

ABSTRACT
The mammalian target of rapamycin complex 1 (mTORC1) controls cell growth and anabolic metabolism and is a critical host factor activated by human cytomegalovirus (HCMV) for successful infection. The multifunctional HCMV protein pUL38 previously has been reported to activate mTORC1 by binding to and antagonizing tuberous sclerosis complex protein 2 (TSC2) (J. N. Moorman et al., Cell Host Microbe 3:253–262, 2008, http://dx.doi.org/10.1016/j.chom.2008.03.002). pUL38 also plays a role in blocking endoplasmic reticulum stress-induced cell death during HCMV infection. In this study, we showed that a mutant pUL38 lacking the N-terminal 24 amino acids (pHA-UL3825–331) was fully functional in suppressing cell death during infection. Interestingly, pHA-UL3825–331 lost the ability to interact with TSC2 but retained the ability to activate mTORC1, although to a lesser extent than full-length pHA-UL38. Recombinant virus expressing pHA-UL3825–331 replicated with ∼10-fold less efficiency than the wild-type virus at a low multiplicity of infection (MOI), but it grew similarly well at a high MOI, suggesting an MOI-dependent importance of pUL38-TSC2 interaction in supporting virus propagation. Site-directed mutational analysis identified a TQ motif at amino acid residues 23 and 24 as critical for pUL38 interaction with TSC2. Importantly, when expressed in isolation, the TQ/AA substitution mutant pHA-UL38 TQ/AA was capable of activating mTORC1 just like pHA-UL3825–331. We also created TSC2-null U373-MG cell lines by CRISPR genome editing and showed that pUL38 was capable of further increasing mTORC1 activity in TSC2-null cells. Therefore, this study identified the residues important for pUL38-TSC2 interaction and demonstrated that pUL38 can activate mTORC1 in both TSC2-dependent and -independent manners.
IMPORTANCE HCMV, like other viruses, depends exclusively on its host cell to propagate. Therefore, it has developed methods to protect against host stress responses and to usurp cellular processes to complete its life cycle. mTORC1 is believed to be important for virus replication, and HCMV maintains high mTORC1 activity despite the stressful cellular environment associated with infection. mTORC1 inhibitors suppressed HCMV replication in vitro and reduced the incidence of HCMV reactivation in transplant recipients. We demonstrated that mTORC1 was activated by HCMV protein pUL38 in both TSC2-dependent and TSC2-independent manners. The pUL38-independent mode of mTORC1 activation also has been reported. These novel findings suggest the evolution of sophisticated approaches whereby HCMV activates mTORC1, indicating its importance in the biology and pathogenesis of HCMV.
INTRODUCTION
Human cytomegalovirus (HCMV) is a member of the betaherpesvirus family with broad cell tropism. It is capable of escaping the immune surveillance and persists as a lifelong latent and recurrent infection in the host (1, 2). HCMV infection in adults and healthy people normally is asymptomatic or causes mild illness, but it can cause severe and life-threatening diseases in immunocompromised individuals, and importantly, HCMV congenital infection is a leading cause of birth defects (3).
Viruses consist of lipid-enveloped or unenveloped protein shells and encapsulated genomes but lack the metabolic enzymes or cellular machineries needed to complete their life cycle. Thus, successful virus replication relies exclusively on their ability to manipulate and exploit host cell processes and resources. The mammalian target of rapamycin complex 1 (mTORC1), which plays a central role in the regulation of protein translation and anabolic metabolism, is a major target of virus manipulation. Viruses have evolved diverse mechanisms to activate this important cellular pathway by targeting mTORC1 or its up- or downstream components (4, 5). For example, adenovirus E4 open reading frame 1 (6, 7) and EBV LMP2A (8) stimulate phosphoinositide 3-kinase (PI3K) signaling and subsequently activate mTORC1. The M-T5 protein of rabbit myxoma virus activates Akt (9), an mTORC1-positive regulator downstream of PI3K, while herpes simplex virus US3 mimics Akt to activate mTORC1 (10) and HCMV immediate-early proteins activate PI3K and Akt (11). Adenovirus E4 ORF4 (7) may activate mTORC1 directly via a mechanism dependent on phosphatase 2A binding. Some RNA viruses and the small DNA virus simian virus 40 (SV40), however, affect phosphorylation of the mTORC1 substrate 4E-BP1 (4, 5, 12), while human papillomavirus (HPV) protein E6 (13) and HCMV pUL38 (14) bind to and inhibit tuberous sclerosis complex protein 2 (TSC2) to activate mTORC1.
TSC2 is a major component of the tuberous sclerosis complex (TSC), consisting of TSC1 (hamartin), TSC2, and TBC1D7 (TBC1 domain family, member 7) (15–17). TSC is located downstream of Akt and functions as a GTPase-activating protein toward Rheb (Ras homolog enriched in brain) (15, 16, 18, 19). Enzymatically activated Rheb GTPase converts its bound GTP to GDP and downregulates mTORC1 activity. Therefore, TSC functions as a negative regulator of mTORC1. Many cellular stresses activate TSC, resulting in inhibition of mTORC1 activity. However, HCMV has been reported to maintain mTORC1 activation regardless of cell stress (20–22). Binding of the viral protein pUL38 to TSC2 and subsequent antagonism of TSC2 function represents one possible mechanism (14).
HCMV UL38 is located within the intron of the UL37 gene and is specific to betaherpesviruses (23). It has equivalents in human herpesvirus 6 and human herpesvirus 7, and UL38-homologous genes are carried by cytomegaloviruses of other species (24–27). The UL38 gene product pUL38 is a multifunctional protein that regulates viral and host gene expression and, in collaboration with other viral proteins pUL29/28 (28, 29), acts as an antiapoptotic molecule during HCMV infection (23, 30), interacts with TSC2, and activates mTORC1 (14). However, the mechanisms whereby pUL38 interacts with TSC2 to activate mTORC1 are unclear. We previously showed that the N-terminal 239 residues of pUL38 were necessary and sufficient to block premature cell death but were unable to activate mTORC1 in isolation, demonstrating that cell death inhibition by pUL38 was independent of its mTORC1 activation domain (31). In the present study, we further defined the TSC2-binding motif of pUL38 and showed that a TSC2-binding-deficient pUL38 mutant still was able to activate mTORC1. These new findings indicate that HCMV has evolved sophisticated mechanisms to maintain mTORC1 activation during infection, which may be essential for the successful completion of the virus life cycle.
