

ABSTRACT
Interaction between gH/gL and the fusion protein gB is likely a conserved feature of the entry mechanism for all herpesviruses. Human cytomegalovirus (HCMV) gH/gL can be bound by gO or by the set of proteins UL128, UL130, and UL131, forming gH/gL/gO and gH/gL/UL128-131. The mechanisms by which these complexes facilitate entry are poorly understood. Mutants lacking UL128-131 replicate well on fibroblasts but fail to enter epithelial/endothelial cells, and this has led to the general assumption that gH/gL/UL128-131 promotes gB-mediated fusion on epithelial/endothelial cells whereas gH/gL/gO provides this function on fibroblasts. This was challenged by observations that gO-null mutants were defective on all of these cell types, suggesting that entry into epithelial/endothelial cells requires both of the gH/gL complexes, but the severe replication defect of the gO mutants precluded detailed analysis. We previously reported that the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope varied dramatically among HCMV strains. Here, we show that strains not only differ in the ratio, but also vary in the total amount of gH/gL in the virion. Cell-type-specific particle-to-PFU ratios of HCMV strains that contained different amounts of gH/gL/gO and gH/gL/UL128-131 were determined. Infection of both fibroblasts and epithelial cells was generally correlated with the abundance of gH/gL/gO, but not with that of gH/gL/UL128-131. The low infectivity of virions rich in gH/gL/UL128-131 but low in gH/gL/gO could be overcome by treatment with the chemical fusogen polyethylene glycol (PEG), strongly arguing that gH/gL/gO provides the conserved herpesvirus gH/gL entry function of promoting gB-mediated fusion for entry into all cell types, whereas gH/gL/UL128-131 acts through a distinct mechanism to allow infection of select cell types.
IMPORTANCE The functions of HCMV gH/gL complexes in entry are unclear. Unlike the well-studied Epstein-Barr virus (EBV), where gH/gL and gH/gL/gp42 complexes both seem capable of promoting gB fusion during entry into different cell types, our studies here suggest that for HCMV, gH/gL/gO promotes gB fusion on all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct, as yet unknown mechanism. To our knowledge, this is the first suggestion of a herpesvirus gH/gL that does not act by promoting gB fusion, which might make HCMV a useful model to study the fundamental mechanisms by which herpesvirus gH/gL regulates gB fusion. Moreover, gH/gL/UL128-131 is a candidate vaccine target. Our findings help to explain the cell-type-dependent virus neutralization exhibited by anti-gH/gL/UL128-131 antibodies and underscore the importance of gH/gL/gO as another important part of vaccine or therapeutic strategies.
INTRODUCTION
Primary infection of healthy adults by human cytomegalovirus (HCMV) is usually subclinical or mildly symptomatic but leads to lifelong persistent or latent infection. Primary infection or reactivation of HCMV in immunocompromised hosts, such those infected with HIV and transplant recipients on antirejection chemotherapies, is associated with significant morbidity and mortality, and maternal transmission of HCMV to the developing fetus across the placenta can result in severe congenital birth defects (1–3). The diverse nature of HCMV-associated disease is likely related to the ability of the virus to infect many cell types in vivo, including epithelial and endothelial cells, fibroblasts, monocytes-macrophages, dendritic cells, hepatocytes, leukocytes, and neurons (4–6). Much research effort has been focused on understanding the mechanisms by which HCMV initiates infection of different cell types (reviewed in reference 7). Most studies have described an entry mechanism dichotomy between fibroblasts and epithelial/endothelial cells. Other relevant cell types, though more difficult to work with in the laboratory, likely fit to one side or the other of the fibroblast-epithelial/endothelial cell entry dichotomy.
Entry of all herpesviruses requires membrane fusion, which is mediated by a conserved core fusion apparatus comprised of glycoprotein B (gB) and gH/gL. Extensive studies of herpes simplex virus (HSV) and Epstein-Barr virus (EBV) have shown that gB has characteristics of a class III viral fusion protein (8–11). The necessary role of gH/gL in herpesvirus membrane fusion is unclear but likely involves direct interaction between gH/gL and gB (12–17). Herpesviruses also encode accessory glycoproteins, such as HSV gD and EBV gp42, which regulate the core fusion apparatus through receptor binding and stable or transient interactions with gH/gL (reviewed in reference 18).
HCMV shares the conserved features of herpesviruses entry, but there are some important unique aspects, especially related to cell tropism. Structural analyses of HCMV gB suggest general mechanistic similarities with other herpesvirus gB homologues (19), and the combination of HCMV gB and gH/gL is necessary and sufficient for membrane fusion in cell-cell fusion experiments (20). The ectodomain of HCMV gH/gL can be bound by either gO or proteins encoded by the genes UL128, UL130, and UL131 (UL128-131), forming the gH/gL/gO and gH/gL/UL128-131 complexes on the virion envelope (21–24). Experiments reported by Zhou et al. indicated the following: (i) extracellular virions contain little, if any, unbound gH/gL; (ii) the ratio of gH/gL/gO to gH/gL/UL128-131 can vary dramatically between different HCMV strains; and (iii) gO and UL128-131 compete for binding of gH/gL (25). The mutually exclusive nature of gH/gL/gO and gH/gL/UL128-131 formation is consistent with recent structural analyses showing that gO and the UL128 protein make a disulfide bond with the same cysteine residue of the gL polypeptide (26). The gH/gL/gO and gH/gL/UL128-131 complexes influence cell tropism at the level of entry, although the mechanisms are not well understood.
The roles of HCMV gH/gL complexes have mostly been studied with deletion mutant approaches. In general, mutants that lack gH/gL/UL128-131 replicate as well as or better than wild-type strains on fibroblasts (21, 27–29). This indicates that gB-mediated fusion with fibroblast membranes can be facilitated by gH/gL/gO. In contrast, gH/gL/UL128-131 mutants fail to infect cell types such as epithelial and endothelial cells and monocytes/macrophages (21, 27–30). These observations have generally been interpreted to mean that gB-mediated fusion with the membranes of epithelial cells (and other cell types for which gH/gL/UL128-131 is necessary for HCMV entry) is facilitated by gH/gL/UL128-131. The resulting model suggests that both gH/gL/gO and gH/gL/UL128-131 provide essentially the same function for entry into different cell types, i.e., promoting gB-mediated membrane fusion. This model was attractive due to analogy with the well-described model of EBV tropism (reviewed in reference 18). EBV gH/gL can exist in the virion envelope as unmodified gH/gL or bound by gp42. For entry into epithelial cells, gH/gL binds specific integrins and promotes gB-mediated membrane fusion. The presence of gp42 on gH/gL blocks integrin binding, and instead, gp42 binds major histocompatibility complex class II (MHC-II), which facilitates entry into B cells. Presumably, both gH/gL and gH/gL/gp42 promote gB-mediated membrane fusion through interactions involving “pro-fusion” surfaces of gH/gL, with gp42 acting as a receptor-adapter to switch cell tropism. While HCMV mutants lacking gH/gL/UL128-131 replicate well on fibroblasts, mutants lacking gH/gL/gO are severely replication impaired on all cell types (31–34). In particular, Wille et al. showed that HCMV virions lacking gH/gL/gO were impaired at the fusion step of entry into fibroblasts and epithelial and endothelial cells, despite containing elevated amounts of gH/gL/UL128-131 (34). Together, the results of deletion mutant approaches seem to suggest that gH/gL/gO is sufficient for entry into fibroblasts but that entry into epithelial/endothelial cells requires both gH/gL/gO and gH/gL/UL128-131.
