

Summary
Uridylation of RNA species represents an emerging theme in post-transcriptional gene regulation. In the microRNA pathway, such modifications regulate small RNA biogenesis and stability in plants, worms, and mammals. Here, we report Tailor, an uridylyltransferase that is required for the majority of 3′ end modifications of microRNAs in Drosophila and predominantly targets precursor hairpins. Uridylation modulates the characteristic two-nucleotide 3′ overhang of microRNA hairpins, which regulates processing by Dicer-1 and destabilizes RNA hairpins. Tailor preferentially uridylates mirtron hairpins, thereby impeding the production of non-canonical microRNAs. Mirtron selectivity is explained by primary sequence specificity of Tailor, selecting substrates ending with a 3′ guanosine. In contrast to mirtrons, conserved Drosophila precursor microRNAs are significantly depleted in 3′ guanosine, thereby escaping regulatory uridylation. Our data support the hypothesis that evolutionary adaptation to Tailor-directed uridylation shapes the nucleotide composition of precursor microRNA 3′ ends. Hence, hairpin uridylation may serve as a barrier for the de novo creation of microRNAs in Drosophila.
Graphical Abstract
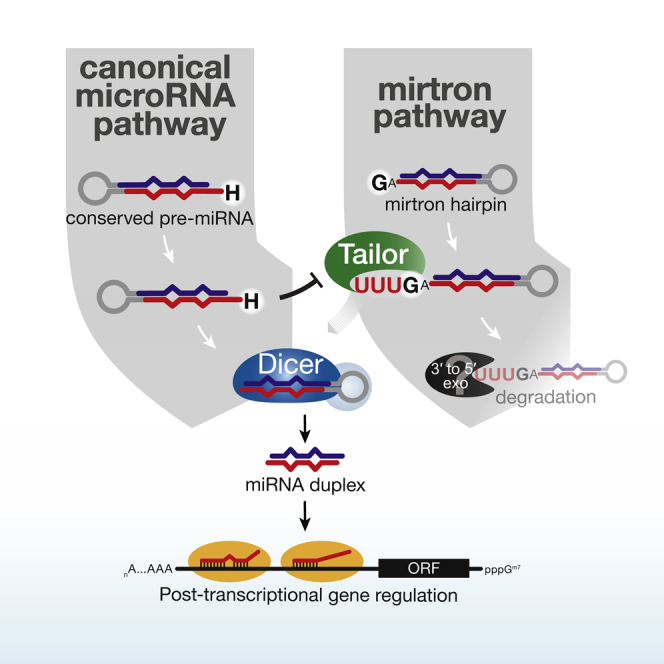
Highlights
-
•
Tailor is a small RNA uridylyltransferase in Drosophila
-
•
Tailor uridylates pre-miRNAs and regulates miRNA maturation
-
•
Tailor prevents the maturation of non-canonical miRNAs, i.e., mirtrons
-
•
Tailor may act as a barrier for the de novo creation of miRNAs
Reimão-Pinto et al. report a small RNA-specific terminal uridylyltransferase in Drosophila, which acts on precursor miRNAs to regulate miRNA maturation and may serve as a barrier for the de novo creation of miRNAs in flies.
Introduction
MicroRNAs (miRNAs) mediate post-transcriptional gene silencing in plants and animals and control organismal development, physiology, and disease (Bartel, 2009). Most miRNAs derive from hairpin-containing transcripts (primary miRNAs; pri-miRNAs) that are sequentially processed by RNase III enzymes into mature small RNAs (Kim et al., 2009). First, Drosha liberates an ∼60 nt hairpin (precursor miRNA; pre-miRNA) from the pri-miRNA transcript in the nucleus (Lee et al., 2003; Gregory et al., 2004; Denli et al., 2004). Upon Exportin-5-directed export of pre-miRNAs from the nucleus to the cytoplasm, Dicer (Dcr-1 in flies) converts pre-miRNAs into mature ∼22-nt small RNA duplexes that are subsequently loaded into an Argonaute (Ago) protein (Ago1 in flies) (Lee et al., 2004; Okamura et al., 2004), where they guide post-transcriptional silencing of complementary mRNA (Huntzinger and Izaurralde, 2011).
Alternative pathways bypass Drosha or Dicer processing for the production of non-canonical miRNAs (Yang and Lai, 2011). The most prevalent example is the splicing of short introns followed by lariat debranching, which produces Drosha-independent pre-miRNA-like hairpins, referred to as mirtrons (Okamura et al., 2007; Ruby et al., 2007). Mirtron hairpins are exported by Exportin-5 and serve as substrates for Dicer, generating mature miRNAs that are functionally indistinguishable from products of canonical miRNA biogenesis (Okamura et al., 2007; Ruby et al., 2007; Babiarz et al., 2008; Berezikov et al., 2010). While numerous mirtrons have been annotated, they rarely accumulate to levels that compare to canonical miRNAs (Chung et al., 2011), and mirtrons more frequently appear and disappear in evolution compared to canonical miRNAs (Berezikov et al., 2010). The spurious contribution of mirtrons to small RNA profiles is apparently at odds with the conservation of the mirtron pathway in flies, worms, and mammals (Chung et al., 2011; Ladewig et al., 2012; Westholm and Lai, 2011). This raises the question of whether mechanisms exist that dampen the contribution of non-canonical pathways to the pool of small RNAs.
Perhaps because of its enormous regulatory potential, miRNA-mediated gene regulation is tightly controlled (Ha and Kim, 2014). At the post-transcriptional level, the addition of uridine(s) to the 3′ end of miRNAs and their precursors recently emerged as a hallmark for the regulation of miRNA biogenesis and turnover (Kim et al., 2010; Ameres and Zamore, 2013). Small RNA-modifying terminal uridylyltransferases (TUTases) have been reported in plants and animals (Scott and Norbury, 2013). In Arabidopsis, the methyltransferase HEN1 protects miRNAs and siRNAs from HESO 1-directed uridylation, a modification that destabilizes miRNAs (Li et al., 2005; Ren et al., 2012; Zhao et al., 2012). In animals, more versatile functions of uridylation have been described: in worms and mammals, the RNA-binding protein Lin28 recruits the TUTase ZCCHC11/PUP2 to pre-let-7, resulting in pre-miRNA oligouridylation (Heo et al., 2009; Hagan et al., 2009; Lehrbach et al., 2009). Pre-let-7 uridylation prevents dicing and triggers degradation by the exonuclease Dis3L2 (Chang et al., 2013). In the absence of Lin28, the non-processive addition of a single uridine to a selected class of pre-miRNA by the TUTases ZCCHC11 and ZCCHC6 enhances dicing in mammalian cells because it restores the two-nucleotide 3′ overhangs of pre-miRNAs (Heo et al., 2012). A similar mechanism may also contribute to the identification of defective pre-miRNAs that lack intact 3′ overhangs and trigger their destruction via the exosome (Liu et al., 2014). Finally, destabilization of mature miRNAs upon binding to highly complementary targets is associated with small RNA uridylation (and adenylation) in flies and mammals (Ameres et al., 2010; Xie et al., 2012). While diverse mechanisms employ TUTases in the regulation of small RNAs, their overall impact on shaping the miRNA repertoire remains unclear. In flies, no miRNA-specific TUTase has been identified. Nevertheless, meta-analyses of small RNA libraries from flies indicate that—like in mammals—miRNAs are frequently subjected to post-transcriptional uridylation in Drosophila (Berezikov et al., 2011; Westholm et al., 2012).
Here, we report the origin, molecular mechanism, and consequences of miRNA uridylation in flies.
Results
Frequent Uridylation of Selected miRNAs in Drosophila
To systematically characterize 3′ terminal post-transcriptional modifications of miRNAs we performed high-throughput sequencing of 18- to 30-nt small RNAs of Drosophila S2 cells and adult male flies. Reads were mapped to the genome and separated into genome-matching reads (GM; mapping perfectly to the genome) and prefix-matching reads (PM; containing one or more non-genome-matching nucleotide additions to the 3′ end) (Figure 1A). For all abundantly expressed miRNAs, prefix-matching reads were readily detectable, albeit to varying extent (Figure 1B).
