

Summary
β-adrenergic signaling pathways mediate key aspects of cardiac function. Its dysregulation is associated with a range of cardiac diseases, including dilated cardiomyopathy (DCM). Previously, we established an iPSC model of familial DCM from patients with a mutation in TNNT2, a sarcomeric protein. Here, we found that the β-adrenergic agonist isoproterenol induced mature β-adrenergic signaling in iPSC-derived cardiomyocytes (iPSC-CMs), but that this pathway was blunted in DCM iPSC-CMs. Although expression levels of several β-adrenergic signaling components were unaltered between control and DCM iPSC-CMs, we found that phosphodiesterases (PDE) 2A and PDE3A were upregulated in DCM iPSC-CMs, and that PDE2A was also upregulated in DCM patient tissue. We further discovered increased nuclear localization of mutant TNNT2 and epigenetic modifications of PDE genes in both DCM iPSC-CMs and patient tissue. Notably, pharmacologic inhibition of PDE2A and PDE3A restored cAMP levels and ameliorated the impaired β-adrenergic signaling of in DCM iPSC-CMs, suggesting therapeutic potential.
Graphical abstract
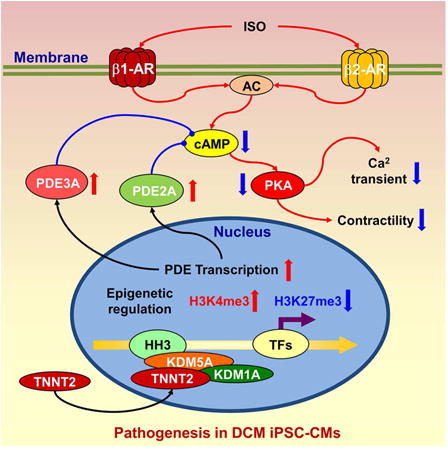
Introduction
Dilated cardiomyopathy (DCM) is a common myocardial disorder characterized by ventricular chamber enlargement and systolic dysfunction (Maron et al., 2006). DCM gives rise to sudden cardiac death, hypertension, and heart failure, and contributes significantly to health care costs. Recent studies have shown that more than 40% of DCM is caused by mutations in genes that encode sarcomeric, cytoskeletal, mitochondrial, calcium handling, or nuclear membrane proteins (Burkett and Hershberger, 2005; Morita et al., 2005). Accordingly, multiple molecular mechanisms, including loosened mechanical linkage of the extracellular matrix to the cytoskeleton (Lapidos et al., 2004), disarrangement of Z-disc protein elements (Knoll et al., 2002), decreased myofilament calcium sensitivity (Kamisago et al., 2000), ion channel abnormalities (Bienengraeber et al., 2004), and remodeled intracellular calcium handling (Schmitt et al., 2003) have been reported to underlie the decreased systolic contractile function of cardiac muscle in DCM. However, the heterogeneous etiologies underlying DCM also have limited our understanding of the respective roles of such factors in the long-term pathogenesis of DCM.
Ever since the discovery of the 4 key reprogramming factors by Yamanaka et al. (Takahashi and Yamanaka, 2006), significant strides have been made in deriving cardiomyocytes from human originated stem cells (Burridge et al., 2012; Takahashi et al., 2007; Yu et al., 2007). These advances have enabled disease modeling and development of regenerative medicine approaches for cardiac diseases (Chong et al., 2014; Lan et al., 2013; Liang et al., 2013; Sun et al., 2012; Wang et al., 2014). Human iPSC-derived cardiomyocytes (iPSC-CMs) have been shown to recapitulate morphological and functional properties of native cardiomyocytes. However, few studies have evaluated the platform's ability to recapitulate signaling pathways, molecular pathophysiology, and underlying transcriptional regulation in diseased cardiomyocytes. The ability to generate iPSC-CMs from patients carrying known or novel mutations, coupled with the feasibility of introducing specific modifications to their genome, presents an unprecedented opportunity to investigate pathogenic mutations and identify new treatments for the diseases they cause. Thus, uncovering novel mechanism of DCM in stem cell-derived cardiomyocyte models will greatly contribute to our understanding of the application of stem cell based disease models in both basic scientific and translational research.
It is well known that β-adrenergic signaling pathways mediate the inotropic and chronotropic regulation of cardiac function, and release reserved pumping power to meet the increased demand for heart output under stress (Rockman et al., 2002; Xiang and Kobilka, 2003). Moreover, abnormalities in β-adrenergic signaling are associated with certain cardiomyopathies such as DCM (Cho et al., 1999), cardiac hypertrophy (Engelhardt et al., 1999), and heart failure (Lohse et al., 2003; Post et al., 1999). Clinically, β-blockers are commonly prescribed for hypertension, arrhythmia, and heart failure. Therefore, improving the understanding of β-adrenergic signaling in iPSC-CMs and its regulation in DCM is scientifically and clinically significant because it can elucidate pathophysiologic mechanism of the disorder and identify new treatments for DCM.
In the present study, we focused on β-adrenergic signaling pathway development in iPSC-CMs, measured their responses to β-adrenergic activation, and investigated their receptor subtype dependence at different maturation stages. Then, by comparing control (Ctrl) and DCM iPSC-CMs, we demonstrated impaired β-adrenergic signaling and contractile function in DCM iPSC-CMs. Expression profiles showed a significant up-regulation of PDE subtypes in DCM iPSC-CMs, which could restrict cAMP signaling evoked by β-adrenergic activation. Further functional assays confirmed that DCM iPSC-CMs regain their reactivity to β-agonist stimulation after subtype-specific blockade of phosphodiesterases (PDE) 2A and 3A. Finally, chromatin immunoprecipitation (ChiP) studies suggest nuclear TNNT2 may contribute to novel epigenetic mechanisms that underlie DCM pathogenesis.
Results
Differentiation of iPSC-CMs was accompanied by specific regulation of β-adrenergic signaling related proteins
To investigate the maturation of β-adrenergic signaling pathways in iPSC-CMs, we used iPSC lines from 3 healthy volunteers (Supplemental Table 1). Genetic screening showed no known mutations related to familial heart diseases. All of the iPSC lines were identified by positive immunostaining for multiple ESC-like markers such as SSEA-4, TRA-1-81, Oct4, Sox2, Nanog, and Klf4 (Supplemental Figure 1A). The pluripotent nature of iPSC lines was further demonstrated by their potential to form all three germ layers in vivo (Supplemental Figure 1B). Beating iPSC-CMs were differentiated and purified as described (Lian et al., 2012) (Supplemental Video 1). Fluorescence-activated cell sorting (FACS) and immunostaining illustrated typical properties of cardiomyocyte lineages in these iPSC-CMs (Supplemental Figure 1 C to 1E).