Characterization of the mechanisms by which HCMV gH/gL/gO and gH/gL/UL128-131 facilitate cell-type-dependent entry using deletion mutants has been limited, largely due to the severe replication defects associated with gO deletions and the lack of an adequate complementation system. The studies described here capitalize on our previously reported observations that the amounts of gH/gL complexes in the HCMV virion envelope can vary dramatically among strains and that the ratio of the two gH/gL complexes can be manipulated through suppression of the UL128-131 proteins. Here, we report further characterizations of strain variability in the abundance of gH/gL complexes in the virion envelope and relate these differences to the efficiencies of entry in different cell types. Comparisons of genetically identical HCMV preparations enriched in either gH/gL/gO or gH/gL/UL128-131 suggest a model in which gH/gL/gO provides the herpesvirus core fusion gH/gL function of promoting gB-mediated membrane fusion for entry into all cell types, whereas gH/gL/UL128-131 facilitates entry into select cell types, such as epithelial cells, through a distinct, yet necessary, mechanism.
MATERIALS AND METHODS
Cell lines.
Primary human foreskin fibroblasts (HFF) (Life Technologies), MRC-5 fibroblasts (ATCC CCL-171), and HFFtet (which express the tetracycline [Tet] repressor protein [35]; provided by Richard Stanton [Cardiff University, Cardiff, United Kingdom]) were grown in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 6% heat-inactivated fetal bovine serum (FBS) (Rocky Mountain Biologicals, Inc., Missoula, MT, USA) and 6% bovine growth serum (BGS) (Rocky Mountain Biologicals, Inc., Missoula, MT, USA). The retinal pigment epithelial cell line ARPE-19 (American Type Culture Collection, Manassas, VA, USA) was grown in a 1:1 dilution mixture of DMEM and Ham's F-12 medium (DMEM–F-12) (Life Technologies) supplemented with 10% FBS.
HCMV.
All human cytomegalovirus (HCMV) strains were derived from bacterial artificial chromosome (BAC) clones. BAC clone TR was provided by Jay Nelson (Oregon Health and Sciences University, Portland, OR, USA) (36). BAC clone TB40/e (BAC-4) (TB) was provided by Christian Sinzger (University of Ulm, Ulm, Germany) (37). BAC clone Merlin (ME) (pAL1393), which contains tetracycline operator sequences within the transcriptional promoter of UL130 and UL131, was provided by Richard Stanton (Cardiff University, Cardiff, United Kingdom) (35). Infectious HCMV was recovered by electroporation of BAC DNA into MRC-5 fibroblasts, as described by Wille et al. (34). Cell-free HCMV stocks were produced by infecting HFF or HFFtet at 2 PFU per cell. At 8 to 10 days postinfection (p.i.) (when cells were still visually intact), culture supernatants were harvested, and cellular contaminants were removed by centrifugation at 1,000 × g for 10 min and again at 6,000 × g for 10 min. Stocks were judged cell free by the lack of calnexin and actin in Western blot analyses and stored at −80°C. The number of PFU was determined by plaque assay on triplicate HFF or ARPE-19 cultures. Freeze/thaw cycles were avoided.
Antibodies.
Monoclonal antibodies (MAbs) specific for HCMV major capsid protein (MCP) 28-4 and gB 27-156 were provided by Bill Britt (University of Alabama, Birmingham, AL, USA) (38, 39). Anti-UL128 MAb 4B10 was provided by Tom Shenk (Princeton University, Princeton, NJ, USA) (24). Rabbit polyclonal antipeptide antibodies directed against HCMV gH/gL, UL130, and UL131 were provided by David Johnson (Oregon Health and Sciences University, Portland, OR, USA) (40). Rabbit polyclonal antipeptide antibodies directed against MEgO were described previously (25).
Western blotting.
Cell-free virions from culture supernatants (as described above) were concentrated by centrifugation at 50,000 × g for 1 h and resuspended in 2% SDS in 20 mM Tris-buffered saline (TBS) (pH 6.8). Insoluble material was removed by centrifugation at 16,000 × g for 30 min, and the cleared extracts were heated to 95°C for 10 min. For reducing blots, extracts were adjusted to 25 mM dithiothreitol (DTT). Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Whatman) in a buffer containing 10 mM NaHCO3 and 3 mM Na2CO3 (pH 9.9) plus 10% methanol. The transferred proteins were probed with MAbs or rabbit polyclonal antibodies specific for HCMV proteins, followed by horseradish peroxidase-conjugated secondary antibodies; chemiluminescence was detected using a Bio-Rad ChemiDoc MP imaging system. Band densities were quantified using Bio-Rad Image Lab v5.1 or ImageJ v1.48 software.
qPCR.
Cell-free HCMV stock (200 μl) was first treated with DNase I to remove DNA not protected within viral capsids. Capsids were disrupted by using the viral lysis buffer and proteinase K provided in the PureLink Viral RNA/DNA minikit (Life Technologies). Viral DNA was eluted in 50 μl RNase-free water. A region within UL83 conserved among ME, TR, and TB was chosen as the amplicon, and genomes were quantified by real-time quantitative PCR (qPCR) using SYBR green dye (Bio-Rad) and the forward primer TGGTCACCTATCACCTGCAT and the reverse primer GAAAGAGCCCGACGTCTACT. PCR samples were made to a total well volume of 20 μl with 1× SYBR green master mix (Bio-Rad), a 1:100 dilution of viral DNA, and 500 nM forward and reverse primers. PCR products were detected using the MyiQ real-time PCR detection system (Bio-Rad). The qPCR results were compared to a plasmid pTRUL83 standard and then calculated as genomes per milliliter. The efficiency of DNase I treatment was tested in independent experiments by spiking plasmid DNA. For each experiment, two independent viral DNA extractions and three independent qPCRs were performed.
Determination of PFU.
Cell monolayers were infected with cell-free HCMV or HCMV-infected cells (infectious centers), and the cell culture medium was replaced with a 1:1 mixture of 2× DMEM and 1.2% SeaPlaque agarose to prevent the spread of progeny virus by diffusion. Plaques were counted by light microscopy 3 weeks later.
Antibody neutralization of HCMV.
Anti-UL130 and anti-UL131 rabbit sera diluted 1:100 (vol/vol) were added to culture medium containing cell-free HCMV. The mixture was incubated at room temperature for 1 h and then added to HFF or ARPE-19 cultures.
PEG treatment.
Cell-free HCMV was adsorbed to cells for 2 h at 10°C under 800 × g centrifugation or at 37°C without centrifugation. Unbound virus was removed by two cold phosphate-buffered saline (PBS) washes, and cells were subsequently treated with 44% polyethylene glycol (PEG) (prewarmed to 37°C) for 30 s, followed by 10 washes with warm culture medium to remove the PEG. The cultures were incubated at 37°C until immunofluorescence (IF) analysis was performed. PEG 6000 (Fluka) was prepared as a 60% (wt/wt) solution in PBS and diluted with PBS to 44%.
Immunofluorescence.
HCMV-infected cells were fixed with 2% formaldehyde in PBS for 15 min, followed by three PBS washes. The fixed cells were incubated for 1 h in IF buffer (0.5% Triton X-100, 0.5% deoxycholate, 1% bovine serum albumin [BSA], 0.05% sodium azide in PBS). To visualize HCMV-infected cells, anti-IE1/IE2 MAb P63-27 was used at 1:50 in IF buffer, and Alexa 488-conjugated goat anti-mouse antibodies were diluted 1:1,000 in IF buffer. DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) was used to visualize total cells.
RESULTS
HCMV strains vary in both the ratio of gH/gL/gO to gH/gL/UL128-131 and the total amount of gH/gL complexes.