Figure 1.

Post-transcriptional Modifications of Small RNAs in Drosophila
(A) Identification of post-transcriptional modifications in small RNA libraries. Genome-matching (GM) reads map perfectly to the genome; prefix-matching (PM) reads contain non-genome-matching nucleotides at the 3′ end (red).
(B) Abundance of genome-matching and prefix-matching miRNAs (in parts per million; ppm) in small RNA libraries generated from S2 cells (left) or adult male flies (right). S2 cell data represented as mean of three biological replicates ± SD. miRNAs with > 5% PM reads are indicated in black.
(C) Classification of miRNAs according to Argonaute loading (miR and miR∗), location in the pre-miRNA hairpin (5p- and 3p-miRNA), and mechanism of biogenesis (canonical miRNAs and mirtrons).
(D) Tailing analysis of small RNA deep sequencing datasets from S2 cells (left panel) and whole male flies (right panel). The fraction of tailed reads was calculated for each miRNA and represented as Tukey boxplots for all miRNAs (black boxes) or subsets of miRNAs as classified in (C) (white boxes). The number of miRNAs for each subset is indicated. p values (Mann-Whitney test) are indicated.
(E) Non-genome-matching nucleotide additions to miRNAs consist mostly of uridine and adenine. The nucleotide composition of non-genome-matching additions was determined for each miRNA and averaged across all miRNAs. See also Figure S1.
Three criteria classify miRNAs: the relative accumulation of the two strands of a miRNA duplex (miR versus miR∗), the origin from the 5′ or 3′ arm of a pre-miRNA (5p- versus 3p-miRNA), and the mode of biogenesis by Drosha processing (canonical miRNA) or splicing and debranching (mirtron) (Figure 1C). While the extent to which miRNAs exhibited non-genome-matching nucleotide additions was not consistently different between miR and miR∗ species, we observed a significantly higher frequency of post-transcriptional modifications at 3p- compared to 5p-miRNAs in both S2 cells and whole male flies. This suggested that pre-miRNAs might be substrates for post-transcriptional modification, which subsequently propagate into mature miRNAs. With a median tailing frequency of close to 10%, mirtrons exhibited the highest modification status, significantly higher compared to canonical miRNAs (p < 1 × 10−3 in S2 cells and p < 5 × 10−4 in whole male flies; Figure 1D). Finally, we determined the nucleotide identity of non-genome-matching additions (Figure 1E): addition of uridine (48% in S2 cells, 52% in flies) and adenine (34% in S2 cells, 42% in flies) dominated over cytosine (10% in S2 cells, 4% in flies) and guanine (8% in S2 cells, 2% in flies). Notably, adenylation was distributed broadly across miRNAs, irrespective of the degree to which these were modified, while uridylation was enriched in highly modified miRNAs (Figure 1E). Frequent uridylation of selected miRNAs suggested a specific targeting strategy for this type of post-transcriptional modification. The predominant class of such miRNAs consists of mirtrons, which are frequently modified (Figure 1D) and predominantly carry uridine modifications (Figure S1D).
The Cytoplasmic TNTase CG1091 Is Required for miRNA Uridylation and Normal Fertility in Flies
The template-independent incorporation of ribonucleoside monophosphate to the 3′ hydroxyl end of RNAs is catalyzed by RNA-specific TNTases. Members of this enzyme family exhibit a characteristic domain architecture consisting of a TNTase domain (NT_PAP_TUTase), often paired with a PAP/25A-associated domain (PAP_assoc) (Norbury, 2013). Based on domain homology search, we identified seven putative TNTases in the Drosophila genome, five of which were expressed in S2 cells (Figure 2A). We depleted candidate enzymes by RNAi in S2 cells followed by detection of the abundantly expressed miR-184 by high-resolution northern hybridization. Although it is not among the most frequently modified miRNAs (∼3%; Figure S1A), miR-184 produced higher-molecular-weight signals in northern hybridization experiments that reflected post-transcriptional modifications in high-throughput sequencing datasets (Figure 2B). Among the tested candidate TNTases, only depletion of CG1091 affected miR-184 tailing (Figure 2C), suggesting that CG1091 is required for the post-transcriptional modification of miR-184.
Figure 2.

The Cytoplasmic TNTase CG1091/Tailor Is Required for miRNA Uridylation and Normal Fertility in Flies
(A) Domain organization of known and putative TNTases in flies based on InterPro database. The characteristic domain structure consists of a nucleotidyltransferase domain (red box) and a PAP/25A-associated domain (blue). Expression in S2 cells was based on modENCODE mRNA sequencing (Cherbas et al., 2011).
(B) Post-transcriptional modification of miR-184-3p in S2 cells is detectable by high-resolution northern hybridization. Higher-molecular-weight bands of miR-184 (> 23 nt) correspond to prefix-matching reads (red) as evidenced by small RNA sequencing.
(C) CG1091 is required for post-transcriptional modification of miR-184. Upon depletion of the indicated candidate TNTases in S2 cells by RNAi miR-184 was detected by high-resolution northern hybridization. Double-stranded RNA targeting green fluorescent protein (GFP) or luciferase (LUC), and untreated S2 cells served as controls. 2S rRNA served as loading control.
(D) Schematic representation of CG1091 gene and RNA transcripts. CG1091-RB is predominantly expressed in S2 cells (see Figure S2C). The region targeted by dsRNA for depletion of CG1091 by RNAi in S2 cells, the location of a 7-bp deletion introduced by CRISPR/Cas9 genome editing in flies (CG1091c4-1/6), and a piggy-bac insertion into the coding sequence of CG1091 (CG1091f05717) are indicated.
(E) RNAi in S2 cells and CG1091c4-1/6 in flies depleted CG1091 protein to levels that are not detectable by western blotting. Actin represents loading control. Asterisks indicate non-specific signal in lysates of male flies.
(F) Depletion of CG1091 affects tailing of miRNAs in S2 cells (left) and in flies (right). The fraction at which each miRNA is modified is plotted. S2 cell data represented as mean of three independent biological replicates ± SD. miRNAs that show a significant depletion in post-transcriptional modifications are indicated in gray (p < 0.05, Student’s t test; FDR < 0.1; Benjamini and Hochberg, 1995). A > 2-fold reduction in tailing is indicated in red.
(G) Pair-wise comparison of miRNA tailing status revealed a statistically significant decrease upon depletion of CG1091 in S2 cells and in flies. p values (Wilcoxon matched-pairs signed rank test) are indicated.
(H) Depletion of CG1091 impacts miRNA uridylation. Abundance and composition of post-transcriptionally added nucleotides was determined for each miRNA upon CG1091 depletion, normalized to control samples (control dsRNA in S2 cells and w1118 in flies), and averaged across all miRNAs.
(I) Upon CG1091 depletion, mirtrons are significantly more depleted in non-genome-matching nucleotide additions when compared to canonical miRNAs in both S2 cells and in flies. Change in tailing was determined for the indicated classes of miRNAs. p values (Mann-Whitney test) are indicated.
(J and K) Depletion of CG1091 in flies affects male (J) and female (K) fertility. Fertility was determined in CG1091c4-1/c4-6 and CG1091c4-6/f05717 flies and compared to control (w1118) flies. p values (Student’s t test) are indicated. Data represented as mean ± SD.
(L) In S2 cells, Myc-tagged CG1091 localizes to the cytoplasm. FLAG-Myc-tagged CG1091 expression was driven by an Actin5C promoter (pAFMW-CG1091) upon transient transfection in S2 cells, followed by immunostaining (anti-Myc) and imaging. Single color channel images show total inversions for DAPI and Myc-CG1091. Scale bar = 5 μm. See also Figure S2.