MATERIALS AND METHODS
Plasmids and reagents.
LentiCRISPR (plasmid 49535; Addgene) is a lentiviral vector expressing cas9 and single guide RNA (sgRNA) (32). The following lentiviral overexpression vectors were derived from pLKO.DCMV.tetO (33). pLKO-HA-UL38, expressing the UL38 open reading frame, and pLKO-HA-UL3825–331, expressing a pUL38 segment from amino acid 25 to amino acid 331, both carried a hemagglutinin (HA) epitope at their amino termini. pLKO-Flag-UL38 and pLKO-Flag-UL3825–331 both carried a 3× Flag tag at their N termini. Each pair of amino acids between amino acids 13 and 24 of pUL38 were mutated to alanine by overlapping PCR and cloned into pLKO.DCMV.tetO vector, namely, pLKO-HA-UL38 SL/AA, pLKO-HA-UL38 LD/AA, pLKO-HA-UL38 E/A, pLKO-HA-UL38 EW/AA, pLKO-HA-UL38 RQ/AA, and pLKO-HA-UL38 TQ/AA. Primers for introducing these mutations are listed in Table 1. pLKO-Flag-GFP expressed the green fluorescent protein (GFP) gene with a 3× Flag tag at its N terminus. pLKO-TSC2 expressed the TSC2 consensus coding sequence (CDS), and pLKO-Flag-TSC2 expressed TSC2 with a 3× Flag tag at its N terminus.
TABLE 1.
Primers used to introduce mutations into the UL38 coding sequence
| Mutant type | Primer sequencea |
|---|---|
| SL/AA | 5′-CCACTCGGCCTCGTCCAGCGCGGCCATGATAGCGGCAGTGCTATG-3′ |
| 5′-GCCGCGCTGGACGAGGCCGAGTGG-3′ | |
| LD/AA | 5′-TCTGTCGCCACTCGGCCTCGGCCGCCAGGCTCATGATAGCGGCA-3′ |
| 5′-GCGGCCGAGGCCGAGTGGCGACAGA-3′ | |
| E/A | 5′-GCGTCTGTCGCCACTCGGCCGCGTCCAGCAGGCTCATGATAGC-3′ |
| 5′-GCGGCCGAGTGGCGACAGACGC-3′ | |
| EW/AA | 5′-CGTCCATCTGCGTCTGTCGCGCCGCGGCCTCGTCCAGCAGGC-3′ |
| 5′-GCGGCGCGACAGACGCAGATGGACG-3′ | |
| RQ/AA | 5′-CCCCCACGTCCATCTGCGTCGCTGCCCACTCGGCCTCGTCCA-3′ |
| 5′-GCAGCGACGCAGATGGACGTGGGGG-3′ | |
| TQ/AA | 5′-TCAGTCCCCCCACGTCCATCGCCGCCTGTCGCCACTCGGCCT-3′ |
| 5′-GCGGCGATGGACGTGGGGGGACTGA-3′ |
Underlined letters indicate nucleotide substitutions introduced into the UL38 coding sequence.
Primary antibodies used in the present study included anti-actin (AC15; Abcam); anti-immediate-early protein IE1/2 (a generous gift from Jay Nelson, Oregon Health & Science University); anti-IE1 (23), anti-pUL38 (23), anti-pp28 (34), and anti-pp71 (35) (gifts from Thomas Shenk, Princeton University); anti-ribosomal S6 kinase (S6K) (Proteintech); anti-cleaved poly(ADP-ribose) polymerase (PARP) (Cell Signaling); anti-phosphorylated S6K (Thr-389) (Cell Signaling); anti-HA and anti-Flag (Abmart); anti-TSC2 (1824-1; Abcam); and anti-TSC1 (4906; Cell Signaling).
Viruses and cell lines.
Primary human foreskin fibroblasts (HF), human glioblastoma-astrocytoma cell line U373-MG (kindly provided by James Alwine, University of Pennsylvania), and HEK293T cells were maintained at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS). HEK293T cells were transfected with the indicated plasmid together with the packaging plasmids 9.2ΔR and VSV-G to produce lentivirus stocks. HF cells or U373-MG cells were transduced with lentivirus supplemented with 5 μg/ml Polybrene and selected with 2 μg/ml puromycin (Sigma-Aldrich) to generate a pool of cells expressing the protein of interest.
Bacterial artificial chromosome (BAC)-HCMV clones were created and used to make recombinant HCMV. HA-UL38, HA-UL3812–331, HA-UL3825–331, and HA-UL3835–331 were amplified by PCR, and we inserted them into the BAC pAD-GFP to replace the full-length UL38 by linear recombination in the bacterial strain SW105 as previously reported (36). The primers used for linear recombination are available upon request. pADinHA-UL38, pADtrunHA-UL3812–331, pADtrunHA-UL3825–331, and pADtrunHA-UL3835–331 were used to reconstitute ADinHA-UL38, ADtrun HA-UL3812–331, ADtrun HA-UL3825–331, and ADtrunHA-UL3835–331 mutant viruses in which the N-terminal amino acid residues of pUL38 were deleted as indicated. pAD-GFP was used to produce wild-type virus (ADwt), and pADpmUL38, a recombinant BAC derived from pAD-GFP in which the expression of pUL38 was disrupted, was used to produce ADpmUL38 as previously reported (23, 30).
To reconstitute the virus, 2 μg of the BAC DNA and 1 μg of the pp71-overexpressing plasmid were transfected into HF cells as described previously (37). Twenty-four hours later, culture medium was replaced. The supernatant was collected when ∼100% of the cell monolayer was lysed.
Viral growth analysis.
HF cells or HF cells expressing a protein of interest were seeded in 12-well plates overnight, and cells were incubated with recombinant HCMV for 1 h at an MOI of 0.01 or 3. The inoculum was removed, the monolayer was washed with phosphate-buffered saline (PBS), and fresh medium was added. Medium from infected cells was harvested at the indicated times. The virus titer was determined by 50% tissue culture infectious dose (TCID50) assay in HF or HF-HA-UL38 cells.
Protein analysis.
Proteins were analyzed by immunoblotting as previously described (30). Briefly, cells were washed with phosphate-buffered saline (PBS) and lysed in the protein sample buffer supplemented with protease inhibitor cocktails (PIC; Roche) and phosphatase inhibitor cocktail IV (PHIC; BioVision, Inc.). Proteins were resolved by electrophoresis and transferred to a polyvinylidene difluoride membrane, and then they were hybridized with primary antibodies and horseradish peroxidase-conjugated secondary antibodies and visualized by using enhanced chemiluminescence (ECL) reagents (Bio-Rad).
