggests that the pathways that control its expression in this cell type are different from those of the other two genes 43.
In our model system, diversity in the transcriptional responses of Tnf, Ngal, Kim-1 and Icam-1 to I/R and LPS were further highlighted by differences in epigenetic alterations at these loci (Fig.8 and S16). For example, Pol II recruitment at both Tnf and Ngal closely followed changes in mRNA levels (Fig.3) but unlike induced histone acetylation at Tnf, there was little or no change in histone acetylation at the Ngal gene at the time-points examined (Fig. 8 and S16). Similarly, Kim-1 and Icam1 exhibited more modest increases in histone acetylation compared to the robust Tnf response. These results demonstrate that transcription could be upregulated with or without increased histone acetylation in a gene-specific manner. Our findings in AKI are consistent with similar observations in other systems, for example, during erythroid differentiation, GATA factor-induced transcription occurs while permissive chromatin modifications remain unchanged 44. In contrast to permissive histone acetylation processes, there was greater AKI-induced reduction in repressive marks at Ngal, Kim-1 and Icam-1 compared to Tnf gene (Fig.8 and S16), suggesting an alternative mechanism for the observed upregulation of these three genes. Our integrative analysis encompassing multiple injury models, temporal profiling, and detailed epigenetic interrogation illustrates the rich diversity of transcriptional control in AKI, and highlights common and major differences in regulation of gene expression in this complex syndrome (Fig.8 and S16).
There are several limitations in the current study. We assessed only one time point post endotoxin injection and therefore did not capture potential changes in LPS-induced Kim-1 expression later in the course of evolving injury. Similarly if we had examined more time points post I/R we may have noted additional temporal changes at the Icam-1 locus. Although we investigated twenty different transcription and epigenetic events—a more comprehensive survey than any previous AKI epigenetics study—the number of known epigenetic marks is significantly larger than our examined set. Further, we studied only a select group of candidate AKI genes and not a genome-wide screen (Fig.1). These limitations reflect remaining technical challenges preparing large number of renal cortex samples for ChIP assays. It is anticipated that with further advancement in epigenetic platforms combined with next generation sequencing, these technical bottlenecks can be overcome. In this context, our general approach can serve as ground-work for future AKI studies using the next generation epigenetic technologies.
How does the observed epigenetic heterogeneity in AKI advance research and ultimately help us improve clinical outcomes of this syndrome? First, these studies provide further evidence that epigenetic mechanisms play a key role in AKI2. Second, the rapid decline of renal function in AKI followed by structural changes 9 poses a challenge in initially identifying the culprits of injury and then instituting appropriately-timed therapies tailored to specific patients. Our study design allowed us to demonstrate that several epigenetic alterations at different genes exhibited distinct time-dependent changes following AKI (e.g. H4pSer1). Thus, extending our integrated approach to urine nucleic acids and chromatin analysis 45-47 may help identify appropriate time windows for AKI treatment. Third, epigenetic modifications are reversible, providing means for pharmacologic manipulation of gene expression. Therefore, epigenetic enzymes mediating detrimental changes in transcription can be targeted to modulate the severity of renal injury.
Conclusion
The striking diversity of epigenetic portraits of I/R- and endotoxin-induced renal genes observed is this study suggests that different signaling pathways drive recruitment of distinct sets of epigenetic enzymes to modify chromatin structures of candidate AKI genes. Thus, these observations open up an exciting possibility that this epigenetic heterogeneity can be exploited to devise therapeutic interventions to target groups of AKI-associated genes with similar epigenetic profiles, while minimizing off target side effects.
Methods
Reagents
Bovine serum albumin (BSA), phosphate buffered saline (PBS), salmon sperm DNA, and protein A were from Sigma, and proteinase K was from Invitrogen. Matrix ChIP-MeDIP 96-well polypropylene plates were from Bioexpress. Formaldehyde, ethanol, NaCl, EDTA, Triton X-100, NP-40, Tris, leupeptin, PMSF, p-nitrophenyl phosphate, NaF, Na3VO4, Na2MoO4 and β-glycerophosphate were from Sigma. The antibodies were commercially available and are listed in Table 1.
