

Abstract
The mitochondrial pathway to apoptosis is a major pathway of physiological cell death in vertebrates. The mitochondrial cell death pathway commences when apoptogenic molecules present between the outer and inner mitochondrial membranes are released into the cytosol by mitochondrial outer membrane permeabilization (MOMP). BCL-2 family members are the sentinels of MOMP in the mitochondrial apoptotic pathway; the pro-apoptotic B cell lymphoma (BCL)-2 proteins, BCL-2 associated x protein and BCL-2 antagonist killer 1 induce MOMP whereas the anti-apoptotic BCL-2 proteins, BCL-2, BCL-xl and myeloid cell leukaemia 1 prevent MOMP from occurring. The release of pro-apoptotic factors such as cytochrome c from mitochondria leads to formation of a multimeric complex known as the apoptosome and initiates caspase activation cascades. These pathways are important for normal cellular homeostasis and play key roles in the pathogenesis of many diseases. In this review, we will provide a brief overview of the mitochondrial death pathway and focus on a selection of diseases whose pathogenesis involves the mitochondrial death pathway and we will examine the various pharmacological approaches that target this pathway.
Keywords: mitochondrial permeability transition, apoptosis, Bcl-2, caspases, mitochondrial outer membrane permeabilization
Introduction
Mitochondria and cell death
-
Mitochondrial outer membrane permeabilization (MOMP): point of no return
MOMP by BCL-2 family proteins
-
MOMP by permeability transition pore
Regulation of MOMP: many ways to skin the cat
Role of calcium in MOMP
ROS-induced MOMP
Role of caspases in MOMP
Other regulators of MOMP
Mitochondrial IMS: poison cabinet
-
Mitochondrial pathway of cell death and disease pathogenesis
Ischemia/reperfusion
Neurodegenerative disorders
Cancer
Mitochondrial encephalomyopathies
Others
-
Therapeutic strategies that promote MOMP and cell death
-
Targeting the BCL-2 family
BCL-2 antisense-based strategies
BAX-delivery vector
BH3 mimetic peptides
Natural and synthetic BH3 mimetic drugs
-
Targeting mitochondria directly: mitochondriotoxic compounds inducing mitochondrial membrane permeabilization
Peptide derivatives
Small molecules
Cationic lipophilic agents
Bypassing the mitochondria: mitochondrial pro-apoptotic factors as chemotherapeutic agents
-
-
Therapeutic strategies that inhibit MOMP and cell death
Cyclosporin A and the inhibition of MPT
Novel CsA analogues and other inhibitors of the pore
Preconditioning of the heart protects by sparing mitochondria
Pharmacological IPC mimetics
Minocycline
Inhibitors of PARP and the prevention of DNA damage-mediated mitochondrial damage
Conclusion and future directions
Introduction
The integrity of mitochondrial function is fundamental to cell life. Mitochondria are involved in many processes essential for cell survival, such as energy production, redox control, calcium homeostasis and a number of metabolic and biosynthetic pathways. Mitochondrial calcium uptake plays a major role in influencing cell signalling and in the regulation of mitochondrial function, while excessive mitochondrial calcium accumulation has been implicated in disease. In addition, mitochondria often play an essential role in cell life and cell death decisions. The mitochondrial genome is prone to damage due to increased exposure to reactive oxygen species (ROS) and inadequate repair mechanisms, and therefore genetic damage can lead to impaired electron transport capability and increased free radical generation. These have been linked to the development of neurodegenerative and cardiovascular diseases. Dysfunctional mitochondrial cell death pathways are central players in a wide range of pathological conditions as diverse as cancer, diabetes, obesity, ischemia/reperfusion injury and neurodegenerative disorders such as Parkinson’s (PD) and Alzheimer’s diseases. It is a long and growing list. Hence, it is not surprising that unravelling the mitochondrial pathway of cell death is one of the actively pursued research frontiers of biomedicine. As there are already many reviews on the mitochondrial death pathways, we will discuss some of the fundamental attributes of mitochondrial death pathways and focus on the pharmacological approaches that can be used to target these pathways.
Mitochondria and cell death
The mitochondrial ‘intrinsic’ pathway and the death receptor ‘extrinsic’ pathway are the two principal pathways leading to apoptosis, both of which converge on caspase activation (Fig. 1) [1–3]. Caspases are a family of cysteine proteases that, upon activation, cleave specific substrates, leading to the demise of the cell. Based on the order of activation in cell death pathways, caspases are divided into two major groups: initiator caspases and executioner caspases [4]. The subset of caspases that cleave substrates to produce the typical biochemical changes associated with apoptosis are known as executioner caspases, which in mammals are caspases-3, -6 and -7 [5]. Executioner caspases have a short pro-domain and are in turn activated by apical initiator caspases (caspases-2, -8, -9 and -10). Initiator caspases possess long pro-domains that contain one of the two characteristic protein–protein interaction motifs: the death effector domain or the caspase recruitment domain (CARD) and are involved in interacting with the upstream adapter molecules. The initiator caspases are activated by prodomain-mediated dimerization of the zymogens followed by autocatalytic processing [6]. The initiator caspase for the mitochondrial pathway is caspase-9, whereas the initiator caspases for the death receptor pathway are caspases-8 and -10 [7]. Both pathways share the effector caspases (caspases-3, -6 and -7) which cleave cellular substrates leading to apoptotic cell death. Furthermore, caspase-2 is a long prodomain containing initiator caspase involved in stress-induced apoptosis [8]. A protein complex, named PIDDosome has been shown to mediate the activation of caspase-2 in response to genotoxic stimuli [9]. In addition to caspase-2, the PIDDosome contains the p53-induced protein with a death domain (PIDD) and an adapter protein, called RAIDD. Caspase-2 has also been suggested to induce mitochondrial cytochrome c release through a mechanism not yet understood [10, 11]. Caspases-1, -4, -5 and -11 function primarily in the processing of inflammatory cytokines through another proteolytic platform called the inflammosome [12].
Figure 1.
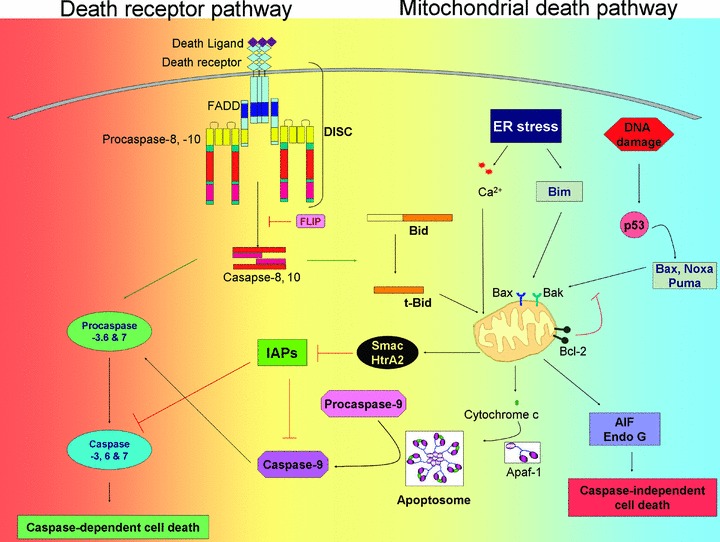
Schematic representation of the extrinsic and intrinsic apoptotic pathway. In the extrinsic pathway, ligation of receptor to death receptor causes DISC formation leading to caspase-8 activation. In type I cells caspase-8 directly cleaves caspase-3, which starts the death cascade. In type II cells an additional amplification loop is required, which involves tBid-mediated cytochrome c release from mitochondria followed by apoptosome formation. In the intrinsic pathway, stress signals from a variety of insults are sensed by BH3-only pro-apoptotic proteins and communicated to multidomain pro-apoptotic and anti-apoptotic BCL-2 proteins. The functional interplay of the proteins ultimately results in the activation of BAX and BAK at target organelles such as mitochondria and ER, which participate in apoptosis by releasing apoptogenic factors. Cytosolic cytochrome c triggers the formation of apoptosome, followed by activation of caspases-9 and -3. Function of caspase can be modulated on several levels. Activation of caspases at the DISC is inhibited by c-FLIP proteins; activation of effector caspases is inhibited by IAPs (see text for details). Smac/DIABLO and HtrA2/Omi neutralize the inhibition of caspases by IAPs. Smac/DIABLO, HtrA2/Omi, AIF and endo G may also initiate a caspase-independent cell death pathway. Abbreviations: FADD, Fas-associated death domain; DISC, death-inducing signalling complex; BAK, BCL-2 antagonist/killer; BAX, BCL-2-associated X protein; BCL-2, B-cell lymphoma 2 protein; IAPs; inhibitor of apoptosis proteins; Apaf-1; apoptosis protease activating factor1 and AIF, apoptosis-inducing factor.
Death signals originating from cellular stress, including radiation, oxidative stress, genotoxic stress and chemotherapeutic drugs, activate an intrinsic apoptotic pathway that is mediated largely by the mitochondria. Mitochondrial release of cytochrome c into the cytoplasm induces the formation of a multiprotein complex called the apoptosome. Apoptosome contains among others cytochrome c, pro-caspase-9 and the adaptor protein Apaf-1 [13] and, supports the activation of caspase-9 through enforced multimerization, which in turn cleaves and activates the effector caspase-3 resulting in the subsequent degradation of cellular death substrates (Fig. 1) [14]. In addition to cytochrome c, other mitochondrial proteins released during apoptosis have been identified over the past decade and these include second mitochondria-derived activator of caspase/direct inhibitor of apoptosis protein (IAP) binding protein with a low pI, (Smac/DIABLO), endonucle-ase G (Endo G), apoptosis-inducing factor (AIF), HtrA2/Omi [15], as well as Hsp60, Hsp10 and adenylate kinase [16] (Fig. 1). Some intermembrane space (IMS) proteins have essential survival functions in mitochondria and a well-established lethal function in the cytosol.
In the extrinsic pathway, ligation of death receptors (a subset of the tumour necrosis factor [TNF] receptor [TNFR] family, including TNFR-1, CD95, TNF-related apoptosis-inducing ligand receptors-1 and -2 [TRAIL-R1 and -R2] and DR3/TRAMP) causes the recruitment and oligomerization of the adapter molecule Fas-associated death domain (FADD) within the death-inducing signalling complex (DISC) [17]. The oligomerized FADD binds pro-caspases-8 and -10, causing their homodimerization and activation (Fig. 1) [18–22]. Depending on the cell type, activated caspase-8 induces apoptosis by two different signalling pathways [23]. In type I cells, large amounts of active caspase-8 formed at the DISC directly induces activation of pro-caspase-3 independently of mitochondria [24]. In type II cells, probably only a low amount of active caspase-8 is generated that is unable to activate sufficient amounts of pro-caspase-3 directly to a level that would be sufficient to execute apoptosis. In these cells, caspase-8 cleaves the ‘Bcl-2 homology (BH) 3-only protein’ Bid, generating an active fragment (tBid) that activates the mitochondrial death pathway (Fig. 1) [25, 26]. In this manner, the death signal may be amplified through formation and activation of the apoptosome which contributes to effector caspase activation.
Mitochondrial outer membrane permeabilization (MOMP): point of no return
During apoptosis in vertebrate cells, the process of MOMP appears to represent a point-of-no-return for many cell types as it appears to commit a cell to death regardless of caspase activity [27]. Why, and how, does MOMP commit a cell to die? MOMP is lethal because it leads to the release of caspase-activating molecules, caspase-independent death effectors and metabolic failure in the mitochondria. Majority of cells appear to be committed to die following MOMP because it leads not only to the activation of the well-established caspase-mediated apoptotic pathway, but, should there be a failure of its execution through insufficient caspase activation, a parallel, caspase-independent cell death pathway is set in motion that is controlled by HtrA2/Omi, AIF and Endo G. However, sympathetic neurons that are induced to undergo apoptosis by withdrawal of nerve growth factors (NGF) can recover upon addition of NGF, even after the release of cytochrome c, provided that caspase activation is blocked [28, 29]. This is due to the fact that neuronal cells express low levels of Apaf-1 and, sufficient levels of endogenous caspase inhibitors such as X-linked IAP (XIAP) to block the ability of cytochrome c to induce apoptosis [30]. The other factors that contribute to cell demise following MOMP are general decline in mitochondrial function. The most important function of mitochondria is the generation of ATP through the process of oxidative phosphorylation. Dissipation of the ΔΨm is a general feature of apoptosis, irrespective of cell type and of the apoptotic stimuli (for a review, see [31]). It has been demonstrated that a reduction in ΔΨm follows within minutes after the release of cytochrome c, but in the absence of caspase activity, mitochondria can regenerate ΔΨm and maintain ATP generation [32]. However, loss of mitochondrial energy production will lead to cell death unless another source of energy is available to the cell. If we are to target the mitochondrial death pathway for disorders connected with apoptosis dysregulation, it will be essential to have a detailed understanding of MOMP and its regulation. The mechanisms of MOMP have been controversial, and there are two principal hypotheses: in the first, MOMP is regulated by the BCL-2 family of proteins, and in the second, by the permeability transition pore (PTP) (Fig. 2) [33].
Figure 2.
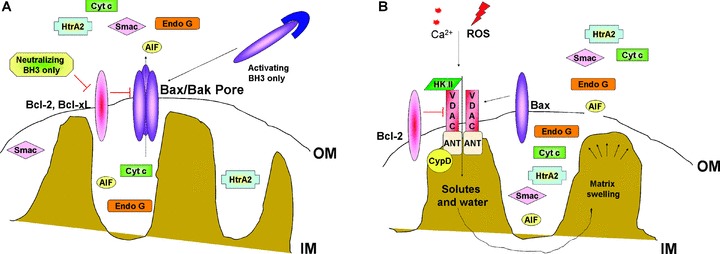
Molecular mechanisms of MOMP. (A) According to first model, the pro-apoptotic members of BCL-2 family BAX and BAK form a multimeric pore across the outer mitochondrial membrane upon activation by BH3-only proteins. This channel mediates the release of apoptogenic factors from IMS. (B) According to second model, opening of voltage-gated channel results in mitochondrial matrix swelling and rupture of MOM, releasing IMS proteins in the cytosol. Abbreviations: ANT, adenine nucleotide translocator; BAK, BCL-2 antagonist/killer; BAX, BCL-2-associated X protein; BCL-2, B-cell lymphoma 2 protein; BH3, BCL-2 homology domain 3; CypD, cyclophilin D; HK, hexokinase; IM, mitochondrial inner membrane; OM, mitochondrial outer membrane and VDAC, voltage-dependent anion channel.
MOMP by BCL-2 family proteins
The first model considers MOMP as a process that is essentially intrinsic to the outer membrane and controlled by members of the BCL-2 family of proteins to promote or prevent the formation of large, protein permeable pores (Fig. 2). The BCL-2 family of proteins is divided into three groups, based on the presence of BCL-2 homology domains (BH1–4 domains) [34]. The multidomain, anti-apoptotic BCL-2 proteins (e.g. BCL-2, BCL-w, BCL-xl[BCL-2 related gene, long isoform], A1 and MCL-1 [myeloid cell leukaemia 1]] contain BH domains 1–4 and are generally localized to various intracellular membranes such as outer mitochondrial membrane, endoplasmic reticulum (ER) membrane and nuclear membrane [35]. These proteins are thought to function within the apoptotic pathway by directly binding and inhibiting the pro-apoptotic BCL-2 proteins.
The pro-apoptotic members of the family are divided into two groups: the multidomain pro-apoptotic molecules and the BH3-only proteins. The multidomain molecules (e.g. BAK [BCL-2 antagonist killer 1] and BAX [BCL-2 associated x protein]) contain BH 1–3 domains and are believed to permeabilize the outer mitochondrial membrane by forming oligomeric pores (megachannels) that allow the release of apoptogenic molecules from the intermembrane space [36]. The BH3-only proteins (e.g. BAD [BCL-2 antagonist of cell death], BID [BCL-2 interacting domain death agonist], BIK [BCL-2 interacting killer], BIM [BCL-2 interacting mediator of cell death], BMF [BCL-2 modifying factor], bNIP3 [BCL-2/adenovirus E1B 19-KD protein interacting protein 3], HRK [Harakiri], NOXA and PUMA [p53 up-regulated modulator of apoptosis]), function by physical interactions with the other BCL-2 family members either resulting in inhibition of the anti-apoptotic members, or activation of the pro-apoptotic multidomain members [36]. The BH3-only pro-apoptotic proteins are sentinels that sense apoptotic signals and communicate with the multidomain anti-apoptotic and pro-apoptotic molecules to shift their balance towards promotion of death. In what is generally referred to as the ‘rheostat’ model, cell survival is determined by the balance among the anti-apoptotic BCL-2 family proteins such as MCL-1, BCL-xl or BCL-2, and pro-apoptotic members [33]. Activation of BAX and BAK during apoptosis involves multiple conformational changes that are accompanied by homo-oligomerization and insertion into the membrane. Oligomerization of BAX and BAK at the outer mitochondrial membrane is a crucial step in MOMP [37]. Indeed, structural and biophysical studies using synthetic lipid bilayers and vesicles support the intrinsic pore-forming capacity of several BCL-2 family proteins, including BAX, BCL-2 and BCL-xl[38–41]. Studies using vesicles formed from isolated mitochondrial outer membrane (MOM) have shown that BCL-2–family proteins can regulate the permeability of the MOM in the absence of interior structures of the mitochondria; moreover, many features of this process of membrane permeabilization can be reproduced using defined liposomes and recombinant BCL-2–family proteins [38, 39]. Although several BCL-2–family proteins possess ion channel activity in lipid bilayers, only the multidomain pro-apoptotic proteins BAX and BAK can render membranes permeable to cytochrome c or larger macromolecules. However, these cell-free systems do not recapitulate all the complexity of the permeabilization process as it occurs in the cell; other proteins of the MOM could modulate or potentiate the function of BAX and BAK. Furthermore, the nature and consequence of protein--protein interactions among members of the BCL-2 family are still not clearly understood. BAX and BAK are thought to homo-oligomerize to induce MOM permeabilization. The BH3-only proteins regulate MOM permeabilization by a combination of release of BAX/BAK from their inhibitory complexes with anti-apoptotic counterparts including BCL-2 and MCL-1 as well as direct activation of BAX/BAK to promote their conformational change, membrane insertion and oligomerization. For a detailed account of regulation of MOMP by BCL-2 family members refer to the recent review by Green and colleagues [33].
MOMP by permeability transition pore
The second prominent model for MOMP is based on a phenomenon known as the mitochondrial permeability transition (MPT) (Fig. 2). PT involves the permeabilization of the inner mitochondrial membrane (IMM) to solutes with a molecular mass of less than 1500 Da, this results in the loss of the mitochondrial membrane potential (ΔΨm), mitochondrial swelling, and as observed in vitro, rupture of the outer mitochondrial membrane [42]. The pore allowing the release of the matrix solutes is the PT-pore (PTP) which is thought to be composed of at least three proteins, the voltage-dependent anion channel (VDAC) located in the outer mitochondrial membrane, the adenine nucleotide translocator (ANT), a specific ATP/ADP transporter located in the IMM and cyclophilin D (CypD), a chaperone with peptidylprolyl isomerase (PPIase) activity. CypD is directly associated with ANT in the matrix of mitochondria [43]. The PTP complex is thought to span the contact sites between the inner and outer mitochondrial membranes.