Immunoprecipitation.
Immunoprecipitations were done either in HF cells infected with HCMV at an MOI of 3 at 72 hpi or in HEK293T cells transfected with plasmids of interest. Cell samples were lysed in lysis buffer, containing 120 mM NaCl, 40 mM HEPES, 1 mM EDTA, 0.3% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), PIC, and PhIC, for 1 h and centrifuged at 16,000 × g for 15 min at 4°C. The supernatants were incubated with Flag antibody-conjugated beads (Sigma-Aldrich) for 1 h at room temperature in rotation, washed with wash buffer (120 mM NaCl, 40 mM HEPES, 1 mM EDTA, 0.3% CHAPS), and eluted with 200 μg/ml flag peptide. The eluents were analyzed by immunoblotting.
Assays for mTORC1 activation.
Immunoblotting of phosphorylated S6K was used in this study as the readout for mTORC1 activation. To determine mTORC1 activation in infected cells, HF cells were infected with various HCMV at an MOI of 3 and collected at various hours postinfection for immunoblot analysis. To determine mTORC1 activation in expressing cells, HF cells or U373-MG cells overexpressing proteins of interest were serum starved for 72 h or 48 h, respectively. Cell lysates were collected for immunoblot analysis.
shRNA-mediated TSC2 knockdown in HF cells.
The control (shCtrl) short hairpin RNA (shRNA) and TSC2-targeting shRNA (shTSC2-1 or shTSC2-2) expressing lentiviral vectors were constructed based on the PLKO.1 vector. The shRNA targeting sequences are 5′-CAA CAA GAT GAA GAG CAC CAA-3′ (shCtrl), 5′-CAA TGA GTC ACA GTC CTT TGA-3′ (shTSC2-1), and 5′-GGA TTA CCC TTC CAA CGA AGA-3′ (shTSC2-2). Lentivirus preparation and transduction were the same as those described above for viruses and cell lines. Pools of HF cells stably expressing the respective shRNAs were used as control or TSC2 knockdown cells.
Generation of TSC2 knockout U373-MG cell lines.
Genome-engineering experiments using the CRISPR-Cas9 system were performed as described previously (38). Briefly, two sgRNAs targeting TSC2 were cloned into lentiCRISPR vector. Lentivirus was produced in HEK293T cells. U373-MG cells were transduced with lentivirus and selected with 2 μg/ml puromycin. TSC2 knockout cell clones were isolated by limited serial dilution. Finally, TSC2 knockout cell clones were confirmed by sequencing and immunoblot analysis. The sgRNA sequences targeting TSC2 were 5′-CAC CGT GGC CTC AAC AAT CGC ATC-3′ (site 1) and 5′-CAC CGA GCA CGC AGT GGA AGC ACT C-3′ (site 2) (32).
RESULTS
Construction and analysis of serial N-terminal-truncated mutants of HCMV pUL38.
We previously identified the domains or motifs responsible for the distinct activities of HCMV protein pUL38 by mutagenesis (31). Deletion of the first 56 residues was associated with rapid degradation of the protein, making it hard to dissect the structural and functional relationships of this region. Therefore, we generated mutant viruses expressing small, serial N-terminal deletions of pUL38, with the aim of obtaining stable mutant proteins with altered functions in the context of HCMV infection. Mutations were introduced into the virus genome using a bacterial artificial chromosome (BAC) recombineering technique (36). Recombinant virus ADinHA-UL38 carried the entire UL38 open reading frame (ORF) with an HA tag on its N terminus, ADtrunHA-UL3812–331 carried the UL38 ORF lacking the 11 N-terminal amino acids, ADtrunHA-UL3825–331 carried the UL38 ORF lacking the 24 N-terminal amino acids, and ADtrunHA-UL3835–331 carried the UL38 ORF lacking the 34 N-terminal amino acids. All of the truncated variants of pUL38 were tagged with HA at their N termini to facilitate protein detection. The parental HCMV-BAC clone pAD-GFP, derived from the HCMV AD169 strain (23), was used to produce wild-type virus (ADwt), which was used as a positive control. The integrity of the recombinant BAC clones was examined by digestion with EcoRI and BamHI (Fig. 1A and B). The digestion patterns were consistent with the predictions, indicating that the recombinant BAC clones carried the intact viral genome and the precise modifications at the correct loci.
FIG 1.

Construction and analysis of recombinant HCMV expressing serial N-terminal truncations of pUL38. EcoRI (A) and BamHI (B) restriction digestion analysis of recombinant BAC-HCMV clones of pADinHA-UL38 and its mutants. White dots indicate differences between the parental BAC clone pAD-GFP and pADinHA-UL38 or its mutants. Molecular size markers (in kilobases) are indicated. (C) Growth of wild-type virus (ADwt), ADinHA-UL38, and serial N-terminal-truncated mutants, including ADtrunHA-UL3812–331, ADtrunHA-UL3825–331, and ADtrunHA-UL3835–331, was examined in fibroblasts at a multiplicity of infection (MOI) of 0.01. Supernatants of infected cells were collected on different days postinfection as indicated and analyzed for virus progeny by TCID50 assay.
The recombinant viruses were reconstituted, and multistep viral growth assays were performed to detect the effects of the serial pUL38 N-terminal truncations on viral growth. ADinHA-UL38 and ADtrunHA-UL3812–331 replication was similar to that of the wild-type virus, ADwt (Fig. 1C). However, ADtrunHA-UL3825–331 replication was about 10-fold lower than that of the wild-type virus, and replication was almost absent from ADtrunHA-UL3835–331. The growth defect of ADtrunHA-UL3825–331 and the wild-type replication kinetics of ADtrunHA-UL3812–331 indicated the importance of amino acids 12 to 24. In addition, the dramatic growth defect of ADtrunHA-UL3835–331 suggested that this variant lost pUL38 function and that amino acids 25 to 34 were necessary for functional pUL38 and viral growth.