For the expression profiling of genes related to β-adrenergic signaling during maturation, total RNA was extracted from iPSC lines and at days 12, 30, and 60 of differentiation. The cDNA libraries were constructed and then subjected to RNA-seq analysis. Most components of the β-adrenergic signaling apparatus such as β2 adrenergic receptor (ADRB2), adenylate cyclase (ADCY) 5 and 6, and PDE4D and PDE5A (Figure 1A) were sharply up-regulated in iPSC-CMs compared to iPSC lines. RNA-seq results were further verified by qPCR. Although the expression of ADRB2 was increased by 7.37±0.74-fold at day 12 of differentiation, no significant changes in β1 adrenergic receptors (ADRB1) expression were seen prior to day 30 (Figure 1B and 1C). Expression of alpha adrenergic receptors varied: ADRA1A was mildly up-regulated by 1.81±0.21-fold at day 60 of differentiation. ADRA1B was significantly up-regulated by 15.08±1.31-fold at day 12 of differentiation, while ADRA1D expression was negligible (Supplemental Figure 1F to 1H). The expression of adenylyl cyclase subtypes (ADCY5/6) increased significantly to a level comparable to that of adult human left ventricle (LV) tissues after 12-30 days of differentiation (Figure 1D and E). Throughout differentiation, PDE subtypes underwent distinct alterations in expression: PDE2A expression was decreased sharply to 1.85±0.06% of its original level after 12 days of differentiation and remained low (Figure 1F), while the levels of both PDE4D and PDE5A rose more than 40-fold by day 60 of maturation (Figure 1G, Supplemental Figure 1I). No significant up-regulation of PDE3A was observed until day 60 of differentiation (Supplemental Figure 1J). The expression levels of key calcium-handling proteins (e.g., PLN, CASQ2, LCC, RyR2 and SERCA2a) and other β-adrenergic signaling related proteins (e.g., PRKACA, CAMIIA and CAMIID) showed significant up-regulation during differentiation (Supplemental Figure 1K to 1R). No change was observed for the Gi subunit expression (Supplemental Figure 1S), while Gs subunit expression level decreased throughout differentiation (Supplemental Figure 1T).
Figure 1. Expression profiling of β-adrenergic signaling proteins during differentiation and maturation of iPSC-CMs.
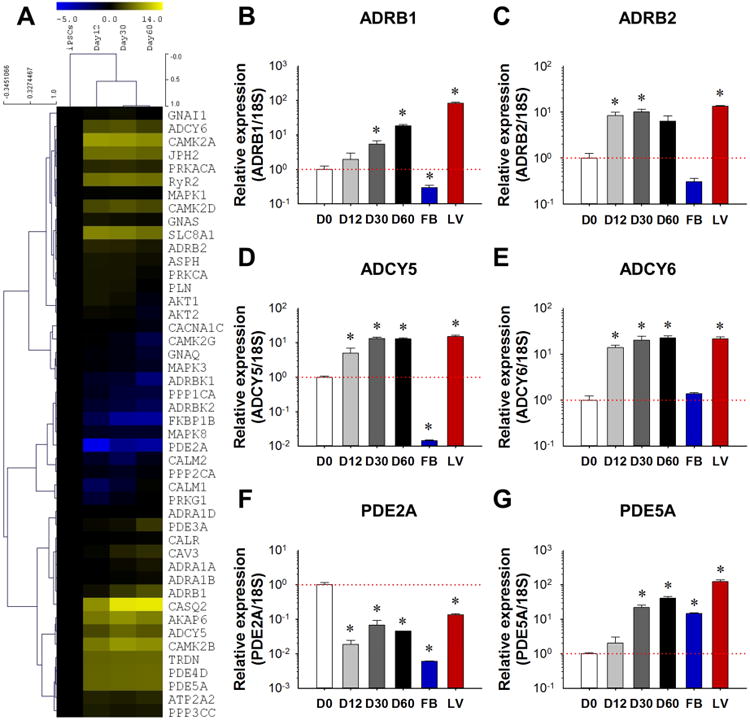
(A) RNA-seq heat map representation of gene expression level in undifferentiated iPSCs, and in day 12, day 30, and day 60 iPSC-CMs after differentiation. (B-G) Real-time PCR verification of the expression levels of ADRB1, ADRB2, ADCY5, ADCY6, PDE2A, and PDE5A, with n=3 cell lines at each time point from each group. Human fibroblast cells (FBs) were used as a negative control and healthy human left ventricle tissue sample (LV) was used as a positive control. *P<0.05 vs. D0 by one way ANOVA (Holm-Sidak method). See also Supplemental Figure 1, Supplemental Table 1 and Supplemental Video1.
β-adrenergic stimulation induced chronotropic responses in iPSC-CMs
In order to test the effects of β-adrenergic stimulation, iPSC-CMs were next treated with 1 μM isoproterenol (ISO) and spontaneous calcium transients were analyzed (Figure 2A and 2B). Human iPSC-CMs at day 30 and day 60 of differentiation exhibited similar beating rates (35.7±1.4 and 34.5±0.7 bpm) at basal level, increasing to 84.2±1.9 and 58.7±3.0 bpm after ISO treatment (Figure 2C). The transient decay Tau in day 30 and day 60 iPSC-CMs were curtailed by 50.5±2.6% and 19.7±4.7%, respectively, by β-adrenergic activation suggesting a possible β-adrenergic signaling-dependent activation of calcium recycling in iPSC-CMs (Figure 2D). Likewise, time to peak in both groups decreased by 47.4±2.4% and 19.8±2.8%, respectively (Figure 2E). Interestingly, transient amplitude was not increased after ISO treatment at different stages of differentiation (Supplemental Figure 2A), yet other temporal parameters such as transient duration 90 and 50 were accelerated (Supplemental Figure 2B and 2C). We also observed that pre-treatment with the non-selective β-blocker propranolol eliminated the effect of ISO in iPSC-CMs, indicating that the ISO signaling in these cells was indeed conducted via β-adrenergic receptors (ARs) (data not shown).
Figure 2. Functional regulation by β-adrenergic signaling in iPSC-CMs.
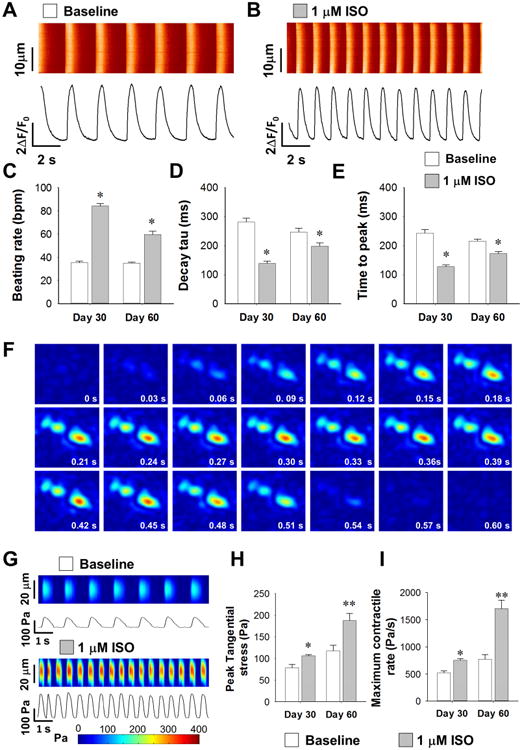
(A-B) Representative calcium imaging recording traces in WT iPSC-CMs before and after ISO treatment. (C-E) Statistics of calcium handling properties such as beating rate (C), decay tau (D), and time to peak (E) with and without ISO at different maturation stages with n>35 iPSC-CMs from 3 lines in each group. *P<0.05 vs. baseline group in each time point by two way ANOVA (Holm-Sidak method). (F) Representative temporal series of contractility distribution pattern throughout one contraction cycle of WT iPSC-CMs. (G) Representative contractility profiling before and after ISO treatment, Pa: Pascal. (H-I) Measurements on contraction parameters showing increased peak tangential force (H) and maximum contract rate (I) with n>25 iPSC-CMs from 3 lines in each group. **P<0.01 vs. baseline untreated group by Student's t-test. See also Supplemental Figure 2, Supplemental Table 5 and Supplemental Video 2.