It has long been thought that VDAC, ANT and CypD play an essential role in PT, but convincing evidence is still lacking to conclude that these are both necessary and sufficient to induce PT. There is evidence to suggest that VDAC is a component of the PT pore [44, 45]. A direct role of the VDAC in PT has been demonstrated in studies using specific anti-VDAC antibodies. Two polyclonal anti-VDAC antibodies, which recognize different VDAC epitopes and inhibit its activity in liposomes, have been shown to inhibit the Ca2+-induced PT, supporting a crucial role for VDAC in this process [46]. However, mitochondria isolated from VDAC1-deficient cells undergo PT normally, suggesting that VDAC1 is not important for this process [47]. However, this result could have been due to compensation for VDAC1 deficiency by other isoforms, including VDAC2 and VDAC3. This question was recently addressed by Baines and colleagues by using fibroblasts from a triple knockout mouse lacking Vdac1, Vdac2 and Vdac3. They demonstrated that mitochondria from Vdac1–Vdac3-null mice exhibited a Ca2+- and oxidative stress-induced PT that was indistinguishable from wild-type mitochondria. Furthermore, wild-type and Vdac1-Vdac3 deficient mitochondria and cells exhibited equivalent cytochrome c release, caspase cleavage and cell death in response to the pro-death BCL-2 family members BAX and BID [48]. These results suggest that VDACs are dispensable for both PT and BCL-2 family member-driven cell death. Regarding the role of ANT in PT, biochemical isolation and reconstitution of the PTP in liposomes suggested that these protein complexes consist of ANT in the inner membrane and VDAC in the outer membrane [49, 50]. However, liver mitochondria from mice lacking both ANT1 and ANT2 still underwent PT, [51]. This finding suggests that ANT1/2 only play a limited role, if any, in PT or it is possible that deficiency of ANT1/2 was compensated by other channel protein(s). The lack of an important role for ANT in PT corroborates the observation that mitochondria isolated from yeast lacking ANT can undergo PT-like changes, including loss of membrane potential and swelling in response to ethanol [52]. If ANT is not involved in PT, modulation of PT by ANT ligands such as bongkrekic acid or atractyloside suggests a role for a yet unidentified, ANT-like inner membrane channel(s) in PT. The role of CypD in PT was initially suggested by the finding that PT is blocked by cyclosporine A (CsA) a known inhibitor of the PPIase activity of cyclophilins. It has been demonstrated that CypD-deficient mitochondria isolated from the livers of CypD deficient mice do not undergo the CsA-sensitive PT in response to a variety of inducers, including Ca2+, atractyloside and H2O2[53, 54]. However, cells isolated from CypD-deficient mice, such as thymocytes, mouse embryonic fibroblasts (MEFs) and hepatocytes, undergo apoptosis normally in response to various stimuli, including etoposide, staurosporine and TNF-α[53, 55]. The inhibitory effect of CsA on apoptosis might need to be re-evaluated because the concentration of CsA used in these experiments was relatively high which could have had a secondary effect on apoptosis. It is also possible that BAX/BAK megachannels were responsible for the induction of apoptosis in the CypD deficient cells. More studies are needed to elucidate the molecular nature of PT pore complex, especially the role of the accessory proteins that have been detected associated with the PTP such as hexokinases-I and -II (HK) [56]. This protein interacts directly with VDAC in the outer membrane of mitochondria [57]. Its displacement from VDAC is necessary for BAX binding and cell death induction [58, 59]. Hence, BAX as well as other BCL-2 family members can, depending of the physiological state of the cell, interact with PTP components in the MOM. Finally there is a strong possibility that the PT pore complex may not be a single entity but that multiple proteins can adopt a pore function. For instance, recent work has implicated the mitochondrial phosphate carrier as a component as well as a potential independent pore involved in PT [60].
In normal conditions the PTP exists in a state of low conductance that may be subject to transient flicker during inositol 1, 3, 4-trisphosphate-mediated Ca2+ mobilization from nearby ER sacs. However, when excessive amounts of Ca2+ are released from the ER that overloads the mitochondria, the pore transitions to a high-conductance state [61]. This passage from low to high conductance is irreversible and strictly depends on the saturation of the calcium-binding sites of the PTP [61]. The high-conductance conformation allows free diffusion of water and ions between the cytosol and the matrix, causing collapse of ΔΨm, uncoupling of oxidative phosphorylation and swelling of the mitochondrial matrix [42, 43]. This may lead to rupture of the MOM and consequent release of IMS proteins (Fig. 2). Despite the lack of evidence for PT as a mechanism of apoptotic death in general, it may be an important therapeutic target for inducing PT-mediated cell death selectively in certain cell types. Regardless of the mechanism, MOMP is a crucial step for many pathways that induce apoptosis and disruption of this event is likely to affect cell fate.
Regulation of MOMP: many ways to skin the cat
Many death signals originating from cellular stress activate an intrinsic pathway of apoptosis which is mediated by the mitochondria. MOMP is an important step in the intrinsic pathway. Multiple distinct signalling pathways converge on MOMP.
Role of calcium in MOMP
Ca2+ is one of the key regulators of not only cell survival but also cell death in response to a variety of cellular signals. The pro-apoptotic effects of Ca are mediated by a diverse range of Ca2+-sensitive factors that are compartmentalized in various intracellular organelles including the ER and mitochondria [62]. The ER is a complex organelle composed of membrane sheets that enclose the nuclear envelope and an elaborate interconnected tubular network in the cytosol. The ER can be an initiator of apoptosis when accumulation of unfolded proteins or inhibition of the ER-Golgi transport results in the ER stress response [63]. Ca2+-dependent stimuli at the ER induce apoptosis through a mitochondrial pathway including mitochondrial dysfunction, cytochrome c release and caspase activation [64]. Both BCL-2 and BAX can localize to the ER and they can modulate Ca2+ fluxes [65]. Overexpressed BCL-2 reduces resting ER Ca2+ concentration and the extent of capacitative Ca2+ entry, pointing to a specific role of BCL-2 at the ER in the control of cell death or setting the threshold of sensitivity to pro-apoptotic stimuli [66]. When large quantities of Ca2+ accumulate in the mitochondrial matrix, Ca2+ interacts with CypD and other components of the PTP to induce opening of the pore [61, 67]. Furthermore, the rise in mitochondrial Ca2+ stimulates the generation of ROS and free fatty acids that also promote the opening of the PTP [68, 69]. Recent studies have demonstrated that the regulated process of mitochondrial fusion and fission controls the spatiotemporal properties of mitochondrial Ca2+ responses and thus, the physiological and pathological consequences of increased Ca2+ concentration in the cytosol and Ca2+ taken up by mitochondria [70]. Two proteins involved in the mitochondrial fission machinery, Drp1 and hFis1, have an antagonistic effect on BCL-2 [70]. Drp1, with the assistance of hFis1, sensitizes cells to PT by reducing mitochondrial Ca2+ retention.
ROS-induced MOMP
All mammals use O2 for energy production. Oxidation is the loss of an electron by a substance. Under normal metabolic conditions, the electron-transporting complexes I, II, III and IV of the mitochondrial respiratory chain plus the non-redox H+-translocating complex, the ATP synthase (also called complex V, F0F1-ATP synthase) together with co-enzyme Q and cytochrome c carry out the process of terminal oxidation. The respiratory enzyme complexes transfer electrons from the reducing equivalents NADH or FADH2 to O2, while transporting protons across the IMM. The total proton-motive force across the IMM is the sum of the mitochondrial membrane electrical potential and the H+-concentration gradient (ApH+). This proton-motive force is used to drive protons from the intermembrane space into the matrix through the ATP synthase. The ATP synthase uses the energy of the H+ flow to synthesize ATP from ADP and Pi. The energy of the electrochemical proton gradient is also used to import proteins into the mitochondria and to regulate metabolite transport across the mitochondrial membrane. A small percentage of the total O2 consumed by terminal oxidation even in healthy tissues becomes ROS, such as superoxide (O2∼), hydrogen peroxide (H2O2) and hydroxyl radical (OH−) [71]. This ROS production occurs primarily in complex I (NADH dehydrogenase) and complex III (ubiquinone-cytochrome c reductase) and the accumulation of ROS has been shown to occur predominantly in the IMS [72, 73]. ROS can attack DNA, proteins, lipids and carbohydrates and cause DNA strand breaks, protein oxidation and lipid peroxidation. Polyunsaturated fatty acid residues in phospholipids are especially sensitive to oxidation [74]. Mitochondrial lipid peroxidation products can impair the barrier function of membranes by either directly interacting with the protein and/or indirectly interacting with the lipid moieties in the membrane [75]. Oxidative damage to DNA causes modification of the purine and pyrimidine bases and can result single or double strand-breaks [76]. mtDNA is especially susceptible to damage by ROS owing to its close proximity to the electron transport chain, the major locus for free-radical production and the lack of protective histones. The amino acids tyrosine, histidine, arginine, lysine and proline are particularly vulnerable to ROS modification, which can lead to gain or loss of receptor activity, enzyme function and signal transduction pathways [77, 78]. ROS can lead to oxidative damage and inactivation of the iron-sulphur (Fe-S) proteins in the mitochondria such as aconitases, complex I NADH dehydrogenase and succinate dehydrogenase [79]. Oxidative stress has been shown to markedly sensitize mitochondria toward MOMP [80]. Oxidative stress is a key factor in many diseases, including neurodegenerative diseases such as PD. For example, in models of PD, inhibition of complex I with MPP+ induces cell death and 6-hydroxydopamine causes oxidative stress that is linked to MOMP and release of IMS proteins including cytochrome c and Smac/DIABLO [81, 82]. Similarly, cardiolipin, an anionic phospholipid located in the IMM and therefore exposed to the oxidizing environment of the IMS [73], can be readily oxidized, which reduces its capacity to bind cytochrome c and increases the level of soluble cytochrome c within the IMS [83, 84].
Role of caspases in MOMP
Previous studies have shown that incubation of isolated mitochondria with recombinant human caspases promotes MOM permeabilization and release of cytochrome c and Smac/DIABLO into the cytosol [85, 86]. Pro-caspase-2 efficiently inserts into the mitochondrial membranes and triggers the release of cytochrome c bound to cardiolipin [87]. One study has shown that active caspase-3 can enter the mitochondria and cleave NDUF1, a component of complex I of the respiratory chain. Cleavage of NDUF1 by caspase-3 reduced electron transport by complexes I and II by up to 88% and 94%, respectively [88]. Mutation of the caspase-3 cleavage site in NDUF1 could preserve mitochondrial functions during apoptosis and delay plasma membrane events associated with caspase activation, including loss of plasma membrane integrity and externalization of phosphatidylserine. Interestingly, treatment with zVAD-fmk, a pan-caspase inhibitor, preserved electron transport chain functionality but failed to inhibit cytochrome c release. Additional studies on intact cells and isolated mitochondria have shown that zVAD-fmk was able to inhibit the release of Smac/DIABLO, HtrA2/Omi, AIF and Endo G, but could not inhibit the release of cytochrome c[89]. Embryonic fibroblasts and thymocytes derived from mice lacking both caspases-3 and -7 exhibited resistance to drugs that induce the intrinsic (mitochondrial) and extrinsic (membrane death receptor) pathways to apoptosis [90]. In all conditions studied, the cells displayed a pronounced delay in cytochrome c release and translation of BAX to the outer membrane. The mitochondrial membrane potential was unaffected. Overall, the results of this study suggest that caspases-3 and -7 are important mediators for mitochondrial events in apoptosis [90]. However, it remains unclear how a cytosolic protease can cross both mitochondrial membranes to cleave a matrix-exposed subunit embedded within IMM.
Other regulators of MOMP
The tumour suppressor p53 acts, in part, to induce apoptosis by inducing expression of the BH3-only protein, PUMA and PUMA-deficient cells display a resistance to p53 mediated apoptosis. However, p53 can trigger MOMP and apoptosis in the absence of transcription, and this can occur through direct activation of BAX or BAK or through sequestration of BCL-2 and BCL-xl to block their activity [91]. Resolving the role of p53 at the mitochondria versus its role in the nucleus as a transcription factor will be important in understanding the apoptotic function of p53. An emerging theme is one of nuclear proteins and nuclear factors functioning in the cytosol through direct interactions with BCL-2 family proteins. Ku70, involved in DNA repair, can inhibit BAX [92]. Another nuclear protein, TR3, binds BCL-2 and perhaps promotes MOMP through this interaction [93]. Histone 1.2 released from the nucleus upon X-ray–induced DNA damage can trigger MOMP perhaps through an interaction with BCL-2 family members [94]. Finally, ADP-ribose polymers which are formed extensively in response to DNA damage and are released from the nucleus have been shown to mediate BAX-dependent MOMP [95].
Mitochondrial IMS: poison cabinet
Irrespective of its mechanisms, MOMP can seal the point of no return for the cell by the release of several apoptogenic molecules such as cytochrome c, Smac/DIABLO, Endo G, AIF and HtrA2/Omi [15]. Some of these proteins have cytotoxic activities due to caspase-dependent and -independent processes.
The release of cytochrome c into the cytoplasm, in the presence of dATP induces the formation of the Apaf-1-containing macromolecular platform called the apoptosome that activates caspase-9 [13]. Mature caspase-9 remains bound to the apoptosome, recruiting and activating executioner caspase-3 and/or caspase-7 [14]. The release of cytochrome c has often been considered the point of no return in cell death since cytochrome c participates in the mitochondrial electron-transport chain, using its haem group as a redox intermediate to shuttle electrons between complex III and complex IV and is responsible for the generation of the ΔΨm [96]. The loss of mitochondrial cytochrome c has also been associated with enhanced ROS formation [97]. Cytochrome c has two distinct functions in the cell: (i) under normal physiological conditions it is required within mitochondria for maintenance of mitochondrial electron transport chain and (ii) under apoptotic conditions it is released from mitochondria and has an apoptotic role in the cytoplasm. Thus, the release of cytochrome c has a strong impact on cell fate determination because in addition to the activation of the caspase cascade, in the absence of cytochrome c, mitochondrial respiration and the control of ROS formation are impaired [98].
The absence of the apoptosome inhibits the execution of the apoptotic process in many systems. Genetic studies have confirmed the importance of the apoptosome in the intrinsic apoptotic pathway. Apaf-1 null and caspase-9 null mice display brain malformation due to impaired neuronal apoptosis [99–102]. Lys72 of cytochrome c is essential for the stability of the interaction between cytochrome c and Apaf-1. Lys72Ala knock-in mice recapitulate the embryonic lethality and brain developmental defects of Apaf-1 knockout and caspase-9 knockout mice [103]. Apaf-1 failed to oligomerize in Lys72Ala-cytochrome c-mutant cells following an apoptotic stimulus. Mouse embryonic fibroblasts from Lys72Ala-mutant mice failed to activate caspases-3 and -9 and thus were resistant to several apoptotic stimuli [103]. The phenotypic similarity between the Lys72Ala knock-in mice and Apaf-1- and caspase-9-knockout mice suggests that these molecules are equally important for the apoptotic function of mitochondria in cytochrome c release, apoptosome formation and caspase-9 activation. Several studies based on cryo-electron microscopy have been carried out to identify the apoptosome structure. They have revealed a wheel-like complex made up of seven Apaf-1 molecules [104–106]. In the apoptosome, the CARD domains are located at the central hub, where pro-caspase-9 binds, whereas the WD40 repeats form Y-shaped tails at the end of the spokes.
Many factors are involved in apoptosome formation and regulation [107]. Heat shock protein 90 and 70, induced by several toxic stimuli; prevent apoptosome formation by binding to Apaf-1 and inhibiting its oligomerization [108, 109]. Inhibition of cytochrome c release, e.g. by Hsp27, prevents induction of apoptosis in a model of PD [81]. Tumour up-regulated CARD-containing antagonist of caspase-9 binds pro-caspase-9 by its CARD domain, thereby preventing its interaction with Apaf-1 [110, 111]. Phosphorylation of caspase-9 by extracellular signal-regulated kinase (ERK) 1/2 at Threonine-125 prevents proteolytic processing and activation of pro-caspase-9 without affecting its recruitment to apoptosome [112]. The oncoprotein, prothymosin-A, inhibits the formation of the apoptosome, while tumour suppressor putative HLA-DR-associated proteins facilitate apoptosome-mediated pro-caspase-9 activation [113].
IAPs are involved in the regulation of apoptosome function and are characterized by the presence of the baculoviral IAP repeat (BIR) domain [114]. XIAP regulates activity of initiator and effector caspases through different mechanisms [115]. An active effector caspase, such as caspase-7, exists as a homodimer and contains two active sites, one on each monomer. The active site of caspase-7 can be tightly bound by a short peptide sequence in the linker region preceding the BIR2 domain of XIAP [116]. This binding blocks substrate entry resulting in the inhibition of caspase-7 [116]. An initiator caspase, such as caspase-9 exists as monomer and BIR3 of XIAP interacts with caspase-9 monomer, thereby trapping caspase-9 in its monomeric state [117]. XIAP-mediated steric hindrance thereby prevents homodimerization-induced activation of caspase-9 and retains a caspase-9 monomer in its inactive state [117]. The Ring domain which has an E3 ubiquitin ligase activity promotes ubiquitination and subsequent degradation of pro-caspases-3 and -9 [118, 119]. However, recent studies have revealed that unlike XIAP, other IAPs are unable to inhibit caspases at physiological concentrations. In particular, cIAP1 and cIAP2 are able to bind, but not to inhibit caspases, probably because of the lack of critical amino acids required for caspase inhibition [120]. However, the two mitochondrial IMS proteins namely Smac/DIABLO and HtrA2/OMI appear to interact with and inhibit IAPs [121–125]. All IAP binding proteins share a conserved four-residue IAP binding motif (IBM) at their N-terminus that allows them to bind IAPs. Smac/DIABLO and HtrA2/Omi are nuclear encoded and synthesized as precursor proteins of 239 and 458 amino acid residues containing an N-terminal mitochondrial localization signal (MLS) [122–125]. The amino acid residues from 1–55 in Smac/DIABLO and 1–133 in HtrA2/Omi comprise the MLS. Upon mitochondrial import, the MLS is removed by proteolysis, exposing the IBM at the N-terminus of mature Smac/DIABLO and HtrA2/Omi [122–125]. Smac/DIABLO knockout mice are viable, grow normally into adulthood and do not exhibit any histological abnormalities [126]. HtrA2/Omi, has an important role within the mitochondria and the proteolytic activity of HtrA2/Omi is required to maintain mitochondrial function. The binding of Smac/DIABLO and HtrA2/Omi to IAPs can promote cell death by releasing the inhibitory activity of IAPs on caspase activation and caspase activities. Unlike Smac/DIABLO, HtrA2/Omi possesses serine protease in addition to IAP binding property and it can promote cell death independent of the cellular caspase activity. Indeed expression of cytosolic protease active HtrA2/Omi has been shown to induce cell death in Apaf minus/− and caspase-9minus/− mouse embryo fibroblasts [123]. Further inhibition of cellular caspases with zVAD-FMK, XIAP, XIAP-BIR3, or dominant negative caspase-9 does not affect the ability of cytosolic protease active HtrA2/Omi to kill the cells [123]. c-IAP1 and c-IAP2 have been shown to interact with (TNF-α receptor associated factors) TRAF molecules, the proteins that mediate signal transduction pathway induced by TNF receptor-like proteins [127, 128]. It remains to be elucidated whether Smac/DIABLO and HtrA2/Omi could regulate TNF signalling by removing IAPs from TRAFs.