Zhou et al. (25) analyzed gH/gL complexes of different HCMV strains by nonreducing Western blotting. Since the main goal of those experiments was to compare the ratios of gH/gL/gO to gH/gL/UL128-131 among strains, the gel loads were adjusted to equalize the total amounts of gH/gL. TR and TB virions were found to contain gH/gL mostly in the form of gH/gL/gO, whereas ME had mostly gH/gL/UL128-131 (25). The experiments made use of a recombinant ME in which the assembly of gH/gL/UL128-131 could be reduced by the tetracycline-repressor protein (TetR), which resulted in ME virions containing mostly gH/gL/gO (denoted Merlin-trimer [ME-T]) (25, 35). To further characterize the amounts of gH/gL complexes among HCMV strains, extracellular virions of TB, TR, ME, and ME-T (ME virions produced in TetR-expressing cells) were analyzed by Western blotting under reducing conditions, and the gel loads were normalized to major capsid protein to allow comparison of the amounts of the different glycoproteins per virion (Fig. 1). All strains contained comparable amounts of gB, but the amounts of gH/gL varied dramatically. TB contained the most total gH/gL, followed by TR, ME, and ME-T (Fig. 1A). Despite the small amount of gH/gL, ME virions contained dramatically more UL131 (a marker of the gH/gL/UL128-131 complex) (Fig. 1B). Note that in Fig. 1B, the gel loads were adjusted to allow visualization of the smaller amounts of UL131 in TB and TR virions, suggesting that the difference in the level of UL131 between ME and the other viruses was greater than 10-fold.
FIG 1.
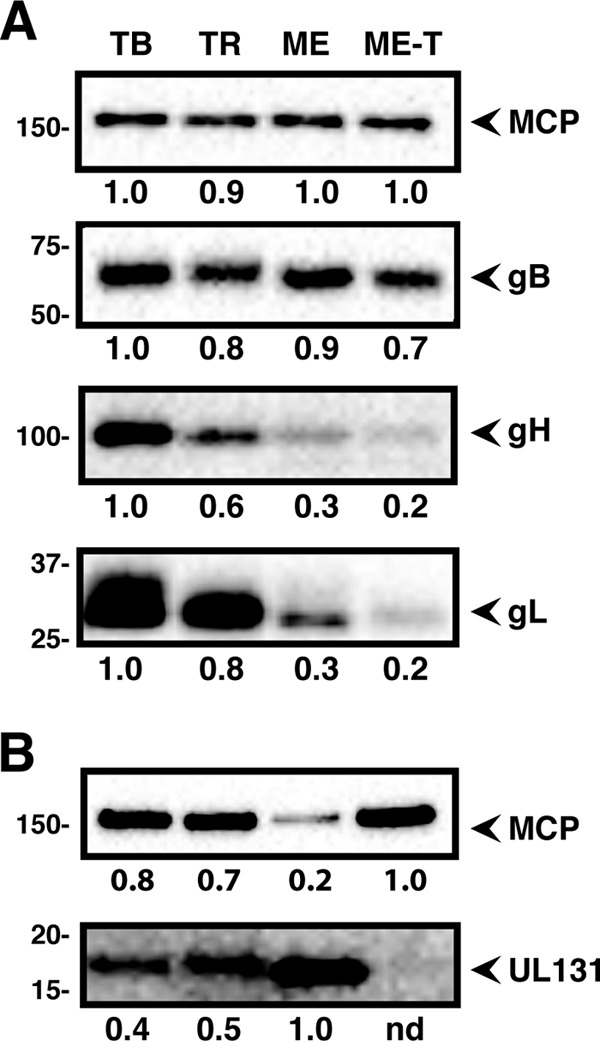
Quantitative comparison of glycoproteins in the virion envelopes of different HCMV strains. Cell-free virions of HCMV TB, TR, ME, or ME-T were separated by reducing SDS-PAGE and analyzed by Western blotting with antibodies specific for MCP, the 55-kDa portion of gB, gH, gL, or UL131. Mass markers (kilodaltons) are shown on the left. The gel loads were normalized to equal MCP (A) or to 5-fold less MCP for ME (B). The numbers below the blots indicate band density analysis, normalized to the most intense band on the same blot. nd, not detected.
UL128, UL130, and gO were not included in the above-mentioned reducing-gel analyses because amino acid polymorphisms between strains affect the cross-reactivities of available antibodies (25). Thus, HCMV gH/gL complexes were analyzed by nonreducing Western blotting, probing for the antigen common to both complexes, gL (Fig. 2). Consistent with analyses reported by Zhou et al., the vast majority of gH/gL in TB, TR, and ME-T virions migrated at a size indicative of disulfide-linked gH/gL/gO, whereas most of the gH/gL in ME virions was at the size of disulfide-linked gH/gL/UL128 (Fig. 2A). Note that UL130 and UL131 are not disulfide linked to gH/gL/UL128-131 and are thus removed by SDS-PAGE (24, 40). Unbound, or free, gH/gL comprised a very small fraction of the total gH/gL for all strains. Whether this represents spontaneous reduction of disulfide bonds in the extract samples or is indicative of small amounts of free gH/gL in virions is not clear. Band density analyses of the three gH/gL species on nonreducing blots corresponded well with the quantitation of total gH and gL on reducing blots (compare Fig. 1A and 2).
FIG 2.
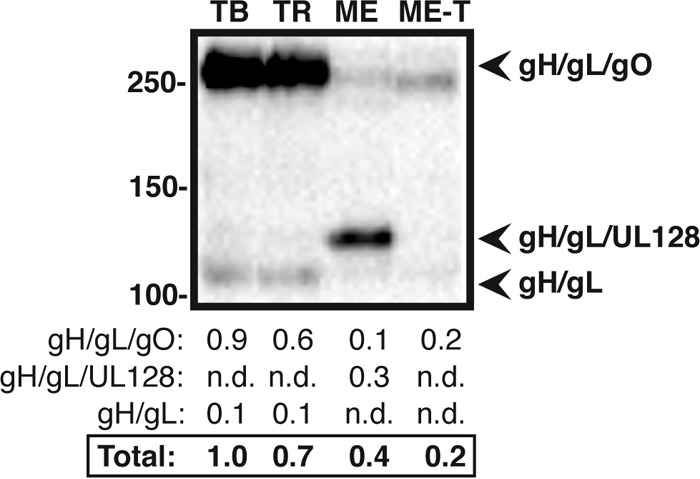
Quantitative comparison of gH/gL complexes in the virion envelopes of different HCMV strains. Cell-free virions of HCMV TB, TR, ME, or ME-T were separated by nonreducing SDS-PAGE and analyzed by Western blotting with antibodies specific for gL. Mass markers (kilodaltons) are shown on the left. The gel loads were normalized to equal MCP, as in Fig. 1A (not shown). The numbers below the blot indicate analysis of band density for each species of gH/gL. nd, not detected.
The small amounts of gH/gL complexes in ME and ME-T virions made quantitation in these viruses difficult to perform on the same blots as TB and TR. Thus, ME and ME-T virions were further analyzed on separate nonreducing gels with increased gel loads to allow detection in a quantifiable range (Fig. 3). Furthermore, since ME and ME-T are genetically identical viruses, the virions could also be directly compared using anti-MEgO and anti-UL128 antibodies as Western blot probes (25). As in the previous analyses, ME virions contained more total gH/gL than ME-T virions, although at these increased loads, the difference was less pronounced. This likely reflected the limitations of band density quantitation on the lower end of the spectrum in Fig. 1 and 2. Comparison of the amounts of gH/gL/gO and gH/gL/UL128-131 in ME and ME-T virions by probing blots for either the common antigen (gL) or the unique antigen (gO or UL128) confirmed that ME-T virions contained more gH/gL/gO than ME virions, whereas ME virions had more gH/gL/UL128-131 than ME-T virions (compare analogous bands in Fig. 3A, B, and C). Together, these analyses indicated that strains of HCMV vary widely in the total amount of gH/gL complexes in the virion envelope and also vary in whether that gH/gL is in the form of gH/gL/gO or gH/gL/UL128-131.