β-adrenergic stimulation induced inotropic responses in iPSC-CMs
As β-adrenergic activation accelerated the calcium handling in iPSC-CMs, we wonder if it can also increase contractile force in these cells. Using a hydrogel-based traction force microscopy (TFM) imaging assay (Supplemental Video 2), we next measured the contractile force of iPSC-CMs with or without ISO treatment at different stages of maturation (Figure 2F and 2G). ISO treatment increased the peak contractile force and the maximum contractile rate by 35.0±3.5% and 44.9±5.0%, respectively, in day 30 iPSC-CMs, and by 59.3±12.9% and 120.6±19.0%, respectively, in day 60 iPSC-CMs (Figure 2H and 2I). Furthermore, ISO-treated iPSC-CMs demonstrated positive functional regulation in other contractile parameters such as time to peak, contractile duration 90 and 50, half rising and decay time, beating rate, and maximum decay rate at both maturation stages (Supplemental Figure 2D to 2K). By analyzing all the readouts in the contractility assay, we found that ISO treatment induced a right-shift on the distribution of peak contractile force (Supplemental Figure 2L to 2O). Collectively, these data suggested that the maturation of iPSC-CMs was accompanied by the formation of functional β-adrenergic signaling pathways, which can induce inotropic modification of contractile function.
β2 adrenergic receptor dominates the response of iPSC-CMs to β-adrenergic stimulation at early stages of iPSC-CM differentiation
In adult cardiomyocytes, β-adrenergic signaling is conducted by β1 and β2 ARs (Bristow et al., 1986; Brodde, 1991; del Monte et al., 1993). To date, the subtype selectivity of β-AR-dependent signaling has not been well defined in iPSC-CMs. To address the issue, we employed β1 (CGP-20712A) and β2-specific blockers (ICI-118551) to selectively isolate the contribution of each β-AR subtypes in iPSC-CMs (Figure 3A). Calcium transient amplitude was not affected by selective activation of either β1 or β2 ARs (Supplemental Figure 3A). Interestingly, while iPSC-CMs at day 30 only responded to β2 receptor activation (e.g., CGP-20712A + ISO), both β1 and β2 ARs contributed significantly to the functional regulation in day 60 iPSC-CMs (Figure 3B and 3C, Supplemental Figure 3B), indicating a switch of β-AR subtype dependence at different maturation stages of iPSC-CMs.
Figure 3. Subtype dependence of β-adrenergic signaling in different maturation stages of iPSC-CMs.
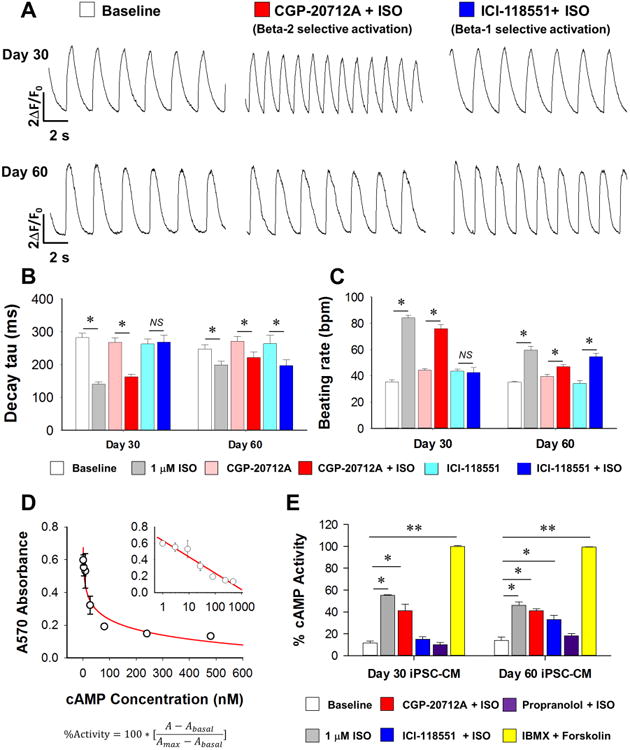
(A) Representative calcium handling traces of day 30 and day 60 iPSC-CMs at baseline after specific β2 AR activation (CGP-20712A + ISO) and after specific β1 AR activation (ICI-118551 + ISO). (B-C) Statistics of calcium handling properties of day 30 and day 60 iPSC-CMs in baseline, ISO treated, CGP-20712A, CGP-20712A + ISO, ICI-118551, and ICI-118551 + ISO groups with n>30 iPSC-CMs from 3 lines in each time point from each group. *P<0.05 vs. baseline group in each time point by two way ANOVA (Holm-Sidak method). (D) Standard curve for ELISA-based cAMP assay, with final results presented in the form of percentage activity, as shown in the formula. (E) cAMP assay assessment of cAMP generation in iPSC-CMs upon different drug treatment at day 30 and day 60 after differentiation. Data were from 6 independent experiments using 3 lines of iPSC-CMs. *P<0.05 and **P<0.01 vs baseline group by one way ANOVA (Holm-Sidak method). See also Supplemental Figure 3.
We also tested the receptor subtype dependence of β-adrenergic stimulation in contractility assays. Our results showed that β2 AR blockade with ICI-118551 completely eliminated the effect of ISO on peak tangential stress and beating rate in the day 30 group, whereas in the day 60 group, neither β1 nor β2 AR blockers inhibited responses to β-adrenergic stimulation (Supplemental Figure 3C and 3D). In further support of this observation, an ELISA-based cAMP assay also showed that at day 30, β2 activation (e.g., CGP-20712A + ISO) but not β1 activation (e.g., ICI-118551 + ISO) induced significant cAMP elevation. By contrast, abundant cAMP was generated by both β1 and β2 activation in day 60 iPSC-CMs (Figure 3D and 3E). These functional results are in line with our β-AR expression profiles, with β2 AR showing a relatively higher expression at early stages of maturation. These findings also independently suggest a dynamic regulation of the receptor dependence of β-adrenergic signaling pathways during the maturation of iPSC-CMs.
DCM iPSC-CMs exhibit impaired response to β-adrenergic stimulation
As abnormal β-adrenergic regulation in cardiac diseases has been well documented (Lohse et al., 2003; Post et al., 1999), we next examined whether DCM iPSC-CMs can also recapitulate this dysfunction. To address this question, we differentiated iPSC-CMs from both familial control (Ctrl) and DCM (TNNT2 R173W) groups as described (Sun et al., 2012). Flow cytometry assays indicated that the efficiency of iPSC-CM differentiation in n = 3 Ctrl and n = 3 DCM patients was >90% (Supplemental Figure 4A to 4C). Successful differentiation was further confirmed by immunostaining of cardiac-specific markers such as TNNT2 and α-actinin (Supplemental Figure 4D and 4F). Furthermore, detailed analysis of myofilament protein arrangement revealed an abnormal pattern of sarcomere structure in DCM iPSC-CMs, but not in Ctrl iPSC-CMs (Supplemental Figure 4E and 4G, 4H to 4K). According to our baseline data, most of the experiments in DCM study were carried out using day 60 iPSC-CMs while they developed more matured beta-adrenergic signaling.