The neurodegenerative phenotype of mice lacking HtrA2/Omi or expressing the enzymatically inactive protein as in Mnd2 mutant mice indicates that the protease activity of HtrA2/Omi has a protective role in the mitochondria of neuronal cells [129, 130]. The phenotype of HtrA2/Omi deficient mice and Mnd2 mutant mice resemble the clinical manifestations of PD. Moreover, single nucleotide polymorphisms in the HtrA2/Omi gene that cause missense mutations (A141S and G399S) and affect the enzymatic activity of the protease have been associated with the development of PD in human beings [131]. The protease activity of HtrA2/Omi is up-regulated in the presence of peptides corresponding to carboxy terminus of presenilin-1 that bind to their PDZ domains suggesting a link between HtrA2/Omi and Alzheimer’s disease [132].
During apoptotic conditions following OMM permeabilization, Endo G and AIF are released from mitochondria. However, release of Endo G and AIF is compromised in Apaf-1 deficient cells and can also be inhibited by broad caspase inhibitor zVAD-fmk. These observations suggest that release of Endo G and AIF into the cytosol requires caspase activation downstream of MOMP [89]. Once in the cytosol they translocate to the nucleus and affect chromatin in a caspase-independent way [15]. Endo G was identified by mass spectrometry in the supernatant fraction of tBid-treated mouse liver mitochondria, as a protein that induces caspase-independent DNA fragmentation in purified HeLa nuclei [133]. Although early reports suggested that the knock-out of Endo G is embryonic lethal, it has been recently demonstrated that this phenotype was due to the disruption of an adjacent gene, and that Endo G knock-out mice can develop to adulthood without obvious abnormalities [134, 135]. AIF is an NADH oxidase with a local redox activity that is required for the correct assembly and/or function of the respiratory chain. Upon MMP, AIF is released into the cytosol and translocates to the nucleus, where it promotes chromatin condensation and DNA degradation independently of caspases [136, 137]. Like HtrA2/Omi, AIF also plays an important role in mitochondrial homeostasis in healthy cells. Harlequin mutant mice, that express only 20% of the AIF levels of their WT counterpart due to a retroviral insertion in the first intron of the AIF gene develop neurodegeneration (with ataxia owing to cerebellar atrophia) and blindness because of retinal degeneration [138]. Muscle-specific knockout of AIF leads to severe mitochondrial dysfunction, skeletal muscle atrophy and dilated cardiomyopathy [139]. However, as with AIF null mice, distinguishing between the true apoptotic role and the vital mitochondrial function of AIF remains a challenge.
Mitochondrial pathway of cell death and disease pathogenesis
It is evident that defects in the apoptotic machinery or aberrations in apoptotic responses to death signals can contribute to various human diseases. MOMP-dependent apoptosis is involved in major pathologies, with far-reaching medical and pharmaceutical implications. Mitochondria are involved in several known human diseases, including ischemia-reperfusion injury of the heart, ischemic and traumatic brain damage, muscular dystrophy caused by collagen VI deficiency, amyotrophic lateral sclerosis (ALS), acetaminophen-induced hepatotoxicity, hepatocarcinogenesis induced by 2-acetylaminofluorene and death receptor induced hepatitis [140].
Ischemia/reperfusion
Ischemia is the process whereby the blood supply of an organ is interrupted and results in cell death, most likely due to disruption of cellular energy metabolism such as loss of ATP. Injury following reoxygenation may be due in part to the formation of ROS [141] and mitochondrial calcium overload both of which, have the capacity to induce opening of the MTP pore (permeability transition pore complex [PTPC]). PTPC opening has been observed in many models of ischemia/reperfusion [142], and cytochrome c release has been observed following reperfusion of the ischemic brain [143]. In many cases, the cell death is preventable by agents that act at the level of the mitochondria. For example, treatments that are known to prevent PTPC opening seem to protect tissue from damage [144]. The majority of available evidence concerns the immunosuppressive compound CsA, thought to act by binding to the CypD component of the PTPC (discussed later). Therefore, mitochondrial apoptosis pathway may a play an important role in reperfusion injury. By preventing mitochondrial cell death, the damage can be minimized and greater organ function retained.
Neurodegenerative disorders
Apoptotic pathways and specifically MOMP is involved in the pathophysiology of widespread and devastating neurodegenerative disorders (reviewed in [145]). Imaging studies of postmortem brain tissue have revealed apoptotic nuclei in patients with Alzheimer’s disease [146], ALS [147], Huntington’s disease (HD) [148] and PD [149].
Alzheimer’s disease is characterized by a general decrease in cognitive ability, especially short-term memory [150]. In addition to postmortem nuclei suggestive of apoptosis, certain proteins with apoptotic potential were modulated in the brains of Alzheimer’s patients [150]. BCL-2 expression was decreased and BAX was up-regulated in neurons with neurofibrillary tangles [151]. Reduced complexes II, III and IV activity is seen postmortem in AD brains [152]. Presenilins are genes that were originally isolated due to their ability to induce an early-onset form of familial AD. Mutations in presenilin-1 (PS-1) increase neuronal sensitivity to ischemia, hyperosmotic shock and calcium overload [153]. PS-1 and PS-2 have been demonstrated to bind anti-apoptotic BCL-xl and at least PS-2 can modulate mitochondrial apoptosis, presumably through inactivation of anti-apoptotic BCL-2 family members [154].
PD is characterized clinically by bradykinesia, rigidity and tremor, which correlates histologically with a loss of dopaminergic neurons in the substantia nigra pars compacta [155]. Impairment of complex I and subsequent oxidative stress have been widely demonstrated in experimental models of PD and in postmortem PD samples [156]. In neuronal culture, dopamine (DA) has been shown to result in apoptotic cell death in a dose-dependent manner [157]. Moreover, BCL-2 overexpression, seen in vivoin surviving DA neurons in the substantia nigra [158], is also able in vitro to abrogate DA-induced cell death, suggesting mitochondrial involvement in the death process [157].
HD is a dominantly inherited neuromotor disease characterized by involuntary, hyperkinetic movements, retardation of voluntary movements and cognitive impairment [159]. The disease is caused by expanded CAG repeats within the huntingtin gene, and degeneration of neurons in specific brain regions [159]. The importance of apoptosis in HD was suggested by experiments wherein expression of full length human huntingtin containing 48 or 89 CAG repeats: (a) caused JNK activation and apoptosis in a rat hippocampal neuronal cell line (HN33) and (b) demonstrated clinical disease in transgenic mice [160]. Mitochondrial depolarization and apoptosis was blocked by CsA treatment, implicating involvement of the ANT subunit of the PTPC. Further implicating mitochondrial involvement in HD, a primate model of HD is generated by 3-nitropropionic acid, a respiratory chain complex II poison [161].
ALS is a disease characterized by selective and progressive degeneration of upper and lower motor neurons, progressive muscle weakness and paralysis [162]. The pathophysiology of ∼20% of familial ALS is known to result from mutations of the superoxide dismutase-1 gene, whose overexpression in neuronal cells is sufficient to trigger apoptosis, and is inhibited by BCL-2 and caspase inhibitors [163, 164]. A proportion of SOD1 has been shown to localized to mitochondrial intermembrane space [165, 166] and matrix [167]. These findings support the hypothesis that mutant SOD1 may damage mitochondrial function and integrity directly, from inside the mitochondria. Caspase inhibition and BCL-2 expression are known to delay the onset and mortality of disease in mouse models of ALS [168, 169].
Cancer
Tumorigenesis can be aided by defects in apoptotic pathways enhancing cell survival and transformation, by giving the cells more time to accumulate genetic alterations that deregulate proliferation and provide growth advantage. The cancerous cells must evade or blunt their apoptotic response to survive and form tumours. Genes encoding important players of mitochondrial apoptosis are involved in the development of cancer. The two best examples are the p53 tumour suppressor and members of the BCL-2 protein family. The p53 tumour suppressor gene is mutated in the majority of human cancers [170], possibly owing to the diverse roles it serves in the cell. As ‘guardian’ of genome integrity p53 is capable of inducing cell cycle arrest and senescence as well as apoptosis [171]. p53−/minus; mice were predisposed to tumour development [172], and there was less evidence of apoptosis seen in situ in developed tumours. p53 is capable of inducing apoptosis through a variety of mechanisms, at least three of which involve the mitochondrial pathway [173]. First, the transcription of several pro-apoptotic genes, including the BCL-2 family members BAX, PUMA and NOXA, have been shown to be induced by p53 following genetic damage [174–176]. Second, there are reports suggesting that p53 induces the production of ROS that can stimulate mitochondrial apoptosis [177, 178]. Third, p53 can act through the permeabilization of the mitochondrial outer membrane, and the release of intermembrane proteins [91, 179].
Mitochondrial encephalomyopathies
Mitochondrial encephalomyopathies are a set of clinically diverse diseases that result from either inherited or spontaneous mutations in mtDNA which lead to altered function of the proteins or RNA molecules that normally reside in mitochondria [180]. Most of these mutations disrupt members of the electron transport chain, thus leading to defects in oxidative phosphorylation. The varied clinical presentations evident in those with the same mutation, and the varying mutations that present with the same clinical syndrome suggest that both genetic background and extra-genetic factors [181, 182] play a role in disease pathology. In addition, different mitochondria within the same cell can genetically complement one another, and thus the phenotype resulting from mtDNA mutations depends also on the percentage of mitochondria in a given cell that carry the mutation [182]. A recent study indicated that apoptotic cell death was dramatically increased in muscle biopsies from patients carrying mtDNA mutations in genes encoding bioenergetic proteins relative to those carrying mutations in structural genes [183]. Defects in oxidative phosphorylation have been demonstrated in many other neurodegenerative diseases, suggesting a mitochondrial role. These include: (i) Leber’s optic atrophy [184] and (ii) idiopathic dystonia [185], both with defects in respiratory chain complex I and (iii) some forms of hereditary spastic paraplegia, caused by the mitochondrial metalloproteinase paraplegin [186].
Others
Many viruses have acquired the capacity to intercept or to activate the principal signal transduction pathways leading to cell death [187, 188]. For example, certain viruses kill the host cell by inducing MOMP, while others prevent MOMP to allow propagation of the virus. The HIV protein, viral accessory protein R (Vpr) induces MOMP. The amino acids 52–96 of Vpr directly interacts with ANT and VDAC, thereby triggering MMP associated with ΔΨm loss, IMS proteins release, and caspase cascade activation [189]. When added in vitro to purified mouse liver mitochondria, a synthetic Vpr-derived peptide (Vpr52–96) induced large amplitude swelling. This effect could be prevented by BCL-2 as well as by pharmacological agents targeting ANT or VDAC [189]. Importantly, mutation in one of the arginine residues (R77Q) that is required for the interaction of Vpr with ANT [190] is associated with a reduced risk of developing AIDS. In contrast, the cytomegalovirus encodes several proteins that subvert host cell functions in order to favour viral propagation [191]. One of the best characterized among these factors is viral mitochondria-localized inhibitor of apoptosis (vMIA). vMIA has been shown to inhibit apoptosis triggered by different stimuli, including ligation of death receptors and exposure to cytotoxic agents. vMIA exerts its anti-apoptotic activity predominantly by inhibiting MMP at the level of mitochondria [191]. Several oncogenic viruses encode MOMP inhibitory proteins and in human beings, such proteins may contribute to the formation of virally-induced lymphomas or Kaposi’s sarcoma. Open reading frame (ORF)16 of human herpesvirus 8 encodes the so-called Kaposi sarcoma-associated BCL-2, a polypeptide of 175 residues that shares limited (15–20%) overall sequence identity with other BCL-2 family proteins [192]. HVS ORF16 has been shown to interact with BAX and BAK to inhibit virus-induced apoptosis [193].
Therapeutic strategies that promote MOMP and cell death
There are a number of drugs that are aimed to induce apoptosis by targeting components of the mitochondrial pathway to induce MOMP. A summary of the agents that promote MOMP and cell death can be found in Table 1.
Table 1.
Drugs promoting the mitochondrial membrane permeabilization
| Target | Drug | Type of compound | Remarks |
|---|---|---|---|
| Anti-apoptotic BCL-2 proteins | Oblimersen Sodium | Antisense BCL-2 oligonu-cleotide | Down-regulation of BCL-2 protein level phase III clinical trial in chronic lymphocytic leukaemia and melanoma |
| Antimicyn A | BH3 mimetic | Pan inhibitor of anti-apoptotic BCL-2 proteins | |
| Gossypol | BH3 mimetic | Pan inhibitor of anti-apoptotic BCL-2 proteins phase I/II clinical trials in metastatic breast cancer and glioblastome multiforme | |
| Porpullogallin | BH3 mimetic | Pan inhibitor of anti-apoptotic BCL-2 proteins | |
| Chelerythrine | BH3 mimetic | Inhibits BCL-xL by binding at the BH groove | |
| Sanguinarine | BH3 mimetic | Inhibits BCL-xL by binding at the BH1 region | |
| ABT-737 | BH3 mimetic | Inhibits BCL-2, BCL-xL and BCL-w | |
| Mitochondria | TEAM-VP | Chimeric peptide | Induces MMP viaVDAC and ANT interaction |
| Mastoparan | Peptide | Induces MMP viaVDAC interaction | |
| pCoxlV, 3-22aa | Peptide | Induces MMP viaCoxIV interaction | |
| (KLAKLAK)2 | Peptide | Direct inducer of MMP | |
| HXK2VBD | Peptide | Sensitizes to BAX-dependent apoptotis via inhibition of Hexokinase (HK) II/VDAC interaction | |
| 2-chloro-2′- deoxyadenosine; 2-chloro-2′-ara-fluorodeoxyadenosine | Deoxyadenosine analogue | Induces opening MPT pores | |
| Jasmonates | Jasmonic acid | Induces swelling in mitochondria in a PTPC-mediated manner | |
| Peripheral-type benzodiazepine receptor ligands | Small molecules | Induces MMP | |
| Lonidamine | Indazole-carboxylic acid | Inducer of MMP through ANT conformational change phase II/III clinical trials for metastatic breast, non-small cell lung, ovarian cancer and glioblastoma multiforme | |
| Lamellarins | Marine pyrrole alkaloids | Disruption of the inner mitochondrial transmembrane potential in a MPT-dependent manner | |
| Arsenite | Small molecule | Inducer of MPT pores phase II clinical trials for multiple myeloma | |
| Avicins | Triterpenoid saponins | Inducers of MMP | |
| Pyridinium derivative F16 | Cationic lipophilic | Accumultes in mitochordria; inducer of MMP | |
| MKT-077 | Cationic lipophilic | Accumulates in mitochondira; inducer of MMP; effects on mitochondrial DNA | |
| Verteporfm | Photo-activable agent | Inducer of MMP | |
| CM oro methyl-X-ro s amine | Photo-activable agent | Inducer of mitochondrial depolarization and swelling | |
| Hypericin | Photo-activable agent | Induces detachment of HKs from mitochondria | |
| Pc4 | Photo-activable agent | Induces degradation of BCL-2 |
VIMP: mitochondrial membane permeabilization and MPT: mitochondrial permeability transition.
Targeting the BCL-2 family
The ratio of the levels of pro-survival and pro-apoptotic members of the BCL-2 protein family is thought to be an important regulatory factor for determining mitochondrial integrity and regulates the sensitivity of mammalian cells to apoptotic stimuli. This information has, in the early days of apoptosis research, led to the consideration of BCL-2 family members as possible therapeutic targets for diseases with deregulated apoptosis (Fig. 3) [194–197]. The following section describes various approaches developed around the members of BCL-2 family.
Figure 3.
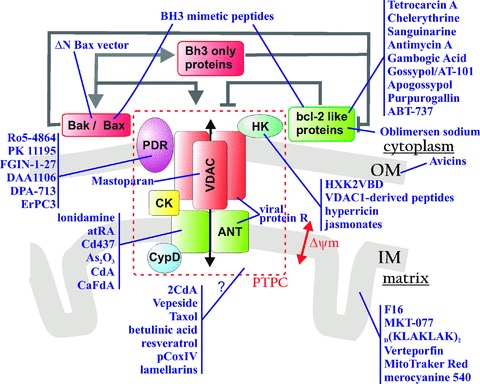
Therapeutic agents acting on mitochondria to promote cell death. Pharmacological inducers of cell death and their target molecules are shown. Please refer to the text for additional detail. Abbreviations: ANT, adenine nucleotide translocator; BAK, BCL-2 antagonist/killer; BAX, BCL-2-associated X protein; BCL-2, B-cell lymphoma 2 protein; BH3, BCL-2 homology domain 3; CK, creatine kinase; CypD, cyclophilin D; HK, hexokinase; IM, mitochondrial inner membrane; OM, mitochondrial outer membrane; PBR, peripheral-type benzodiazepine receptor; PTPC, permeability transition pore complex and VDAC, voltage-dependent anion channel.
BCL-2 antisense-based strategies
BCL-2 (B-cell lymphoma 2) is an oncoprotein, which was originally identified as the t(14;18) chromosomal translocation found in the majority of human follicular lymphomas [194]. BCL-2 plays a critical role in inhibiting mitochondria-dependent apoptotic cell death. Its pathologic over-expression observed in many tumour types identified BCL-2 as a possible drug target in the early 1990s. Because BCL-2 is an intracellular protein lacking intrinsic catalytic function, its inhibition by neutralizing antibodies or small molecule drugs are not viable options. On the other hand, in several preclinical and clinical studies antisense BCL-2 therapy combined with chemotherapy has proven to be beneficial in various tumour types. Antisense BCL-2 (AS BCL-2; G3139, oblimersen sodium, Genasense, Genta, Berkeley Heights, NJ, UDA) is an 18-bp phos-phorothioate oligonucleotide targeting the first six codons of BCL-2 mRNA. In preclinical studies, the treatment with antisense BCL-2 in combination with an anticancer drug decreased BCL-2 expression and enhanced the mitochondria-dependent apoptosis pathway leading to cell death [198–200]. Oblimersen sodium has advanced through clinical trials, including phase III with tolerable side effects [201] (http://www.clinicaltrial.com). However, the demonstration of efficacy of antisense BCL-2 in those trials has been variable. In chronic lymphocytic leukaemia, combined oblimersen, fludarabine and cyclophosphamide showed improved major responses in patients, whereas oblimersen and dacarbazine combination therapy in patients with metastatic melanoma or oblimersen and dexamethasone combination therapy in patient with myeloma provided no significant benefit in overall survival. In addition, in these clinical trials the down-regulation of BCL-2 was not observed with any high frequency in tumour cells [201]. In fact, in addition to its antisense effect, several non-antisense effects need to be considered when analyzing the therapeutic efficacy of antisense BCL-2 observed in clinical trials. Production of ROS, interferon (IFN)-γ production and immunostimulatory action through a cytosine-phosphate-guanosine motif in the antisense oligodeoxynucleotides might contribute to the antitumour effects [200, 202]. Thus, the efficacy of BCL-2 antisense strategies has not been overwhelming, and approval of oblimersen as an anticancer agent remains in doubt.