FIG 3.
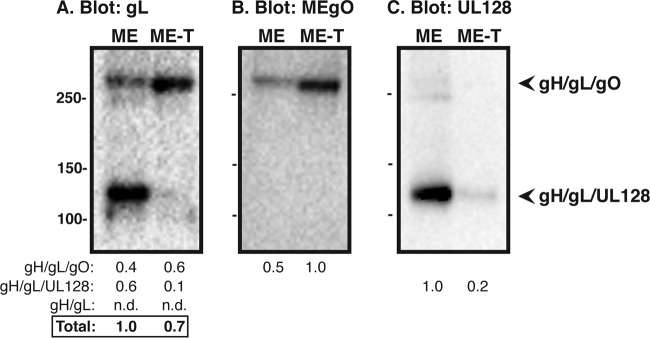
Quantitative comparison of gH/gL complexes in the virion envelope of ME or ME-T. Cell-free virions were separated by nonreducing SDS-PAGE and analyzed by Western blotting with antibodies specific for gL (A), MEgO (B), or UL128 (C). Mass markers (kilodaltons) are shown on the far left. The gel loads were normalized to equal MCP (not shown). The numbers below the blots indicate band density analysis normalized to the most intense band within the same blot. nd, not detected.
Particle-to-PFU ratio analysis of strains of HCMV that contain different amounts of gH/gL complexes.
One model of HCMV tropism suggests that gH/gL/gO and gH/gL/UL128-131 serve mechanistically analogous roles for entry into fibroblasts and epithelial cells, respectively. However, that model is inconsistent with observations that a gO-null mutant TR was impaired for entry into fibroblasts and epithelial cells (34). To help resolve this issue, we reasoned that the particle-to-PFU ratio of HCMV on either fibroblasts or epithelial cells should be influenced by the amounts of the gH/gL complexes in the virion envelope. Thus, cell-free virions of TB, TR, ME, and ME-T were quantitated by real-time qPCR for viral genomes, the titers were determined by plaque assay on both fibroblasts and epithelial cells, and the particle-to-PFU ratio was calculated (Fig. 4).
FIG 4.

Particle-to-PFU analysis of different HCMV strains. Cell-free stocks of TB, TR, ME, or ME-T virions were analyzed by qPCR to quantitate genome-containing virions, and PFU were determined on fibroblast (A) or epithelial cell (B) cultures. The particle-to-PFU ratio was calculated as the ratio of the number of genomes per milliliter to the number of PFU per milliliter. The averages and standard deviations of three experiments are plotted. The brackets above the bars indicate the results of a 2-tailed, unpaired Student t test. *, P = 0.04 to 0.05; **, P < 0.04.
Particle-to-PFU ratio measurements on fibroblasts varied widely between strains and were statistically significant in all comparisons (Fig. 4A). The extremes were TB and ME at 11 and 5,825, respectively, which correlated with the large and small amounts, respectively, of gH/gL/gO detected in these strains (Fig. 2). The values for TR and ME-T were intermediate (217 and 83, respectively), which generally correlated with their intermediate amounts of gH/gL/gO relative to TB and ME. Notably, however, TR contained considerably more gH/gL/gO than ME-T but was 2.6-fold less infectious than ME-T. It is possible that this relatively modest difference might indicate that the level of gH/gL/gO in ME-T represents a minimum threshold. Additionally, it might reflect other genetic differences between the strains that affect plaque formation. Notably, ME-T was 70-fold more infectious than ME on fibroblasts (Fig. 4A). Since ME-T and ME are genetically identical viruses, this difference likely reflects the direct effect of the different amounts of gH/gL/gO and gH/gL/UL128-131 on entry into these cells.
Particle-to-PFU ratios were generally higher on epithelial cells, consistent with previous observations that these cells are more difficult to infect with HCMV than are fibroblasts, perhaps reflecting the more complicated route of entry, which involves internalization followed by fusion from within endosomes (27). TB was the most infectious on epithelial cells (419 particles/PFU), followed by ME-T (1,250 particles/PFU), ME (10,529 particles/PFU), and TR (325,330 particles/PFU) (Fig. 4B). All comparisons were statistically significant. Given that ME virions contained dramatically more gH/gL/UL128-131 than the other viruses, the observation that ME was nearly 10-fold less infectious than ME-T and 25-fold less infectious than TB on epithelial cells was inconsistent with the model that gH/gL/UL128-131 performs the conserved herpesvirus gH/gL function of promoting gB-mediated fusion on these cells. Rather, the comparatively poor infectivity of ME on both fibroblasts and epithelial cells is more consistent with the hypothesis that gH/gL/gO provides the core fusion function for entry into all cell types and that gH/gL/UL128-131 provides a mechanistically distinct additional function necessary for entry into epithelial cells. However, the particle-to-PFU ratio of TR on epithelial cells was several log-fold higher than any of the other viruses, despite being relatively rich in gH/gL/gO. This discrepancy was likely due in part to the fact that plaque formation is a function not only of initial entry of cell-free virions, but also of replication and the spread of progeny.
To address postentry differences in plaque formation among HCMV strains, an infectious-center experiment was performed. Fibroblasts or epithelial cells were infected with TB, TR, or ME, and at 2 days p.i., the cells were detached from the culture dishes and replated onto monolayers of the homologous cell type, and the resultant plaques were counted (Fig. 5). Note that since ME-T virions were produced by propagating ME in cells expressing TetR to repress transcription from the UL128 locus (25, 35), they are genetically identical to ME virions. Thus, there was no logical distinction to be made between ME and ME-T in these infectious-center experiments. On fibroblasts, TB, TR, and ME were all comparably efficient at forming plaques from infectious centers (40 to 60 plaques per 100 infectious centers seeded) (Fig. 5A). On epithelial cells, TB and ME formed plaques from infectious centers with efficiencies similar to those on fibroblasts, but TR was notably less efficient, yielding only 10 plaques per 100 infectious centers (Fig. 5B). In interpreting these results, it is important to note that gO-null TR was severely impaired for entry but more efficient at cell-cell spread in these epithelial cells than the parental wild-type TR (34), suggesting a mechanistic distinction between entry and spread on these cells. Thus, the poor ability of TR to form plaques on epithelial cells from infectious centers helps to explain the apparently high particle-to-PFU ratio of TR on these cells and is also consistent with reports indicating that HCMV tropism is not solely determined at the level of entry (41, 42).
FIG 5.