Day 60 iPSC-CMs from both groups were challenged with ISO (Figure 4A to 4D). We found that the same dose of ISO induced larger effects on transient decay Tau in Ctrl iPSC-CMs than in DCM iPSC-CMs (Figure 4E). ISO boosted the spontaneous beating rate of Ctrl iPSC-CMs by 70.3±6.8% (P < 0.001), whereas an increase of only 18.6±3.9% was observed in DCM cells (P=0.035) (Figure 4F). In addition, fluorescence resonance energy transfer (FRET)-based protein kinase A (PKA) activity imaging confirmed that ISO stimulation induced lower levels of PKA activity in DCM iPSC-CMs in comparison to Ctrl iPSC-CMs (Figure 5A to 5C). Moreover, contractility assays (Figure 5D and 5E) confirmed that β-adrenergic signaling-induced inotropic and chronotropic augmentation in DCM iPSC-CMs was greatly impaired compared to that seen in Ctrl iPSC-CMs. While ISO induced a 59.4±7.7% increase of peak tangential stress and 83.1±9.1% increase of maximum contraction rate in the Ctrl group, ISO failed to improve the contractile force of DCM iPSC-CMs and only induced a 36.7±4.5% increase in their maximum contraction rate (Figure 5F and 5G). Other readouts for contractile function also showed the impaired responsiveness to β-adrenergic activation in DCM iPSC-CMs (Supplemental Figure 5A to 5F). These results taken en masse suggest that DCM iPSC-CMs can recapitulate the disease-like β-adrenergic regulation phenotypes seen in the intact diseased heart (Cho et al., 1999). Interestingly, a similar development process of β-adrenergic signaling is at work in both Ctrl and DCM iPSC-CMs, since the β2-AR dominance at day 30 also shift more toward β1-AR and β2-AR co-dominance at day 60 in the DCM as we observed in Ctrl cells (Supplemental Figure 5G to 5J, Supplemental Table 2).
Figure 4. Impaired β-adrenergic signaling response of calcium handling properties in DCM iPSC-CMs.
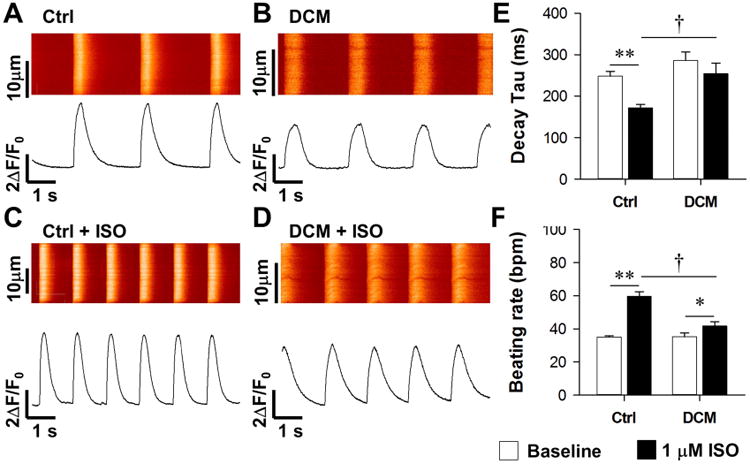
(A-B) Representative recording of spontaneous calcium transient of Ctrl (A) and DCM iPSC-CMs (B) at baseline. (C-D) Representative recording of calcium transient in Ctrl (C) and DCM iPSC-CMs (D) after 1 μM ISO treatment. (E-G) Statistics of calcium handling parameters (E, time to peak; F, decay Tau; G, beating rate) in Ctrl and DCM iPSC-CMs before and after ISO treatment with n>30 iPSC-CMs in each group. **P<0.01 vs. baseline and †P<0.05 vs. WT in each group by two way ANOVA (Holm-Sidak method). See also Supplemental Figure 4 and Supplemental Table 2.
Figure 5. DCM iPSC-CMs exhibit smaller increases in PKA signaling as well as impaired inotropic and chronotropic functional regulation upon β-adrenergic stimulation.
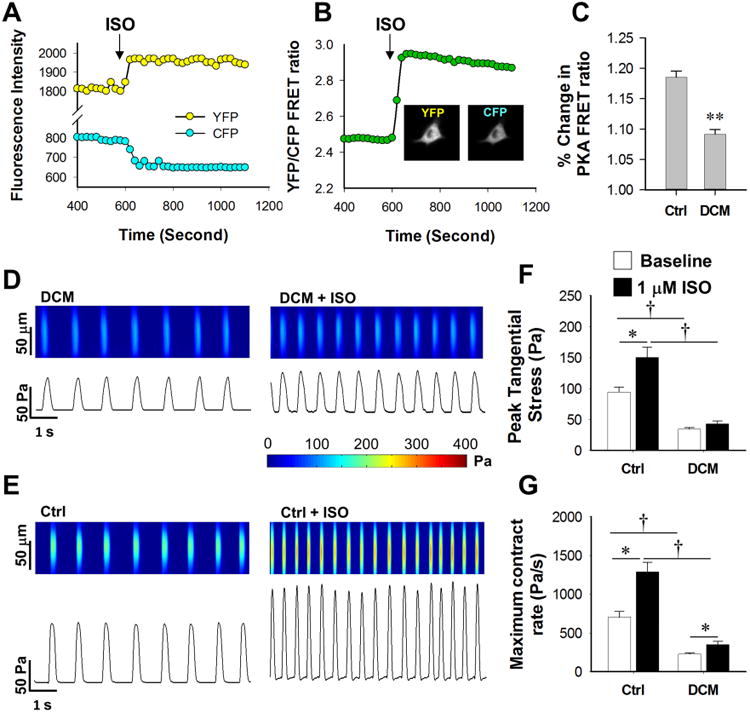
(A) Representative tracing of living cell PKA activity imaging based on FRET; arrow indicates application of ISO. (B) YFP/CFP FRET ratio profile shows an increase in signaling ratio after ISO treatment; inserted panels show original false color recording of a single iPSC-CM in both YFP and CFP channels. (C) DCM iPSC-CMs show compromised responsiveness in cAMP generation upon ISO treatment compared to Ctrl iPSC-CMs with n>20 iPSC-CMs in each group, **P<0.01 vs Ctrl group by Student's T-test. (D-E) Representative recording of tangential stress generated by spontaneous contraction of both Ctrl (D) and DCM iPSC-CMs (E) at baseline. (F-G) Statistics of peak tangential stress (F) and maximum contract rate (G) in both Ctrl and DCM iPSC-CMs before and after ISO treatment with n>25 iPSC-CMs in each group. *P<0.05 vs. baseline and †P<0.05 vs. Ctrl line in each group by two way ANOVA (Holm-Sidak method). See also Supplemental Figure 5 and Supplemental Table 5.
PDE2A and PDE3A expressions are up-regulated in DCM iPSC-CMs
In order to uncover the molecular basis of the “desensitized” β-adrenergic signaling pattern in DCM iPSC-CMs, we next utilized microarray analysis to examine the whole transcriptomes of both DCM and Ctrl groups. The mRNA levels of the main components of the β-adrenergic signaling protein apparatus were compared (Figure 6A). Interestingly, while the expression levels of β1 and β2 ARs were comparable in day 60 DCM and Ctrl groups, several members from the PDE family showed subtype-specific alterations in DCM iPSC-CMs. In cardiomyocytes, the PDEs hydrolyze phosphodiester bonds of cyclic nucleotides and regulate the distribution, duration, and amplitude of cyclic nucleotide signaling (Jeon et al., 2005). Real-time PCR showed no significant differences between PDE levels in undifferentiated DCM versus Ctrl iPSCs, while the expression levels of PDE2A, PDE3A, and PDE5A were increased by 680±129%, 77±12%, and 76±13% in day 30, and increased by 1681±95%, 367±14%, and 97±20% in day 60 DCM iPSC-CMs compared to Ctrl iPSC-CMs. By contrast, PDE4D showed a 48±5% decrease in day 60 DCM iPSC-CMs (Figure 6B to 6D, Supplemental Figure 6 A). Expression levels of ADRB1 and ADRB2 were comparable between DCM and Ctrl iPSC lines, and in day 30 and day 60 DCM and Ctrl iPSC-CMs (Supplemental Figure 6B and 6C). Expression profile comparison of other key maturation related genes from Ctrl and DCM iPSC-CMs at both maturation stages were summarized in Supplemental Table 3. Taken together, these results clearly demonstrated subtype-specific expression regulation of PDEs in DCM iPSC-CMs.