BAX-delivery vector
BAX is one of the pro-apoptotic factors that belong to the BCL-2 family, and its overexpression leads to apoptosis in a wide variety of mammalian cells [203]. BAX, which normally resides in the cytoplasm, translocates to mitochondria in response to apoptotic stimuli, promotes MOMP and elicits the release of pro-apoptotic factors from the intermembrane space [33]. Adenovirus-mediated BAX overexpression is capable of inducing cell death in vitroand in vivo by engaging the mitochondrial pathway [204–206]. Through its BH3 domain, BAX forms homodimers to promote apoptosis but also forms heterodimers with BCL-2 and BCL-xl, which silences its function. Likewise, adenovirus-mediated gene delivery of an amino-terminal truncated version of BAX (ΔN BAX: corresponding to amino acid 112–192 of full-length BAX), that cannot be suppressed by the anti-apoptotic BCL-2 family members has been generated. Interestingly, ΔN BAX exhibited a significantly stronger suppression of tumour growth than full-length BAX, suggesting that the truncated version of BAX may provide a better alternative for gene therapy trials [207].
BH3 mimetic peptides
Pro-apoptotic multidomain members of the BCL-2 family (BAK, BAX) are activated by BH3-only proteins (such as BAD, BID, BIM, PUMA, NOXA) to induce mitochondrial apoptotic death events, and BH3 domains are necessary and sufficient for this effect [208]. The interaction between these BCL-2 family members is primarily mediated through the amphipathic α-helices of their BH3 domains [209]. BH3-only proteins act in two ways, either by inactivation of the anti-apoptotic BCL-2 proteins and displacement of BAX (as seen with BAD) or by direct activation of BAX and BAK (as observed with BID and BIM). BH3 mimetic peptides derived not only from BH3-only proteins, such as BAD and BID but also from the multidomain BAX and BAK have been generated [210]. BH3 peptides longer than 14 amino acids can retain an α-helical structure and some biological activities [209]. For example, BH3 mimetic peptides induce oligomerization of BAX and BAK, permeabilization of MOM, and release of cytochrome c[211, 212].
In principle, peptides containing BH3 domain sequences should be explored as pharmaceutical lead molecules. However, their use as therapeutic agents is limited by their unfavourable pharmacological properties, including poor cellular permeability, bioavailability, solubility and metabolic stability in vivo. Several methods have been tried to overcome these limitations. BH3 peptides have been tagged with peptide transduction domains from Drosophila antennapedia protein, human immunodeficiency virus-1 trans-activating (TAT) protein or an arginine homopolymer (R8) transduction domain [211, 212]. These different approaches enhanced the intracellular uptake of BH3 peptides. A chemical strategy, termed hydrocarbon stapling, was also explored, and resulted in maintenance of the α-helical conformation, increased stability, cell-permeability, increased affinity to multidomain BCL-2 member pockets and improved pharmacological properties [209].
Natural and synthetic BH3 mimetic drugs
Natural compounds such as tetrocarcin A, a second metabolite derived from Actinomyces spp. [213], chelerythrine [214, 215] and sanguinarine which are plant benzophenanthridine alkaloids, antimycin A, a Streptomyces-derived inhibitor of ubiquinone–cytochrome c oxidoreductase at the mitochondrial respiration chain [216], gambogic acid derived from the gamboges resin of the tree Garcinia hanburyi[217], and certain polyphenols such as gossypol, apogossypol (compounds from cotton seed extracts) [218] and purpurogallin (a natural compound extracted from Quercus sp. nutgall) [218] promote death by binding BCL-2 and BCL-xl and inhibiting their anti-apoptotic functions (Fig. 3).
Computational molecular docking analysis predicted that antimycin A targets the BH3-binding pocket of the anti-apoptotic BCL-2 family molecules [216]. NMR binding studies with BCL-xl revealed that gossypol and purpurogallin also compete for the BH3-binding pocket [218]. Whereas some of these compounds act as ‘true BH3 mimetics’, others appear to inhibit anti-apoptotic BCL-2 family members without targeting the BH3-binding pocket per se. Indeed, chelerythrine and sanguinarine bind separately at the BH groove and BH1 region of BCL-xl respectively, as opposed to the BH3 binding cleft which is targeted by other known inhibitors of BCL-xl[215].
At the moment, of all the above mentioned natural BH3 mimetic small molecules, only gossypol, in an oral form (AT-101) has advanced into clinical trials (phase I/II) for the treatment of patients with refractory metastatic breast cancer [219] and for the treatment of patients with Glioblastoma Multiforme in combination with the alkylating agent temozolomide with or without radiation therapy (http://www.clinicaltrial.gov).
A recent highlight in the field is the development of ABT-737. ABT-737 is a cell permeating, synthetic BH3 mimetic that was designed by Oltersdorf et al. [220] using a NMR structure-based approach to target the BH3-binding groove on BCL-xl. It binds with high affinity in the subnanomolar range to BCL-2 and BCL-w. However, despite its high affinity for BCL-2, BCL-xl and BCL-w many cell types proved refractory to ABT-737. It appeared that the resistance reflects ABT-737’s inability to target another pro-survival BCL-2 relative, MCL-1. Subsequently, down-regulation of Mcl-1 by several strategies was shown to confer sensitivity to ABT-737 [221]. Numerous studies have evaluated the merit of ABT-737 in triggering apoptosis via the mitochondrial pathways in cancer cell lines and mouse xenograft models [222]. Collectively these studies indicate that ABT-737 as a single agent shows strong potency against a variety of tumour types such as lymphoma, leukaemia, multiple myeloma and small-cell lung cancer and that when used in combination therapy it may help to overcome drug resistance phenotypes in additional tumour types [200, 220, 223]. The BH3 mimetic has also recently been shown to sensitise cancer cells to TRAIL [224] and to induce IMM permeabilization and mitochondrial swelling reminiscent of MTP in chronic leukaemia cells [225]. Even if the preclinical data strongly support a rationale for clinical trials with ABT-737, the compound has not yet entered clinical trials [226].
Targeting mitochondria directly: mitochondriotoxic compounds inducing mitochondrial membrane permeabilization
Shortly after the discovery that MOMP is a ‘point of no return’[227] and that once it occurs cells die, mitochondria have become an attractive target to induce apoptosis (Fig. 3). In addition, the rich repertoire of mitochondrial proteins and the essential requirement of the membrane permeability barrier for proper organelle function offer an array of drug development opportunities. To date more than 20 mitochondriotoxic compounds acting directly on the mitochondria to induce cell death have been described and some of them have already been validated pre-clinically and entered clinical trials. These compounds can be classified according to their chemical nature into three main groups: peptide derivatives, small molecules and cationic lipophilic agents.
Peptide derivatives
Peptides derived from viral proteins [228], from proteins of the PTP and natural as well as synthetic peptides have been shown to be able to kill mammalian cells by triggering MOMP. The human immunodeficiency virus, HIV-1 encoded apoptogenic protein Vpr induces MMP via interactions with VDAC and ANT [190]. The chimeric peptide TEAM-VP using the MMP-inducing sequence derived from Vpr and a tumour blood vessel RGD-like ‘homing’ motif has been engineered. This virus-derived mitochondriotoxic compound targets mitochondria of angiogenic endothelial cells to induce MOMP and the release of mitochondrial apoptogenic molecules resulting in apoptosis [229].
Mastoparan, a peptide isolated from wasp venom (as well as its derivative mitoparan) has an α-helical structure and possesses positive charges clustered on one side of the helix. Mastoparan is the first peptide known to induce MOMP via interaction with VDAC in a CsA-regulated mechanism [230, 231]. A second amphipathic peptide, the signal sequence of cytochrome oxidase subunit IV from Neurospora crassa (pCoxIV, amino acids 3–22), which targets subunit IV to its mitochondrial location has also been shown to increase the permeability of isolated mitochondria [232].
A peptide structurally similar to Mastoparan, dklaklakklak-LAK or (KLAKLAK)2 (K = lysine, L = alanine and A = leucine) has recently been shown to disrupt mitochondrial membranes and directly permeabilize this organelle [233]. Moreover, when fused to targeting peptides that interact with surface receptors expressed on angiogenic endothelial cells in tumours (with the tumour blood vessel RGD-like ‘homing’ motif) [233] or on prostate vasculature (with the prostate-homing phage, SMSIARL) [234], or even with a peptide that inhibits the ErbB-2 receptor kinase [235], these chimeric peptides are translocated to the mitochondria where they induce MOMP especially in the targeted cell population. The ErbB-2 receptor kinase inhibiting peptide also exhibited ability to reduce tumour growth in HER-2-overexpressing human mammary xenografts established in SCID mice [235]. These studies provide a proof of concept for the strategy of targeting mitochondriotoxic hybrid molecules to cancer cells, primarily via the recognition by various surface receptors.
For over 70 years, it has been known that tumour cells exhibit a high rate of glycolysis. The high glycolytic rate is now known to be due in part to the greatly increased expression of HKs in transformed cells. HK isoforms I and II bind to VDAC and by this means interfere with the ability of BAX to interact with mitochondria and thereby cell death [58, 236]. A cell-permeable peptide analogue of HK-II VDAC binding domain (HXK2VBD) peptide fused to the internalization sequence of the Antennapedia homeoprotein has been shown to inhibit HK localization to the mitochondrion. HXK2VBD sensitizes cells to a BAX-dependent apoptosis inducer.
Therefore, it seems that interference with the binding of HK-I to mitochondria by VDAC1-derived peptides may offer a novel strategy by which to potentiate the efficacy of conventional chemotherapeutic agents [237].
The jasmonates are a group of plant hormones which help regulate plant growth and development. Jasmonates include jasmonic acid and its esters, such as methyl jasmonate (MeJa). MeJa induces death in cancer cells, while being selectively inactive towards non-transformed cells [238]. MeJa acts directly on mitochondria derived from cancer cells in a PTPC-mediated manner [239]. MeJa binds to human HK isoforms I and disrupt its interaction with VDAC, causing the inhibition of glycolysis and the induction of MOMP [240]. MeJa has already been shown to have selective anticancer activity in preclinical studies [241–243], and this finding may stimulate the development of a novel class of small anticancer compounds that inhibit the HK-VDAC interaction.
Small molecules
Conventional chemotherapeutic agents, such as 2-chloro-2′-deoxyadenosine (2CdA) [244], topoisomerase II inhibitor etoposide (Vepeside, VP16) [245] which affects DNA replication, and paclitaxel (Taxol, TM) [246] which disrupts microtubule assembly can disrupt the integrity of mitochondria and induce cell death via the opening of the MTP pore. However, the high doses required to obtain this effect raise the question of whether these drugs in a clinical setup can induce cell death via MOMP.
Synthetic ligands of the peripheral-type benzodiazepine receptor (PBR) such as the benzodiazepines (Ro5–4864), isoquinoline carboxamides (PK 11195), indoleacetamides (FGIN-1–27), phenoxyphenyl-acetamides (DAA1106), pyrazolopyrimidines (DPA-713) [247] and erucylphosphohomocholine (ErPC3) [248] can induce apoptosis or sensitize cells to apoptosis induction as demonstrated by the drop in the mitochondrial transmembrane potential and an increased mitochondrial release of cytochrome c and Smac/DIABLO proteins. These compounds can overcome the cytoprotective effects of BCL-2 or BCL-xl[249, 250]. Consistently many experimental studies in vitro and also in SCID mice transplanted with human tumour cells, suggest that PBR ligands might be good candidates as chemotherapeutic, or at least chemosensitizing, agents [249, 250]. Whether the apoptogenic or chemosensitizing effects of the above-mentioned agents are truly due to a direct effect on mitochondria, is at least in some cases, a matter of debate. For instance, the doses of PBR ligands required to obtain cytotoxic effects are several orders of magnitude higher than the Kd of the high-affinity PBR and in certain cases their cytotoxicity even appeared to be unrelated to PBR expression [251].
A number of agents, for example lonidamine [252, 253], all-trans-retinoic acid [254], the synthetic retinoid RAR-7 ligand 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid (CD437) [252, 255], arsenic trioxide (As2O3) [252], 2CdA Cladribine and 2-choloro-2′-ara-fluorodeoxyadenosine (CaFdA) [244], have been reported to induce a conformational change in the ANT leading to mitochondrial channel formation. Lonidamine enhances the apoptotic response to cisplatin, cyclophosphamide, doxorubicin, paclitaxel, melphalan and γ-irradiation both in vivo and in vitro[253]. Lonidamine kills a wide range of tumour cells in vitro and in animal models. This drug in combination therapy is currently being tested in phase II/III trials for metastatic breast [256], non-small cell lung cancer [257], ovarian cancer [258] and glioblastoma [259] with encouraging results so far. Arsenite, the trivalent inorganic salt formed from arsenic trioxide, which is used to treat acute promyelocytic leukaemia [260] and has entered phase II clinical trials for multiple myeloma [261], causes glutathione depletion, induces PTP opening and its effect is prevented by BCL-2. 2CdA and 2-choloro-2′-ara-fluorodeoxyadenosine (CaFdA) drugs are clinically used for the treatment of indolent lym-phoproliferative diseases, they disrupt the integrity of mitochondria and induce the release of pro-apoptotic mitochondrial proteins from these organelles [244, 262].
A number of agents that act on mitochondria possess a steroid-like core structure. For instance, this applies to the above mentioned ANT ligands CD437 [252, 255], all-trans-retinoic acid [254] as well avicins [263], betulinic acid [264] and resveratrol [265]. Betulinic acid, a naturally occurring pentacyclic triterpenoid, induces apoptosis in tumour cells through the mitochondrial pathway. Combined treatment with betulinic acid and anticancer drugs acted in concert to induce loss of mitochondrial membrane potential and the release of cytochrome c and Smac from mitochondria. Isolated mitochondria from different cell types are permeabilized by betulinic acid, and this effect is prevented by CsA, the ANT ligand bongkrekate, as well as by BCL-2 overexpression [264]. It is unclear, however, through which receptor (if any) betulinic acid acts on mitochondria. Avicins, a novel plant-derived metabolite reduces energy metabolism in tumour cells by targeting the outer mitochondrial membrane and pushing cells towards the apoptotic pathway by permeabilization of the outer mitochondrial membrane [263]. The effect of Resveratrol (3,5,4′-trihydroxy-trans-stilbene), a phytoalexin found in grapes and other plant food, is more complex as it has been shown to be able to both promote [266] and inhibit [267] the mitochondrial death pathways. Resveratrol treatment of isolated mitochondria also led to depolarization, suggesting that the drug may target mitochondria directly [265, 266]. However, as in the case with PBR ligands, it is always difficult to ascertain in an in vivo context whether mitochondria are targeted directly by the above drugs or whether mitochondrial damage is an event that is secondary to their interaction with other cellular targets.
Lamellarins are a large family of marine alkaloids, with potential anticancer activities, isolated from diverse marine organisms, mainly ascidians and sponges. The best known member in the series is lamellarin D (lam D), first regarded as a conventional topoisomerase I poison [268], it has been shown promote an MPT-dependent release of cytochrome c and AIF from isolated mitochondria [269, 270]. A few synthetic analogues of lam D have been selected as preclinical drug candidates based on their in vivo efficacy against a panel tumour xenograft models and acceptable absorption, distribution, metabolism and excretion profiles. One of the prominent candidates is the amino derivative PM031379 which has revealed little toxicity toward non tumour cells in vitro[269] and displays potent anticancer activities in vivo in a human colon tumour xenograft model [269].
Cationic lipophilic agents
The lipid composition of the IMM is very different from that of other intracellular membranes. Lipophilic cations can cross cellular membranes and accumulate in mitochondria driven by the mitochondrial membrane potential to induce MOMP (Fig. 3). Because ΔΨm is often higher in malignant cells, lipophilic cations may selectively accumulate in their mitochondria, sparing the organelles of normal cells [271]. Several types of cancer cells have been described to accumulate such agents, e.g. rhodamine 123, to a higher level than normal cells [272]. Attempts have been made to use cationic lipophilic toxins as mitochondriotoxic agents. For instance, the pyridinium derivative F16 accumulates in the mitochondria and inhibits growth of human breast cancer cell lines [273]. MKT-077, a cationic rhodacyanine dye, is selectively toxic to cancer cells in vitro and in vivo[274]. MKT-077 activity has been associated with an effect on mitochondrial membranes and mitochondrial DNA [275] The antitumour effect of MKT-077 in xenografted mice models along with its selective accumulation in tumour mitochondria prompted its evaluation in the clinic. Renal toxicity encountered during the phase I trial, stopped further development of MKT-077 [276]. Another cationic ampholyte is the previously mentioned a-helical peptide d(klaklak)2 which can directly permeabilize mitochondria. Furthermore, lipophilic cations may be employed as specific carriers, to selectively deliver toxins to mitochondria of cancer cells. An attempt was made in the early 1980s to complex platinum(II) tetrachlorodianion to rhodamine 123. The platinum-rhodamine 123 complex displayed some degree of selectivity towards transformed cells [277].
Finally, a promising class of photoactivatable antitumour agents is being developed for mitochondrion-targeted chemotherapy. Photodynamic therapy involves the treatment of tumours in which visible light is used to activate a photosensitizer. Mitochondrial membranes have been identified as an important intracellular target for singlet oxygen produced during the photochemical pathway. Verteporfin, a porphyrin-derived photosensitizer, similarly causes mitochondrial membrane permeabilization irrespective of BCL-2 or BCL-xl overexpression [278]. The photoproduct of merocyanine 540 triggers cytochrome c release from isolated mitochondria and promotes apoptosis [279].
Photoactivation also enhances the mitochondrial toxicity of the cationic rhodacyanine MKT-077 [274]. Another cationic lipophilic dye, chloromethyl-X-rosamine (MitoTraker Red), a mitochondrion-selective fluorescent probe, has a strong photosensitising action. Photo-irradiation of intact cells loaded with MitoTraker Red induces depolarization of the IMM and swelling of mitochondria, subsequently resulting in apoptosis [280]. The photosensitizer hypericin detaches HKs from mitochondria [281], whereas the phthalocyanine photosensitizer Pc4 causes the photochemical destruction of BCL-2 [282, 283].
Bypassing the mitochondria: mitochondrial pro-apoptotic factors as chemotherapeutic agents
MOMP results in the release of a plethora of cell death effectors from the intermembrane space. Such factors include Smac/DIABLO, a protein that exerts much of its pro-apoptotic function by neutralizing IAPs that function as caspase inhibitors. Another pro-apoptotic factor released from permeabilized mitochondria is the AIF, a mitochondrial flavoprotein that translocates to the nucleus where it contributes to chromatin degradation [284, 285].