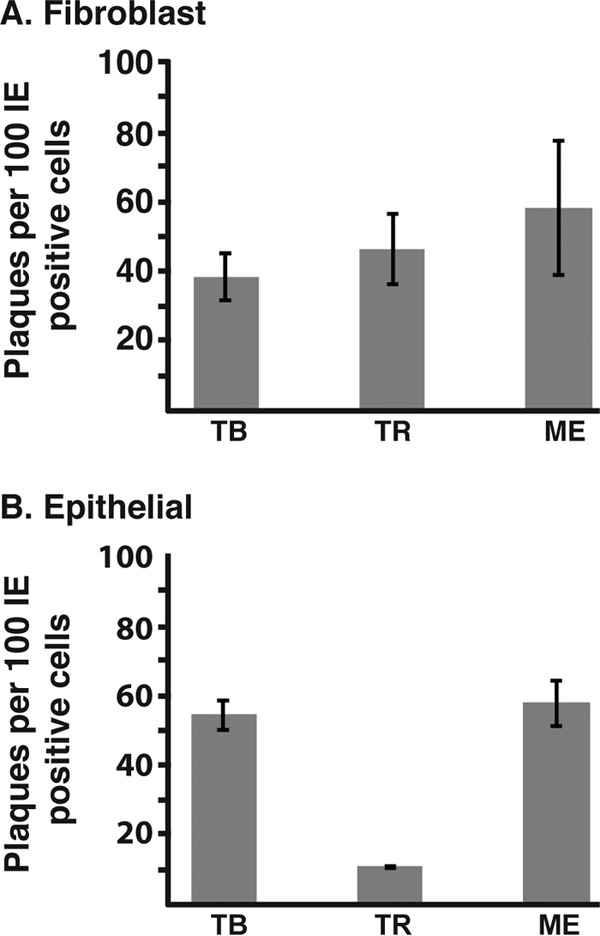
HCMV plaque formation from infectious centers. Replicate cultures of fibroblasts (A) or epithelial cells (B) were infected with TB, TR, or ME. At 2 days p.i., one set of cultures was fixed, and the fraction of infected cells was quantified by immunofluorescence detection of immediate-early gene expression. Cells in replicate cultures were detached and counted, and an amount of cells corresponding to 100 IE-positive cells were plated onto cultures of the homologous cell type. Plaques were allowed to develop and then counted. The averages and standard deviations from the results of three experiments are plotted.
Perhaps the most surprising result of the particle-to-PFU analysis presented in Fig. 4 was that ME-T was approximately 10-fold more efficient than ME on epithelial cells (Fig. 4B). Western blot analysis clearly demonstrated that ME-T virions contain mostly gH/gL/gO and very little gH/gL/UL128-131, whereas ME contains abundant gH/gL/UL128-131 and smaller amounts of gH/gL/gO (Fig. 1 to 3). It is possible that these differences in gH/gL complexes affected the fundamental mechanism or route of entry for ME and ME-T into these cells. To test this, antibody neutralization experiments were performed using anti-UL130 and anti-UL131, which have been shown previously to block HCMV infection of epithelial cells, but not fibroblasts (43) (Fig. 6). Anti-UL130/131 antibodies blocked infection of both ME and ME-T on epithelial cells, but not on fibroblasts, indicating that for both viruses, infection of epithelial cells, but not fibroblasts, was gH/gL/UL128-131 dependent. These results indicate that (i) ME-T virions were not completely devoid of gH/gL/UL128-131 and (ii) the enhanced infectivity of ME-T on epithelial cells was not due to an aberrant, gH/gL/UL128-131-independent process. Taken together, these analyses strongly contradict the model that gH/gL/gO and gH/gL/UL128-131 serve mechanistically analogous roles for entry of HCMV into fibroblasts and epithelial cells, respectively. Instead, it seems more likely that the two complexes act through distinct mechanisms to facilitate entry into these cell types.
FIG 6.
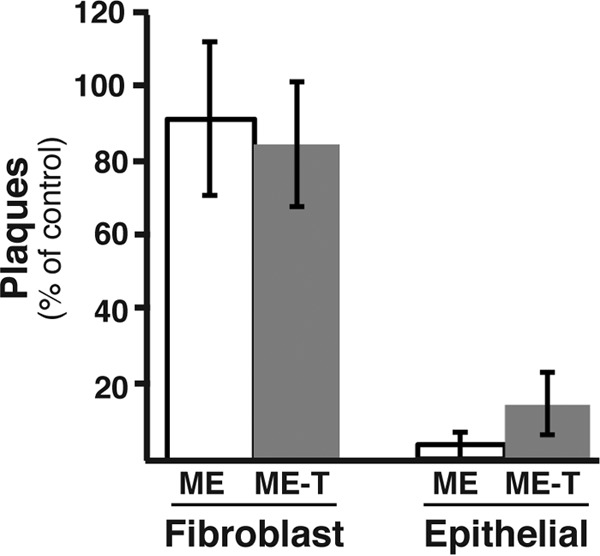
Neutralization of HCMV by UL130- and UL131-specific antibodies. Cell-free ME or ME-T was plated on fibroblast or epithelial cell cultures in the presence of anti-UL130 and anti-UL131 antibodies, and the number of resultant plaques was determined. The average numbers of plaques as percentages of the control (no antibody) and standard deviations from the results of three experiments are plotted. For each experiment, the number of plaques under control conditions ranged from 80 to 120.
The particle-to-PFU ratio difference between ME and ME-T reflects the fusion step of entry.
Since ME and ME-T are genetically identical viruses, their postentry replication and spread characteristics are identical in cells that do not express TetR (25, 35). Thus, the observed differences in particle-to-PFU ratios (Fig. 4) were likely indicative of differences in the entry process as a direct result of the amounts of gH/gL/gO and gH/gL/UL128-131 in the virion envelope, likely acting on the ability of these viruses to accomplish the fusion step of entry. To test this hypothesis, equivalent numbers of ME or ME-T virion particles were adsorbed onto fibroblasts or epithelial cells, and then both viruses were provided with an equivalent surrogate fusion mechanism, PEG treatment (Fig. 7) (27).
FIG 7.

Effects of PEG on infection by gH/gL/UL128-131-rich and gH/gL/gO-rich HCMV. Cell-free ME or ME-T virions were plated on fibroblast (A) or epithelial cell (B) cultures at 150 particles per cell and then treated with either PBS (− PEG) or PEG (+ PEG). The cells were fixed 48 h later and analyzed by immunofluorescence to detect HCMV IE gene expression. Representative microscope fields are shown (left), and the average percentages of cells infected among 6 microscope fields (>80 cells each) are plotted (right). The error bars represent standard deviations.
As expected from the particle-to-PFU analyses, ME-T efficiently infected fibroblasts, but ME did not (66% and 2.5%, respectively) (Fig. 7A). PEG treatment had no significant effect on the number of cells infected with ME-T, suggesting that the endogenous fusion mechanism of ME-T on these cells was similar to the efficiency of PEG-induced fusion. In contrast, PEG resulted in an approximately 20-fold increase in the numbers of cells infected with ME, bringing the percentage of cells infected with ME very near the values for ME-T. These results suggested that ME virions were poorly infectious on fibroblasts, largely due to an inefficient fusion mechanism.
At the dose of virus used in these experiments (150 particles per cell), neither ME nor ME-T infected quantifiable numbers of epithelial cells (Fig. 7B). This was consistent with the much higher particle-to-PFU ratio measured on these cells than on fibroblasts (Fig. 4). PEG treatment resulted in similar numbers of infected cells for both ME and ME-T, consistent with the notion that the high particle-to-PFU ratio of ME compared to ME-T reflected a difference in the abilities of these viruses to accomplish the fusion step of entry, not later stages of the replication cycle. However, because of the relatively high particle-to-PFU ratios for both ME and ME-T on these cells (1,250 and 10,529, respectively) (Fig. 4B), it was possible that the 150 particles per cell used in these experiments did not reflect the replication-competent virions in each population. To address this caveat, another set of experiments were performed in which 10 or 30 PFU were plated on epithelial cells, virus entry was induced with PEG, and the number of resultant plaques was determined (Fig. 8). PEG had no significant effect on the numbers of ME-T plaques, suggesting that the endogenous entry machinery of replication-competent ME-T virions was as efficient as PEG-induced entry. In contrast, PEG treatment resulted in plaque numbers 4- to 5-fold greater than the inputs of ME (Fig. 8). This represents a recovery of approximately half of the particle-to-PFU difference between ME and ME-T on these cells (Fig. 4B). The remaining half may reflect the limits of PEG efficiency. These results strongly suggest that the comparatively poor infectivity of ME on epithelial cells is due to an inability to accomplish the fusion stage of entry, despite their elevated levels of gH/gL/UL128-131.
FIG 8.