Animal model of AKI
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Washington and Fred Hutchinson Cancer Research Center. CD 1 mice (Charles River Laboratories, Wilmington, MA; 6-10 weeks of age, male; 30-35 gms), were maintained under routine vivarium conditions. The experimental protocol in this study is shown in Fig.S1. To study mechanisms of renal endotoxin hyperresponsiveness, we have used an I/R model 27 where one kidney of each animal is subjected to ischemia/reperfusion with subsequent treatment with either saline (I/R) or LPS (I/R+LPS). In brief, mice were anesthetized with pentobarbital and subjected to a midline abdominal incision under sterile conditions. Left renal ischemia was induced with an atraumatic microvascular clamp applied to the renal pedicle. After 30 min of unilateral renal artery occlusion, the clamp was released and reperfusion of the entire kidney was assessed visually (by loss of global cyanosis). A 30 min ischemic insult was selected for study because it induces moderately severe ischemic kidney damage 48. One, twenty-four or seventy-two hours after I/R injury, mice received a tail vein injection of either LPS (2 mg/kg; 0111:B4; l-2630; Sigma, St. Louis, MO; in 80 μl of saline) or saline. Two hours after injection, mice were re-anesthetized, the abdominal cavity was opened, the kidneys were extracted, and renal cortical samples were cut from the kidneys with a razor blade and rapidly frozen at -70°C. The mice died within 30 sec due to exsanguinations. Thus, the timing of sacrifice relative to I/R was 3, 26 and 74 hrs, and 2 hrs post either LPS or saline treatment. As previously documented 28 the right non-ischemic, contralateral (control) kidney recapitulates what is seen in sham operated kidneys and hence served as an internal control. Histology for I/R and LPS models of AKI have previously been evaluated 48-50
RNA Extraction and cDNA Synthesis
RNA was extracted from tissue fragments using Trizol reagent as per the manufacturer's protocol. To synthesize cDNA, 400 ng of Trizol-extracted total RNA was reverse transcribed with 200 units MMLV reverse transcriptase (Invitrogen) and oligo dT primers in 10μl reactions in 96-well microplates. RT reactions were diluted 100-fold prior to running qPCR 51.
Chromatin preparation and multiplex Matrix ChIP platform
The multiplex microplate Matrix ChIP method was previously described 29-30. Briefly, for ChIP assays, tissue fragments (10-20 mg) were cross-linked with formaldehyde, and chromatin was sheared using Diagenode Bioruptor. ChIP assays were done using protein A-coated 96-well polypropylene microplates as described before 29. 1-2 μl of eluted DNA was used in 2-4 μl real-time PCR reactions (ABI7900HT). All PCR reactions were run in quadruplicates. PCR calibration curves were generated for each primer pair from a dilution series of total mouse genomic DNA. The PCR primer efficiency curve was fit to cycle threshold (Ct) versus log(DNA concentration) using an r-squared best fit. DNA concentration values for each ChIP and input DNA sample were calculated from their respective average Ct values. Final results are expressed as fraction of input DNA 30. Matrix ChIP PCR primers to 5′ end of genes used in this study are shown in Table S2 and list of antibodies in Table S3 (supplement).
Statistics and visualization
To acquire, store and analyze large data sets generated by the high throughput Matrix ChIP platform, we developed a novel graphical method (GraphGrid). Pair-wise statistically significant differences are represented by the size of a circle for each comparison made with a small circle representing p<0.05, a large circle indicating p<0.01 and no circle implying non-significance. GraphGrid uses a two-tailed Student's t-test to compute p-values 15.
Cluster analysis
All RT-qPCR gene expression values during injury conditions (I/R, LPS, I/R+LPS) were normalized to their uninjured, saline-treated control values and log-transformed. PCR primers are listed in Table S1 (supplement). We applied hierarchical clustering to log-transformed relative expression values using Euclidian distance measure and grouped genes based on their expression patterns across injury conditions 52. Heatmaps were also generated using similarly normalized and log-transformed values for Pol II levels and the various epigenetic marks under different AKI conditions.
Supplementary Material
Key points of the study.
Different but overlapping groups of genes respond to I/R and endotoxin treatments in kidneys.
Transcription plays a role in AKI-induced Tnf (I/R and endotoxin), Ngal (I/R and endotoxin), Kim-1 (I/R) and Icam-1 (endotoxin) expression.
Epigenetic changes at AKI genes share similarities but also have major differences.
Epigenetic alterations at different genes exhibit distinct time-dependent changes.
Heterogeneity of epigenetic changes at AKI genes indicate involvement of several epigenetic pathways that could potentially be exploited pharmacologically through rational-drug design to alter the course and improve clinical outcomes of this syndrome.
Acknowledgments
This work was supported by NIH R01 DK083310, R01 DK094934, RO1 DK38432, R21 GM111439, R21 DK09881, R33 CA191135.