The binding of Smac/DIABLO to IAPs is mediated by a relatively short stretch of amino acids located in the N-terminus of the protein [286]. Several studies have shown that overexpression of Smac/DIABLO sensitizes neoplastic cells to apoptotic death [287]. These findings prompted the development of peptides derived from the NH2-terminus of Smac/DIABLO and small molecules that mimic Smac/DIABLO functions which could potentially be used in cancer therapy. NH2-terminal peptides of Smac/DIABLO fused to Drosophila antennapaedia penetrating sequences enhance apoptosis mediated by different anti-neoplastic agents in breast cancer [288, 289] and glioblastoma cell lines [290, 291]. Similarly, a small Smac-mimic compound was able to increase the apoptotic effects of death stimuli [291]. It is interesting to note that this small molecule induces apoptosis by itself in MDA-MB-231 breast cancer cells, which have high expression levels of XIAP and c-IAP1. In contrast, it only sensitizes MDA-MB-453 (human breast cancer cell line) and T47D cells, which have low IAP expression, to apoptotic triggers [289]. The sensitizing effects of Smac peptidomimetics and small Smac-mimic compounds have also been demonstrated in vivo using malignant glioblastoma xenograft mouse models [289, 292]. Taken together, these results show that the NH2-terminal Smac/DIABLO derivatives and small molecules that mimic its function could be useful as adjuvant therapy in tumours and await clinical trials.
An alternative strategy is to exploit the pro-apoptotic properties of AIF. In fact, microinjection of recombinant AIF is sufficient to induce hallmarks of apoptosis [285, 293]. Different AIF delivery strategies are under development. Recently, a chimeric protein containing an ErbB-2-specific antibody and the cytotoxic moiety of AIF has been shown to direct AIF to ErbB-2-expressing cancer cells, causing their demise [294]. BZL101 is a drug derived from Scutellaria barbatae that has been shown to induce the translocation of AIF to the nucleus and the induction of cell death. Recent clinical trials have documented its effectiveness in treating patients with advanced breast cancers [295]. Likewise, the novel cationic amphiphilic compound atiprimod has been shown to exert antimyeloma effects in mouse experiments and its ability to kill mantle cell lymphoma cells has been attributed to the activation of the AIF-dependent pathway [296].
Therapeutic strategies that inhibit MOMP and cell death
Although mitochondria are an important target for drugs designed to kill target cells, especially in cancer chemotherapy, considerable work has also focused on protecting mitochondria in an effort to prevent cells from dying in stress situations such as during ischemia/reperfusion injury, stroke and drug-induced damage. Indeed, a number of pharmacological agents that directly or indirectly protect mitochondria (Table 2) have been discovered and show promising therapeutic applications (Fig. 4).
Table 2.
Drugs inhibiting the mitochondrial membrane permeabilization
| Target | Drug | Type of compound | Remarks |
|---|---|---|---|
| MPT pores | Cyclosporin A | Small molecule | Prevents opening of MPT pores via cyclophilin D (CypD) interaction. Side toxicity due to interaction with other cyclophilins |
| NIM811 | Small molecule | Prevents opening of MPT pores via CypD interaction. More specific than cyclosporine A (CsA). | |
| Sanglifehrin A | Small molecule | Prevents opening of MPT pores via CypD interaction. Binds to CypD at different sites from CsA | |
| Adenosine | IPC mimetics | Induces desensitization of MPT by causing IPC | |
| Bradykinin | IPC mimetics | Induces desensitization of MPT by causing IPC | |
| Opioids | IPC mimetics | Induces desensitization of MPT by causing IPC | |
| Diazoxide | IPC mimetics | Mimics IPC, induces desensitization of MPT by opening mitoKATP | |
| Pinacidil | IPC mimetics | Mimics IPC, induces desensitization of MPT by opening mitoKATP | |
| Nicoradil | IPC mimetics | Mimics IPC, induces desensitization of MPT by opening mitoKATP | |
| Isofluorane | Anaesthetic | Induces IPC-like condition (APC), inhibits complex I and glycogen synthase kinase 3 p, preserves mitochondrial functions. | |
| Minocycline | Semi-synthetic antibiotic | Inhibits MPT by decreasing mitochondrial Ca2+ uptake |
MPT: mitochondrial permeability transition; IPC: ischemic precondition. mitoKATP : mitochondrial ATP-sensitive K-channels and APC: anaesthetic precondition.
Figure 4.
Therapeutic agents acting on mitochondria to prevent cell death. Pharmacological compounds and their target molecules are shown. Please refer to the text for additional detail. Abbreviations: CsA, cyclosporine A; SfA, sanglifehrin A; PTPC, permeability transition pore complex; AIF, apoptosis inducing factor; tAIF, truncated AIF and ROS, reactive oxygen species.
Cyclosporin A and the inhibition of MPT
Work dating back to the early 1990s showed that CsA alleviated ischemic liver injury in vivo[297]. In these early studies, the link between hepatoprotection and the prevention of PT was not investigated, and CypD-independent effects of CsA cannot be ruled out. However, subsequent work in perfused hearts subjected to anoxic injury convincingly demonstrated that the ability of CsA to prevent pore opening was responsible for its protective effect on the heart [298]. Likewise, CsA was reported to protect isolated rat hepatocytes from prooxidant-induced cell death as a result of its pore blocking activity in mitochondria [299]. Subsequent in vivo and in vitro studies have identified a contribution of the PTP in CD95- and TNFR-1-induced liver injury [300]. These two death receptors are important mediators of inflammatory liver injury during septic shock and immune-dependent drug-induced hepatotoxicity. CsA has been shown in these cases to alleviate the injury, although the cytoprotective effect was generally more consistent under in vivo conditions as compared to the more variable cytoprotection achieved in liver cells grown in culture [247]. CsA was also found to significantly protect rats [301] and mice [302, 303] from acetaminophen (paracetamol)-induced liver injury. The protection correlated with a diminution of mitochondrial GSH depletion, cytochrome c release and mitochondrial swelling after their isolation [303]. Similarly, studies in liver slices and cultured or freshly isolated hepatocytes have found that the MPT inhibitor CsA (but not FK506, an immunosuppressive drug that does not block MPT) protected from paracetamol-induced loss of viability [301, 304–306], and in two studies [305, 306] the use of calcein acetoxymethyl ester in conjunction with a mitochondrial membrane potential probe, confirmed CsA-sensitive PT pore opening in hepatocytes exposed to paracetamol. CsA has also shown promising neuroprotective effects and is currently under clinical investigation against traumatic brain injury [307]. Indeed, preclinical work has shown that CsA (but not the inactive FK506) reduces cortical damage after traumatic brain injury in mice and rats even when administered after injury [308].
Novel CsA analogues and other inhibitors of the pore
The toxicity of CsA and its non-specificity towards multiple cyclophilins has hampered its wide-spread use to test the role of PT in different pathologies. The search for potent and more specific CsA analogues has led to the identification of NIM811 (N-methyl-4-isoleucine cyclosporine). This cyclic polypeptide is as potent as CsA in blocking the pore but is selective for CypD and has no immunosuppressive properties [309]. NIM811 has been used with success to block PTP opening and prevent cell death in a number of experimental settings, including experimental liver transplantation [310] and spinal cord injury [311]. Another potent inhibitor of the pore is sanglifehrin A. This compound is a macrolide produced by actinomycetes that binds to CypD at a different site from CsA [312] and that has been shown to have cytoprotective effects in the reperfused heart [313]. Another pharmacological inhibitor of PT with potential therapeutic applications is 5-(benzylsulfonyl)-4-bromo-2-methyl-3(2H)-pyridazinone [314]. However, further work is required to ascertain its usefulness as a PT blocker.
Preconditioning of the heart protects by sparing mitochondria
Ischemic preconditioning (IPC) has long been known to provide substantial protection to the myocardium. By exposing the heart to one or more brief cycles of sub-lethal ischemia, this organ is protected from subsequent more prolonged and lethal ischemic insult as first reported by [315]. Mitochondria play a key role in IPC, and they have been shown to respond to IPC with diminished calcium sequestration [316–318], improved respiration [319, 320] and desensitization to PT [313, 321]. Not surprisingly, considerable efforts have been devoted to the study of the role of PT in IPC. The sensitivity of cardiac mitochondria to undergo PT is consistently decreased after IPC [313, 321]. However, this is normally only observed in cells and intact tissues, not with mitochondria isolated from IPC myocardium. This suggests that IPC affects PT indirectly and involves key upstream signalling events that target the mitochondria. The signalling molecules that have been linked to IPC and inhibition of PT include among others protein kinase Cɛ, nitric oxide, protein kinase G, protein kinase B (Akt), glycogen synthase kinase 3p, AMPK, mitochondrial ATP sensitive K-channels and connexion 43. A full review of their individual contributions to IPC would be beyond the scope of this publication and the reader is referred to an excellent recent review on this topic [322]. Therefore, we will only focus on the pathways that have been identified as potential targets for pharmacological intervention, with the rationale that pharmacological induction of IPC could be used to protect the heart and possibly other organs such as liver and brain from ischemia/reperfusion damage.
Pharmacological IPC mimetics
A number of physiological ligands to plasma membrane receptors are released during ischemia and these are now known to play a key role in IPC (Fig. 4). These include adenosine, bradykinin and opioids and appear to provide the initial signal for the pathways leading to IPC. A proposed pathway involves receptor-mediated activation of phosphatidylinositol 3-kinase followed by the activation of Akt and endothelial nitric oxide synthase, causing it to produce nitric oxide and downstream activation of protein kinase G, which in turn causes the opening of the mitochondrial ATP-sensitive K-channels (mitoKATP) through a yet to be defined phosphorylation step [323]. In support of this pathway, the pharmacological inhibition of protein kinase c by KT-5823 prevented the desensitization of PT in IPC [324]. There is also evidence for the involvement of mitochondrial protein kinase Cɛ in the effect of IPC on mitochondria, either through signalling downstream of cell surface receptors or through ROS-mediated activation [325]. A direct physical interaction between protein kinase Cɛ and mitoKATP has been proposed [323, 324]. The evidence for mitoKATP being responsible for the manifestation of IPC stems to a large extent from pharmacological studies. Indeed, agents such as diazoxide and pinacidil have been shown to mimic IPC by opening mitoKATP[326, 327]. More recently, a similar mechanism of action on mitochondria through mitoKATP opening was reported with nicorandil [328] and bepridil [329]. The exact link between mitoKATP opening and desensitization of PT is unclear but may involve a mild uncoupling of the mitochondria leading to decreased mitochondrial Ca2+ levels and decreased ROS production. However, one of the key remaining issues is that neither the pharmacological tools that were used to implicate mitoKATP in IPC are not specific for this channel nor is the exact molecular identity of mitoKATP known at present.
Volatile anaesthetics such as isoflurane have also been known to induce an IPC-like condition known as anaesthetic preconditioning (APC) [330]. Like IPC, APC involves the preservation of mitochondrial function in the cell following ischemia/reperfusion injury. For example, isoflurane and sevoflurane have been reported to inhibit complex I of the respiratory chain [331] and to lead to mild uncoupling of the mitochondria [332] and the generation of ROS by complex III [333]. The opening of mitoKATP and mitoKCa2+ by the anaesthetics appears to play an important role in the mechanism of protection and the observed prevention of PT during subsequent conditions of ischemia/reperfusion [334]. Isoflurane has also been shown in the heart to inhibit glycogen synthase kinase 3β[335] which has been shown to play a role in controlling the mitochondrial pathway of apoptosis. Recent phosphoproteomic studies on mitochondria isolated from isoflurane preconditioned hearts have identified Tyr194 as a novel phosphorylation site on ANT1 [336]. Although this report demonstrated a role for this phosphorylation event in mitochondrial bioenergetics in yeast, its role in APC remains to be determined. In the brain, APC induced by isoflurane may additionally involve the up-regulation of the anti-apoptotic BCL-2 protein [337].
Minocycline
The semi-synthetic tetracycline antibiotic minocycline has been found in a large number of studies to provide protection from neurodegeneration and ischemic neuronal injury [338], CD95-mediated hepatic injury [131] and hypoxic/ischemic renal and hepatic injury [310, 339]. Different mechanisms involving mitochondria have been proposed by which minocycline may exert cytoprotection and these include decreased mitochondrial release of pro-apoptotic factors such as cytochrome c[340], up-regulation of anti-apoptotic BCL-2 [291] and inhibition of PT [310, 341]. The latter was attributed to minocycline decreasing mitochondrial Ca2+ uptake [310, 341]. However, the direct targeting of mitochondria by minocycline and the involvement PT in its protective effect have recently been challenged in preference to its anti-inflammatory properties [342] and effects on poly(ADP-ribose)polymerases (PARP) [343] (see below).
Inhibitors of PARP and the prevention of DNA damage-mediated mitochondrial damage
PARP are nuclear enzymes that are responsible for the formation of poly(ADP-ribose) (PAR) polymers from NAD+ on nuclear proteins to regulate gene expression, chromatin configuration and metabolism in response to DNA damage, ROS and ischemic injury [344]. Although the mechanism of PARP overactivation-induced cell death was initially attributed to excessive NAD and ATP consumption [345], there is recent evidence that suggests that PAR itself can act as a death signal. This has been observed in response to excitotoxic neuronal damage, N-methyl-N′-nitro-N-nitrosoguanidine-induced DNA damage or hydrogen peroxide treatment [95]. The target for PAR has been identified to be AIF which is released from mitochondria to translocate to the nucleus to induce DNA fragmentation, chromatin condensation and caspase-independent cell death. The release of active AIF from mitochondria occurs in response to PAR-mediated activation of calpain and the proteolytic processing of native AIF to its truncated form [95]. This event also requires BAX translocation to mitochondria and its activation (but not that of BAK) to induce MOMP [95]. The identification of PARP as a key trigger of cell death has prompted a number of preclinical and clinical studies on pharmacological inhibitors of PARP in an effort to prevent neuronal and cardiac cell death. For example, the PARP inhibitor PJ34 has been reported to be neuroprotective in a model of permanent focal cerebral ischemia in mice [346]. Other PARP inhibitors such as benzamide, nicotinamide, GPI-15427 and 4 amino 1,8-napthalimide has also been shown to be protective in various in vivo models of neuronal damage [347–350]. However, further studies are required to confirm that the protection afforded by PARP inhibitors in these studies indeed involves the inhibition of the PARP-PAR-AIF pathway.
Conclusion and future directions
Deregulation of the mitochondrial apoptosis pathway with a causative or contributing role in many diseases has become increasingly evident. Emerging knowledge about molecular mechanisms of apoptosis has revealed a plethora of potential drug discovery targets. Structural analysis of apoptotic proteins and studies of their biochemical mechanisms have suggested strategies for lead generation resulting in numerous novel chemical entities with mechanism-based activities. The mitochondrial apoptosis pathway holds great promise as a target for therapeutic intervention. There is ample evidence showing that apoptosis can be potently activated with the use of compounds targeting key protein components of the mitochondria cell death pathway. Whether or not these routes and targets are suitable to block the proliferation of tumour cells remain to be seen. The relatively low rate of clinical entry associated with these molecules is related to the lack of specificity, low efficacy, or development of drug resistance. These issues are being addressed as our understanding of the field evolves. However, pharmacological control of mitochondria is not without risk for the physiology of normal cells as, mitochondria play a critical role in supplying the cell with the bulk of its ATP needs via oxidative phosphorylation and any cell type or tissue with a high aerobic energy requirement is likely to be affected when this organelle is dysfunctional. Therefore, due to the frequent implication of mitochondria in cardiac and neurodegenerative disorders, the impact of mitochondria-targeted anticancer agents on normal physiology of cardiovascular system and central nervous system will need a special attention.
Acknowledgments
We apologize to authors whose primary references could not be cited due to space limitations. We thank Drs. Sandra Healy and Alesandro Natoni for critical reading of the manuscript.