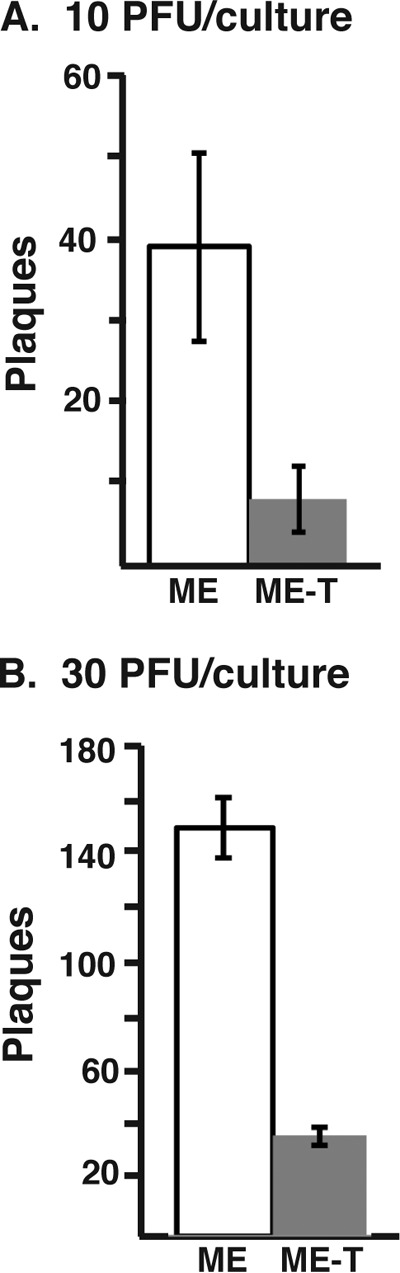
Effects of PEG on HCMV plaque formation on epithelial cells. Ten PFU (A) or 30 PFU (B) of ME or ME-T was plated on epithelial cell cultures and treated with PEG, and the number of resultant plaques was determined. The average numbers of plaques and standard deviations from three experiments are plotted.
DISCUSSION
Specific infectivity (also referred to as plating efficiency) was among the earliest established parameters of virus characterization and has been generally expressed as some unit of physical quantitation of virus (e.g., grams of protein or nucleic acid, number of capsids, or number of genomes) per unit of virus infectivity (e.g., PFU, 50% lethal/infectious dose [LD50/ID50], or 50% tissue culture infectious dose [TCID50]) (44–46). High particle-to-PFU ratios are common and reflect the fact that some viral particles in a given preparation are defective and incapable of initiating or completing a replication cycle. However, irrespective of bona fide defective virions, high specific-infectivity ratios also reflect the generally low probability that any given replication-competent virion within a population will successfully accomplish a measurable replication event in an inherently time-limited assay. Indeed, Thomas et al. reported that high particle-to-PFU ratios of HIV in part reflect the poor odds that any given virion will even attach to a host cell (47). It has been suggested that the envelope composition of HCMV virions within a population is heterogeneous (48, 49), and it is plausible that this phenomenon contributes to high and variable particle-to-PFU ratios through stochastic probability effects.
Early analyses of the specific infectivity of HCMV involved counting particles by electron microscopy, and particle-to-PFU ratios ranging from approximately 102 to 108 were reported (50–52). The wide ranges and high values were likely due in part to variable numbers of dense bodies (DB) and noninfectious enveloped particles (NIEPs), which lack genomes and therefore cannot be PFU (53–55). Our use of quantitative PCR avoided particles that lack genomes, and consequently, the values obtained were log orders lower, more consistent between independent samples of the same virus, and consistent with previous uses of this approach (56).
While the particle-to-PFU measurements reported here were highly reproducible for any given strain of HCMV, there were large differences between strains and between cell types. The values on epithelial cells were considerably higher than on fibroblasts for all strains analyzed. This indicates that the susceptibility and permissiveness of different cell types to HCMV is not a simple “yes-no” binary but, rather, a continuum that likely reflects complex differences, including receptor availabilities and the mechanism of entry and spread on different cell types. Indeed, entry of HCMV into fibroblasts likely occurs through fusion at the cell surface, whereas entry into other cell types, such as epithelial cells, likely involves internalization of the virus by endocytosis-like processes and subsequent fusion from within endosomes (27, 57, 58). Additionally, the large differences between strains of HCMV observed likely have multilocus genetic underpinnings, since plaque formation is a function of every stage of the replication cycle.
Comparative Western blot analyses indicated considerable variation in the amounts of gH/gL complexes in the virion envelope between strains of HCMV. TB and TR were found to contain gH/gL mostly in the form of gH/gL/gO and comparatively less gH/gL/UL128-131, whereas ME virions contained much less total gH/gL, and that was mostly in the form of gH/gL/UL128-131. These results were consistent with our previous data (25). Importantly, despite the overall small amount of gH/gL in ME, the amount of gH/gL/UL128-131 was dramatically more than was detected in TB or TR virions. While the HCMV virions analyzed here were produced in fibroblasts, Murrell et al. produced virions in epithelial cells and found that TR contained amounts of gH/gL/UL128-131 comparable to those in ME and that TB had noticeably less (59). These discrepancies suggest that factors of the host cell type might influence the assembly of HCMV gH/gL complexes. Furthermore, our analyses suggest that that the gH/gL deficit in ME represents a deficit of gH/gL/gO. This was reversed by Tet repression of gH/gL/UL128-131, resulting in ME virions that contained mostly gH/gL/gO (25) (Fig. 3). This virus was denoted ME-T. These characterizations of gH/gL complexes among HCMV strains allowed studies of the mechanisms by which gH/gL/gO and gH/gL/UL128-131 facilitate entry into different cell types.
Characterization of gH/gL/gO and gH/gL/UL128-131 mechanisms has relied largely on the analysis of UL128-131 and gO deletion mutants (21, 27–29, 31–34). UL128-131 mutants replicate well on fibroblasts, allowing them to be easily propagated and clearly characterized for their defect(s) on select cells, such as epithelial and endothelial cells. In contrast, gO mutants are severely impaired on all cell types. This suggested a fundamental role for gH/gL/gO in entry, but without a complementation system, further characterization of the mechanism has been difficult, since the rare infection events observed likely represent nonphysiological mechanisms. Our use of ME virions enriched for either gH/gL/UL128-131 or gH/gL/gO (ME and ME-T, respectively, avoided many of the caveats associated with null mutants. Since the viruses are not devoid of either gH/gL/gO or gH/gL/UL128-131, they are theoretically capable of employing physiologically relevant viral entry mechanisms on all cell types, albeit with different efficiencies. Furthermore, since they are genetically identical viruses, after the initial entry event, replication and plaque spread should be identical. Indeed, plaque sizes and morphologies were indistinguishable on all cells lacking TetR expression (35). The higher particle-to-PFU ratio of ME than of ME-T on both fibroblasts and epithelial cells indicated that a greater fraction of virions in ME stocks failed to form plaques. Experiments involving PEG-induced fusion indicated that these ME virions failed largely because they could not accomplish the fusion event of entry. The fate of nonfusing virions is unclear but may include endocytosis and destruction within the lysosome. Regardless, these results are inconsistent with the model that gH/gL/gO and gH/gL/UL128-131 are each sufficient as a herpesvirus core fusion gH/gL for entry into fibroblasts and epithelial cells, respectively. Rather, it is more likely that only gH/gL/gO performs the conserved herpesvirus gH/gL function of promoting gB-mediated membrane fusion during entry into all cell types and gH/gL/UL128-131 provides a distinct, yet necessary, function for entry into select cell types, such as epithelial cells.