Figure 6. Subtype specific blockade of PDE rescue impaired beta signaling and contractility in DCM iPSC-CMs.
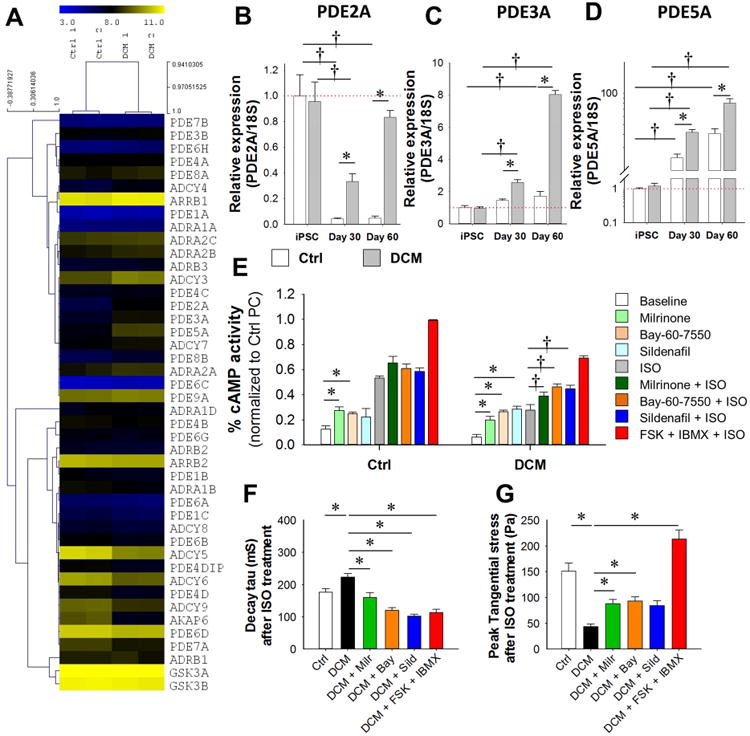
(A) Heat map profiling of expression level of β-adrenergic related genes in Ctrl and DCM iPSC-CMs by microarray data. (B-D) Real-time PCR analysis of mRNA expression level of PDE2A (B), PDE3A (C) and PDE5A (D) in iPSCs, day 30 and day 60 iPSC-CMs from both Ctrl and DCM groups. *P<0.05 vs. Ctrl group and †P<0.05 vs. iPSC group by two way ANOVA (Holm-Sidak method). N=3 different lines for each bar. (E) ELISA-based cAMP assay assessment of β-adrenergic responsiveness in Ctrl and DCM iPSC-CMs with or without PDE blocker treatments. Data from 6 independent experiments. *P<0.05 vs baseline group and †P<0.05 vs ISO group by one way ANOVA (Holm-Sidak method). (F) Spontaneous calcium transient decay Tau of Ctrl and DCM iPSC-CMs following different PDE blocker treatments with n>25 iPSC-CMs in each group. *P<0.05 vs DCM group by one way ANOVA (Holm-Sidak method). (G) Assessment of the peak tangential contractile force regulation by ISO in Ctrl and DCM iPSC-CMs pretreated by different PDE blockers with n>25 iPSC-CMs in each group. *P<0.05 vs DCM group by one way ANOVA (Holm-Sidak method). See also Supplemental Figure 6 and Supplemental Table 3.
Subtype specific inhibition of PDEs rescues the β-adrenergic signaling response in DCM iPSC-CMs
Since both PDE2A and PDE3A hydrolyze cAMP (Baillie, 2009), we hypothesized that the elevated expression of PDE2A and PDE3A in DCM iPSC-CMs might contribute to lower cAMP production and weakened β-adrenergic responses upon ISO treatment. To test this hypothesis, DCM and Ctrl iPSC-CMs were pre-treated with PDE blockers (PDE2A: 100 nM Bay-60-7550; PDE3A: 10 μM milrinone; PDE5A: 10 μM sildenafil) for 15 min before challenge with 1 μM ISO, and the resultant levels of cAMP were measured. Readings were normalized to the positive controls (10 μM IBMX + 10 μM Forskolin) in Ctrl group.
We noticed that PDE2A or PDE3A inhibition significantly enhanced the response to β-adrenergic stimulation in both Ctrl and DCM iPSC-CMs (Figure 6E). Interestingly, although PDE5A is cGMP specific, the treatment of PDE5A blocker enhanced cAMP generation in DCM iPSC-CMs upon ISO treatment (Figure 6E). Also, FRET-based imaging of PKA activity showed that blocking either PDE2A or PDE3A could recover PKA FRET signal in DCM iPSC-CMs (Supplemental Figure 6D).
We then examined if treatment by PDE blockers could relieve the impaired functional outputs in DCM iPSC-CMs. By calcium imaging and TFM study, our results showed pre-inhibition of PDE2A or PDE3A slightly improved calcium cycling in DCM iPSC-CMs at baseline, and induced a much more robust functional enhancement in DCM iPSC-CMs compared to Ctrl groups (Supplemental Figure 6E to 6G). Thus, PDE2A/3A selective inhibition restored the impaired β-adrenergic signaling in DCM iPSC-CMs (Figure 6F and 6G, Supplemental Figure 6H to 6J). These results suggest that the functional impairment of β-adrenergic signaling in DCM iPSC-CMs could be ameliorated by subtype selective repression of the observed over-expressed PDE2A and PDE3A.
Epigenetic modifications contribute to over-expression of PDE2/3a during maturation of DCM iPSC-CMs
To uncover the underlying reasons for the up-regulated expression of PDE2A/3A in DCM iPSC-CMs, we next examined epigenetic modulation of PDE family members by chromatin immunoprecipitation (ChIP) (Figure 7A). We observed no significant differences in the histone markers for activation (H3K4me3) and repression (H3K27me3) in PDE2A gene in Ctrl and DCM iPSC cells (Figure 7B and 7C). However, compared to Ctrl cells, the level of the activation marker H3K4me3 in the regions of PDE2A-R1 was increased by 200±35% and 284±29% in day 30 and day 60 DCM iPSC-CMs, respectively. In PDE2A-R2, H3K4me3 marker was increased by 186±28% and 484±66% in day 30 and day 60 DCM iPSC-CMs, respectively. By contrast, repression marker H3K27me3 was decreased by 54±6% and 67±6% in PDE2A-R1 and PDE2A-R2, respectively, in day 60 DCM iPSC-CMs compared to Ctrl cells. However, in day 30 DCM iPSC-CMs, H3K27me3 marker was unchanged in PDE2A-R1, and decreased by 42±2% in PDE2A-R2 (Figure 7B and 7C). Measurement of additional histone markers such as H3K26me3 and H3K27AC in multiple regions of PDE2A also shows general epigenetic activation of PDE2A expression (Supplemental Figure 7A to 7D), which was supported by the observation of increased PDE2A protein levels (Figure 7D). We also examined histone marker modifications in the PDE3A and PDE5A genes (Supplemental Figure 7E and 7F). Again, no differences were detected between Ctrl and DCM iPSCs. However, in accordance with the increased protein expression of PDE3A in DCM iPSC-CMs and tissues (Supplemental Figure 7G), the activation marker in the PDE3A-R1 was increased by 298±9.5% and 448±75% in day 30 and 60 DCM iPSC-CMs, respectively, whereas the repressive marker was decreased by 68±8.1% and 69±6.2%, in day 30 and 60 DCM iPSC-CMs, respectively, compared to Ctrl group (Supplemental Figure 7H). Measurement of additional histone markers in other PDE3A gene regions showed similar increases in activation markers and decreases in repressive markers (Supplemental 7H to 7J). For PDE5A, both the activation marker H3K4me3 and repressive marker H3K27me3 were down-regulated in DCM iPSC-CMs (Supplemental Figure 7K); no significant difference was detected in marker H3K36me3, while H3K27AC showed a slight increase in DCM cells. These results suggest a much more complex picture on regulation of the PDE5A gene.