To characterize the global effect of CG1091 on miRNA modifications we performed high-throughput sequencing of 18- to 30-nt small RNAs from S2 cells depleted of CG1091 by RNAi, as well as adult male flies, carrying two independently generated frame-shift deletions in the third exon of CG1091 (CG1091c4-1 and CG1091c4-6; Figure S2A), introduced by CRISPR/Cas9 (Figure 2D). Depletion of CG1091 was confirmed by western blot analysis (Figure 2E). Upon depletion of CG1091, the frequency at which miRNAs contained non-genome-matching nucleotide additions was significantly reduced in S2 cells when compared to control RNAi (p < 10−4; Wilcoxon matched pairs signed rank test) or untreated cells (p < 3 × 10−4), and in whole male flies homozygous for CG1091c-4-1/6 when compared to heterozygous siblings (p < 10−4) or w1118 flies (p < 10−4) (Figures 2F and 2G and Figure S2B). In S2 cells, we detected a statistically significant reduction in the post-transcriptional modification of 48 out of 79 miRNAs (p < 0.05, FDR < 0.1). For 19 of these, tailing was reduced by more than 2-fold (red dots, Figure 2F). In whole male flies, 39 out of 107 miRNAs were consistently reduced in tailing by more than 2-fold in the two independent CG1091c4 allele-carrying flies (Figures 2F and S2B).
Analysis of the nucleotide identity in miRNA 3′ tails revealed that CG1091 is required for the majority of miRNA uridylation (Figure 2H): upon depletion of CG1091 in S2 cells, uridine-containing extensions decreased by 2.6-fold when compared to the control, while all other nucleotide additions stayed unchanged. Similarly, uridylation decreased by 1.9-fold in CG1091c4-1/6 heterozygous, and by 4.2-fold in homozygous, flies when compared to w1118 control flies. Furthermore, depletion of CG1091 affected adenylation, although to a lesser extent (1.2- and 1.4-fold decrease in CG1091−/+ and CG1091−/−, respectively), while the addition of cytidine or guanosine was not affected. Note that under CG1091 depletion conditions we still detected residual miRNA uridylation above background, indicating that CG1091 may not be the sole small RNA-uridylating enzyme. CG1091 acts preferentially on mirtrons, which were significantly more depleted in post-transcriptional modifications compared to canonical miRNAs, both in S2 cells (p < 10−4) and in flies (p < 0.03) (Figure 2I).
While CG1091 is ubiquitously expressed, mRNA levels are highest in the ovary and testis (Graveley et al., 2011), prompting us to investigate any loss-of-function effects on fertility. We observed a significant defect in male (p < 10−4; Figure 2J) and female (p < 0.01; Figure 2K) fertility in CG1091c4-1/c4-6 mutant flies when compared to control flies (w1118). In females, fertility defects were not due to impaired fecundity, since mutant and control flies laid similar numbers of eggs (Figure 2K, top graph). We confirmed the observed phenotypes in trans-heterozygous flies, carrying one CRISPR allele (CG1091c4-6) over a publicly available piggy-bac transposon insertion in the coding sequence of tailor (CG1091f05717; Figure 2D) (Thibault et al., 2004), excluding any secondary perturbations linked to the use of CRISPR/Cas9.
Finally, expression of Myc-tagged CG1091 in S2 cells revealed predominant—if not exclusive—localization of CG1091 dispersed throughout the cytoplasm, without detectable signal in the nucleus (Figure 2L and Figures S2D and S2E).
In conclusion, the cytoplasmic, putative TNTase CG1091 is required for normal fertility in flies and is responsible for the majority of uridylation events associated with miRNAs in S2 cells and in flies, with a particular specificity for mirtron uridylation. Because it mediates the selective uridylation of a subset of miRNAs, we refer to CG1091 as Tailor.
Tailor Regulates miRNA Abundance and Function and Prevents the Accumulation of Mirtrons
Post-transcriptional uridylation can trigger RNA destabilization (Scott and Norbury, 2013). We therefore tested if Tailor depletion impacts miRNA abundance. In S2 cells depleted of Tailor, more than 40% of all miRNAs (32 out of 79 miRNAs) exhibited a statistically significant change in abundance (p < 0.05, FDR < 0.1) when compared to control RNAi conditions (Figure 3A). The majority of these (28 out of 32) increased in abundance, suggesting Tailor generally has a negative impact on miRNA accumulation. Mirtrons exhibited a significantly higher accumulation upon Tailor depletion compared to canonical miRNAs (p < 10−4; Figure 3B), suggesting that Tailor prevents mirton accumulation.
Figure 3.

Tailor-Dependent Uridylation Impacts Mature miRNA Levels and Prevents Mirtron Accumulation
(A) Greater than 40% of all miRNAs are significantly changed in abundance upon Tailor depletion in S2 cells (p < 0.05, Student’s t test; FDR < 0.1; Benjamini and Hochberg, 1995). Data represented as mean ± SD.
(B) Mirtrons significantly increase in abundance upon depletion of Tailor. p values (Mann-Whitney test) are indicated.
(C) Changes in miRNA abundances correlate with post-transcriptional modifications. The fraction at which miRNAs are tailed in control-dsRNA-treated S2 cells is indicated for miRNAs that do, or do not, change significantly in abundance upon depletion of Tailor. p value (Mann-Whitney test) is indicated.
(D) Changes in miRNA abundance correlate with Tailor-directed tailing. Changes in miRNA tailing between Tailor-depleted and control-dsRNA-treated S2 cells is shown for miRNAs that do, or do not, change significantly in abundance upon depletion of Tailor. p value (Mann-Whitney test) is indicated. See also Figure S3.
miRNAs that significantly changed in abundance upon depletion of Tailor were more frequently tailed under unperturbed conditions (p < 1 × 10−3; Figure 3C and Figure S3A), and tailing of these miRNAs was significantly more dependent on Tailor (p < 0.05; Figure 3D) when compared to miRNAs that remain unchanged in abundance. We concluded that Tailor-mediated uridylation regulates the abundance of miRNAs and prevents the accumulation of mirtrons.
Notably, four miRNAs decreased in abundance upon depletion of Tailor (Figure 3A), including miR-184. We confirmed this effect by northern hybridization, where miR-184 showed a significant, 1.5-fold decrease in abundance upon depletion of Tailor in S2 cells (Figures S3B and S3C). In selected cases Tailor therefore promoted the accumulation of miRNAs.
Finally, high-throughput mRNA sequencing revealed that the observed changes in miRNA levels upon depletion of Tailor significantly impact miRNA-mediated gene regulation (Figures S3D–S3I).
Taken together, Tailor regulates the abundance and function of mature miRNAs and prevents the accumulation of mirtrons.
Tailor-Directed Modification of Pre-miRNA 3′ Ends Regulates miRNA Maturation
To understand how Tailor regulates miRNA abundance we performed gain-of-function experiments by generating clonal S2 cells stably expressing Tailor (CG1091OE) and monitored miRNA modification and abundance by northern hybridization experiments. Among six tested miRNAs, five significantly changed in abundance upon Tailor expression when compared to control S2 cells (p < 0.05; Figures 4A and 4B): two increased (1.4-fold for both, miR-8-3p and bantam-3p), and three decreased in abundance (2.3-fold for miR-184-3p, 1.2-fold for miR-33-5p, and 3.3-fold for miR-252-5p). But only a subset of the deregulated miRNAs—the ones derived from the 3p arm of the respective pre-miRNA—showed tailing signatures, manifested as higher-molecular-weight bands in northern hybridization experiments (Figure 4A). This indicated that Tailor modifies pre-miRNAs. We tested this hypothesis by analyzing pre-miRNAs in northern hybridization experiments: upon depletion of Dcr-1 by RNAi (enriching pre-miRNAs to detectable amounts) we observed an accumulation of higher-molecular-weight isoforms of both pre-miR-184 and pre-bantam in CG1091OE cells compared to control S2 cells (Figure 4C). While both pre-miRNAs showed tailing signals, this had differential consequences on the accumulation of the respective mature miRNA species—an increase of bantam-3p and a decrease of miR-184-3p (Figure 4B). Accurate 2-nt 3′ overhangs are hallmarks for pre-miRNA processing by Dicer proteins: changes in pre-miRNA 3′ overhangs impact pre-miRNA processing by Dicer (Tsutsumi et al., 2011). To test if this may explain the observed effects, we cloned and Sanger sequenced pre-miR-184 and pre-bantam to characterize Tailor-directed changes in pre-miRNA 3′ ends (Newman et al., 2011). In control S2 cells, 87% of all pre-miR-184 reads mapped to the predicted 3′ end, producing a 2-nt- 3′ overhang, while the accuracy decreased in CG1091OE cells to 66%, with 14% containing elongated 3-nt 3′ overhangs (Figure 4D). To test if changes in 3′ end accuracy impact processing, we performed in vitro dicing assays (Tsutsumi et al., 2011): affinity-purified Dcr-1 processed pre-miR-184 with 3-nt 3′ overhang at significantly lower rates when compared to canonical 2-nt 3′ overhangs (p < 10−4; Figures 4E and S4A–S4C), explaining the observed decrease in miR-184-3p in CG1091OE cells.