Given that pUL38 can inhibit premature cell death and antagonize TSC2 to facilitate HCMV replication, we determined if the truncated mutant proteins pUL3812–331, pUL3825–331, and pUL3835–331 retained the ability to block cell death during virus infection. Immunoblot analysis showed that pUL3812–331 was accumulated to a slightly higher level than HA-tagged pUL38 and pUL3825–331 protein was expressed at a level similar to that of HA-tagged pUL38, but pUL3835–331 was almost undetectable during infection (Fig. 2A). As expected, ADinHA-UL38 inhibited premature cell death, unlike ADpmUL38 (23, 30), a mutant virus that does not express pUL38 (Fig. 2B). The cell death morphology is indicated by more round floating cells and fewer cells adhering to the dishes. ADtrunHA-UL3812–331 and ADtrunHA-UL3825–331 also inhibited cell death, while ADtrunHA-UL3835–331 induced cell death late postinfection (Fig. 2B). Consistent with these results, PARP cleavage was inhibited in HF cells infected with ADinHA-UL38, ADtrunHA-UL3812–331, and ADtrunHA-UL3825–331 but induced in cells infected with ADtrunHA-UL3835–331 and ADpmUL38 (Fig. 2A). Thus, the phenotype of ADtrunHA-UL3835–331 virus mimics that of ADpmUL38, probably as a result of the loss of pUL38 protein expression. Therefore, in the rest of the manuscript, we focused on pUL3825–331, which was sufficiently expressed and blocked cell death (Fig. 2) but resulted in a 10-fold reduction in virus growth (Fig. 1C).
FIG 2.
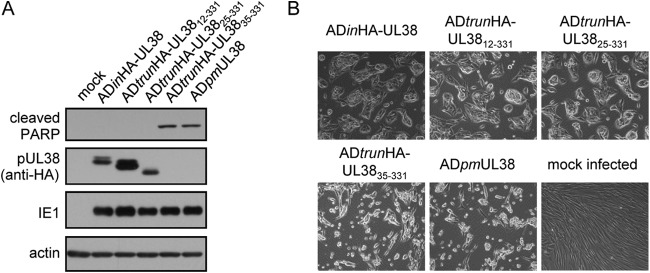
Ability of various UL38-truncated mutants to inhibit cell death. (A) Normal HF cells were infected with ADinHA-UL38, ADtrunHA-UL3812–331, ADtrunHA-UL3825–331, ADtrunHA-UL3835–331, and ADpmUL38 viruses at a multiplicity of infection (MOI) of 3. Cell samples were collected to examine the cleavage of PARP at 72 h postinfection (hpi). (B) HF cells were infected with different viruses as indicated at an MOI of 3 as described for panel A. Cells were analyzed for morphological changes by phase-contrast microscopy at 72 h postinfection. ADtrunHA-UL3835–331 and ADpmUL38 infection leaded to more rounded and floating cells and fewer spindle-shaped cells adhering to the dishes compared to the others, which indicates the cell death morphology.
pUL3825–331 induces S6K phosphorylation without binding to TSC2 during HCMV infection.
We previously showed that pUL38 suppressed cell death independently of its ability to induce mTORC1 activation (31). ADtrunHA-UL3825–331 virus retained the ability to block cell death, but virus growth was reduced by about 10-fold. Therefore, we determined if the N-terminal-truncated mutants of pUL38 retained the ability to activate mTORC1. We used S6K phosphorylation (phos-S6K) as an indicator of mTORC1 activation, as described previously (31). The ADpmUL38 mutant virus induced apparent S6K phosphorylation, consistent with our previous report that HCMV could activate mTORC1 in a pUL38-independent manner (31). However, ADinHA-UL38 and ADtrunHA-UL3812–331 viruses induced S6K phosphorylation to much greater levels, indicating that pUL38 was required for full mTORC1 activation and that pUL3812–331 was fully capable of activating mTORC1 (Fig. 3A). ADtrunHA-UL3825–331 virus induced S6K phosphorylation at levels greater than those by ADpmUL38 but lower than those by ADinHA-UL38, suggesting only partial pUL38-dependent activation of mTORC1 (Fig. 3A). pUL38 was reported to induce mTORC1 activation through binding to TSC2 (14). Therefore, we examined the interactions of pUL38 and its mutants with TSC2 during virus infection (Fig. 3B). As expected, HA-tagged full-length pUL38 (HA-UL38) and pUL3812–331 pulled down TSC2 effectively. However, pUL3825–331 failed to pull down TSC2 in the assay, despite being able to induce S6K phosphorylation.
FIG 3.

ADtrunHA-UL3825–331 was able to induce S6K phosphorylation without binding to TSC2 during HCMV infection. (A) HF cells were mock infected or were infected with ADinHA-UL38, ADtrunHA-UL3812–331, ADtrunHA-UL3825–331, or ADpmUL38 virus at a multiplicity of infection of 3. mTORC1 activity was examined by detecting the level of phos-S6K at 24 h postinfection (hpi). The numbers indicate the phosphorylation rate of S6K. The ratio of phos-S6K to S6K in mock-infected cells was set as 1. (B) HF cells infected with the indicated viruses were collected at 72 hpi. Cell lysates then were subjected to immunoprecipitation with protein A beads incubated with HA antibody. (C) HF cells that were mock infected or infected with ADinHA-UL38 or ADtrunHA-UL3825–331 were tested for mTORC1 activity at different times postinfection. (D) Expression levels of viral immediate-early, early, and late proteins were detected in HF cells that were mock infected or were infected with ADinHA-UL38 or ADtrunHA-UL3825–331 at different times postinfection, as indicated. (E) Growth of ADinHA-UL38 and the N-terminal-truncated mutant virus ADtrunHA-UL3825–331 was examined in HFs at a multiplicity of infection (MOI) of 3 or 0.01. Supernatants of infected cells were collected on different days postinfection as indicated and analyzed for virus production by TCID50 assay.
We characterized the ADtrunHA-UL3825–331 mutant virus further by comparing mTORC1 activation and viral protein expression of HA-UL38 and HA-UL3825–331 at multiple time points. Both ADinHA-UL38 virus and ADtrunHA-UL3825–331 mutant virus induced S6K phosphorylation, but ADtrunHA-UL3825–331 mutant virus caused less S6K phosphorylation at all time points than ADinHA-UL38 virus infection (Fig. 3C). Therefore, reduced mTORC1 activation by the ADtrunHA-UL3825–331 mutant virus was not time dependent, suggesting that during infection pUL38 was able to activate mTORC1 independently of TSC2 binding but that maximal mTORC1 activation required its interaction with TSC2. Viral immediate-early protein (IE1/2) and early protein (pUL38) expression levels were similar during infection with mutant ADtrunHA-UL3825–331 and ADinHA-UL38 viruses (Fig. 3D). The expression levels of late proteins (pp28 and pp71) were slightly reduced during ADtrunHA-UL3825–331 mutant virus infection compared with those for ADinHA-UL38 infection (Fig. 3D). We further compared the virus growth of ADtrunHA-UL3825–331 and ADinHA-UL38 at high or low multiplicity of infection (MOI). As shown in Fig. 3E, ADtrunHA-UL3825–331 grew almost as well as the wild-type virus expressing the full-length pHA-UL38 at an MOI of 3 but produced ∼10-fold less progeny virus at an MOI of 0.01. The data indicate that the N-terminal 24 amino acids of pUL38, its interaction with TSC2, and the full activation of mTORC1 are required for optimal virus growth in an MOI-dependent manner.
pUL3825–331 activates mTORC1 independently of TSC2 binding when expressed in isolation.