Figure 7. Epigenetic regulation underlying the PDE expression pattern in DCM iPSC-CMs and DCM heart tissues.
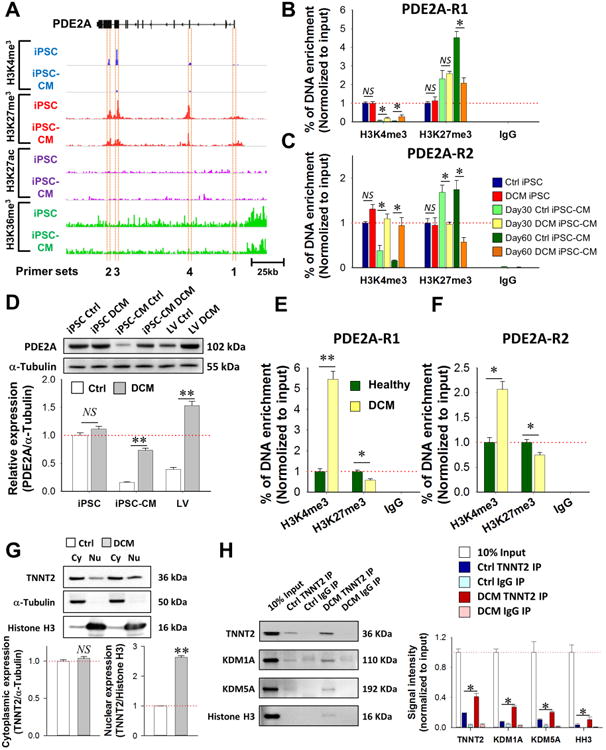
(A) Designing of ChIP primers in PDE2A gene structure based on key active and repressive histone marker regions of iPSC-CMs. (B-C) ChIP-qPCR measurement of histone marker modification levels at region 1 (B) and region 2 (C) of PDE2A gene in iPSC and iPSC-CM cells from both Ctrl and DCM group. *P<0.05 in two way ANOVA (Holm-Sidak method). (D) Western blot assessment of PDE2A protein expression in iPSCs, iPSC-CMs, and LV tissue samples from both Ctrl and DCM groups (n = 3 different lines for cell lines, n = 4 in both healthy and DCM patients). **P<0.01 vs. Ctrl in two way ANOVA (Holm-Sidak method). (E-F) Quantification of active and repressive histone marker of PDE2A -R1 (E) and region 2 (F) in left ventricle tissue of healthy individuals (n=3) and DCM patients (n=3). *P<0.05 and **P<0.01 vs. healthy group in two way ANOVA (Holm-Sidak method). (G) Western blot analysis of the sub-cellular distribution of TNNT2 in Ctrl and DCM iPSC-CMs. **P<0.01 vs Ctrl group by Student's t-test. N = 3 cell lines in both Ctrl and DCM groups. (H) Co-immunoprecipitation analysis of the TNNT2-interacting proteins in nucleus from both Ctrl and DCM iPSC-CMs. *P<0.05 vs Ctrl TNNT2 IP group in two way ANOVA (Holm-Sidak method). See also Supplemental Figure 7 and Supplemental Table 1, Table 4.
We next examined the epigenetic status of the PDE2A/3A/5A gene in human cardiac tissues obtained from 5 healthy individuals and 4 DCM patients undergoing cardiac surgeries. Western blot confirmed PDE2A up-regulation in DCM patients (Figure 7D). Interestingly, PDE2A-R1 and PDE2A-R2 in DCM hearts showed 446±38% and 160±16% of increase in activation marks and 42±6.7% and 25±4.8% of decrease in repressive marks compared to healthy tissues (Figure 7E and 7F). The PDE3A gene in DCM patient tissues demonstrated similar patterns as PDE2A (Supplemental Figure 7L). For PDE5A, there was no significant change in the activation marker, but the repressive histone marker level was decreased (Supplemental Figure 7M). These findings are congruent with our observations in DCM iPSC-CMs, and support the importance of the underlying epigenetic regulatory mechanisms that lead to altered expression levels of PDE subtypes in DCM iPSC-CMs.
One question that remains unclear is how the mutation in TNNT2 affects the epigenetic modification of histones. It has been reported that a fraction of TNNT2 was localized in the nuclei of adult cardiomyocytes (Bergmann et al., 2009), and TNNT2 is predicted to contain a strong nuclear localization signal (NLS), which the R173W mutation may alter (Supplemental Figure 7N). The function of this nuclear TNNT2 is unknown. In our experiments, both Western blot (Figure 7G) and immunostaining (Supplemental Figure 7O and 7P) show that mutated TNNT2 is more likely to be located in the nuclei of iPSC-CMs. We used a TNNT2-specific antibody to isolate potential TNNT2-interacting proteins from cardiomyocyte nuclear extracts. These samples were subjected to mass spectrometry analysis (Supplemental Table 4). Among these proteins, several of them were related to our current study: histone H3, KDM1A and KDM5A. KDM1A (also known as LSD1) and KDM5A (also known as JARID1) are both histone demethylases, which could lead to demethylation of H3K4 (Chaturvedi et al., 2012; Schenk et al., 2012). Our results indicate that in nucleus of cardiomyocyte, TNNT2 may interact with these key histone demethylases and affect epigenetic modification. In DCM iPSC-CMs, increased nuclear TNNT2 content may induce stronger interactions with KDM1A and KDM5A, and lead to abnormal distribution of these key enzymes (or influence their enzymatic activities), resulting in enhanced active epigenetic markers in the PDE2A and PDE3A genes. Indeed, further co-immunoprecipitation (Co-IP) experiments confirmed the increased interaction of TNNT2 with KDM1A, KDM5A and histone H3 in DCM iPSC-CMs compared to Ctrl group (Figure 7H).
Collectively, our results demonstrate that undifferentiated iPSC lines from both DCM and Ctrl groups showed no differences in epigenetic modifications on key genes. However, similar epigenetic activation of PDE2A and 3A was observed in both mature DCM iPSC-CMs and DCM cardiac tissues. Mechanistic studies indicate a potential role of mutated TNNT2 in epigenetic regulation, and support an “acquired epigenetic pattern” during cardiomyocyte formation that leading to PDE subtype dysregulation in DCM pathogenesis.
Discussion
In summary, the current study focused on the development of β-adrenergic signaling in Ctrl and DCM iPSC-CMs. Human iPSC-CMs are utilized for cardiovascular disease modeling and drug screening (Itzhaki et al., 2011; Lan et al., 2013; Liang et al., 2013; Sun et al., 2012). However, the degree of conservation of β-adrenergic signaling in iPSC-CMs compared to in vivo cardiomyocytes was previously unclear. To our knowledge, this is the first study to investigate β-adrenergic signaling during the differentiation and maturation of iPSC-CMs. Our results show that despite some immature features, β-adrenergic signaling can induce inotropic and chronotropic regulation of contractile function in iPSC-CMs.