Figure 4.

Tailor Regulates miRNA Abundance by Changing the Accuracy of Pre-miRNA 3′ Overhangs in Gain-of-Function Experiments
(A) Expression of Tailor in S2 cells (CG1091OE) changes the abundance and tailing status of mature miRNAs when compared to untreated S2 cells in northern hybridization experiments. MicroRNAs originating from the 3p or 5p arm of pre-miRNAs are indicated. 2S rRNA represents loading control.
(B) Quantification of three independent biological replicates of (A). Relative abundance of each miRNA in CG1091OE compared to untreated S2 cells is shown. p values (Student’s t test) are indicated. Data presented as mean ± SD.
(C) Tailor modifies pre-miRNAs. The indicated pre-miRNAs from CG1091OE or untreated S2 cells were detected by northern hybridization. Dcr-1 was depleted by RNAi to increase pre-miRNA signals.
(D and F) Tailor changes the accuracy of pre-miRNA 3′ ends. Sanger sequencing results of the indicated number of clones report the 3′ end of pre-miRNA-mapping reads. Genome-matching (GM, black) and prefix-matching (PM, red) reads are depicted. The fraction of reads mapping to the predicted 2-nt 3′ overhang is shown.
(E and G) Efficient Dcr-1-directed pre-miRNA processing requires accurate 2-nt 3′ overhangs. In vitro dicing assays employed SBP-tagged affinity-purified Dcr-1 and the indicated 5′ radiolabelled synthetic pre-miRNAs. Data presented as mean ± SD. p values (Student’s t test) are indicated. See Figure S4 for primary data. See also Figure S4.
For pre-bantam we observed the opposite: while only 25% of cloned pre-bantam mapped to the predicted 2-nt 3′ overhang in S2 cells (> 40% of all reads supported a blunt 3′ end structure), the accuracy increased in CG1091OE cells, where 53% of all reads mapped to the predicted 3′ end (Figure 4F). In vitro, Dcr-1-directed processing of pre-bantam was 5-fold more efficient in the presence of a 2-nt 3′ overhang when compared to a blunt end (p < 10−3; Figures 4G and S4D–S4F), explaining the increase in mature bantam-3p in CG1091OE cells (Figure 4B).
We concluded that Tailor-mediated uridylation of pre-miRNAs changes the accuracy of 3′ ends, which impacts the rates at which mature miRNAs are produced by Dcr-1, ultimately regulating the abundance of mature miRNAs.
Tailor-Dependent Uridylation Destabilizes Pre-miRNAs and Prevents Hairpin Dicing
The conclusion that Tailor uridylates pre-miRNAs, as proposed by gain-of-function experiments, warranted further experimental support under conditions where Tailor is expressed at physiological levels. To this end, we cloned 40- to 100-nt-long RNAs from S2 cells using an unbiased cloning approach. As expected, the majority of ∼78 Mio reads in these libraries mapped to abundant, non-coding RNA species that overlapped in size with pre-miRNAs, i.e., tRNA, snoRNA, and rRNA (Figure 5A). While only less than 0.1% of all reads mapped to pre-miRNAs, we recovered sufficient depth (> 70,000 reads) for an unbiased analysis of pre-miRNAs: using a cutoff of > 7 reads, we detected 89 pre-miRNAs, representing the precursors of 73 (50 miR and 23 miR∗) out of 79 (55 miR and 24 miR∗) abundantly expressed mature miRNAs. Among the remaining six, three mapped to duplicated miRNA loci and could not be uniquely assigned to one locus in small RNA sequencing datasets (miR-13-b1, miR-276b, and miR-2c); two derived from an unusual, 97-nt-long precursor (miR-998-5p and miR-998-3p), which may have been excluded by size selection; and one was present in small RNA libraries only at low levels (< 250 ppm in small RNA libraries; miR-263a). This analysis confirms that cloning of 40- to 100-nt RNAs at sufficient depth recovers the cellular pre-miRNA pool at near saturation. This conclusion was further supported by the fact that we recovered 38 pre-miRNAs (in average > 50 reads), which were not detected in mature small RNA libraries (Table S1).
Figure 5.

Pre-miRNAs, Particularly Mirtron Hairpins, Are Physiological Substrates for Tailor-Directed Destabilization through Tailing
(A) Mapping results of high-throughput sequencing of 40- to 100-nt RNAs from S2 cells.
(B) Abundance of genome-matching and prefix-matching pre-miRNAs in S2 cells. Data presented as mean of three independent biological replicates ± SD. Pre-miRNAs with highest fraction of prefix-matching reads (> 5%) are indicated in black.
(C) Mirtrons are significantly more tailed compared to canonical pre-miRNAs. The fraction at which pre-miRNAs are tailed is shown for all miRNAs and the indicated classes of pre-miRNAs. The number of pre-miRNAs in each group is indicated. p value (Mann-Whitney test) is indicated.
(D) Pre-miRNA tails consist mostly of uridine and adenine. The nucleotide composition of non-genome-matching additions was determined for each pre-miRNA and averaged across all pre-miRNAs.
(E) Depletion of Tailor by RNAi affects tailing of pre-miRNAs in S2 cells. The fraction at which each pre-miRNA is modified is plotted. miRNAs that show > 2-fold reduction in tailing are indicated in black.
(F) Depletion of Tailor by RNAi in S2 cells impacts pre-miRNA tailing. p values (Wilcoxon matched-pairs signed rank test) are indicated.
(G) Depletion of Tailor by RNAi impedes addition of uridine, but not any other nucleotides, to pre-miRNAs. Abundance and composition of nucleotide additions was determined for each pre-miRNA, normalized to control treatment, and averaged across all pre-miRNAs.
(H) Mirtron-hairpin uridylation requires Tailor. Change in tailing upon Tailor depletion in S2 cells by RNAi was determined for classified pre-miRNAs and normalized to control treatment. p value is indicated (Mann-Whitney test).
(I) Tailor-directed uridylation destabilizes pre-miRNAs. Upon Tailor depletion, the levels of pre-miRNAs that are tailed by Tailor under unperturbed conditions increase significantly. p values are indicated (Mann-Whitney test).
(J) Uridylation inhibits pre-miRNA dicing. In vitro dicing assays employed SBP-tagged affinity-purified Dcr-1 and the indicated 5′ radiolabelled synthetic pre-miRNA.
(K) Quantification of data shown in (J). Data presented as mean ± SD. p values are indicated (Student’s t test). See also Figure S5 and Table S1.
For further analysis we focused on the 58 pre-miRNAs that were sequenced at sufficient depth to reproducibly recover species with non-genome-matching 3′ additions (Figures 5B and S5A–S5D). Among these, mirtrons exhibited a significantly higher fraction of non-genome-matching additions compared to canonical pre-miRNAs (p < 10−4; Figure 5C). Like in mature small RNAs, uridylation (42%) and adenylation (35%) dominated post-transcriptional modifications in pre-miRNAs over cytosine (17%) and guanosine additions (5%) (Figure 5D). Uridylation was likewise enriched in heavily modified pre-miRNAs (Figure 5D) and dominated modifications associated with mirtrons (62%; Figure S5E). Finally, we detected a statistically significant correlation of pre-miRNA tailing and mature miRNA-3p (Pearson’s correlation coefficient r = 0.49; p < 3 × 10−3), but not -5p, modifications (r = 0.08, n.s.), indicating that a significant fraction of post-transcriptional modifications detected in mature 3p-miRNAs originated from pre-miRNA tailing (Figure S5F).