The previous study demonstrates that pUL38 is sufficient to mediate TSC2-dependent activation of mTORC1 in isolation (14). We were interested in determining whether pUL3825–331 also could mediate TSC2-independent activation of mTORC1 in the absence of any other viral proteins. To test this, we created HF cells stably expressing empty vector, pHA-UL38, or pHA-UL3825–331. These cells were serum starved for 3 days and then collected to test for mTORC1 activity by detecting the levels of phos-S6K. Immunoblot analysis showed that pHA-UL38 was capable of inducing high levels of phos-S6K, indicating high mTORC1 activation, consistent with previous results (Fig. 4A) (31). Importantly, pHA-UL3825–331 also was able to induce relatively high levels of phos-S6K compared to those of the empty vector control, although they were lower than those for pHA-UL38, suggesting that the mutant protein retained the ability to activate mTORC1. Furthermore, when coexpressed, full-length pUL38 and TSC2 could pull down each other while pUL3825–331 (either HA or Flag-tagged) failed to do so (Fig. 4B and C). pUL3825–331 did not pull down TSC1, a protein complexed with TSC2 to negatively regulate mTORC1, either (Fig. 4B). These results suggest that amino acids 1 to 24 are essential for the interaction between pUL38 and TSC2, and that pUL38, lacking these amino acids, still was able to activate mTORC1 outside the context of HCMV infection.
FIG 4.

pUL3825–331 activates mTORC1 when expressed alone. (A) HF cells expressing empty vector (vector), pHA-UL38 (HA-UL38), or pHA-UL3825–331 (UL3825–331) were generated by lentivirus transduction. Different HF cells were serum starved for 72 h before collecting the cell lysates. mTORC1 activity was examined by detecting the levels of S6K phosphorylation. (B) The interaction between pUL38, pUL3825–331, and TSC2 or TSC1 was investigated by cotransfecting 293T cells with TSC2 and Flag-GFP-, pFlag-UL38-, or pFlag-UL3825–331-expressing plasmid, and cell samples were collected 48 h later for immunoprecipitation with anti-Flag beads. WCL, whole-cell lysates. (C) The inability of pHA-UL3825–331 binding to TSC2 was confirmed by reciprocal coimmunoprecipitation. 293T cells were cotransfected with Flag-GFP and pHA-UL38-, Flag-TSC2 and pHA-UL38-, or Flag-TSC2 and pHA-UL3825–331-expressing plasmid, and cell samples were collected 48 h later and subjected to immunoprecipitation with anti-Flag beads.
A TQ motif of pUL38 is required for pUL38-TSC2 interaction.
As pHA-UL3812–331 interacts with TSC2 but pHA-UL3825–331 failed to do so (Fig. 3B), we hypothesized that the TSC2-binding motif was located among residues 13 to 24 of pUL38. Therefore, we mapped the key amino acids required for TSC2 binding by constructing six recombinant clones of pUL38, sequentially replacing two neighboring amino acids at a time to alanines among residues 13 to 24. The mutants were SL/AA, LD/AA, E/A, EW/AA, RQ/AA, and TQ/AA, all of which were tagged with HA at their N termini to facilitate protein detection (Fig. 5A). Coimmunoprecipitation analysis showed that the mutants SL/AA, LD/AA, EW/AA, and RQ/AA still were able to combine with TSC2 (Fig. 5B). However, the E/A mutant demonstrated reduced binding to TSC2, while the TQ/AA mutant completely lost the ability to interact with TSC2 (Fig. 5B). These data indicate that the E residue is important and the TQ motif is necessary for pUL38 binding to TSC2. We tested if the TQ mutant that failed to bind to TSC2 was able to activate mTORC1. As shown in Fig. 5C, the levels of phos-S6K were increased after expressing pHA-UL38 TQ/AA and pHA-UL3825–331 in HFs, although phosphorylation was less than that in cells expressing full-length HA-UL38 (Fig. 5C). As expected, when cells were treated with rapamycin, an mTORC1-specific allosteric inhibitor, the phos-S6K induced by pUL38 isoforms was completely blocked (Fig. 5D). These data demonstrated that the TQ motif was required for pUL38 to interact with TSC2, and that mTORC1 could be activated in a TSC2-binding-independent manner by pUL38 in isolation.
FIG 5.

TQ motif of pUL38 is a binding site for TSC2. (A) Schematic of pUL38 with different mutations. The binding site of pUL38 for TSC2 was mapped by mutating two neighboring amino acids between residues 13 and 24 at a time. The sequences between 13 and 24 of wild-type pUL38 and its mutants are indicated in the diagram. Mutant amino acids are in gray. (B) 293T cells were cotransfected with Flag-TSC2 and different pUL38 mutants, including SL/AA, LD/AA, E/A, EW/AA, RQ/AA, and TQ/AA. Cotransfection of pHA-UL38 and Flag-TSC2 or Flag-GFP were used as positive and negative controls, respectively. Cell lysates were subjected to immunoprecipitation with anti-Flag beads 48 h after transfection. (C) TQ/AA mutant was able to activate mTORC1. HF cells stably expressing empty vector (vector), pHA-UL38, pHA-UL3825–331 (UL3825–331), and pHA-UL38 TQ/AA (TQ/AA) were created by lentivirus transduction. The cells then were serum starved for 72 h and collected to detect mTORC1 activity. (D) Rapamycin sensitivity of S6K phosphorylation induced by pUL38 variants. HF cells stably expressing empty vector (vector), pHA-UL38, pHA-UL3825–331 (UL3825–331), and pHA-UL38 TQ/AA (TQ/AA) were serum starved for 72 h with or without rapamycin. Cell lysates were collected to detect mTORC1 activity. Long, long exposure; short, short exposure.