Footnotes
Statement of Competing Financial Interests: None to declare.
References
- 1.Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nat Rev Nephrol. 2011;7:209–217. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bomsztyk K, Denisenko O. Epigenetic alterations in acute kidney injury. Semin Nephrol. 2013;33:327–340. doi: 10.1016/j.semnephrol.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012;82:516–524. doi: 10.1038/ki.2012.208. [DOI] [PubMed] [Google Scholar]
- 4.Lameire NH, Bagga A, Cruz D, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382:170–179. doi: 10.1016/S0140-6736(13)60647-9. [DOI] [PubMed] [Google Scholar]
- 5.Chertow GM, Burdick E, Honour M, et al. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 6.Coca SG. Long-term outcomes of acute kidney injury. Curr Opin Nephrol Hypertens. 2010;19:266–272. doi: 10.1097/MNH.0b013e3283375538. [DOI] [PubMed] [Google Scholar]
- 7.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015 doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly KJ. Acute renal failure: much more than a kidney disease. Semin Nephrol. 2006;26:105–113. doi: 10.1016/j.semnephrol.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alge JL, Arthur JM. Biomarkers of AKI: A Review of Mechanistic Relevance and Potential Therapeutic Implications. Clin J Am Soc Nephrol. 2014 doi: 10.2215/CJN.12191213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grigoryev DN, Liu M, Hassoun HT, et al. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol. 2008;19:547–558. doi: 10.1681/ASN.2007040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonventre JV, Vaidya VS, Schmouder R, et al. Next-generation biomarkers for detecting kidney toxicity. Nat Biotechnol. 2010;28:436–440. doi: 10.1038/nbt0510-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Bomsztyk K, Flanagin S, Mar D, et al. Synchronous Recruitment of Epigenetic Modifiers to Endotoxin Synergistically Activated Tnf-alpha Gene in Acute Kidney Injury. PLoS One. 2013;8:e70322. doi: 10.1371/journal.pone.0070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikula M, Bomsztyk K. Direct recruitment of ERK cascade components to inducible genes is regulated by the heterogeneous nuclear ribonucleoprotein (HnRNP) K. J Biol Chem. 2011;286:9763–9775. doi: 10.1074/jbc.M110.213330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson JD, Leboeuf RC, Bomsztyk K. Direct recruitment of insulin receptor and ERK signaling cascade to insulin-inducible gene loci. Diabetes. 2011;60:127–137. doi: 10.2337/db09-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Zarjou A, Traylor AM, et al. In vivo regulation of the heme oxygenase-1 gene in humanized transgenic mice. Kidney Int. 2012;82:278–291. doi: 10.1038/ki.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramesh G, Zhang B, Uematsu S, et al. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am J Physiol Renal Physiol. 2007;293:F325–332. doi: 10.1152/ajprenal.00158.2007. [DOI] [PubMed] [Google Scholar]
- 20.Matejovic M, Chvojka J, Radej J, et al. Sepsis and acute kidney injury are bidirectional. Contrib Nephrol. 2011;174:78–88. doi: 10.1159/000329239. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs R, Honore PM, Joannes-Boyau O, et al. Septic acute kidney injury: the culprit is inflammatory apoptosis rather than ischemic necrosis. Blood Purif. 2011;32:262–265. doi: 10.1159/000330244. [DOI] [PubMed] [Google Scholar]
- 22.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 23.Chvojka J, Sykora R, Karvunidis T, et al. New developments in septic acute kidney injury. Physiol Res. 2010;59:859–869. doi: 10.33549/physiolres.931936. [DOI] [PubMed] [Google Scholar]
- 24.Bagshaw SM, George C, Bellomo R. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care. 2008;12:R47. doi: 10.1186/cc6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. 2015;11:88–101. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 26.Nath KA. Renal response to repeated exposure to endotoxin: implications for acute kidney injury. Kidney Int. 2007;71:477–479. doi: 10.1038/sj.ki.5002150. [DOI] [PubMed] [Google Scholar]
- 27.Zager RA, Johnson AC, Lund S, et al. Acute renal failure: determinants and characteristics of the injury-induced hyperinflammatory response. Am J Physiol Renal Physiol. 2006;291:F546–556. doi: 10.1152/ajprenal.00072.2006. [DOI] [PubMed] [Google Scholar]