To test if Tailor mediates post-transcriptional uridylation of pre-miRNAs we sequenced 40- to 100-nt RNAs from S2 cells depleted of Tailor by RNAi. Tailor depletion caused a significant decrease in tailed pre-miRNAs when compared to control RNAi (p < 0.04; Figures 5E and 5F) or untreated S2 cells (p < 0.03; Figure 5F). Among the non-genome-matching additions to pre-miRNAs, solely uridylation decreased by more than 3-fold upon Tailor depletion when compared to S2 cells treated with control dsRNA (Figure 5G). In comparison to canonical pre-miRNAs, mirtrons were significantly more depleted in post-transcriptional modifications (p < 0.03; Figure 5H).
Finally, we tested the consequences of loss of uridylation on pre-miRNA abundance. We found a significant increase in pre-miRNA levels upon depletion of Tailor for the top 15 (p < 0.02), top 10 (p < 0.02), and top 5 (p < 0.009) Tailor-modified pre-miRNAs (Figure 5J). To determine the impact of uridylation on pre-miRNA processing, we determined in vitro dicing rates of pre-miR-1003, a mirtron that was highly modified in pre-miRNA and mature miRNA libraries (Figures 1B, 2F, and 5B) and was significantly upregulated in its mature form upon depletion of Tailor (Figure 3A): addition of a single uridine to pre-miR-1003 decreased dicing efficiency by more than 2-fold (p < 10−4), and di- or tri-uridylation reduced processing to almost undetectable levels (p < 10−8 and p < 10−11, respectively; Figures 5J and 5K). Furthermore, pre-miRNA tailing correlated significantly with the observed change in mature miRNA abundance upon depletion of Tailor (Pearson’s r = 0.51, p < 10−4; Figures S5G and S5H), with a correlation coefficient that was higher than what we observed for the correlation of mature miRNA tailing with change in mature miRNA abundance (Pearson’s r = 0.23, p < 0.05; Figure S3A). Together, these data establish pre-miRNAs—in particular mirtron-hairpins—as major targets for Tailor-dependent uridylation, a modification that destabilizes pre-miRNAs and prevents efficient hairpin dicing.
Tailor Is a Bona Fide, RNA-Specific TUTase with Unique Targeting Properties
For biochemical characterization we immunopurified FLAG-tagged Tailor upon expression in S2 cells and confirmed enzyme purity by western blotting (Figure S6B). When incubated with a 22-nt, 5′ radiolabelled RNA in the presence of ribonucleotide triphosphates (rNTPs), Tailor extended the length of the substrate RNA by up to ∼10 nucleotides within 5 min. This activity was abolished when we replaced one of three highly conserved aspartates in the conserved catalytic pol β superfamily motif with alanine (Figures S6A and S6C). In the presence of individual rNTPs, Tailor catalyzed the efficient incorporation of uridine and—to a lesser extent—cytidine and adenosine, but rarely guanosine (Figures 6A, S6D, and S6E). Tailor-directed terminal nucleotidyl transfer is consistent with the previously proposed two-divalent-cation-dependent catalytic mechanism of TNTases (Martin et al., 2008): the divalent cation-chelating agent EDTA inhibited, while addition of 5 mM excess Mg2+ rescued, tailing activity (Figure 6B). Together, these experiments classify Tailor as a bona fide RNA-specific TUTase.
Figure 6.

Tailor Is a Bona Fide TUTase with Unique Targeting Properties
(A) Immunopurified Tailor exhibits TNTase activity. FLAG-tagged Tailor was expressed in S2 cells, immunopurified, and incubated with a 22-nt, 5′ radiolabelled RNA in the presence of the indicated ribonucleotide triphosphate.
(B) Tailor-directed nucleotide transfer requires Mg2+. Tailing reactions were performed using a 5′ radiolabeled substrate RNA described in (A) in the presence of rNTPs. Addition of EDTA inhibited the tailing reaction, whereas excess Mg2+ rescued the activity.
(C–G) High-throughput biochemical characterization of Tailor-directed RNA tailing. A 37-nt RNA substrate containing four random nucleotides at the 3′ end was subjected to in vitro tailing reactions using immunopurified Tailor for 2 or 5 min, in the presence of rNTP, followed by 3′ adaptor ligation and high-throughput sequencing.
(C) Validation of high-throughput tailing assay. Product of in vitro tailing reactions was resolved by denaturing PAGE (left) or analyzed by high-throughput sequencing (right). Sequencing reveals high selectivity for UTP incorporation despite even concentration of rNTPs in the tailing reaction.
(D) Tailor-directed tailing efficiency is not influenced by substrate secondary structures. Substrates were binned according to secondary structure stability (effective free energy; EFE) and analyzed for the fraction at which each substrate was tailed, averaged across two time points (2 and 5 min). Median (white line), inner quartile range (IQR; dark gray area), and 1.5 IQR of lower and upper quartile (light gray area) are shown. No statistically significant difference was detected.
(E) Substrate secondary structures impact Tailor processivity. Representation as described in (D), but showing for each substrate the mean length of the tail added to each substrate. Stars indicate significantly different tail length of individual EFE groups (Mann-Whitney test).
(F) Tailor exhibits primary sequence specificity. The nucleotide identity of the four random 3′ nucleotides (irrespective of their position) in the indicated substrate RNAs is shown.
(G) Tailor exhibits specificity for the 3′-terminal nucleotide. Cumulative distributions of the relative fraction tailed for the indicated substrate RNAs is shown. See also Figure S6.
To dissect the enzymatic properties of Tailor in more detail, we developed a high-throughput biochemical assay for the characterization of RNA-specific TNTases. To this end we employed as a substrate a 37-nt RNA containing four random 3′ nucleotides, incubated it with immunopurified Tailor for 2 min or 5 min in the presence of equal concentrations of rNTPs, and subjected the product to high-throughput sequencing (Figure S6F). Analysis of the resulting libraries (∼7 Mio reads per library; Figure S6H) revealed that the tailing product exhibited a length distribution similar to the one observed in denaturing PAGE (Figure 6C). This confirmed a quantitative recapitulation of the tailing reaction by sequencing, and that Tailor catalyzes the incorporation of nucleotides to the 3′ end of an RNA substrate. Notably, Tailor exhibited nearly exclusive selectivity for uridine incorporation: less than 0.02% and 0.25% of all incorporated nucleotides consisted of a nucleotide other than uridine at 2 and 5 min, respectively. The ability of Tailor to efficiently catalyze the incorporation of cytosine or adenosine in the sole presence of CTP or ATP (see Figures S6D and S6E) is therefore apparently without relevance in the presence of UTP.
The randomized 3′ end of the substrate RNA enabled us to simultaneously analyze 256 different substrates, all of which were recovered, and their abundance stayed constant over the course of the assay (Figure S6G). We first determined the secondary structure stability of each substrate, binned them into effective free energy groups (EFE groups) from highly structured (group 1) to single-stranded (group 6), and tested each group for a significant over- or underrepresentation in the mean fraction at which it was tailed (Figure 6D) (note that the substrate was designed to avoid secondary structures not involving the random 3′ nucleotides; see Figures S6I and S6J). This showed that Tailor acted on RNA substrates, irrespective of whether the 3′ end is embedded in secondary structures. In contrast, the number of nucleotides that were added was strongly affected by secondary structures: structured RNAs (EFE groups 1–3) contained significantly shorter tails (p < 10−4; Mann-Whitney test), whereas single-stranded substrates were tailed significantly longer (EFE group 6; p < 10−4) (Figure 6E). We concluded that substrate secondary structures impact the processivity of Tailor-directed uridylation.