References
- 1.Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113:753–7. doi: 10.1242/jcs.113.5.753. [DOI] [PubMed] [Google Scholar]
- 2.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samali A, Zhivotovsky B, Jones D, Nagata S, Orrenius S. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999;6:495–6. doi: 10.1038/sj.cdd.4400520. [DOI] [PubMed] [Google Scholar]
- 4.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;28:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 5.Salvesen GS. Caspases and apoptosis. Essays Biochem. 2002;38:9–19. doi: 10.1042/bse0380009. [DOI] [PubMed] [Google Scholar]
- 6.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;9:443–6. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Wang J. Initiator caspases in apoptosis signaling pathways. Apoptosis. 2002;7:313–9. doi: 10.1023/a:1016167228059. [DOI] [PubMed] [Google Scholar]
- 8.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial per-meabilization. Science. 2002;297:1352–4. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 9.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem. 2002;277:13430–7. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 11.Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277:29803–9. doi: 10.1074/jbc.M204185200. [DOI] [PubMed] [Google Scholar]
- 12.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 14.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 15.Saelens X, Festjens N, Vande Walle L, Van Gurp M, Van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–74. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 16.Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999;18:2040–8. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005;118:265–7. doi: 10.1242/jcs.01610. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA. 2001;98:13884–8. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002;21:4520–30. doi: 10.1093/emboj/cdf441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–3. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 21.Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–20. doi: 10.1016/s1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 22.Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, Krammer PH, Walczak H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. doi: 10.1016/s1074-7613(00)80211-3. [DOI] [PubMed] [Google Scholar]
- 23.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–87. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stennicke HR, Jürgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084–90. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- 25.Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273:16589–94. doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 27.Green DR, Amarante-Mendes GP. The point of no return: mitochondria, caspases, and the commitment to cell death. Results Probl Cell Differ. 1998;24:45–61. doi: 10.1007/978-3-540-69185-3_3. [DOI] [PubMed] [Google Scholar]
- 28.Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 29.Martinou I, Desagher S, Eskes R, Antonsson B, André E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–9. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potts PR, Singh S, Knezek M, Thompson CB, Deshmukh M. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J Cell Biol. 2003;163:789–99. doi: 10.1083/jcb.200307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 32.Waterhouse NJ, Goldstein JC, Von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319–28. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization. Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 35.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 36.Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–63. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- 37.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–42. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 39.Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, Newmeyer DD. The proapoptotic proteins, Bid and Bax, cause a limited per-meabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–22. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Saez AJ, Mingarro I, Perez-Paya E, Salgado J. Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry. 2004;43:10930–43. doi: 10.1021/bi036044c. [DOI] [PubMed] [Google Scholar]
- 41.Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–8. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 43.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 44.Szabo I, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett. 1993;330:201–5. doi: 10.1016/0014-5793(93)80273-w. [DOI] [PubMed] [Google Scholar]
- 45.Szabo I, De Pinto V, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electro-physiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett. 1993;330:206–10. doi: 10.1016/0014-5793(93)80274-x. [DOI] [PubMed] [Google Scholar]
- 46.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol. 2001;152:237–50. doi: 10.1083/jcb.152.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim Biophys Acta. 2006;1757:590–5. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 48.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mito-chondrial-dependent cell death. Nat Cell Biol. 2007;9:550–5. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–35. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 50.Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–90. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–5. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimizu S, Shinohara Y, Tsujimoto Y. Bax and Bcl-xL independently regulate apoptotic changes of yeast mitochondria that require VDAC but not adenine nucleotide translocator. Oncogene. 2000;19:4309–18. doi: 10.1038/sj.onc.1203788. [DOI] [PubMed] [Google Scholar]
- 53.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 54.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 55.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 56.Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–6. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 57.Nakashima RA. Hexokinase-binding properties of the mitochondrial VDAC protein: inhibition by DCCD and location of putative DCCD-binding sites. J Bioenerg Biomembr. 1989;21:461–70. doi: 10.1007/BF00762518. [DOI] [PubMed] [Google Scholar]
- 58.Galluzzi L, Kepp O, Tajeddine N, Kroemer G. Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene. 2008;27:4633–5. doi: 10.1038/onc.2008.114. [DOI] [PubMed] [Google Scholar]
- 59.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40:171–82. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–23. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- 62.Hajnoczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–54. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- 63.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–99. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 65.Distelhorst CW, Shore GC. Bcl-2 and calcium: controversy beneath the surface. Oncogene. 2004;23:2875–80. doi: 10.1038/sj.onc.1207519. [DOI] [PubMed] [Google Scholar]
- 66.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–16. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–18. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- 68.Hansson MJ, Månsson R, Morota S, Uchino H, Kallur T, Sumi T, Ishii N, Shimazu M, Keep MF, Jegorov A, Elmér E. Calcium-induced generation of reactive oxygen species in brain mitochondria is mediated by permeability transition. Free Radic Biol Med. 2008;45:284–94. doi: 10.1016/j.freeradbiomed.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 69.Petersen A, Castilho RF, Hansson O, Wieloch T, Brundin P. Oxidative stress, mitochondrial permeability transition and activation of caspases in calcium ionophore A23187-induced death of cultured striatal neurons. Brain Res. 2000;857:20–9. doi: 10.1016/s0006-8993(99)02320-3. [DOI] [PubMed] [Google Scholar]
- 70.Kong D, Xu L, Yu Y, Zhu W, Andrews DW, Yoon Y, Kuo TH. Regulation of Ca2+ -induced permeability transition by Bcl-2 is antagonized by Drpl and hFis1. Mol Cell Biochem. 2005;272:187–99. doi: 10.1007/s11010-005-7323-3. [DOI] [PubMed] [Google Scholar]
- 71.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–14. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 72.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu J, Dong L, Outten CE. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J Biol Chem. 2008;283:29126–34. doi: 10.1074/jbc.M803028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bucher JR, Tien M, Aust SD. The requirement for ferric in the initiation of lipid peroxidation by chelated ferrous iron. Biochem Biophys Res Commun. 1983;111:777–84. doi: 10.1016/0006-291x(83)91366-9. [DOI] [PubMed] [Google Scholar]
- 75.Chen JJ, Bertrand H, Yu BP. Inhibition of adenine nucleotide translocator by lipid peroxidation products. Free Radic Biol Med. 1995;19:583–90. doi: 10.1016/0891-5849(95)00066-7. [DOI] [PubMed] [Google Scholar]
- 76.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–12. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 77.Stadtman ER, Oliver CN. Metal-catalyzed oxidation of proteins. Physiological consequences. J Biol Chem. 1991;266:2005–8. [PubMed] [Google Scholar]
- 78.Starke-Reed PE, Oliver CN. Protein oxidation and proteolysis during aging and oxidative stress. Arch Biochem Biophys. 1989;275:559–67. doi: 10.1016/0003-9861(89)90402-5. [DOI] [PubMed] [Google Scholar]
- 79.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem. 1990;265:16330–6. [PubMed] [Google Scholar]
- 80.Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996;378:150–2. doi: 10.1016/0014-5793(95)01449-7. [DOI] [PubMed] [Google Scholar]
- 81.Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxy-dopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun. 2005;327:801–10. doi: 10.1016/j.bbrc.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 82.Quigney DJ, Gorman AM, Samali A. Heat shock protects PC12 cells against MPP+ toxicity. Brain Res. 2003;993:133–9. doi: 10.1016/j.brainres.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 83.Shidoji Y, Hayashi K, Komura S, Ohishi N, Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun. 1999;264:343–7. doi: 10.1006/bbrc.1999.1410. [DOI] [PubMed] [Google Scholar]
- 84.Shidoji Y, Komura S, Ohishi N, Yagi K. Interaction between cytochrome c and oxidized mitochondrial lipids. Subcell Biochem. 2002;36:19–37. [PubMed] [Google Scholar]
- 85.Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Rèmy R, Xie ZH, Reed JC, Kroemer G. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med. 1998;187:1261–71. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marzo I, Susin SA, Petit PX, Ravagnan L, Brenner C, Larochette N, Zamzami N, Kroemer G. Caspases disrupt mitochondrial membrane barrier function. FEBS Lett. 1998;427:198–202. doi: 10.1016/s0014-5793(98)00424-4. [DOI] [PubMed] [Google Scholar]
- 87.Enoksson M, Robertson JD, Gogvadze V, Bu P, Kropotov A, Zhivotovsky B, Orrenius S. Caspase-2 permeabilizes the outer mitochondrial membrane and disrupts the binding of cytochrome c to anionic phospholipids. J Biol Chem. 2004;279:49575–8. doi: 10.1074/jbc.C400374200. [DOI] [PubMed] [Google Scholar]
- 88.Ricci JE, Muñoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–86. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 89.Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003;22:4385–99. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–51. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 92.Amsel AD, Rathaus M, Kronman N, Cohen HY. Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc Natl Acad Sci USA. 2008;105:5117–22. doi: 10.1073/pnas.0706700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–40. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 94.Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y, Zhang L, Yoneda Y, Fujii Y, Skoultchi AI, Tsujimoto Y. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell. 2003;114:673–88. doi: 10.1016/s0092-8674(03)00719-0. [DOI] [PubMed] [Google Scholar]
- 95.Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–62. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Solary E, Giordanetto F, Kroemer G. Re-examining the role of cytochrome c in cell death. Nat Genet. 2008;40:379–80. doi: 10.1038/ng0408-379. [DOI] [PubMed] [Google Scholar]
- 97.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–4. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 98.Ow YL, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–42. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 99.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–37. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 100.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, De La Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–52. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 101.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–37. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 102.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–50. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 103.Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, Yeh WC, Ohashi P, Wang X, Mak TW. Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell. 2005;121:579–91. doi: 10.1016/j.cell.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 104.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–32. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 105.Yu X, Acehan D, Ménétret JF, Booth CR, Ludtke SJ, Riedl SJ, Shi Y, Wang X, Akey CW. A structure of the human apoptosome at 12.8 A resolution provides insights into this cell death platform. Structure. 2005;13:1725–35. doi: 10.1016/j.str.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 106.Yu X, Wang L, Acehan D, Wang X, Akey CW. Three-dimensional structure of a double apoptosome formed by the Drosophila Apaf-1 related killer. J Mol Biol. 2006;355:577–89. doi: 10.1016/j.jmb.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 107.Kim HE, Jiang X, Du F, Wang X. PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol Cell. 2008;30:239–47. doi: 10.1016/j.molcel.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 108.Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476–83. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 109.Kurokawa M, Zhao C, Reya T, Kornbluth S. Inhibition of apoptosome formation by suppression of Hsp90beta phosphorylation in tyrosine kinase-induced leukemias. Mol Cell Biol. 2008;28:5494–506. doi: 10.1128/MCB.00265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yamamoto M, Torigoe T, Kamiguchi K, Hirohashi Y, Nakanishi K, Nabeta C, Asanuma H, Tsuruma T, Sato T, Hata F, Ohmura T, Yamaguchi K, Kurotaki T, Hirata K, Sato N. A novel isoform of TUCAN is overexpressed in human cancer tissues and suppresses both caspase-8-and caspase-9-mediated apoptosis. Cancer Res. 2005;65:8706–14. doi: 10.1158/0008-5472.CAN-04-4649. [DOI] [PubMed] [Google Scholar]
- 111.Pathan N, Marusawa H, Krajewska M, Matsuzawa S, Kim H, Okada K, Torii S, Kitada S, Krajewski S, Welsh K, Pio F, Godzik A, Reed JC. TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J Biol Chem. 2001;276:32220–9. doi: 10.1074/jbc.M100433200. [DOI] [PubMed] [Google Scholar]
- 112.Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5:647–54. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- 113.Jiang X, Kim HE, Shu H, Zhao Y, Zhang H, Kofron J, Donnelly J, Burns D, Ng SC, Rosenberg S, Wang X. Distinctive roles of PHAP proteins and prothymosin-alpha in a death regulatory pathway. Science. 2003;299:223–6. doi: 10.1126/science.1076807. [DOI] [PubMed] [Google Scholar]
- 114.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–23. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tenev T, Zachariou A, Wilson R, Ditzel M, Meier P. IAPs are functionally nonequivalent and regulate effector caspases through distinct mechanisms. Nat Cell Biol. 2005;7:70–7. doi: 10.1038/ncb1204. [DOI] [PubMed] [Google Scholar]
- 116.Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Crystal structure of a procaspase-7 zymogen: mechanisms of activation and substrate binding. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 117.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11:519–27. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 118.Vaux DL, Silke J. IAPs–the ubiquitin connection. Cell Death Differ. 2005;12:1205–7. doi: 10.1038/sj.cdd.4401696. [DOI] [PubMed] [Google Scholar]
- 119.Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–97. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 120.Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–60. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- 121.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 122.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 123.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis proteincas-pase interaction. J Biol Chem. 2002;277:432–8. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 124.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–44. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 125.Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ, Vaux DL. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem. 2002;277:445–54. doi: 10.1074/jbc.M109891200. [DOI] [PubMed] [Google Scholar]
- 126.Okada H, Suh WK, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW, Wang X, Mak TW. Generation and characterization of Smac/DIABLO-deficient mice. Mol Cell Biol. 2002;22:3509–17. doi: 10.1128/MCB.22.10.3509-3517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–52. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 128.Uren AG, Pakusch M, Hawkins CJ, Puls KL, Vaux DL. Cloning and expression of apoptosis inhibitory protein homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor-associated factors. Proc Natl Acad Sci USA. 1996;93:4974–8. doi: 10.1073/pnas.93.10.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnóczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–7. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- 130.Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–62. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Müller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Krüger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14:2099–111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 132.Gupta S, Singh R, Datta P, Zhang Z, Orr C, Lu Z, Dubois G, Zervos AS, Meisler MH, Srinivasula SM, Fernandes-Alnemri T, Alnemri ES. The C-terminal tail of presenilin regulates Omi/HtrA2 protease activity. J Biol Chem. 2004;279:45844–54. doi: 10.1074/jbc.M404940200. [DOI] [PubMed] [Google Scholar]
- 133.Van Loo G, Schotte P, Van Gurp M, Demol H, Hoorelbeke B, Gevaert K, Rodriguez I, Ruiz-Carrillo A, Vandekerckhove J, Declercq W, Beyaert R, Vandenabeele P. Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001;8:1136–42. doi: 10.1038/sj.cdd.4400944. [DOI] [PubMed] [Google Scholar]
- 134.Zhang J, Dong M, Li L, Fan Y, Pathre P, Dong J, Lou D, Wells JM, Olivares-Villagómez D, Van Kaer L, Wang X, Xu M. Endonuclease G is required for early embryogenesis and normal apoptosis in mice. Proc Natl Acad Sci USA. 2003;100:15782–7. doi: 10.1073/pnas.2636393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Irvine RA, Adachi N, Shibata DK, Cassell GD, Yu K, Karanjawala ZE, Hsieh CL, Lieber MR. Generation and characterization of endonuclease G null mice. Mol Cell Biol. 2005;25:294–302. doi: 10.1128/MCB.25.1.294-302.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–23. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- 137.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prévost MC, Leber B, Andrews D, Penninger J, Kroemer G. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–39. [PubMed] [Google Scholar]
- 138.Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–74. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 139.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–91. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 140.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 141.Skulachev VP. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol Aspects Med. 1999;20:139–84. doi: 10.1016/s0098-2997(99)00008-4. [DOI] [PubMed] [Google Scholar]
- 142.Leducq N, Delmas-Beauvieux MC, Bourdel-Marchasson I, Dufour S, Gallis JL, Canioni P, Diolez P. Mitochondrial permeability transition during hypothermic to normothermic reperfusion in rat liver demonstrated by the protective effect of cyclosporin A. Biochem J. 1998;336:501–6. doi: 10.1042/bj3360501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pérez-Pinzón MA, Xu GP, Born J, Lorenzo J, Busto R, Rosenthal M, Sick TJ. Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J Cereb Blood Flow Metab. 1999;19:39–43. doi: 10.1097/00004647-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 144.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–72. [PubMed] [Google Scholar]
- 145.Beal MF. Mitochondria and neurodegeneration. Novartis Found Symp. 2007;287:183–92. doi: 10.1002/9780470725207.ch13. [DOI] [PubMed] [Google Scholar]
- 146.Li WP, Chan WY, Lai HW, Yew DT. Terminal dUTP nick end labeling (TUNEL) positive cells in the different regions of the brain in normal aging and Alzheimer patients. J Mol Neurosci. 1997;8:75–82. doi: 10.1007/BF02736774. [DOI] [PubMed] [Google Scholar]
- 147.Yoshiyama Y, Yamada T, Asanuma K, Asahi T. Apoptosis related antigen, Le(Y) and nick-end labeling are positive in spinal motor neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1994;88:207–11. doi: 10.1007/BF00293395. [DOI] [PubMed] [Google Scholar]