The exceedingly high particle-to-PFU ratio of TR on epithelial cells was seemingly incongruent with the model of distinct functions for gH/gL complexes, since TR virions contained amounts of gH/gL/gO and gH/gL/gUL128-131 comparable to those in TB and contained more of both gH/gL complexes than ME-T virions, yet both TB and ME-T were vastly more efficient at plaque formation than TR. However, as noted above, plaque formation reflects the entire entry-replication-spread cycle. Analysis of the efficiency of postentry replication and spread indicated that TR was much less efficient at progressing from entry to form an observable plaque on epithelial cells than was TB or ME (Fig. 5). Furthermore, plaques on epithelial cells that were observed for TR were notably smaller than those for TB and ME. Thus, it seems likely that the poor plaque formation of TR on epithelial cells reflects genetic differences between strain loci other than gO and UL128-131 and underscores observations that HCMV tropism is not only determined by entry mechanisms (41, 42).
The model of distinct mechanisms for HCMV gH/gL complexes fits well with other recent observations of CMV entry and spread. First, HCMV entry into epithelial cells likely involves membrane fusion from within low-pH endosomes (27, 57, 58). Straschewski et al. and Nogalski et al. characterized the interaction of HCMV with monocytes, a cell type for which infection is also gH/gL/UL128-131 dependent (30, 60). They found positive correlation between the level of gH/gL/UL128-131 in the virion envelope and the intensity of Src kinase activation following virion attachment (60). Thus, one possibility is that signaling through gH/gL/UL128-131 receptors might affect the nature of the endosome in a way that fosters subsequent membrane fusion mediated by gB and gH/gL/gO. Second, while data presented here indicate that gH/gL/gO is important for infection of all cell types by cell-free HCMV, analyses of gO mutants suggested that cell-to-cell spread on epithelial or endothelial cells and monolayers does not require gH/gL/gO (33, 34). Together, these observations are consistent with a recent in vivo study involving murine CMV, suggesting that gH/gL/gO is important for initial infection of mice but less important for subsequent spread to distal organs and tissues (61).
The proposed mechanistic distinction between gH/gL/gO and gH/gL/UL128-131 may provide a unique opportunity to study the general principles through which the herpesvirus gH/gL promotes fusion by gB (i.e., the core fusion function of gH/gL). Herpesvirus fusion has been most extensively studied for HSV, which has only one form of gH/gL, and EBV, which has two, gH/gL and gH/gL/gp42 (reviewed in reference 18). In the case of EBV, both complexes appear to perform the conserved function of promoting gB-mediated membrane fusion for entry into either epithelial cells or B cells, indicating that gp42 does not cover or otherwise alter the pro-fusion surfaces of gH/gL. Fusion by HCMV also likely involves interactions between gB and surfaces of gH/gL (19, 20). However, data presented here suggest that gH/gL does not promote gB fusion when the UL128-131 proteins are bound. This could be due to differential allosteric effects on gH/gL conformation as the result of gO versus UL128-131 binding, or it could be that gO and UL128-131 physically cover different surfaces of gH/gL. Recent cryo-electron microscopy (cryo-EM) analyses reported by Ciferri et al. indicated that gO and UL128-131 bind the same face of gH/gL, but the data provided neither support nor contradict either possibility (26). It is also noteworthy that Ciferri et al. suggested that gH/gL/UL128-131 was capable of facilitating gB-mediated membrane fusion in cell-cell fusion experiments. However, these data were difficult to interpret, because in the same experiments, unbound gH/gL (lacking UL128-131 or gO) failed to promote gB-mediated fusion. This was an important discrepancy with the results of Vanarsdall et al., showing that gH/gL alone was capable of promoting gB fusion in the same type of cell-cell fusion experiment (20, 26).
Several recent studies have characterized the antibody response to the pentameric gH/gL/UL128-131 with an aim of vaccine development (62–64). Pentamer vaccines clearly elicit antibodies that are specific to epitopes that are contained on the UL128-131 protein surfaces themselves or present on the gH/gL surfaces that are allosterically dependent on UL128-131 (i,e., pentamer-specific antibodies), as well as antibodies that react to surfaces of gH/gL that are independent of the UL128-131 proteins (i.e., gH/gL-specific antibodies). Pentamer-specific antibodies powerfully neutralize HCMV infection of epithelial cells but show little to no neutralizing potency on fibroblasts. In contrast, the gH/gL-specific antibodies have significantly lower neutralizing potency but block infection of both fibroblasts and epithelial cells with comparable potencies. In the context of the model of distinct mechanisms proposed here, it seems likely that the pentamer-specific antibodies interfere with the binding of the pentamer to cell-type-specific receptors and thus block the subsequent signaling cascades that enhance infection of select cell types, whereas the gH/gL-specific antibodies block the role of gH/gL/gO in promoting gB fusion on all cell types. The disparity in potency between pentamer-specific and gH/gL-specific antibodies may be due to any of several non-mutually exclusive factors, including (i) the differential abundances of gH/gL/gO and gH/gL/UL128-131 in the virion envelope; (ii) the apparent fact that infection of epithelial cells can likely be inhibited at either of two independent mechanisms, internalization or fusion, compared to only fusion on fibroblasts; and (iii) the fact that the relevant gH/gL-specific epitopes may be differentially displayed due to different allosteric effects of UL128-131 binding compared to gO binding or the presence of the extensive gO glycan network. The last possibility seems especially plausible, given that the gH/gL-specific antibodies described have been raised against either gH/gL alone or gH/gL/UL128-131 (i.e., in the absence of gO).
In summary, the observations reported here and elsewhere strongly argue that the fusion event of entry of HCMV into all cell types is mediated by gB and gH/gL/gO. On some cell types, such as epithelial cells, efficient fusion must be preceded by a process mediated by the pentameric gH/gL/UL128-131 complex. This model of entry and tropism may have important implications for the development of HCMV vaccines, as well as broader implications for understanding the principles of what the herpesvirus gH/gL does to promote gB fusion.
ACKNOWLEDGMENTS
We are grateful to Bill Britt, David Johnson, Jay Nelson, Tom Shenk, Christian Sinzger, and Richard Stanton for generously supplying HCMV BAC clones, antibodies, and cell lines, as indicated in Materials and Methods, and members of the Ryckman laboratory for support and insightful discussions.
B.J.R. is supported by grants from the National Institutes of Health (5-R01-AI097274-02 and PG20GM103546).
The experiments were designed by B.J.R., M.Z., and J.-M.L. and performed by M.Z. and J.-M.L. The data were analyzed and the manuscript was prepared by B.J.R. and M.Z.