Finally, we analyzed the impact of primary sequence on tailing efficiency: we found that highly tailed species were enriched in guanosine (Figure 6F). Analysis of each individual position of the randomized four nucleotides revealed that only the 3′ terminal position significantly discriminates tailing efficiency (Figure 6G): while 3′ adenosine or 3′ cytidine substrates were significantly less frequently tailed (p < 10−4), a 3′ terminal uridine significantly enhanced the tailing reaction (p < 10−4), and 3′ guanosine-containing substrates were tailed most efficiently (p < 10−4) at both time points (Figures 6G and S6L). While additional 3′-terminal dinucleotide analysis indicated a trend toward more frequent tailing of substrates ending in AG-3′ (as well as UG-3′ and AU-3′), this further increase in tailing was not statistically significant when compared to the effect of the 3′-terminal nucleotide alone (Figures S6M and S6N).
In summary, high-throughput biochemical characterization of Tailor uncovered substrate structure-dependent processivity and primary sequence specificity, with a preference for RNAs ending in 3′ guanosine (and 3′ uridine). Our data revealed unique targeting properties of a previously uncharacterized RNA-specific TUTase.
Tailor Uridylates Pre-miRNAs Ending in 3′G
To assess primary sequence specificity in Tailor-directed modification of miRNAs, we analyzed the position preceding the non-genome-matching tail in (small) RNA libraries. In both S2 cells and in flies we detected an enrichment of 3′G in Tailor-modified miRNAs (Figure 7A): notably, this effect was most pronounced in pre-miRNAs, the major Tailor substrates.
Figure 7.

Tailor Targets Non-conserved Hairpins Ending in 3′G to Confine miRNA Production
(A) Tailor directs uridylation of miRNAs ending in 3′G. The nucleotide identity preceding non-genome-matching nucleotides of mature miRNAs (left panel) and pre-miRNAs (middle panel) in S2 cells and mature miRNAs in 0- to 5-day-old male whole flies (right panel) is shown. The number of analyzed (pre-) miRNAs is indicated.
(B) Efficient Tailor-directed tailing of the mirtron-hairpin miR-1003 requires a 3′G. 5′ radiolabeled pre-miR-1003 containing the indicated 3′-terminal nucleotide was incubated with immunopurified Tailor for the indicated time, followed by denaturing gel electrophoresis and phosphorimaging.
(C) Quantification of three independent replicates of the experiment shown in (B). Data presented as mean ± SD.
(D) Conserved pre-miRNAs, but not recently evolved hairpins, are depleted in 3′ guanosine. Nucleotide frequency at the 3′ terminal position of high-confidence Drosophila melanogaster pre-miRNAs annotated in mirBase as well as the overall nucleotide frequency in the fly genome is depicted.
(E) Relative nucleotide frequency of the data shown in (D). p values (binomial test) are indicated.
(F) Non-conserved pre-miRNAs are more efficiently tailed compared to conserved hairpins. Fraction tailed for pre-miRNAs in S2 cells is shown. p value is indicated (Mann-Whitney test).
(G) miRNAs that were detected by pre-miRNA—but not mature miRNA—cloning in S2 cells are enriched in non-conserved hairpins. Black bars represent conserved, and white bars non-conserved, pre-miRNAs. The number of hairpins in each dataset is indicated.
(H) Non-conserved miRNAs are significantly upregulated upon depletion of Tailor in S2 cells when compared to conserved miRNAs. p value is indicated (Mann-Whitney test). See also Figure S7.
Selective tailing of substrates ending in 3′G also explained the high degree of Tailor-dependent uridylation of mirtron hairpins, whose 3′ ends are defined by the splice acceptor consensus sequence, ending in 3′ΑG. We confirmed this by in vitro tailing assays using a synthetic precursor of the mirtron miR-1003. While 65% ± 2% of pre-miR-1003 with wild-type sequence (ending in 3′G) was tailed within 15 min, changing the terminal nucleotide to 3′U caused a significant decrease in tailing efficiency (35% ± 2% after 15 min; p < 1 × 10−4), and mutations to 3′A or C nearly abolished mirtron tailing (9.5% ± 1.8% and 9.3% ± 1.7% tailed, respectively, after 15 min; p < 1 × 10−5) (Figures 7B and 7C). Single-stranded miR-1003 exhibited a similar 3′ nucleotide preference, albeit with an overall reduced tailing efficiency: while Tailor modified 65% of wild-type pre-miR-1003, only 31% of mature single-stranded miR-1003 was modified within 15 min (Figures S7A and S7B). The intrinsic substrate preference of Tailor for pre-miRNA substrates ending in 3′G therefore explains why mirtron hairpins are preferentially uridylated in flies.
Conserved Pre-miRNAs, but Not Newly Emerging Hairpins, Are Depleted in 3′G
While more than 30 mirtrons have been identified in flies, only few produce abundant, mature miRNAs, perhaps because a mechanism exists that prevents the efficient conversion of such hairpins into small RNAs (Figure 5) (Chung et al., 2011; Westholm and Lai, 2011). We speculated that uridylation of hairpins, such as mirtrons, may represent a regulatory threshold for the accumulation of de novo-generated Dicer substrates. If this were true, one would expect an adaptation to regulatory uridylation in conserved miRNAs. To test this, we investigated the 3′ terminal genome-matching nucleotide of Drosophila pre-miRNAs, expecting that the hallmark of Tailor substrates—a 3′ terminal G—would be underrepresented in conserved pre-miRNAs. We investigated the 3′ end of 143 Drosophila melanogaster miRNAs annotated with high confidence in mirBase, for which sufficient experimental evidence exists to indubitably annotate the 3′ end of the underlying pre-miRNA (Kozomara and Griffiths-Jones, 2014). When compared to the nucleotide distribution in the Drosophila genome, we detected a statistically significant underrepresentation of guanosine at the 3′ end of pre-miRNAs (21% in the genome compared to 15% in pre-miRNA 3′ ends; p < 0.02; binomial test) (Figures 7D and 7E). This depletion was more pronounced in the 108 pre-miRNAs that are conserved among all sequenced Drosophila species, with only 12% of all pre-miRNAs ending in 3′G (p < 0.007). In contrast, among the 35 non-conserved pre-miRNAs, 26% contained a 3′ terminal guanosine, a frequency that was not significantly different from the occurrence of guanosine in the Drosophila genome. Therefore, our data support the hypothesis that evolutionary adaptation to pre-miRNA uridylation shapes the nucleotide composition of pre-miRNA 3′ ends and may serve as a barrier for the efficient de novo creation of hairpins that can be converted into mature miRNAs.
Tailor Targets Non-conserved miRNA Hairpins to Repress Their Maturation
To test if non-conserved miRNA hairpins are more susceptible to Tailor-directed suppression, we determined the extent of post-transcriptional modifications in pre-miRNAs with regard to their evolutionary conservation. We observed a statistically significant, higher fraction of post-transcriptional modifications in poorly conserved pre-miRNAs (i.e., found only in Drosophila melanogaster or Sophophorans) in comparison to miRNA hairpins that were conserved among all 12 sequenced Drosophila species (p < 10−4; Figure 7F) (Mohammed et al., 2014). Genome-matching reads of non-conserved hairpins tended to end in 3′G more frequently (42%) than conserved hairpins (18%; Figure S7C), consistent with a Tailor-specific targeting strategy. Notably, 14 out of 18 non-conserved hairpins were only detected as pre-miRNAs, but not in mature small RNA sequencing (Figures 7G and S7D), and the few non-conserved miRNAs that we detected in both pre-miRNA and mature miRNA cloning were significantly more abundant upon depletion of Tailor compared to conserved miRNAs (p < 0.03; Figure 7H).
Together our data support the hypothesis that Tailor targets non-conserved miRNA hairpins and prevents their efficient maturation.