- 148.Thomas LB, Gates DJ, Richfield EK, O’Brien TF, Schweitzer JB, Steindler DA. DNA end labeling (TUNEL) in Huntington’s disease and other neuropathological conditions. Exp Neurol. 1995;133:265–72. doi: 10.1006/exnr.1995.1029. [DOI] [PubMed] [Google Scholar]
- 149.Mochizuki H, Goto K, Mori H, Mizuno Y. Histochemical detection of apoptosis in Parkinson’s disease. J Neurol Sci. 1996;137:120–3. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 150.Newman M, Musgrave IF, Lardelli M. Alzheimer disease: amyloidogenesis, the presenilins and animal models. Biochim Biophys Acta. 2007;1772:285–97. doi: 10.1016/j.bbadis.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 151.MacGibbon GA, Lawlor PA, Sirimanne ES, Walton MR, Connor B, Young D, Williams C, Gluckman P, Faull RL, Hughes P, Dragunow M. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer’s disease hippocampus. Brain Res. 1997;750:223–34. doi: 10.1016/s0006-8993(96)01351-0. [DOI] [PubMed] [Google Scholar]
- 152.Reichmann H, Florke S, Hebenstreit G, Schrubar H, Riederer P. Analyses of energy metabolism and mitochondrial genome in postmortem brain from patients with Alzheimer’s disease. J Neurol. 1993;240:377–80. doi: 10.1007/BF00839971. [DOI] [PubMed] [Google Scholar]
- 153.Mattson MP, Zhu H, Yu J, Kindy MS. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20:1358–64. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Passer BJ, Pellegrini L, Vito P, Ganjei JK, D’Adamio L. Interaction of Alzheimer’s presenilin-1 and presenilin-2 with Bcl-X(L). A potential role in modulating the threshold of cell death. J Biol Chem. 1999;274:24007–13. doi: 10.1074/jbc.274.34.24007. [DOI] [PubMed] [Google Scholar]
- 155.Schulz JB. Update on the pathogenesis of Parkinson’s disease. J Neurol. 2008;255:3–7. doi: 10.1007/s00415-008-5011-4. [DOI] [PubMed] [Google Scholar]
- 156.Vila M, Ramonet D, Perier C. Mitochondrial alterations in Parkinson’s disease: new clues. J Neurochem. 2008;107:317–28. doi: 10.1111/j.1471-4159.2008.05604.x. [DOI] [PubMed] [Google Scholar]
- 157.Lud Cadet J, Harrington B, Ordonez S. Bcl-2 overexpression attenuates dopamine-induced apoptosis in an immortalized neural cell line by suppressing the production of reactive oxygen species. Synapse. 2000;35:228–33. doi: 10.1002/(SICI)1098-2396(20000301)35:3<228::AID-SYN8>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 158.Mogi M, Harada M, Kondo T, Mizuno Y, Narabayashi H, Riederer P, Nagatsu T. bcl-2 protein is increased in the brain from parkinsonian patients. Neurosci Lett. 1996;215:137–9. [PubMed] [Google Scholar]
- 159.Roze E, Saudou F, Caboche J. Pathophysiology of Huntington’s disease: from huntingtin functions to potential treatments. Curr Opin Neurol. 2008;21:497–503. doi: 10.1097/WCO.0b013e328304b692. [DOI] [PubMed] [Google Scholar]
- 160.Liu YF. Expression of polyglutamine-expanded Huntingtin activates the SEK1-JNK pathway and induces apoptosis in a hippocampal neuronal cell line. J Biol Chem. 1998;273:28873–7. doi: 10.1074/jbc.273.44.28873. [DOI] [PubMed] [Google Scholar]
- 161.Brouillet E, Hantraye P. Effects of chronic MPTP and 3-nitropropionic acid in nonhuman primates. Curr Opin Neurol. 1995;8:469–73. doi: 10.1097/00019052-199512000-00014. [DOI] [PubMed] [Google Scholar]
- 162.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–23. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 163.Ghadge GD, Lee JP, Bindokas VP, Jordan J, Ma L, Miller RJ, Roos RP. Mutant superoxide dismutase-1-linked familial amyotrophic lateral sclerosis: molecular mechanisms of neuronal death and protection. J Neurosci. 1997;17:8756–66. doi: 10.1523/JNEUROSCI.17-22-08756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee JP, Palfrey HC, Bindokas VP, Ghadge GD, Ma L, Miller RJ, Roos RP. The role of immunophilins in mutant superoxide dismutase-1linked familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1999;96:3251–6. doi: 10.1073/pnas.96.6.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–33. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 166.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–9. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 167.Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochon-drial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–70. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li M, Ona VO, Guégan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of cas-pase-1 and caspase-3 in an ALS trans-genic mouse model. Science. 2000;288:335–9. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 169.Spooren WP, Hengerer B. DNA laddering and caspase 3-like activity in the spinal cord of a mouse model of familial amy-otrophic lateral sclerosis. Cell Mol Biol. 2000;46:63–9. [PubMed] [Google Scholar]
- 170.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apopto-sis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 171.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–10. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 172.Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996;7:269–78. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 173.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–21. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 175.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–94. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 176.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 177.Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci USA. 1996;93:11848–52. doi: 10.1073/pnas.93.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li PF, Dietz R, Von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apop-tosis blocked by Bcl-2. EMBO J. 1999;18:6027–36. doi: 10.1093/emboj/18.21.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–5. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 180.Mitochondrial Medicine Society’s Committee on Diagnosis. Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Wong LJ, Cohen BH, Naviaux RK. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94:16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, Shohat M, Fischel-Ghodsian N. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syn-dromic deafness. Nat Genet. 1993;4:289–94. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 182.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 183.Mirabella M, Di Giovanni S, Silvestri G, Tonali P, Servidei S. Apoptosis in mitochondrial encephalomyopathies with mitochondrial DNA mutations: a potential pathogenic mechanism. Brain. 2000;123:93–104. doi: 10.1093/brain/123.1.93. [DOI] [PubMed] [Google Scholar]
- 184.Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase sub-unit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA. 1994;91:6206–10. doi: 10.1073/pnas.91.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schapira AH, Warner T, Gash MT, Cleeter MW, Marinho CF, Cooper JM. Complex I function in familial and sporadic dystonia. Ann Neurol. 1997;41:556–9. doi: 10.1002/ana.410410421. [DOI] [PubMed] [Google Scholar]
- 186.Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metallo-protease. Cell. 1998;93:973–83. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 187.Best SM. Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol. 2008;62:171–92. doi: 10.1146/annurev.micro.62.081307.163009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochon-drial apoptosis. PLoS Pathog. 2008;4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jacotot E, Ferri KF, El Hamel C, Brenner C, Druillennec S, Hoebeke J, Rustin P, Métivier D, Lenoir C, Geuskens M, Vieira HL, Loeffler M, Belzacq AS, Briand JP, Zamzami N, Edelman L, Xie ZH, Reed JC, Roques BP, Kroemer G. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J Exp Med. 2001;193:509–19. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci USA. 1999;96:12536–41. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS, Hardwick JM. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci USA. 1997;94:690–4. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nava VE, Cheng EH, Veliuona M, Zou S, Clem RJ, Mayer ML, Hardwick JM. Herpesvirus saimiri encodes a functional homolog of the human bcl-2 oncogene. J Virol. 1997;71:4118–22. doi: 10.1128/jvi.71.5.4118-4122.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–3. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 195.Ho PT, Parkinson DR. Antisense oligonucleotides as therapeutics for malignant diseases. Semin Oncol. 1997;24:187–202. [PubMed] [Google Scholar]
- 196.Miyashita T, Reed JC. bcl-2 gene transfer increases relative resistance of S49.1 and WEHI7.2 lymphoid cells to cell death and DNA fragmentation induced by glucocorticoids and multiple chemotherapeutic drugs. Cancer Res. 1992;52:5407–11. [PubMed] [Google Scholar]
- 197.Sakakura C, Sweeney EA, Shirahama T, Igarashi Y, Hakomori S, Tsujimoto H, Imanishi T, Ogaki M, Ohyama T, Yamazaki J, Hagiwara A, Yamaguchi T, Sawai K, Takahashi T. Overexpression of bax sensitizes breast cancer MCF-7 cells to cisplatin and etoposide. Surg Today. 1997;27:676–9. doi: 10.1007/BF02388231. [DOI] [PubMed] [Google Scholar]
- 198.Kim R, Emi M, Tanabe K, Toge T. Preclinical evaluation of antisense bcl-2 as a chemosensitizer for patients with gastric carcinoma. Cancer. 2004;101:2177–86. doi: 10.1002/cncr.20636. [DOI] [PubMed] [Google Scholar]
- 199.Emi M, Kim R, Tanabe K, Uchida Y, Toge T. Targeted therapy against Bcl-2-related proteins in breast cancer cells. Breast Cancer Res. 2005;7:R940–52. doi: 10.1186/bcr1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ. 2008;15:977–87. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Letai A. Pharmacological manipulation of Bcl-2 family members to control cell death. J Clin Invest. 2005;115:2648–55. doi: 10.1172/JCI26250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kim R, Emi M, Matsuura K, Tanabe K. Antisense and nonantisense effects of anti-sense Bcl-2 on multiple roles of Bcl-2 as a chemosensitizer in cancer therapy. Cancer Gene Ther. 2007;14:1–11. doi: 10.1038/sj.cgt.7700986. [DOI] [PubMed] [Google Scholar]
- 203.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–19. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 204.Kagawa S, Gu J, Swisher SG, Ji L, Roth JA, Lai D, Stephens LC, Fang B. Antitumor effect of adenovirus-mediated Bax gene transfer on p53-sensitive and p53-resistant cancer lines. Cancer Res. 2000;60:1157–61. [PubMed] [Google Scholar]
- 205.Li X, Marani M, Yu J, Nan B, Roth JA, Kagawa S, Fang B, Denner L, Marcelli M. Adenovirus-mediated Bax overexpression for the induction of therapeutic apoptosis in prostate cancer. Cancer Res. 2001;61:186–91. [PubMed] [Google Scholar]
- 206.Arafat WO, Buchsbaum DJ, Gómez-Navarro J, Tawil SA, Olsen C, Xiang J, El-Akad H, Salama AM, Badib AO, Stackhouse MA, Curiel DT. An adenovirus encoding proapoptotic Bax synergistically radiosensitizes malignant glioma. Int J Radiat Oncol Biol Phys. 2003;55:1037–50. doi: 10.1016/s0360-3016(02)04488-7. [DOI] [PubMed] [Google Scholar]
- 207.Usui K, Saijo Y, Narumi K, Koyama S, Maemondo M, Kikuchi T, Tazawa R, Hagiwara K, Ishibashi Y, Ohta S, Nukiwa T. N-terminal deletion augments the cell-death-inducing activity of BAX in adenoviral gene delivery to nonsmall cell lung cancers. Oncogene. 2003;22:2655–63. doi: 10.1038/sj.onc.1206331. [DOI] [PubMed] [Google Scholar]
- 208.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 209.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–70. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Moreau C, Cartron PF, Hunt A, Meflah K, Green DR, Evan G, Vallette FM, Juin P. Minimal BH3 peptides promote cell death by antagonizing anti-apoptotic proteins. J Biol Chem. 2003;278:19426–35. doi: 10.1074/jbc.M209472200. [DOI] [PubMed] [Google Scholar]
- 211.Shangary S, Johnson DE. Peptides derived from BH3 domains of Bcl-2 family members: a comparative analysis of inhibition of Bcl-2, Bcl-x(L) and Bax oligomerization, induction of cytochrome c release, and activation of cell death. Biochemistry. 2002;41:9485–95. doi: 10.1021/bi025605h. [DOI] [PubMed] [Google Scholar]
- 212.Li R, Boehm AL, Miranda MB, Shangary S, Grandis JR, Johnson DE. Targeting antiapoptotic Bcl-2 family members with cell-permeable BH3 peptides induces apoptosis signaling and death in head and neck squamous cell carcinoma cells. Neoplasia. 2007;9:801–11. doi: 10.1593/neo.07394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Nakashima T, Miura M, Hara M. Tetrocarcin A inhibits mitochondrial functions of Bcl-2 and suppresses its anti-apoptotic activity. Cancer Res. 2000;60:1229–35. [PubMed] [Google Scholar]
- 214.Chan SL, Lee MC, Tan KO, Yang LK, Lee AS, Flotow H, Fu NY, Butler MS, Soejarto DD, Buss AD, Yu VC. Identification of chelerythrine as an inhibitor of BclXL function. J Biol Chem. 2003;278:20453–6. doi: 10.1074/jbc.C300138200. [DOI] [PubMed] [Google Scholar]
- 215.Zhang YH, Bhunia A, Wan KF, Lee MC, Chan SL, Yu VC, Mok YK. Chelerythrine and sanguinarine dock at distinct sites on BclXL that are not the classic BH3 binding cleft. J Mol Biol. 2006;364:536–49. doi: 10.1016/j.jmb.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 216.Tzung SP, Kim KM, Basañez G, Giedt CD, Simon J, Zimmerberg J, Zhang KY, Hockenbery DM. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol. 2001;3:183–91. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- 217.Zhai D, Jin C, Shiau CW, Kitada S, Satterthwait AC, Reed JC. Gambogic acid is an antagonist of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2008;7:1639–46. doi: 10.1158/1535-7163.MCT-07-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphe-nols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–64. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 219.Van Poznak C, Seidman AD, Reidenberg MM, Moasser MM, Sklarin N, Van Zee K, Borgen P, Gollub M, Bacotti D, Yao TJ, Bloch R, Ligueros M, Sonenberg M, Norton L, Hudis C. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat. 2001;66:239–48. doi: 10.1023/a:1010686204736. [DOI] [PubMed] [Google Scholar]
- 220.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 221.Van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P, Dyer MJ, Cohen GM. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ. 2008;15:820–30. doi: 10.1038/cdd.2008.25. [DOI] [PubMed] [Google Scholar]
- 223.Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8:5–6. doi: 10.1016/j.ccr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 224.Song JH, Kandasamy K, Kraft AS. ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J Biol Chem. 2008;283:25003–13. doi: 10.1074/jbc.M802511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2008;16:360–7. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 226.Vaux DL. ABT-737, proving to be a great tool even before it is proven in the clinic. Cell Death Differ. 2008;15:807–8. doi: 10.1038/cdd.2008.31. [DOI] [PubMed] [Google Scholar]
- 227.Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8:267–71. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 228.Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G. Viral proteins targeting mitochondria: controlling cell death. Biochim Biophys Acta. 2004;1659:178–89. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 229.Borgne-Sanchez A, Dupont S, Langonné A, Baux L, Lecoeur H, Chauvier D, Lassalle M, Déas O, Briëre JJ, Brabant M, Roux P, Péchoux C, Briand JP, Hoebeke J, Deniaud A, Brenner C, Rustin P, Edelman L, Rebouillat D, Jacotot E. Targeted Vpr-derived peptides reach mitochondria to induce apoptosis of alphaVbeta3-expressing endothelial cells. Cell Death Differ. 2007;14:422–35. doi: 10.1038/sj.cdd.4402018. [DOI] [PubMed] [Google Scholar]
- 230.Shol’ts KF, Aliverdieva DA, Snezhkova LG, Miroshnikov AI, Kotel’nikova AV. [Action of the mastoparan from hornet venom on mitochondria] Dokl Akad Nauk SSSR. 1983;273:747–50. [PubMed] [Google Scholar]
- 231.Pfeiffer DR, Gudz TI, Novgorodov SA, Erdahl WL. The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J Biol Chem. 1995;270:4923–32. doi: 10.1074/jbc.270.9.4923. [DOI] [PubMed] [Google Scholar]
- 232.Sokolove PM, Kinnally KW. A mitochondrial signal peptide from Neurospora crassa increases the permeability of isolated rat liver mitochondria. Arch Biochem Biophys. 1996;336:69–76. doi: 10.1006/abbi.1996.0533. [DOI] [PubMed] [Google Scholar]
- 233.Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–8. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 234.Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, Ellerby HM, Bredesen DE, Pasqualini R, Ruoslahti E. Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci USA. 2002;99:1527–31. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fantin VR, Berardi MJ, Babbe H, Michelman MV, Manning CM, Leder P. A bifunctional targeted peptide that blocks HER-2 tyrosine kinase and disables mitochondrial function in HER-2-positive carcinoma cells. Cancer Res. 2005;65:6891–900. doi: 10.1158/0008-5472.CAN-05-0395. [DOI] [PubMed] [Google Scholar]
- 236.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–8. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 237.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–90. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 238.Goldin N, Heyfets A, Reischer D, Flescher E. Mitochondria-mediated ATP depletion by anti-cancer agents of the jas-monate family. J Bioenerg Biomembr. 2007;39:51–7. doi: 10.1007/s10863-006-9061-y. [DOI] [PubMed] [Google Scholar]
- 239.Rotem R, Heyfets A, Fingrut O, Blickstein D, Shaklai M, Flescher E. Jasmonates: novel anticancer agents acting directly and selectively on human cancer cell mitochondria. Cancer Res. 2005;65:1984–93. doi: 10.1158/0008-5472.CAN-04-3091. [DOI] [PubMed] [Google Scholar]
- 240.Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, Bronner V, Notcovich A, Shoshan-Barmatz V, Flescher E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene. 2008;27:4636–43. doi: 10.1038/onc.2008.108. [DOI] [PubMed] [Google Scholar]
- 241.Fingrut O, Flescher E. Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells. Leukemia. 2002;16:608–16. doi: 10.1038/sj.leu.2402419. [DOI] [PubMed] [Google Scholar]
- 242.Heyfets A, Flescher E. Cooperative cytotoxicity of methyl jasmonate with anti-cancer drugs and 2-deoxy-D-glucose. Cancer Lett. 2007;250:300–10. doi: 10.1016/j.canlet.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 243.Reischer D, Heyfets A, Shimony S, Nordenberg J, Kashman Y, Flescher E. Effects of natural and novel synthetic jas-monates in experimental metastatic melanoma. Br J Pharmacol. 2007;150:738–49. doi: 10.1038/sj.bjp.0707146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Genini D, Adachi S, Chao Q, Rose DW, Carrera CJ, Cottam HB, Carson DA, Leoni LM. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96:3537–43. [PubMed] [Google Scholar]
- 245.Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Distinct pathways for stimulation of cytochrome c release by etopo-side. J Biol Chem. 2000;275:32438–43. doi: 10.1074/jbc.C000518200. [DOI] [PubMed] [Google Scholar]
- 246.Kidd JF, Pilkington MF, Schell MJ, Fogarty KE, Skepper JN, Taylor CW, Thorn P. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem. 2002;277:6504–10. doi: 10.1074/jbc.M106802200. [DOI] [PubMed] [Google Scholar]
- 247.James ML, Selleri S, Kassiou M. Development of ligands for the peripheral benzodiazepine receptor. Curr Med Chem. 2006;13:1991–2001. doi: 10.2174/092986706777584979. [DOI] [PubMed] [Google Scholar]
- 248.Kugler W, Veenman L, Shandalov Y, Leschiner S, Spanier I, Lakomek M, Gavish M. Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell Oncol. 2008;30:435–50. doi: 10.3233/CLO-2008-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hirsch T, Decaudin D, Susin SA, Marchetti P, Larochette N, Resche-Rigon M, Kroemer G. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp Cell Res. 1998;241:426–34. doi: 10.1006/excr.1998.4084. [DOI] [PubMed] [Google Scholar]
- 250.Decaudin D, Castedo M, Nemati F, Beurdeley-Thomas A, De Pinieux G, Caron A, Pouillart P, Wijdenes J, Rouillard D, Kroemer G, Poupon MF. Peripheral benzodiazepine receptor ligands reverse apoptosis resistance of cancer cells in vitro and in vivo. Cancer Res. 2002;62:1388–93. [PubMed] [Google Scholar]
- 251.Hans G, Wislet-Gendebien S, Lallemend F, Robe P, Rogister B, Belachew S, Nguyen L, Malgrange B, Moonen G, Rigo JM. Peripheral benzodiazepine receptor (PBR) ligand cytotoxicity unrelated to PBR expression. Biochem Pharmacol. 2005;69:819–30. doi: 10.1016/j.bcp.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 252.Belzacq AS, El Hamel C, Vieira HL, Cohen I, Haouzi D, Métivier D, Marchetti P, Brenner C, Kroemer G. Adenine nucleotide translocator mediates the mito-chondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene. 2001;20:7579–87. doi: 10.1038/sj.onc.1204953. [DOI] [PubMed] [Google Scholar]
- 253.Ravagnan L, Marzo I, Costantini P, Susin SA, Zamzami N, Petit PX, Hirsch F, Goulbern M, Poupon MF, Miccoli L, Xie Z, Reed JC, Kroemer G. Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene. 1999;18:2537–46. doi: 10.1038/sj.onc.1202625. [DOI] [PubMed] [Google Scholar]
- 254.Notario B, Zamora M, Vinas O, Mampel T. All-trans-retinoic acid binds to and inhibits adenine nucleotide translocase and induces mitochondrial permeability transition. Mol Pharmacol. 2003;63:224–31. doi: 10.1124/mol.63.1.224. [DOI] [PubMed] [Google Scholar]
- 255.Marchetti P, Zamzami N, Joseph B, Schraen-Maschke S, Méreau-Richard C, Costantini P, Métivier D, Susin SA, Kroemer G, Formstecher P. The novel retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid can trigger apoptosis through a mitochondrial pathway independent of the nucleus. Cancer Res. 1999;59:6257–66. [PubMed] [Google Scholar]