β1 and β2 ARs have been found to coexist in isolated single human left ventricular cardiomyocytes (del Monte et al., 1993), where the ratio of β1/β2 ARs is around 70-80%/30-20% in human ventricles (Brodde, 1991; Engelhardt et al., 1996). Activation of both β1 and β2 ARs leads to the enhancement of contractile function, whereas the effect of β1 AR is overwhelmingly dominant in normal adult cardiomyocytes. In iPSC-CMs, however, β1 AR showed a late-onset expression pattern compared to β2 AR. Accordingly, adrenergic regulation of calcium homeostasis and contractile force was dominated by β2 AR in iPSC-CMs at early stages, and then gradually switched to β1 AR during maturation in vitro. In day 60 iPSC-CMs, the contractility was actively regulated by both β1 and β2 adrenergic activation. Interestingly, Khan et al. have also showed dominant expression of β2 AR in mouse and human cardiac progenitor cells (CPCs), while cardiac commitment lead to acquisition of β1 AR expression in CPCs, indicating a similar pattern of β1/β2 AR expression regulation in CPCs and CPC-derived cardiomyocytes (Khan et al., 2013).
β-adrenergic stimulation lead to enhanced systolic and diastolic function through the activation of down-stream effectors by PKA (Kaumann et al., 1999) In adult ventricular cardiomyocytes, β2 AR activation is not coupled to calcium dynamics and contractility (Xiao et al., 1994) probably due to the compartmentalized regulation (Baillie, 2009; Xiang, 2011). However, in iPSC-CMs, β2 signaling could go beyond compartmentalization to directly enhance contractile function. The regulatory role of the β2 AR became less important at later stages of iPSC-CM maturation, probably due to loss of coupling of β2 AR to Gs proteins, as the expression level of Gs dramatically decreased in differentiated iPSC-CMs, whereas the expression of Gi slightly increased throughout maturation (Supplemental Figure 3S and 3T). On the other hand, increased expression level of many PDE family proteins in more matured iPSC-CMs greatly contributed to the localized control of intracellular cAMP signaling, suggesting a general suppression of cAMP signaling in more mature iPSC-CMs that may lead to the milder responses to β-adrenergic stimulation in iPSC-CMs at later maturation stages (Figure 2C to 2E, Figure 3E, Supplemental Figure 3). Another intriguing phenomenon is that β-adrenergic activation cannot further increase calcium transient amplitude, even if the calcium recycling was activated. This might be due to the relatively immature sarcoplasmic reticulum system and calcium handling in iPSC-CMs. As shown in Supplemental Figure 3N and 3O, the expression levels of RyR2 and CASQ2 in iPSC-CMs were ∼100-fold lower than in adult ventricle tissue, indicating very limited space for the β-adrenergic signaling-induced functional enhancement of SR calcium load and release.
Abnormalities of β-adrenergic signaling have been well documented in various cardiomyopathies (Ahmet et al., 2008; Lohse et al., 2003; Lowes et al., 2002). Our observations demonstrate that PDE2A, PDE3A, and PDE5A are up-regulated in diseased cardiomyocytes from both DCM iPSC-CMs and DCM heart tissues. Increased levels of PDE2A and PDE3A could contribute to cAMP hydrolysis activity in DCM iPSC-CMs and thus blunt the β-AR responsiveness. Interestingly, a recent report showed that up-regulated PDE2A in human heart failure suppresses the response of cardiomyocytes to β-adrenergic signaling (Mehel et al., 2013). This result was also confirmed by our findings, suggesting that subtype-specific PDE expression regulation might be a key mechanism in DCM. However, it is worth noticing that unlike prior results reported in DCM and heart failure patient heart, we found that the expression of β-AR subtypes was not significantly changed between DCM and Ctrl iPSC-CMs. It is possible that our iPSC-CM model captured the early stage of DCM pathogenesis, but the automatic onset of epigenetic regulation in DCM cell lines could then lead to the remodeling of β-adrenergic signaling and impaired contractility, even without the change of extracellular adrenaline level and the desensitization/down-regulation of β-ARs.
In recent years, epigenetic regulation has been shown related to cardiomyopathies, including HCM, DCM and diabetic cardiomyopathy (Asrih and Steffens, 2013; Haas et al., 2013). Here, we demonstrate a novel epigenetic mechanism of pathogenesis in DCM iPSC-CM models. Interestingly, in undifferentiated Ctrl and DCM iPSCs, the histone markers showed no difference in PDE2A and PDE3A gene. However, the histone markers pattern was discordant in iPSC-CMs from both groups during differentiation, as we observed increased activation marks and decreased repressive marks on the PDE2A and PDE3A genes in DCM iPSC-CMs. Moreover, measurement of different histone markers at various gene locations shows temporal and regional specific regulation in epigenetic modification throughout the maturation of iPSC-CMs and the development of DCM pathogenesis. Similar trends were found in the cardiac tissue samples from DCM patients and healthy individuals. Such results suggest that the epigenetic modifications on PDE2A and PDE3A contribute to the impaired β-adrenergic signaling in DCM, which was recapitulated during the maturation of our DCM iPSC-CMs.
TNNT2 is well known as a thin filament component that anchors tropomyosin with troponin complex. Although TNNT2 was shown to contain a NLS, the functional implication of nuclear TNNT2 is not understood (Bergmann et al., 2009). Our current study has identified potential TNNT2-interacting nuclear proteins, such as KDM1A and KDM5A. We also showed that increased nuclear localization of mutated TNNT2 may enhance the interaction of TNNT2 with these key epigenetic enzymes, and contribute to the altered epigenetic regulation of key β-adrenergic signaling genes in DCM iPSC-CMs. These findings cast new light on the novel mechanism of pathogenesis of familial DCM with a single gene mutation in TNNT2.
Collectively, the current study characterized the properties of β-adrenergic signaling during in vitro differentiation and maturation of iPSC-CMs, and confirmed active inotropic and chronotropic regulation in these models. It also demonstrated that in DCM iPSC-CMs, subtype-specific epigenetic modifications of PDE-induced up-regulation of PDE2A and PDE3A, leading to compromised β-adrenergic signaling and contractile function. Selective blockade of PDE2A, PDE3A, and PDE5A rescued the calcium handling and contractile force in DCM iPSC-CMs, and potentiated the responsiveness of these cells to β-agonist stimulation, which may be a new clinical target in the treatment of DCM. Moreover, our study indicates that DCM iPSC-CM modeling can recapitulate not only the disease phenotype, but also the pathogenesis process, which will greatly facilitate and deepen our understanding of the underlying mechanisms of DCM.
Experimental Procedures
Cell culture and cardiac differentiation of human iPSCs
All of the protocols for this study were approved by the Stanford University Human Subjects Research Institutional Review Board (IRB). Human iPSC lines were maintained on Matrigel-coated plates (BD Biosciences, San Jose, CA) in Essential 8™ Medium (Gibco®, Life Technology). Human iPSC-CMs were generated as described previously (Lian et al., 2012). Briefly, pluripotent stem cells were treated with 6 μM CHIR99021 (Selleckchem.com) for 2 days, recovered in insulin-minus RPMI+B27 for 24 hr, treated with 5 μM IWR-1 (Sigma) for 2 days, and then insulin minus medium for another 2 days, and finally switched to RPMI+B27 plus insulin medium. Beating cells were observed at day 9-11 after differentiation. iPSC-CMs were re-plated and purified with glucose-free medium treatment for 2-3 rounds. Typically, cultures were more than 90% pure by FACS assessment of TNNT2+ cells after purification. Cultures were maintained in a 5% CO2/air environment.