The site-specific mutants mimic the function of pHA-UL3825–331.
In order to study the role of site-specific mutants (TQ/AA and E/A) in the context of virus infection, we made HF cells stably expressing these mutant proteins, infected them with pUL38-deficient virus ADpmUL38, and then tested virus growth, cell death, and mTORC1 activity. The full-length pUL38-expressing cells (HF-HA-UL38) known to complement ADpmUL38 were used as the positive control, and the cells expressing an empty vector (HF vector) were used as the negative control. As expected, pUL38-deficient virus ADpmUL38 was greatly growth attenuated in control cells (HF vector) but grew efficiently in cells expressing wild-type pUL38 (HF-HA-UL38) (Fig. 6A and B). At a low MOI of 0.01, the growth of pUL38-deficient virus was partially rescued in cells expressing pUL38 mutants (HF-UL3825–331, HF-TQ/AA, and HF-E/A) with an ∼10-fold lower efficiency than that in HF-HA-UL38 cells (Fig. 6A). High-MOI growth curve analysis revealed the virus replicated equally well in all of the pUL38 mutant-expressing cells (Fig. 6B), suggesting that the TSC2 binding of pUL38 and the full activation of mTORC1 are not required for maximum HCMV replication on HFs on a single-step growth curve. The site-specific mutants inhibited cell death (Fig. 6C) and activated mTORC1 (Fig. 6D) similarly to pHA-UL3825–331. Taking these findings together, we showed that the site-specific mutants unable to interact with TSC2 mimic the function of pHA-UL3825–331.
FIG 6.

Site-specific mutants of pUL38 mimic the function of pHA-UL3825–331. (A and B) HF cells expressing empty vector (vector), pHA-UL38, pHA-UL3825–331 (UL3825–331), pHA-UL38 TQ/AA (TQ/AA), and pHA-UL38 E/A (E/A) were created. These HF cells then were infected with ADpmUL38 at a multiplicity of infection (MOI) of 0.01 (A) or 3 (B), respectively. Supernatants of infected cells were collected on different days postinfection as indicated, and virus titers were determined by TCID50 assay. (C and D) These different HF cells were infected with ADpmUL38 at a multiplicity of infection (MOI) of 3. Cell lysates then were collected to examine the cleavage of PARP at 72 h postinfection (hpi) or to examine mTORC1 activity by detecting the level of phos-S6K at 24 hpi.
pUL38 further increases mTORC1 activity in TSC2 knockout cells.
To provide additional evidence that pUL38 was able to activate mTORC1 independently of TSC2 interaction, we tested the ability of pUL38 to increase mTORC1 activity in the absence of TSC2. The CRISPR/Cas9 system (38) was used to knock out TSC2 in U373-MG human glioblastoma cells. To minimize off-target effects, we chose two sgRNAs (Fig. 7A) targeting TSC2 at two different gene locations, which were predicted to have minimal off-target activities by an optimized algorithm (32). Cas9- and TSC2-specific sgRNAs were introduced into U373-MG cells by lentiviral vector transduction, and TSC2-null cell lines were cloned by limited serial dilution, as described in Materials and Methods. Two clones, 1-17 and 2-7, derived from each sgRNA, were validated by sequencing the genome-targeting site (data not shown) and by immunoblotting (Fig. 7B). As shown in Fig. 7B, TSC2 protein expression was completely disrupted in both clones. The level of phos-S6K was dramatically upregulated in both clones compared to levels in cells expressing Cas9 only, consistent with a negative regulatory role of TSC2 on mTORC1. When pUL38 was expressed in TSC2-null cells, phos-S6K was further increased compared to levels in cells expressing empty vector (Fig. 7B and C). pUL3825–331 and pUL38-TQ/AA behaved similarly to pUL38 in TSC2-null cells to further increase mTORC1 (Fig. 7C). To test if this also is true in HFs, we used RNA interference (RNAi) to knock down TSC2. CRISPR-mediated gene knockout requires single-cell cloning, which is not applicable to primary human fibroblasts. As shown in Fig. 7D, shTSC2-2 potently suppressed TSC2 expression, and pUL38 and its variants further increased S6K phosphorylation in the TSC2 knockdown HFs. The results indicate that pUL38 can activate mTORC1 through pathways other than antagonizing TSC2. Overall, we have identified residues in pUL38 critical for its binding to TSC2 and demonstrated that pUL38 was able to activate mTORC1 independently of TSC2 binding.
FIG 7.

pUL38 activates mTORC1 in TSC2-knockout cells. (A) Locations of two chosen sgRNAs targeting the TSC2 genome. The two sites are enlarged and the targeting sequences are indicated with boldface italics. (B) Control and TSC2-null U373-MG cell lines (clones 1-17 and 2-7) were transduced with empty vector (vector) and pHA-UL38-expressing lentivirus, respectively. Cells were serum starved for 48 h and collected to measure mTORC1 activity. Long, long exposure; short, short exposure. (C) The ability of pHA-UL3825–331 (UL3825–331) and pHA-UL38 TQ/AA (TQ/AA) to stimulate mTORC1 activity in TSC2-null U373-MG cells (clone 2-7) was tested by using the same experiment procedures as those for panel B. (D) HFs were transduced with control (shCtrl) or TSC2-specific shRNA (shTSC2-1 and shTSC2-2)-expressing lentiviruses. They then were transduced with empty (vector) or pHA-UL38 (HA-UL38)-, pHA-UL3825–331 (UL3825–331)-, or pHA-UL38 TQ/AA (TQ/AA) expressing vector. The cells were serum starved for 72 h. Cell lysates were collected to detect mTORC1 activity.
DISCUSSION
The importance of mTORC1 activation in the life cycle of HCMV has been demonstrated by in vitro studies (14, 20, 21, 39–43). The therapeutic immune suppression in transplant recipients with mTORC1 inhibitors has significantly reduced the incidence of HCMV reactivation (44, 45), implying an antiviral role of targeting mTORC1 in clinical settings. Moorman et al. discovered that the HCMV protein pUL38 interacts with TSC2 and activates mTORC1 by antagonizing TSC function (14). We previously analyzed the domains and motifs important for pUL38 activities and identified the C-terminal residues from 240 to 277 as critical for pUL38 activation of mTORC1 in isolation (31). However, the truncated mutant pUL381–239, which failed to activate mTORC1 in isolation, still retained the ability to interact with TSC2 to activate mTORC1 in the context of virus infection (31). In this study, we identified residues 12 to 24, and particularly a TQ motif at amino acids 23 and 24, as essential for mediating the interaction between pUL38 and TSC2. Surprisingly, however, the mutant pUL38 that was unable to interact with TSC2 still activated mTORC1, both during virus infection and in isolation. In addition, under a background of TSC2 depletion by CRISPR genome editing, pUL38 increased mTORC1 activity in human U373-MG cells, demonstrating that pUL38 was able to activate mTORC1 by a TSC2-indpendent mechanism. These results and those of previous studies reveal a sophisticated method of mTORC1 upregulation by HCMV at multiple levels employing several different mechanisms, indicating a crucial role for mTORC1 in HCMV infection.