- 28.Zager RA, Johnson AC, Hanson SY, et al. Ischemic proximal tubular injury primes mice to endotoxin-induced TNF-alpha generation and systemic release. Am J Physiol Renal Physiol. 2005;289:F289–297. doi: 10.1152/ajprenal.00023.2005. [DOI] [PubMed] [Google Scholar]
- 29.Yu J, Feng Q, Ruan Y, et al. Microplate-based platform for combined chromatin and DNA methylation immunoprecipitation assays. BMC Mol Biol. 2011;12:49. doi: 10.1186/1471-2199-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanagin S, Nelson JD, Castner DG, et al. Microplate-based chromatin immunoprecipitation method, Matrix ChIP: a platform to study signaling of complex genomic events. Nucleic Acids Res. 2008;36:e17. doi: 10.1093/nar/gkn001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urbschat A, Obermuller N, Haferkamp A. Biomarkers of kidney injury. Biomarkers. 2011;16 Suppl 1:S22–30. doi: 10.3109/1354750X.2011.587129. [DOI] [PubMed] [Google Scholar]
- 32.Hu MC, Moe OW. Klotho as a potential biomarker and therapy for acute kidney injury. Nat Rev Nephrol. 2012;8:423–429. doi: 10.1038/nrneph.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suarez Y, Fernandez-Hernando C, Pober JS, et al. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–1173. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 34.Charlton JR, Portilla D, Okusa MD. A basic science view of acute kidney injury biomarkers. Nephrol Dial Transplant. 2014;29:1301–1311. doi: 10.1093/ndt/gft510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson J, Denisenko O, Bomsztyk K. Profiling RNA polymerase II using the fast chromatin immunoprecipitation method. Methods Mol Biol. 2011;703:219–234. doi: 10.1007/978-1-59745-248-9_15. [DOI] [PubMed] [Google Scholar]
- 36.Nelson JD, Flanagin S, Kawata Y, et al. Transcription of laminin γ1 chain gene in rat mesangial cells: constitutive and inducible RNA polymerase II recruitment and chromatin states. Am J Physiol Renal Physiol. 2008;294:F525–F533. doi: 10.1152/ajprenal.00299.2007. [DOI] [PubMed] [Google Scholar]
- 37.Kizer KO, Phatnani HP, Shibata Y, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wendt KD, Shilatifard A. Packing for the germy: the role of histone H4 Ser1 phosphorylation in chromatin compaction and germ cell development. Genes Dev. 2006;20:2487–2491. doi: 10.1101/gad.1477706. [DOI] [PubMed] [Google Scholar]
- 39.Ho JW, Jung YL, Liu T, et al. Comparative analysis of metazoan chromatin organization. Nature. 2014;512:449–452. doi: 10.1038/nature13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naito M, Zager RA, Bomsztyk K. BRG1 increases transcription of proinflammatory genes in renal ischemia. J Am Soc Nephrol. 2009;20:1787–1796. doi: 10.1681/ASN.2009010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu X, Guo R, Wang Y, et al. The role of ICAM-1 in endotoxin-induced acute renal failure. Am J Physiol Renal Physiol. 2007;293:F1262–1271. doi: 10.1152/ajprenal.00445.2006. [DOI] [PubMed] [Google Scholar]
- 43.Ajay AK, Kim TM, Ramirez-Gonzalez V, et al. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J Am Soc Nephrol. 2014;25:105–118. doi: 10.1681/ASN.2013020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu W, Cheng Y, Keller CA, et al. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. Genome Res. 2011;21:1659–1671. doi: 10.1101/gr.125088.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munshi R, Johnson A, Siew ED, et al. MCP-1 gene activation marks acute kidney injury. J Am Soc Nephrol. 2011;22:165–175. doi: 10.1681/ASN.2010060641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olkhov-Mitsel E, Zdravic D, Kron K, et al. Novel multiplex MethyLight protocol for detection of DNA methylation in patient tissues and bodily fluids. Sci Rep. 2014;4:4432. doi: 10.1038/srep04432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miranda KC, Bond DT, McKee M, et al. Nucleic acids within urinary exosomes/microvesicles are potential biomarkers for renal disease. Kidney Int. 2010;78:191–199. doi: 10.1038/ki.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol. 2011;301:F1334–1345. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zahedi K, Barone S, Kramer DL, et al. The role of spermidine/spermine N1-acetyltransferase in endotoxin-induced acute kidney injury. Am J Physiol Cell Physiol. 2010;299:C164–174. doi: 10.1152/ajpcell.00512.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson JD, Denisenko O, Sova P, et al. Fast chromatin immunoprecipitation assay. Nucleic Acids Res. 2006;34:e2. doi: 10.1093/nar/gnj004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.