Discussion
Post-transcriptional modification by TUTases lies at the core of mechanisms that regulate small RNA biogenesis and stability in plants, worms, and mammals (Ameres and Zamore, 2013). Our study reveals the first miRNA-modifying TUTase in flies—Tailor. Tailor is responsible for the majority of uridylation signatures observed in miRNAs of S2 cells and flies, predominantly acts on precursor hairpins, and is required for the normal accumulation of mature miRNAs. Uridylation generally has a dampening role for miRNA maturation, since Tailor depletion increased the abundance of > 35% of all miRNAs. Much of this effect may be explained by the fact that uridylation inhibits hairpin processing by Dcr-1 and targets pre-miRNAs for decay. Our observations are therefore consistent with a destabilizing role of uridylation in general RNA metabolism (Scott and Norbury, 2013) and resemble the Lin28-TUT4-Dis3L2 pathway in mammalian ES cells, where the 3′-to-5′ exoribonuclease Dis3L2 degrades pre-let-7 following TUT4-directed uridylation (Chang et al., 2013; Faehnle et al., 2014). Both Lin28 (CG17334) and Dis3L2 (CG16940) are conserved in flies and interact with Tailor based on high-throughput mapping of Drosophila protein interactions (Guruharsha et al., 2011), but Lin28 is not expressed in S2 cells, suggesting alternative targeting strategies for pre-miRNA uridylation in these cells.
Tailor-directed uridylation of pre-miRNAs is not always detrimental to miRNA biogenesis; in fact, our data support a model in which Tailor modulates the two-nucleotide 3′ overhangs in pre-miRNAs, which in turn regulates the efficiency at which Dicer converts these substrates into mature miRNAs. While the limited depth of our pre-miRNA sequencing datasets prevented a global validation of this model, it may explain why a few miRNAs require Tailor for their efficient accumulation. Such a proof-reading concept has been proposed to shape pre-miRNA biogenesis in mammals, where the TUTases ZCCHC11 and ZCCHC6 restore inaccurate pre-miRNA 3′ overhangs to facilitate substrate recognition by Dicer (Heo et al., 2012; Liu et al., 2014). How TUTases would be able to sense “defective” pre-miRNA end structures is unclear, but biochemical characterization of Tailor suggests that structured 3′ ends—found in blunt-ended pre-miRNAs—are tailed less processively. Addition of shorter tails (< 2 nt) to structured ends may contribute to pre-miRNA 3′ end restoration. Notably, the effect of Tailor on pre-miRNA maturation is dosage dependent, as exemplified by miR-184: normal accumulation of miR-184 requires physiological TUTase levels, since both depletion and overexpression of Tailor decreases miR-184 levels. In that respect, differential expression of Tailor may be involved in the fine-tuning of miRNA expression levels.
The most intriguing function of Tailor is its role in preventing the maturation of an entire class of miRNAs, namely mirtrons. Selective mirtron uridylation is a consequence of biochemical targeting features that to our knowledge are unique among TUTases. Both in vitro and when inspecting uridylated miRNA hairpins in S2 cells and in flies Tailor exhibited a strong preference for RNA substrates ending in 3′G. This is a hallmark of mirtron-hairpin sequences, whose 3′ ends are defined by the conserved splice acceptor sequence AG-3′. Note that a function of CG1091/Tailor in the regulation of mirtrons was also reported in a companion paper (Bortolamiol-Becet et al., 2015). Why does Tailor prevent mirtron maturation? Among the growing number of alternative pathways that give rise to bona fide small RNAs (Yang and Lai, 2011), the mirtron pathway generates the most numerous miRNAs (Westholm and Lai, 2011). Mirtrons are most prevalent in flies and worms, where intron length distributions match those of canonical pre-miRNAs (Lim and Burge, 2001; Yandell et al., 2006). Because of their length, introns may provide a rich source for the rapid de novo generation of miRNAs, a notion that is supported by the fact that mirtrons evolve more rapidly compared to canonical miRNAs in flies (Berezikov et al., 2010). But the fact that mirtrons are less likely to be maintained in the course of evolution may also suggest that the sudden appearance of large quantities of a novel miRNA may frequently be detrimental, rather than beneficial. In this regard, a regulatory threshold for the production of miRNAs from mirtron hairpins may be useful. Notably, mirtron regulation may be conserved in worms and mammals, where frequent uridylation of mirtrons was described (Westholm et al., 2012).
Consistent with a pronounced negative effect of Tailor-directed uridylation on general miRNA biogenesis, we find that miRNA hairpins tend to evade uridylation in Drosophila because their 3′ ends are significantly depleted in 3′G. This effect is even stronger in conserved pre-miRNAs, while non-conserved, evolutionary “young” hairpins are not depleted in 3′G. Therefore, our data support the hypothesis that evolutionary adaptation to pre-miRNA uridylation shapes the nucleotide composition of pre-miRNA 3′ ends. Hence, hairpin uridylation may serve as a barrier for the de novo creation of miRNAs in Drosophila.
Experimental Procedures
General Methods
Total RNA from flies and S2 cells was purified using the mirVana Kit (Ambion) or TRIzol (Invitrogen). High-resolution northern hybridization was as described (Han et al., 2011). Stable and clonal cultured S2 cell lines were generated as described (Ameres et al., 2010). A monoclonal antibody against Tailor was generated against full-length recombinant protein expressed in E. coli. For further information see Supplemental Experimental Procedures.
RNAi in S2 Cells
S2 cells (2 × 106) were soaked with 10 μg dsRNA for 1 hr in ExpressFive SFM medium (Invitrogen) supplemented with 200 mM glutamine (Invitrogen) in the absence of FBS, followed by the addition of medium containing 10% FBS. Soaking was repeated after 4 days, and cells were harvested and processed at day 7.
RNA Library Construction
Small RNA libraries were constructed as described (Ameres et al., 2010) with modifications detailed in Supplemental Experimental Procedures.
In Vitro Tailing Assay
FLAG-tagged Tailor was immunopurified upon expression in S2 cells. In vitro tailing assays employed 10-nM 5′ radiolabelled substrate RNA and FLAG-Tailor under RNAi assays conditions (Ameres et al., 2010), but omitting the ATP-regeneration system. rNTPs were added to a final concentration of 500 μM each. See Supplemental Experimental Procedures for details.
Data Analysis
Gel images were quantified using ImageQuant TL v7.0 (GE Healthcare). Curve fitting was performed according to the integrated rate law for a first-order reaction in Prism v6.0b (GraphPad).
Bioinformatics Analyses and Statistics
For pre-miRNA conservation analysis, high-confidence miRNAs were extracted from mirBase 21 (Kozomara and Griffiths-Jones, 2014) and curated as described in Supplemental Experimental Procedures. Nucleotide composition of the Drosophila genome dm3 was determined excluding chromosome MT, U, and Uextra.
Mapping of sequencing libraries to Drosophila genome dm3 was performed as described (Ameres et al., 2010). Annotations were derived from FlyBase r5.55_FB2014_01. For details see Supplemental Experimental Procedures.
Statistical tests were performed in Prism v6.0b (GraphPad) or Excel v14.4.3 (Microsoft). For multiple hypothesis testing a false-discovery rate (FDR) threshold was set to < 0.1 according to Benjamini and Hochberg (Benjamini and Hochberg, 1995).
Author Contributions
M.M.R.-P. and S.L.A. conceived and designed the experiments and wrote the manuscript. M.M.R.-P., V.I., R.A.M., I.S., V.A.H., S.F.-L., and S.L.A. performed and analyzed the experiments. T.R.B., J.-H.H., B.R., and S.L.A. performed bioinformatics analyses.
Acknowledgments
We thank Y. Tomari for Dcr-1 expression vector; R. Fukunaga for pre-bantam-transcription template plasmid; P. Duchek for CRISPR/Cas9 genome editing in flies; E.C. Lai for discussions on the evolutionary conservation of pre-miRNAs; Exelixis (Harvard Medical School) for fly stocks; and all members of the S.L.A. lab for input and discussions. Monoclonal antibodies were raised by the MFPL-mAB facility (S. Schuechner and E. Ogris). HTP sequencing was performed at the CSF NGS Unit (http://csf.ac.at/home/). This work was supported by a Boehringer Ingelheim Fonds PhD Fellowship to M.M.R.-P. and the Austrian Science Fund FWF (Y-733-B22 START and W1207-B09) and the European Research Council (ERC-StG-338252 miRLIFE) to S.L.A.