There are several possible explanations for the importance of mTORC1 activation in HCMV replication. First, HCMV is the largest human virus, with a double-stranded DNA genome of over 240 kb, which forms a large virion particle consisting of over 70 virus-encoded structural proteins (46). This suggests that HCMV needs to maintain highly active mTORC1 to support the anabolic metabolism and protein translation required to produce progeny viruses. Second, the high energy and nutrient demands of HCMV replication inevitably result in stresses that could suppress mTORC1 and impair progeny virus production if HCMV is unable to counteract them. Finally, HCMV has a broad cell tropism in vivo and demonstrates at least two forms of infection, namely, lytic and latent infections. Therefore, it may mobilize different mechanisms to fine tune mTORC1 activity for infection of various cell types and forms.
mTORC1 can be activated by at least two parallel pathways, an amino acid-sensitive pathway and a growth factor-stimulated PI3K-Akt signaling pathway. Recent studies have investigated the amino acid-mediated signaling pathway leading to mTORC1 activation. Amino acids accumulated inside the lysosome are sensed by the vacuolar H+ ATPase, which activates the guanine nucleotide exchange factor Ragulator (47, 48). The GTP-bound Rag complex recruits mTORC1 to the lysosomal surface (49, 50), where it meets another small GTPase, Rheb. In its GTP-bound form, Rheb eventually activates mTORC1 (15, 16, 19). Rheb itself is regulated by the parallel PI3K-Akt signaling pathway. TSC2 is a component of the TSC, which is a substrate of Akt in this pathway, and TSC functions as a negative regulator of Rheb and, as a result, of mTORC1 (15, 16, 19).
From the pUL38 aspect, HCMV uses three mechanisms to activate mTORC1: (i) a pUL38-independent mechanism, demonstrated by the fact that mutant virus deficient in pUL38 had increased mTORC1 activity compared to that of mock infection (31), although to a lesser extent than wild-type virus; (ii) a pUL38- and TSC2-dependent mechanism (14); and (iii) a pUL38-dependent but TSC2-independent mechanism. Clippinger et al. discovered that HCMV stimulated the perinuclear localization of mTORC1, and its activation by a dynein-dependent but Rag GTPase-independent mechanism (22, 51), as early as 8 h postinfection (51). This suggests that it represents pUL38-independent activation of mTORC1, given that pUL38 is defined as a viral early protein localized in the nucleus at early times postinfection (23) and is unlikely to be involved in very early events in the cytoplasm. In terms of the TSC2-dependent mechanism, it remains to be elucidated precisely how pUL38 inactivates TSC2. HPV E6 interacts with TSC2 and mediates its protein degradation, suppressing its function and in turn activating mTORC1 (13, 52). pUL38 does not lead to TSC2 degradation or compete with TSC1 binding (14); thus, it likely antagonizes TSC2 activity by affecting its posttranslational modification, which has been reported to be involved in regulating TSC2 function (14).
In this study, we identified a pUL38-dependent but TSC2-independent mechanism of mTORC1 activation. The amino acid-mediated signaling pathway leading to mTORC1 activation was thought to be parallel to the growth factor-induced TSC2-dependent pathway. However, it is unlikely that this represents the mechanism used by pUL38, because this pathway is altered during HCMV infection, and mTORC1 relocation during HCMV infection associated with the amino acid-mediated pathway is a very early event (51); therefore, pUL38 would be unable to be involved for the reasons given in the previous paragraph. Posttranslational modifications of mTORC1 pathway components downstream of TSC2 by various protein kinases, such as p38 and PARK (53), DAPK2 (54), IKKα (55), and TAK1 (56), have been revealed, and these events regulate mTORC1 activity. Further studies are needed to determine if these or other novel mechanisms are involved in pUL38-dependent but TSC2-independent mTORC1 activation.
ACKNOWLEDGMENTS
We thank all members of the Herpesvirus and Molecular Virology Research Unit for helpful discussions, Thomas Shenk for antibodies, Jay Nelson for IE2 antibody, and James Alwine for the U373-MG cell line.
This study was supported by Public Health Service grant RO1CA120768 to D.Y., the National Natural Science Foundation of China (grants 81271836 and 81371826 to Z.Q. and grant 31300148 to B.X.), the CAS Cross-disciplinary Collaborative Teams Program for Science, the Chinese Academy of Sciences “100 Talents” program, and the Knowledge Innovation Program of the Chinese Academy of Sciences and the Science and Technology Commission of Shanghai Municipality (grant 13ZR1445500 to B.X.). D.Y. held an Investigators in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund. B.X. was supported by the Youth Innovation Promotion Association, CAS.
The funders had no role in study design, data collection and analysis, or submission of the manuscript.