REFERENCES
- 1.Britt WJ. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 2.Streblow DN, Orloff SL, Nelson JA. 2007. Acceleration of allograft failure by cytomegalovirus. Curr Opin Immunol 19:577–582. doi: 10.1016/j.coi.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boppana SB, Fowler KB, Pass RF, Rivera LB, Bradford RD, Lakeman FD, Britt WJ. 2005. Congenital cytomegalovirus infection: association between virus burden in infancy and hearing loss. J Pediatr 146:817–823. doi: 10.1016/j.jpeds.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 4.Plachter B, Sinzger C, Jahn G. 1996. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res 46:195–261. doi: 10.1016/S0065-3527(08)60073-1. [DOI] [PubMed] [Google Scholar]
- 5.Sinzger C, Grefte A, Plachter B, Gouw AS, The TH, Jahn G. 1995. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J Gen Virol 76:741–750. doi: 10.1099/0022-1317-76-4-741. [DOI] [PubMed] [Google Scholar]
- 6.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanarsdall AL, Johnson DC. 2012. Human cytomegalovirus entry into cells. Curr Opin Virol 2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 10.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J Virol 87:11332–11345. doi: 10.1128/JVI.01700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns TM, Whitbeck JC, Lou H, Heldwein EE, Chowdary TK, Eisenberg RJ, Cohen GH. 2011. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J Virol 85:6175–6184. doi: 10.1128/JVI.00119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J Virol 85:13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plate AE, Smajlovic J, Jardetzky TS, Longnecker R. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J Virol 83:7678–7689. doi: 10.1128/JVI.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma S, Wisner TW, Johnson DC, Heldwein EE. 2013. HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435:239–249. doi: 10.1016/j.virol.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J Virol 82:11837–11850. doi: 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. doi: 10.1099/vir.0.81921-0. [DOI] [PubMed] [Google Scholar]
- 22.Huber MT, Compton T. 1997. Characterization of a novel third member of the human cytomegalovirus glycoprotein H-glycoprotein L complex. J Virol 71:5391–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Nelson JA, Britt WJ. 1997. Glycoprotein H-related complexes of human cytomegalovirus: identification of a third protein in the gCIII complex. J Virol 71:3090–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M, Yu Q, Wechsler A, Ryckman BJ. 2013. Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope. J Virol 87:9680–9690. doi: 10.1128/JVI.01167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciferri C, Chandramouli S, Donnarumma D, Nikitin PA, Cianfrocco MA, Gerrein R, Feire AL, Barnett SW, Lilja AE, Rappuoli R, Norais N, Settembre EC, Carfi A. 2015. Structural and biochemical studies of HCMV gH/gL/gO and pentamer reveal mutually exclusive cell entry complexes. Proc Natl Acad Sci U S A 112:1767–1772. doi: 10.1073/pnas.1424818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Straschewski S, Patrone M, Walther P, Gallina A, Mertens T, Frascaroli G. 2011. Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J Virol 85:5150–5158. doi: 10.1128/JVI.02100-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol 74:7720–7729. doi: 10.1128/JVI.74.17.7720-7729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang XJ, Adler B, Sampaio KL, Digel M, Jahn G, Ettischer N, Stierhof YD, Scrivano L, Koszinowski U, Mach M, Sinzger C. 2008. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J Virol 82:2802–2812. doi: 10.1128/JVI.01550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, Davison AJ, Wilkinson GW. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 38.Schoppel K, Hassfurther E, Britt W, Ohlin M, Borrebaeck CA, Mach M. 1996. Antibodies specific for the antigenic domain 1 of glycoprotein B (gpUL55) of human cytomegalovirus bind to different substructures. Virology 216:133–145. doi: 10.1006/viro.1996.0040. [DOI] [PubMed] [Google Scholar]
- 39.Chee M, Rudolph SA, Plachter B, Barrell B, Jahn G. 1989. Identification of the major capsid protein gene of human cytomegalovirus. J Virol 63:1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol 82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bughio F, Elliott DA, Goodrum F. 2013. An endothelial cell-specific requirement for the UL133-UL138 locus of human cytomegalovirus for efficient virus maturation. J Virol 87:3062–3075. doi: 10.1128/JVI.02510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. doi: 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saccoccio FM, Sauer AL, Cui X, Armstrong AE, Habib E-SE, Johnson DC, Ryckman BJ, Klingelhutz AJ, Adler SP, McVoy MA. 2011. Peptides from cytomegalovirus UL130 and UL131 proteins induce high titer antibodies that block viral entry into mucosal epithelial cells. Vaccine 29:2705–2711. doi: 10.1016/j.vaccine.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuhn CW, Bancroft JB. 1961. Concentration and specific infectivity changes of alfalfa mosaic virus during systemic infection. Virology 15:281–288. doi: 10.1016/0042-6822(61)90359-2. [DOI] [PubMed] [Google Scholar]
- 45.Bachrach HL, Schwerdt CE. 1954. Purification studies on Lansing poliomyelitis virus. II. Analytical electron microscopic identification of the infectious particle in preparations of high specific infectivity. J Immunol 72:30–38. [PubMed] [Google Scholar]
- 46.Bang FB. 1948. Studies on Newcastle disease virus; characters of the virus itself with particular reference to electron microscopy. J Exp Med 88:251–266. doi: 10.1084/jem.88.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas JA, Ott DE, Gorelick RJ. 2007. Efficiency of human immunodeficiency virus type 1 postentry infection processes: evidence against disproportionate numbers of defective virions. J Virol 81:4367–4370. doi: 10.1128/JVI.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scrivano L, Sinzger C, Nitschko H, Koszinowski UH, Adler B. 2011. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog 7:e1001256. doi: 10.1371/journal.ppat.1001256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, Coelingh KL, Britt WJ. 1995. Human cytomegalovirus neutralizing antibody-resistant phenotype is associated with reduced expression of glycoprotein H. J Virol 69:6047–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benyesh-Melnick M, Probstmeyer F, McCombs R, Brunschwig JP, Vonka V. 1966. Correlation between infectivity and physical virus particles in human cytomegalovirus. J Bacteriol 92:1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith KO, Rasmussen L. 1963. Morphology of cytomegalovirus (salivary gland virus). J Bacteriol 85:1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stinski MF, Mocarski ES, Thomsen DR. 1979. DNA of human cytomegalovirus: size heterogeneity and defectiveness resulting from serial undiluted passage. J Virol 31:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Irmiere A, Gibson W. 1983. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 130:118–133. doi: 10.1016/0042-6822(83)90122-8. [DOI] [PubMed] [Google Scholar]
- 54.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahlqvist J, Mocarski E. 2011. Cytomegalovirus UL103 controls virion and dense body egress. J Virol 85:5125–5135. doi: 10.1128/JVI.01682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heider JA, Bresnahan WA, Shenk TE. 2002. Construction of a rationally designed human cytomegalovirus variant encoding a temperature-sensitive immediate-early 2 protein. Proc Natl Acad Sci U S A 99:3141–3146. doi: 10.1073/pnas.052710599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haspot F, Lavault A, Sinzger C, Laib Sampaio K, Stierhof YD, Pilet P, Bressolette-Bodin C, Halary F. 2012. Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner. PLoS One 7:e34795. doi: 10.1371/journal.pone.0034795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. 2012. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog 8:e1002905. doi: 10.1371/journal.ppat.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murrell I, Tomasec P, Wilkie GS, Dargan DJ, Davison AJ, Stanton RJ. 2013. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J Virol 87:10489–10500. doi: 10.1128/JVI.01546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nogalski MT, Chan GC, Stevenson EV, Collins-McMillen DK, Yurochko AD. 2013. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog 9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lemmermann NA, Krmpotic A, Podlech J, Brizic I, Prager A, Adler H, Karbach A, Wu Y, Jonjic S, Reddehase MJ, Adler B. 2015. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog 11:e1004640. doi: 10.1371/journal.ppat.1004640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wussow F, Yue Y, Martinez J, Deere JD, Longmate J, Herrmann A, Barry PA, Diamond DJ. 2013. A vaccine based on the rhesus cytomegalovirus UL128 complex induces broadly neutralizing antibodies in rhesus macaques. J Virol 87:1322–1332. doi: 10.1128/JVI.01669-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kabanova A, Perez L, Lilleri D, Marcandalli J, Agatic G, Becattini S, Preite S, Fuschillo D, Percivalle E, Sallusto F, Gerna G, Corti D, Lanzavecchia A. 2014. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc Natl Acad Sci U S A 111:17965–17970. doi: 10.1073/pnas.1415310111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wen Y, Monroe J, Linton C, Archer J, Beard CW, Barnett SW, Palladino G, Mason PW, Carfi A, Lilja AE. 2014. Human cytomegalovirus gH/gL/UL128/UL130/UL131A complex elicits potently neutralizing antibodies in mice. Vaccine 32:3796–3804. doi: 10.1016/j.vaccine.2014.05.004. [DOI] [PubMed] [Google Scholar]