Published: July 2, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes seven figures, two tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2015.05.033.
Accession Numbers
The accession numbers for the high-throughput sequencing datasets reported in this paper are GEO: GSE66213 and GEO: GSE67646.
Supplemental Information
References
- Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- Ameres S.L., Horwich M.D., Hung J.H., Xu J., Ghildiyal M., Weng Z., Zamore P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- Berezikov E., Liu N., Flynt A.S., Hodges E., Rooks M., Hannon G.J., Lai E.C. Evolutionary flux of canonical microRNAs and mirtrons in Drosophila. Nat. Genet. 2010;42:6–9. doi: 10.1038/ng0110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E., Robine N., Samsonova A., Westholm J.O., Naqvi A., Hung J.H., Okamura K., Dai Q., Bortolamiol-Becet D., Martin R. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res. 2011;21:203–215. doi: 10.1101/gr.116657.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolamiol-Becet D., Hu F., Jee D., Wen J., Okamura K., Lin C.-J., Ameres S.L., Lai E.C. Selective suppression of the splicing-mediated microRNA pathway by the terminal uridyltransferase Tailor. Mol. Cell. 2015;59:217–228. doi: 10.1016/j.molcel.2015.05.034. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.-M., Triboulet R., Thornton J.E., Gregory R.I. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature. 2013;497:244–248. doi: 10.1038/nature12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L., Willingham A., Zhang D., Yang L., Zou Y., Eads B.D., Carlson J.W., Landolin J.M., Kapranov P., Dumais J. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 2011;21:301–314. doi: 10.1101/gr.112961.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W.J., Agius P., Westholm J.O., Chen M., Okamura K., Robine N., Leslie C.S., Lai E.C. Computational and experimental identification of mirtrons in Drosophila melanogaster and Caenorhabditis elegans. Genome Res. 2011;21:286–300. doi: 10.1101/gr.113050.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli A.M., Tops B.B., Plasterk R.H., Ketting R.F., Hannon G.J. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Faehnle C.R., Walleshauser J., Joshua-Tor L. Mechanism of Dis3l2 substrate recognition in the Lin28-let-7 pathway. Nature. 2014;514:252–256. doi: 10.1038/nature13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B.R., Brooks A.N., Carlson J.W., Duff M.O., Landolin J.M., Yang L., Artieri C.G., van Baren M.J., Boley N., Booth B.W. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory R.I., Yan K.P., Amuthan G., Chendrimada T., Doratotaj B., Cooch N., Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- Guruharsha K.G., Rual J.F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D.Y., Cenaj O. A protein complex network of Drosophila melanogaster. Cell. 2011;147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M., Kim V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- Hagan J.P., Piskounova E., Gregory R.I. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009;16:1021–1025. doi: 10.1038/nsmb.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.W., Hung J.H., Weng Z., Zamore P.D., Ameres S.L. The 3′-to-5′ exoribonuclease Nibbler shapes the 3′ ends of microRNAs bound to Drosophila Argonaute1. Curr. Biol. 2011;21:1878–1887. doi: 10.1016/j.cub.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I., Joo C., Kim Y.K., Ha M., Yoon M.J., Cho J., Yeom K.H., Han J., Kim V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Heo I., Ha M., Lim J., Yoon M.J., Park J.E., Kwon S.C., Chang H., Kim V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012;151:521–532. doi: 10.1016/j.cell.2012.09.022. [DOI] [PubMed] [Google Scholar]
- Huntzinger E., Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Kim V.N., Han J., Siomi M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Kim Y.K., Heo I., Kim V.N. Modifications of small RNAs and their associated proteins. Cell. 2010;143:703–709. doi: 10.1016/j.cell.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladewig E., Okamura K., Flynt A.S., Westholm J.O., Lai E.C. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012;22:1634–1645. doi: 10.1101/gr.133553.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Rådmark O., Kim S., Kim V.N. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y.S., Nakahara K., Pham J.W., Kim K., He Z., Sontheimer E.J., Carthew R.W. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- Lehrbach N.J., Armisen J., Lightfoot H.L., Murfitt K.J., Bugaut A., Balasubramanian S., Miska E.A. LIN-28 and the poly(U) polymerase PUP-2 regulate let-7 microRNA processing in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2009;16:1016–1020. doi: 10.1038/nsmb.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yang Z., Yu B., Liu J., Chen X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005;15:1501–1507. doi: 10.1016/j.cub.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.P., Burge C.B. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. USA. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zheng Q., Vrettos N., Maragkakis M., Alexiou P., Gregory B.D., Mourelatos Z. A MicroRNA precursor surveillance system in quality control of MicroRNA synthesis. Mol. Cell. 2014;55:868–879. doi: 10.1016/j.molcel.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G., Doublié S., Keller W. Determinants of substrate specificity in RNA-dependent nucleotidyl transferases. Biochim. Biophys. Acta. 2008;1779:206–216. doi: 10.1016/j.bbagrm.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J., Siepel A., Lai E.C. Diverse modes of evolutionary emergence and flux of conserved microRNA clusters. RNA. 2014;20:1850–1863. doi: 10.1261/rna.046805.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M.A., Mani V., Hammond S.M. Deep sequencing of microRNA precursors reveals extensive 3′ end modification. RNA. 2011;17:1795–1803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury C.J. Cytoplasmic RNA: a case of the tail wagging the dog. Nat. Rev. Mol. Cell Biol. 2013;14:643–653. doi: 10.1038/nrm3645. [DOI] [PubMed] [Google Scholar]
- Okamura K., Ishizuka A., Siomi H., Siomi M.C. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 2004;18:1655–1666. doi: 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K., Hagen J.W., Duan H., Tyler D.M., Lai E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G., Chen X., Yu B. Uridylation of miRNAs by hen1 suppressor1 in Arabidopsis. Curr. Biol. 2012;22:695–700. doi: 10.1016/j.cub.2012.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby J.G., Jan C.H., Bartel D.P. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D.D., Norbury C.J. RNA decay via 3′ uridylation. Biochim. Biophys. Acta. 2013;1829:654–665. doi: 10.1016/j.bbagrm.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Thibault S.T., Singer M.A., Miyazaki W.Y., Milash B., Dompe N.A., Singh C.M., Buchholz R., Demsky M., Fawcett R., Francis-Lang H.L. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat. Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- Tsutsumi A., Kawamata T., Izumi N., Seitz H., Tomari Y. Recognition of the pre-miRNA structure by Drosophila Dicer-1. Nat. Struct. Mol. Biol. 2011;18:1153–1158. doi: 10.1038/nsmb.2125. [DOI] [PubMed] [Google Scholar]
- Westholm J.O., Lai E.C. Mirtrons: microRNA biogenesis via splicing. Biochimie. 2011;93:1897–1904. doi: 10.1016/j.biochi.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westholm J.O., Ladewig E., Okamura K., Robine N., Lai E.C. Common and distinct patterns of terminal modifications to mirtrons and canonical microRNAs. RNA. 2012;18:177–192. doi: 10.1261/rna.030627.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Ameres S.L., Friedline R., Hung J.H., Zhang Y., Xie Q., Zhong L., Su Q., He R., Li M. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods. 2012;9:403–409. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M., Mungall C.J., Smith C., Prochnik S., Kaminker J., Hartzell G., Lewis S., Rubin G.M. Large-scale trends in the evolution of gene structures within 11 animal genomes. PLoS Comput. Biol. 2006;2:e15. doi: 10.1371/journal.pcbi.0020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.S., Lai E.C. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol. Cell. 2011;43:892–903. doi: 10.1016/j.molcel.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Yu Y., Zhai J., Ramachandran V., Dinh T.T., Meyers B.C., Mo B., Chen X. The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr. Biol. 2012;22:689–694. doi: 10.1016/j.cub.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.