REFERENCES
- 1.Sinclair J. 2008. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J Clin Virol 41:180–185. doi: 10.1016/j.jcv.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Sinclair J, Sissons P. 2006. Latency and reactivation of human cytomegalovirus. J Gen Virol 87:1763–1779. doi: 10.1099/vir.0.81891-0. [DOI] [PubMed] [Google Scholar]
- 3.Mocarski ES Jr, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K–Akt–mTOR signalling pathway. Nat Rev Microbiol 6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh D, Mathews MB, Mohr I. 2013. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol 5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metcalfe C, Ibrahim AE, Graeb M, de la Roche M, Schwarz-Romond T, Fiedler M, Winton DJ, Corfield A, Bienz M. 2010. Dvl2 promotes intestinal length and neoplasia in the ApcMin mouse model for colorectal cancer. Cancer Res 70:6629–6638. doi: 10.1158/0008-5472.CAN-10-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Shea C, Klupsch K, Choi S, Bagus B, Soria C, Shen J, McCormick F, Stokoe D. 2005. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J 24:1211–1221. doi: 10.1038/sj.emboj.7600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moody CA, Scott RS, Amirghahari N, Nathan CO, Young LS, Dawson CW, Sixbey JW. 2005. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J Virol 79:5499–5506. doi: 10.1128/JVI.79.9.5499-5506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werden SJ, Barrett JW, Wang G, Stanford MM, McFadden G. 2007. M-T5, the ankyrin repeat, host range protein of myxoma virus, activates Akt and can be functionally replaced by cellular PIKE-A. J Virol 81:2340–2348. doi: 10.1128/JVI.01310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu Y, Alwine JC. 2002. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and the cellular kinase Akt. J Virol 76:3731–3738. doi: 10.1128/JVI.76.8.3731-3738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Y, Kudchodkar SB, Alwine JC. 2005. Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J Virol 79:6882–6889. doi: 10.1128/JVI.79.11.6882-6889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spangle JM, Munger K. 2010. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J Virol 84:9398–9407. doi: 10.1128/JVI.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. 2008. Human Cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dibble CC, Manning BD. 2013. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. 2013. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans 41:906–912. doi: 10.1042/BST20130041. [DOI] [PubMed] [Google Scholar]
- 17.Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, Manning BD. 2012. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 47:535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwiatkowski DJ, Manning BD. 2005. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14(Spec No 2):R251–258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 19.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kudchodkar SB, Del Prete GQ, Maguire TG, Alwine JC. 2007. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J Virol 81:3649–3651. doi: 10.1128/JVI.02079-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tilton C, Clippinger AJ, Maguire T, Alwine JC. 2011. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J Virol 85:12585–12593. doi: 10.1128/JVI.05572-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clippinger AJ, Maguire TG, Alwine JC. 2011. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J Virol 85:9369–9376. doi: 10.1128/JVI.05102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brocchieri L, Kledal TN, Karlin S, Mocarski ES. 2005. Predicting coding potential from genome sequence: application to betaherpesviruses infecting rats and mice. J Virol 79:7570–7596. doi: 10.1128/JVI.79.12.7570-7596.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, McGeoch DJ, Hayward GS. 2003. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J Gen Virol 84:17–28. doi: 10.1099/vir.0.18606-0. [DOI] [PubMed] [Google Scholar]
- 26.Hansen SG, Strelow LI, Franchi DC, Anders DG, Wong SW. 2003. Complete sequence and genomic analysis of rhesus cytomegalovirus. J Virol 77:6620–6636. doi: 10.1128/JVI.77.12.6620-6636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 70:8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savaryn JP, Reitsma JM, Bigley TM, Halligan BD, Qian Z, Yu D, Terhune SS. 2013. Human cytomegalovirus pUL29/28 and pUL38 repression of p53-regulated p21CIP1 and caspase 1 promoters during infection. J Virol 87:2463–2474. doi: 10.1128/JVI.01926-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog 6:e1000965. doi: 10.1371/journal.ppat.1000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol 83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian Z, Xuan B, Gualberto N, Yu D. 2011. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J Virol 85:9103–9113. doi: 10.1128/JVI.00572-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J Virol 84:3476–3487. doi: 10.1128/JVI.02544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J Virol 77:10594–10605. doi: 10.1128/JVI.77.19.10594-10605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc Natl Acad Sci U S A 97:14506–14511. doi: 10.1073/pnas.97.26.14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. 2004. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol 78:11030–11039. doi: 10.1128/JVI.78.20.11030-11039.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. 2006. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc Natl Acad Sci U S A 103:14182–14187. doi: 10.1073/pnas.0605825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moorman NJ, Shenk T. 2010. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J Virol 84:5260–5269. doi: 10.1128/JVI.02733-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clippinger AJ, Maguire TG, Alwine JC. 2011. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J Virol 85:3930–3939. doi: 10.1128/JVI.01913-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poglitsch M, Weichhart T, Hecking M, Werzowa J, Katholnig K, Antlanger M, Krmpotic A, Jonjic S, Horl WH, Zlabinger GJ, Puchhammer E, Saemann MD. 2012. CMV late phase-induced mTOR activation is essential for efficient virus replication in polarized human macrophages. Am J Transplant 12:1458–1468. doi: 10.1111/j.1600-6143.2012.04002.x. [DOI] [PubMed] [Google Scholar]
- 44.Andrassy J, Hoffmann VS, Rentsch M, Stangl M, Habicht A, Meiser B, Fischereder M, Jauch KW, Guba M. 2012. Is cytomegalovirus prophylaxis dispensable in patients receiving an mTOR inhibitor-based immunosuppression? A systematic review and meta-analysis. Transplantation 94:1208–1217. doi: 10.1097/TP.0b013e3182708e56. [DOI] [PubMed] [Google Scholar]
- 45.Ritta M, Costa C, Solidoro P, Sidoti F, Libertucci D, Boffini M, Rinaldi M, Baldi S, Cavallo R. 2015. Everolimus-based immunosuppressive regimens in lung transplant recipients: impact on CMV infection. Antiviral Res 113:19–26. doi: 10.1016/j.antiviral.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 46.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. 2011. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bar-Peled L, Sabatini DM. 2012. SnapShot: mTORC1 signaling at the lysosomal surface. Cell 151:1390. doi: 10.1016/j.cell.2012.11.038. [DOI] [PubMed] [Google Scholar]
- 50.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clippinger AJ, Alwine JC. 2012. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes Dev 26:2015–2026. doi: 10.1101/gad.196147.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu Z, Hu X, Li Y, Zheng L, Zhou Y, Jiang H, Ning T, Basang Z, Zhang C, Ke Y. 2004. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J Biol Chem 279:35664–35670. doi: 10.1074/jbc.M403385200. [DOI] [PubMed] [Google Scholar]
- 53.Zheng M, Wang YH, Wu XN, Wu SQ, Lu BJ, Dong MQ, Zhang H, Sun P, Lin SC, Guan KL, Han J. 2011. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol 13:263–272. doi: 10.1038/ncb2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ber Y, Shiloh R, Gilad Y, Degani N, Bialik S, Kimchi A. 2015. DAPK2 is a novel regulator of mTORC1 activity and autophagy. Cell Death Differ 22:465–475. doi: 10.1038/cdd.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. 2014. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha). J Biol Chem 289:25227–25240. doi: 10.1074/jbc.M114.554881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin JH, Min SH, Kim SJ, Kim YI, Park J, Lee HK, Yoo OJ. 2013. TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1. Sci Rep 3:1561. doi: 10.1038/srep01561. [DOI] [PMC free article] [PubMed] [Google Scholar]