Immunofluorescence staining
For characterization of differentiated iPSC-CMs, immunofluorescent stains were performed using cardiac troponin T (cTnT, Thermo Scientific Barrington, IL), sarcomeric α-actinin (Clone EA-53, Sigma), and DAPI (Molecular Probes) as previously described (Sun et al., 2012). Labeled cells were examined and imaged by confocal microscope (Carl Zeiss, LSM 510 Meta, Göttingen, Germany) at 20× to 63× objectives as appropriate.
Statistical analysis
For statistical analysis, Student's t-test was used to compare two normally distributed data sets. One-way or two-way ANOVA was used, where appropriate, to compare multiple data sets, and Holm-Sidak or Tukey after-tests were used for all pairwise comparisons, depending on the properties of the data sets. A P value <0.05 was considered to be statistically significant. All data were shown as mean ± standard error mean.
A complete description of methods is available in the Supplemental Experimental Procedures section.
Supplementary Material
Highlights.
β-AR signaling switch from β-2 AR to β-1/2 AR mode during human iPSC-CMs maturation;
Up-regulation of PDE2/3 lead to compromised β-adrenergic regulation in DCM iPSC-CMs;
Epigenetic activation of PDE2/3 is a key molecular event during pathogenesis of DCM;
Nuclear localization of mutated TNNT2 contributes to epigenetic modification in DCM.
Acknowledgments
We would like to thank Andrew Olson from Neuroscience Microscopy Service (NMS) and Jon Mulholland from Cell Sciences Imaging Facility (CSIF) for their help with confocal imaging. We thank Dr. Bhagat Patlolla from Stanford Cardiovascular Institute for his help with human tissue sampling. We would like to thank Chris Adams and Ryan Leib at the Vincent Coates Foundation Stanford University Mass Spectrometry facility for their assistance in mass spectrometry data collection and analysis. We would like to thank Yonglu Che and Tianying Su for their help with MATLAB programming and data analysis. This work was supported by American Heart Association Established Investigator Award 14420025, the National Institutes of Health U01 HL099776, R01 HL113006, R01 HL123968, and R24 HL117756 (JCW).
Footnotes
Author Contribution: H.W. conceived, performed, and interpreted the experiments and wrote the manuscript; J.L. performed epigenetic analysis; L.G.V. and A.J.E. provided advice and performed contractility assay; Q.W. and Y.K.X. provided advice and performed cAMP imaging experiment; F.L. and M.G. performed immnuostaining and FACS sorting; E.M. prepared cells; J.C. analyzed data of RNA-seq and microarray; K.S. provided healthy and diseased cardiac tissue; A.S. and J.D.G. provided experimental advice; D.M.B provided advice on data interpretation and editing of manuscript; J.C.W. conceived idea and provided experimental advice, manuscript writing, and funding support.
Conflict of Interest Disclosures: J.C.W. is a co-founder of Stem Cell Theranostics.
References
- Ahmet I, Krawczyk M, Zhu W, Woo AY, Morrell C, Poosala S, Xiao RP, Lakatta EG, Talan MI. Cardioprotective and survival benefits of long-term combined therapy with beta2 adrenoreceptor (AR) agonist and beta1 AR blocker in dilated cardiomyopathy postmyocardial infarction. J Pharmacol Exp Ther. 2008;325:491–499. doi: 10.1124/jpet.107.135335. [DOI] [PubMed] [Google Scholar]
- Asrih M, Steffens S. Emerging role of epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc Pathol. 2013;22:117–125. doi: 10.1016/j.carpath.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Baillie GS. Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 2009;276:1790–1799. doi: 10.1111/j.1742-4658.2009.06926.x. [DOI] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–387. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S, et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- Brodde OE. Beta 1- and beta 2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev. 1991;43:203–242. [PubMed] [Google Scholar]
- Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–981. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10:16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi CP, Somasundaram B, Singh K, Carpenedo RL, Stanford WL, Dilworth FJ, Brand M. Maintenance of gene silencing by the coordinate action of the H3K9 methyltransferase G9a/KMT1C and the H3K4 demethylase Jarid1a/KDM5A. Proc Natl Acad Sci U S A. 2012;109:18845–18850. doi: 10.1073/pnas.1213951109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MC, Rapacciuolo A, Koch WJ, Kobayashi Y, Jones LR, Rockman HA. Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. J Biol Chem. 1999;274:22251–22256. doi: 10.1074/jbc.274.32.22251. [DOI] [PubMed] [Google Scholar]
- Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–277. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Monte F, Kaumann AJ, Poole-Wilson PA, Wynne DG, Pepper J, Harding SE. Coexistence of functioning beta 1- and beta 2-adrenoceptors in single myocytes from human ventricle. Circulation. 1993;88:854–863. doi: 10.1161/01.cir.88.3.854. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Bohm M, Erdmann E, Lohse MJ. Analysis of beta-adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: progressive reduction of beta 1-adrenergic receptor mRNA in heart failure. J Am Coll Cardiol. 1996;27:146–154. doi: 10.1016/0735-1097(95)00425-4. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med. 2013;5:413–429. doi: 10.1002/emmm.201201553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Jeon YH, Heo YS, Kim CM, Hyun YL, Lee TG, Ro S, Cho JM. Phosphodiesterase: overview of protein structures, potential therapeutic applications and recent progress in drug development. Cell Mol Life Sci. 2005;62:1198–1220. doi: 10.1007/s00018-005-4533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- Kaumann A, Bartel S, Molenaar P, Sanders L, Burrell K, Vetter D, Hempel P, Karczewski P, Krause EG. Activation of beta2-adrenergic receptors hastens relaxation and mediates phosphorylation of phospholamban, troponin I, and C-protein in ventricular myocardium from patients with terminal heart failure. Circulation. 1999;99:65–72. doi: 10.1161/01.cir.99.1.65. [DOI] [PubMed] [Google Scholar]
- Khan M, Mohsin S, Avitabile D, Siddiqi S, Nguyen J, Wallach K, Quijada P, McGregor M, Gude N, Alvarez R, et al. beta-Adrenergic regulation of cardiac progenitor cell death versus survival and proliferation. Circ Res. 2013;112:476–486. doi: 10.1161/CIRCRESAHA.112.280735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848–1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–1691. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346:1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Mehel H, Emons J, Vettel C, Wittköpper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechêne P, et al. Phosphodiesterase-2 Is Up-Regulated in Human Failing Hearts and Blunts β-Adrenergic Responses in Cardiomyocytes. Journal of the American College of Cardiology. 2013;62:1596–1606. doi: 10.1016/j.jacc.2013.05.057. [DOI] [PubMed] [Google Scholar]
- Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–526. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post SR, Hammond HK, Insel PA. Beta-adrenergic receptors and receptor signaling in heart failure. Annu Rev Pharmacol Toxicol. 1999;39:343–360. doi: 10.1146/annurev.pharmtox.39.1.343. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–1532. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ Res. 2011;109:231–244. doi: 10.1161/CIRCRESAHA.110.231340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Hohl C, Altschuld R, Jones L, Livingston B, Ziman B, Tantini B, Lakatta EG. Beta 2-adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J Biol Chem. 1994;269:19151–19156. [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.