- 256.Dogliotti L, Danese S, Berruti A, Zola P, Buniva T, Bottini A, Richiardi G, Moro G, Farris A, Baù MG, Porcile G. Cisplatin, epirubicin, and lonidamine combination regimen as first-line chemotherapy for metastatic breast cancer: a pilot study. Cancer Chemother Pharmacol. 1998;41:333–8. doi: 10.1007/s002800050747. [DOI] [PubMed] [Google Scholar]
- 257.Ianniello GP, De Cataldis G, Comella P, Scarpati MD, Maiorino A, Brancaccio L, Cioffi R, Lombardi A, Carnicelli P, Tinessa V. Cisplatin, epirubicin, and vindesine with or without lonidamine in the treatment of inoperable nonsmall cell lung carcinoma: a multicenter randomized clinical trial. Cancer. 1996;78:63–9. doi: 10.1002/(SICI)1097-0142(19960701)78:1<63::AID-CNCR11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 258.De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L, Guida M, Catino A, Crucitta E, Sambiasi D, Mazzei A. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur J Cancer. 2001;37:364–8. doi: 10.1016/s0959-8049(00)00400-7. [DOI] [PubMed] [Google Scholar]
- 259.Oudard S, Carpentier A, Banu E, Fauchon F, Celerier D, Poupon MF, Dutrillaux B, Andrieu JM, Delattre JY. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2003;63:81–6. doi: 10.1023/a:1023756707900. [DOI] [PubMed] [Google Scholar]
- 260.Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrell RP., Jr Complete remission after treatment of acute promye-locytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341–8. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- 261.Hofmeister CC, Jansak B, Denlinger N, Kraut EH, Benson DM, Farag SS. Phase II clinical trial of arsenic trioxide with liposomal doxorubicin, vincristine, and dexamethasone in newly diagnosed multiple myeloma. Leuk Res. 2008;32:1295–8. doi: 10.1016/j.leukres.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 262.Klopfer A, Hasenjager A, Belka C, Schulze-Osthoff K, Dorken B, Daniel PT. Adenine deoxynucleotides fludarabine and cladribine induce apoptosis in a CD95/Fas receptor, FADD and caspase-8-independent manner by activation of the mitochondrial cell death pathway. Oncogene. 2004;23:9408–18. doi: 10.1038/sj.onc.1207975. [DOI] [PubMed] [Google Scholar]
- 263.Haridas V, Li X, Mizumachi T, Higuchi M, Lemeshko VV, Colombini M, Gutterman JU. Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion. 2007;7:234–40. doi: 10.1016/j.mito.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 264.Fulda S, Debatin KM. Sensitization for anticancer drug-induced apoptosis by betulinic Acid. Neoplasia. 2005;7:162–70. doi: 10.1593/neo.04442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Tinhofer I, Bernhard D, Senfter M, Anether G, Loeffler M, Kroemer G, Kofler R, Csordas A, Greil R. Resveratrol, a tumor-suppressive compound from grapes, induces apoptosis via a novel mitochondrial pathway controlled by Bcl-2. FASEB J. 2001;15:1613–5. doi: 10.1096/fj.00-0675fje. [DOI] [PubMed] [Google Scholar]
- 266.Juan ME, Wenzel U, Daniel H, Planas JM. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem. 2008;56:4813–8. doi: 10.1021/jf800175a. [DOI] [PubMed] [Google Scholar]
- 267.Brito PM, Simoes NF, Almeida LM, Dinis TC. Resveratrol disrupts peroxynitritetriggered mitochondrial apoptotic pathway: a role for Bcl-2. Apoptosis. 2008;13:1043–53. doi: 10.1007/s10495-008-0235-4. [DOI] [PubMed] [Google Scholar]
- 268.Facompré M, Tardy C, Bal-Mahieu C, Colson P, Perez C, Manzanares I, Cuevas C, Bailly C. Lamellarin D: a novel potent inhibitor of topoisomerase I. Cancer Res. 2003;63:7392–9. [PubMed] [Google Scholar]
- 269.Kluza J, Gallego MA, Loyens A, Beauvillain JC, Sousa-Faro JM, Cuevas C, Marchetti P, Bailly C. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin D. Cancer Res. 2006;66:3177–87. doi: 10.1158/0008-5472.CAN-05-1929. [DOI] [PubMed] [Google Scholar]
- 270.Gallego MA, Ballot C, Kluza J, Hajji N, Martoriati A, Castéra L, Cuevas C, Formstecher P, Joseph B, Kroemer G, Bailly C, Marchetti P. Overcoming chemoresistance of non-small cell lung carcinoma through restoration of an AIF-dependent apoptotic pathway. Oncogene. 2008;27:1981–92. doi: 10.1038/sj.onc.1210833. [DOI] [PubMed] [Google Scholar]
- 271.Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–81. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- 272.Lampidis TJ, Bernal SD, Summerhayes IC, Chen LB. Selective toxicity of rhodamine 123 in carcinoma cells in vitro. Cancer Res. 1983;43:716–20. [PubMed] [Google Scholar]
- 273.Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell. 2002;2:29–42. doi: 10.1016/s1535-6108(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 274.Modica-Napolitano JS, Koya K, Weisberg E, Brunelli BT, Li Y, Chen LB. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996;56:544–50. [PubMed] [Google Scholar]
- 275.Chiba Y, Kubota T, Watanabe M, Matsuzaki SW, Otani Y, Teramoto T, Matsumoto Y, Koya K, Kitajima M. MKT-077, localized lipophilic cation: antitumor activity against human tumor xenografts serially transplanted into nude mice. Anticancer Res. 1998;18:1047–52. [PubMed] [Google Scholar]
- 276.Britten CD, Rowinsky EK, Baker SD, Weiss GR, Smith L, Stephenson J, Rothenberg M, Smetzer L, Cramer J, Collins W, Von Hoff DD, Eckhardt SG. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin Cancer Res. 2000;6:42–9. [PubMed] [Google Scholar]
- 277.Teicher BA, Holden SA, Jacobs JL, Abrams MJ, Jones AG. Intracellular distribution of a platinum-rhodamine 123 complex in cis-platinum sensitive and resistant human squamous carcinoma cell lines. Biochem Pharmacol. 1986;35:3365–9. doi: 10.1016/0006-2952(86)90437-5. [DOI] [PubMed] [Google Scholar]
- 278.Belzacq AS, Jacotot E, Vieira HL, Mistro D, Granville DJ, Xie Z, Reed JC, Kroemer G, Brenner C. Apoptosis induction by the photosensitizer verteporfin: identification of mitochondrial adenine nucleotide translocator as a critical target. Cancer Res. 2001;61:1260–4. [PubMed] [Google Scholar]
- 279.Pervaiz S, Seyed MA, Hirpara JL, Clement MV, Loh KW. Purified photoproducts of merocyanine 540 trigger cytochrome C release and caspase 8-dependent apoptosis in human leukemia and melanoma cells. Blood. 1999;93:4096–108. [PubMed] [Google Scholar]
- 280.Minamikawa T, Sriratana A, Williams DA, Bowser DN, Hill JS, Nagley P. Chloromethyl-X-rosamine (MitoTracker Red) photosensitises mitochondria and induces apoptosis in intact human cells. J Cell Sci. 1999;112:2419–30. doi: 10.1242/jcs.112.14.2419. [DOI] [PubMed] [Google Scholar]
- 281.Miccoli L, Beurdeley-Thomas A, De Pinieux G, Sureau F, Oudard S, Dutrillaux B, Poupon MF. Light-induced photoactiva-tion of hypericin affects the energy metabolism of human glioma cells by inhibiting hexokinase bound to mitochondria. Cancer Res. 1998;58:5777–86. Dec 15. [PubMed] [Google Scholar]
- 282.Usuda J, Chiu SM, Murphy ES, Lam M, Nieminen AL, Oleinick NL. Domain-dependent photodamage to Bcl-2. A membrane anchorage region is needed to form the target of phthalocyanine photosensitization. J Biol Chem. 2003;278:2021–9. doi: 10.1074/jbc.M205219200. [DOI] [PubMed] [Google Scholar]
- 283.Ke MS, Xue LY, Feyes DK, Azizuddin K, Baron ED, McCormick TS, Mukhtar H, Panneerselvam A, Schluchter MD, Cooper KD, Oleinick NL, Stevens SR. Apoptosis mechanisms related to the increased sensitivity of Jurkat T-cells vs A431 epidermoid cells to photodynamic therapy with the phthalocyanine Pc 4. Photochem Photobiol. 2008;84:407–14. doi: 10.1111/j.1751-1097.2007.00278.x. [DOI] [PubMed] [Google Scholar]
- 284.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 285.Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prévost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–80. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 287.Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–62. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 288.Arnt CR, Chiorean MV, Heldebrant MP, Gores GJ, Kaufmann SH. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J Biol Chem. 2002;277:44236–43. doi: 10.1074/jbc.M207578200. [DOI] [PubMed] [Google Scholar]
- 289.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24:7381–8. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 290.Mizukawa K, Kawamura A, Sasayama T, Tanaka K, Kamei M, Sasaki M, Kohmura E. Synthetic Smac peptide enhances the effect of etoposide-induced apoptosis in human glioblastoma cell lines. J Neurooncol. 2006;77:247–55. doi: 10.1007/s11060-005-9045-5. [DOI] [PubMed] [Google Scholar]
- 291.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 292.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL-or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002;8:808–15. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 293.Gallego MA, Joseph B, Hemström TH, Tamiji S, Mortier L, Kroemer G, Formstecher P, Zhivotovsky B, Marchetti P. Apoptosis-inducing factor determines the chemoresistance of non-small-cell lung carcinomas. Oncogene. 2004;23:6282–91. doi: 10.1038/sj.onc.1207835. [DOI] [PubMed] [Google Scholar]
- 294.Yu CJ, Jia LT, Meng YL, Zhao J, Zhang Y, Qiu XC, Xu YM, Wen WH, Yao LB, Fan DM, Jin BQ, Chen SY, Yang AG. Selective proapoptotic activity of a secreted recombinant antibody/AIF fusion protein in carcinomas overexpressing HER2. Gene Ther. 2006;13:313–20. doi: 10.1038/sj.gt.3302672. [DOI] [PubMed] [Google Scholar]
- 295.Rugo H, Shtivelman E, Perez A, Vogel C, Franco S, Tan Chiu E, Melisko M, Tagliaferri M, Cohen I, Shoemaker M, Tran Z, Tripathy D. Phase I trial and anti-tumor effects of BZL101 for patients with advanced breast cancer. Breast Cancer Res Treat. 2007;105:17–28. doi: 10.1007/s10549-006-9430-6. [DOI] [PubMed] [Google Scholar]
- 296.Wang M, Zhang L, Han X, Yang J, Qian J, Hong S, Samaniego F, Romaguera J, Yi Q. Atiprimod inhibits the growth of mantle cell lymphoma in vitro and in vivo and induces apoptosis via activating the mitochondrial pathways. Blood. 2007;109:5455–62. doi: 10.1182/blood-2006-12-063958. [DOI] [PubMed] [Google Scholar]
- 297.Rossaro L, Mazzaferro V, Scotti-Foglieni CL, Porter KA, Williams DS, Simplaceanu E, Simplaceanu V, Francavilla A, Starzl TE, Ho C, Naccarato R, Van Thiel DH. Cyclosporine and liver regeneration studied by in vivo 31P nuclear magnetic resonance spectroscopy. Dig Dis Sci. 1991;36:687–92. doi: 10.1007/BF01297039. [DOI] [PubMed] [Google Scholar]
- 298.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kass GE, Juedes MJ, Orrenius S. Cyclosporin A protects hepatocytes against prooxidant-induced cell killing. A study on the role of mitochondrial Ca2+ cycling in cytotoxicity. Biochem Pharmacol. 1992;44:1995–2003. doi: 10.1016/0006-2952(92)90102-o. [DOI] [PubMed] [Google Scholar]
- 300.Soriano ME, Nicolosi L, Bernardi P. Desensitization of the permeability transition pore by cyclosporin a prevents activation of the mitochondrial apoptotic pathway and liver damage by tumor necrosis factor-alpha. J Biol Chem. 2004;279:36803–8. doi: 10.1074/jbc.M405297200. [DOI] [PubMed] [Google Scholar]
- 301.Beales D, McLean AE. Protection in the late stages of paracetamol-induced liver cell injury with fructose, cyslosporin A and trifluoperazine. Toxicology. 1996;107:201–8. doi: 10.1016/0300-483x(95)03262-e. [DOI] [PubMed] [Google Scholar]
- 302.Feldmann G, Haouzi D, Moreau A, Durand-Schneider AM, Bringuier A, Berson A, Mansouri A, Fau D, Pessayre D. Opening of the mitochondrial permeability transition pore causes matrix expansion and outer membrane rupture in Fas-mediated hepatic apoptosis in mice. Hepatology. 2000;31:674–83. doi: 10.1002/hep.510310318. [DOI] [PubMed] [Google Scholar]
- 303.Masubuchi Y, Suda C, Horie T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J Hepatol. 2005;42:110–6. doi: 10.1016/j.jhep.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 304.Haouzi D, Cohen I, Vieira HL, Poncet D, Boya P. Castedo M, Vadrot N, Belzacq AS, Fau D, Brenner C, Feldmann G, Kroemer G. Mitochondrial permeability transition as a novel principle of hepatore-nal toxicity in vivo. Apoptosis. 2002;7:395–405. doi: 10.1023/a:1020026923038. [DOI] [PubMed] [Google Scholar]
- 305.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–9. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 306.Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther. 2005;312:509–16. doi: 10.1124/jpet.104.075945. [DOI] [PubMed] [Google Scholar]
- 307.Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care. 2007;13:134–42. doi: 10.1097/MCC.0b013e3280895d5c. [DOI] [PubMed] [Google Scholar]
- 308.Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–92. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- 309.Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–9. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 310.Theruvath TP, Zhong Z, Pediaditakis P, Ramshesh VK, Currin RT, Tikunov A, Holmuhamedov E, Lemasters JJ. Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology. 2008;47:236–46. doi: 10.1002/hep.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.McEwen ML, Sullivan PG, Springer JE. Pretreatment with the cyclosporin derivative, NIM811, improves the function of synaptic mitochondria following spinal cord contusion in rats. J Neurotrauma. 2007;24:613–24. doi: 10.1089/neu.2006.9969. [DOI] [PubMed] [Google Scholar]
- 312.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–9. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 313.Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–24. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Fuks B, Talaga P, Huart C, Hénichart JP, Bertrand K, Grimée R, Lorent G. In vitro properties of 5-(benzylsulfonyl)-4-bromo-2-methyl-3(2H)-pyridazinone: a novel permeability transition pore inhibitor. Eur J Pharmacol. 2005;519:24–30. doi: 10.1016/j.ejphar.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 315.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 316.Murata M, Akao M, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardiopro-tection. Circ Res. 2001;89:891–8. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- 317.Wang GY, Wu S, Pei JM, Yu XC, Wong TM. Kappa- but not delta-opioid receptors mediate effects of ischemic preconditioning on both infarct and arrhythmia in rats. Am J Physiol Heart Circ Physiol. 2001;280:H384–91. doi: 10.1152/ajpheart.2001.280.1.H384. [DOI] [PubMed] [Google Scholar]
- 318.Holmuhamedov EL, Wang L, Terzic A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol. 1999;519:347–60. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Fryer RM, Eells JT, Hsu AK, Henry MM, Gross GJ. Ischemic preconditioning in rats: role of mitochondrial K(ATP) channel in preservation of mitochondrial function. Am J Physiol Heart Circ Physiol. 2000;278:H305–12. doi: 10.1152/ajpheart.2000.278.1.H305. [DOI] [PubMed] [Google Scholar]
- 320.Iwai T, Tanonaka K, Koshimizu M, Takeo S. Preservation of mitochondrial function by diazoxide during sustained ischaemia in the rat heart. Br J Pharmacol. 2000;129:1219–27. doi: 10.1038/sj.bjp.0703148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841–9. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- 322.Otani H. Ischemic preconditioning: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2008;10:207–47. doi: 10.1089/ars.2007.1679. [DOI] [PubMed] [Google Scholar]
- 323.Costa AD, Quinlan CL, Andrukhiv A, West IC, Jaburek M, Garlid KD. The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am J Physiol Heart Circ Physiol. 2006;290:H406–15. doi: 10.1152/ajpheart.00794.2005. [DOI] [PubMed] [Google Scholar]
- 324.Quinlan CL, Costa AD, Costa CL, Pierre SV, Dos Santos P, Garlid KD. Conditioning the heart induces formation of signalosomes that interact with mitochondria to open mitoKATP channels. Am J Physiol Heart Circ Physiol. 2008;295:H953–H61. doi: 10.1152/ajpheart.00520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. 2006;70:222–30. doi: 10.1016/j.cardiores.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 326.Baines CP, Liu GS, Birincioglu M, Critz SD, Cohen MV, Downey JM. Ischemic preconditioning depends on interaction between mitochondrial KATP channels and actin cytoskeleton. Am J Physiol. 1999;276:H1361–8. doi: 10.1152/ajpheart.1999.276.4.H1361. [DOI] [PubMed] [Google Scholar]
- 327.Liu H, Cala PM, Anderson SE. Ischemic preconditioning: effects on pH, Na and Ca in newborn rabbit hearts during Ischemia/Reperfusion. J Mol Cell Cardiol. 1998;30:685–97. doi: 10.1006/jmcc.1997.0636. [DOI] [PubMed] [Google Scholar]
- 328.Carreira RS, Monteiro P, Kowaltowski AJ, Goncalves LM, Providencia LA. Nicorandil protects cardiac mitochondria against permeability transition induced by ischemia-reperfusion. J Bioenerg Biomembr. 2008;40:95–102. doi: 10.1007/s10863-008-9133-2. [DOI] [PubMed] [Google Scholar]
- 329.Sato T, Costa AD, Saito T, Ogura T, Ishida H, Garlid KD, Nakaya H. Bepridil, an antiarrhythmic drug, opens mitochondrial KATP channels, blocks sarcolemmal KATP channels, and confers cardioprotection. J Pharmacol Exp Ther. 2006;316:182–8. doi: 10.1124/jpet.105.094029. [DOI] [PubMed] [Google Scholar]
- 330.Warltier DC, Al-Wathiqui MH, Kampine JP, Schmeling WT. Recovery of contractile function of stunned myocardium in chronically instrumented dogs is enhanced by halothane or isoflurane. Anesthesiology. 1988;69:552–65. doi: 10.1097/00000542-198810000-00016. [DOI] [PubMed] [Google Scholar]
- 331.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–93. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–90. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- 333.Ludwig LM, Tanaka K, Eells JT, Weihrauch D, Pagel PS, Kersten JR, Warltier DC. Preconditioning by isoflurane is mediated by reactive oxygen species generated from mitochondrial electron transport chain complex III. Anesth Analg. 2004;99:1308–15. doi: 10.1213/01.ANE.0000134804.09484.5D. [DOI] [PubMed] [Google Scholar]
- 334.Marinovic J, Bosnjak ZJ, Stadnicka A. Distinct roles for sarcolemmal and mito-chondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. Anesthesiology. 2006;105:98–104. doi: 10.1097/00000542-200607000-00018. [DOI] [PubMed] [Google Scholar]
- 335.Feng J, Lucchinetti E, Ahuja P, Pasch T, Perriard JC, Zaugg M. Isoflurane post-conditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta. Anesthesiology. 2005;103:987–95. doi: 10.1097/00000542-200511000-00013. [DOI] [PubMed] [Google Scholar]
- 336.Feng J, Zhu M, Schaub MC, Gehrig P, Roschitzki B, Lucchinetti E, Zaugg M. Phosphoproteome analysis of isofluraneprotected heart mitochondria: phosphorylation of adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial function. Cardiovasc Res. 2008;80:20–9. doi: 10.1093/cvr/cvn161. [DOI] [PubMed] [Google Scholar]
- 337.Li L, Peng L, Zuo Z. Isoflurane preconditioning increases B-cell lymphoma-2 expression and reduces cytochrome c release from the mitochondria in the ischemic penumbra of rat brain. Eur J Pharmacol. 2008;586:106–13. doi: 10.1016/j.ejphar.2008.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Thomas M, Le WD. Minocycline: neuroprotective mechanisms in Parkinson’s disease. Curr Pharm Des. 2004;10:679–86. doi: 10.2174/1381612043453162. [DOI] [PubMed] [Google Scholar]
- 339.Kelly KJ, Sutton TA, Weathered N, Ray N, Caldwell EJ, Plotkin Z, Dagher PC. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2004;287:F760–6. doi: 10.1152/ajprenal.00050.2004. [DOI] [PubMed] [Google Scholar]
- 340.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–8. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 341.Fernandez-Gomez FJ, Galindo MF, Gomez-Lazaro M, González-García C, Ceña V, Aguirre N, Jordán J. Involvement of mitochondrial potential and calcium buffering capacity in minocycline cytoprotective actions. Neuroscience. 2005;133:959–67. doi: 10.1016/j.neuroscience.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 342.Mansson R, Hansson MJ, Morota S, Uchino H, Ekdahl CT, Elmer E. Re-evaluation of mitochondrial permeability transition as a primary neuroprotective target of minocycline. Neurobiol Dis. 2007;25:198–205. doi: 10.1016/j.nbd.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 343.Alano CC, Kauppinen TM, Valls AV, Swanson RA. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci USA. 2006;103:9685–90. doi: 10.1073/pnas.0600554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96–103. doi: 10.1016/j.coph.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 345.Schraufstatter IU, Hyslop PA, Hinshaw DB, Spragg RG, Sklar LA, Cochrane CG. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc Natl Acad Sci USA. 1986;83:4908–12. doi: 10.1073/pnas.83.13.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Haddad M, Beray-Berthat V, Coqueran B, Palmier B, Szabo C, Plotkine M, Margaill I. Reduction of hemorrhagic transformation by PJ34, a poly(ADP-ribose)polymerase inhibitor, after permanent focal cerebral ischemia in mice. Eur J Pharmacol. 2008;588:52–7. doi: 10.1016/j.ejphar.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 347.Ji D, Li GY, Osborne NN. Nicotinamide attenuates retinal ischemia and light insults to neurones. Neurochem Int. 2008;52:786–98. doi: 10.1016/j.neuint.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 348.Kumaran D, Udayabanu M, Nair RU, R A, Katyal A. Benzamide protects delayed neuronal death and behavioural impairment in a mouse model of global cerebral ischemia. Behav Brain Res. 2008;192:178–84. doi: 10.1016/j.bbr.2008.03.043. [DOI] [PubMed] [Google Scholar]
- 349.Obrosova IG, Xu W, Lyzogubov VV, Ilnytska O, Mashtalir N, Vareniuk I, Pavlov IA, Zhang J, Slusher B, Drel VR. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic Biol Med. 2008;44:972–81. doi: 10.1016/j.freeradbiomed.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Sharma SS, Kumar A, Kaundal RK. Protective effects of 4-amino1,8-napthalimide, a poly (ADP-ribose) polymerase inhibitor in experimental diabetic neuropathy. Life Sci. 2008;82:570–6. doi: 10.1016/j.lfs.2007.11.031. [DOI] [PubMed] [Google Scholar]