

Summary
Very few gene conversions in mitotic cells are associated with crossovers, suggesting that these events are regulated. This may be important for the maintenance of genetic stability. We have analyzed the relationship between homologous recombination and crossing-over in haploid budding yeast and identified factors involved in the regulation of crossover outcomes. Gene conversions unaccompanied by a crossover appear 30 min before conversions accompanied by exchange, indicating that there are two different repair mechanisms in mitotic cells. Crossovers are rare (5%), but deleting the BLM/WRN homolog, SGS1, or the SRS2 helicase increases crossovers 2- to 3-fold. Overexpressing SRS2 nearly eliminates crossovers, whereas overexpression of RAD51 in srs2Δ cells almost completely eliminates the noncrossover recombination pathway. We suggest Sgs1 and its associated topoisomerase Top3 remove double Holliday junction intermediates from a crossover-producing repair pathway, thereby reducing crossovers. Srs2 promotes the noncrossover synthesis-dependent strand-annealing (SDSA) pathway, apparently by regulating Rad51 binding during strand exchange.
Introduction
Many homologous recombination events are initiated by double-strand breaks (DSBs), but the mechanisms of DSB repair remain poorly understood. Particularly little is known about the way strand invasion intermediates are resolved and how the proportion of gene conversions that are accompanied by crossing-over is regulated. One striking difference between mitotic and meiotic recombination is in the proportion of DSB repair events associated with crossing-over (Pâques and Haber, 1999). The high level and distribution of exchanges accompanying gene conversion in meiotic cells depends on a number of meiosis-specific proteins (Keeney, 2001). In contrast, very few gene conversions in mitotic cells are crossover-associated. In many model organisms including yeast, fruit flies, and mammalian cells, mitotic recombination between homologous sequences is rarely associated with crossovers (Esposito, 1978; Johnson and Jasin, 2000; Malkova et al., 2000; Nassif et al., 1994; Stark and Jasin, 2003; Virgin et al., 2001). Crossing-over may be suppressed to prevent loss of heterozygosity (LOH) and reciprocal translocations, however the link between exchanges and LOH is still debated (Shao et al., 2001; Stark and Jasin, 2003). Thus, the mechanisms of DSB repair in meiotic and mitotic cells may be significantly different. Many meiotic recombination events proceed through the formation and resolution of double Holliday junctions (HJs) (Schwacha and Kleckner, 1995), as envisioned by the DSB repair model of Szostak et al. (1983). In contrast, most mitotic recombination events might proceed via a synthesis dependent strand annealing (SDSA) mechanism, leading predominantly to events without crossing-over (reviewed in Pâques and Haber, 1999). The important studies of Allers and Lichten (2001) showed that the situation is likely to be more complex, as there appear to be two kinetically and genetically distinct mechanisms of DSB repair in meiotic yeast cells, one leading to noncrossovers and one to crossovers.
In humans, genome stability depends on many proteins, including the BLM and WRN helicases, whose budding yeast homolog is Sgs1. Complete or partial loss of function of the human genes leads to increased cancer predisposition and genome instability (Ellis et al., 1995; Goss et al., 2002; Luo et al., 2000; Myung et al., 2001; Shen and Loeb, 2001; Sinclair and Guarente, 1997). One striking phenotype of BLM patients is a greatly elevated level of sister chromatid exchange (reviewed in Hickson et al., 2001). The BLM/Sgs1 proteins of humans and yeast interact with topoisomerase III (Top3) (Fricke et al., 2001; Gangloff et al., 1994; Hu et al., 2001). In Saccharomyces cerevisiae, sgs1Δ partially suppresses the growth defects of top3Δ (Gangloff et al., 1994). Sgs1 has a complex relationship with another 3′ to 5′ helicase, Srs2, whose human homolog is not known.
Both sgs1Δ and srs2Δ mutants exhibit increased spontaneous mitotic recombination in various assays (Aboussekhra et al., 1992; Aguilera and Klein, 1988; Rong et al., 1991; Watt et al., 1996); but in other assays SRS2 has also prorecombinogenic functions (Aylon et al., 2003; Ira and Haber, 2002; Pâques and Haber, 1997; Sugawara et al., 2000). Double-mutant sgs1Δ srs2Δ cells exhibit severe growth defects that are suppressed by deletion of RAD52, RAD51, RAD55, or RAD57 (Gangloff et al., 2000; Shor et al., 2002). Sgs1 and Srs2 are probably involved in processing of recombination intermediates arising spontaneously during DNA replication which become lethal when both helicases are absent (Fabre et al., 2002); however these lesions are unlikely to be DSBs, since RAD52 is required to repair DSBs and rad52Δ rescues srs2Δ sgs1Δ. Srs2 and Sgs1 are also involved in the intra-S and DNA damage G2/M checkpoint response in yeast (Frei and Gasser, 2000; Liberi et al., 2000; Vaze et al., 2002).
Here, we have identified factors involved in regulation of crossover outcomes during gene conversion in mitotic cells. Using an HO endonuclease induced ectopic homologous recombination assay we find two kinetically distinct DSB repair processes, one leading primarily to noncrossovers and one to both crossovers and noncrossovers. Sgs1 and Srs2 are both involved in the suppression of crossing-over in budding yeast, but srs2Δ affects homologous recombination in several ways not seen for sgs1Δ, including a reduction in recombination efficiency and the elimination of the kinetic difference between crossovers and noncrossovers. The data support a model in which that Sgs1 and Top3 remove double Holliday junction (HJ) intermediates from a crossover-producing repair pathway whereas Srs2 specifically promotes a recombination pathway leading to noncrossovers, apparently by regulating Rad51 binding to recombination intermediates.
Results
Deletion of SGS1 or SRS2 Helicases Increases the Frequency of Crossover
The kinetics and frequency of crossover outcomes in mitotic cells were studied in an interchromosomal recombination system (Figure 1A). A DSB within a 2 kb MATa sequence, inserted in chromosome V, is created by a galactose-inducible HO endonuclease. The break is repaired by homologous recombination using a MATa-inc sequence on chromosome III as a donor (Ira and Haber, 2002). The single base-pair mutation in MATa-inc prevents cleavage by HO. Repair of the DSB is by RAD51-dependent gene conversion, which can occur either with or without an accompanying crossover; these outcomes can be distinguished by the sizes of restriction fragments separated on agarose gels (Figure 1B). The frequency of crossing-over was calculated based on the density of bands corresponding to non-crossover and crossover products as described in experimental procedures. Three to four hours following HO induction, most wild-type cells arrest at the G2/M stage, due to activation of the DNA damage checkpoint; recombination is completed by 7 hr (Vaze et al., 2002). In wild-type, logarithmically growing cells, about 5% of gene conversions are associated with crossing-over. The low proportion of crossovers seen by physical analysis of DNA from a large population of cells was confirmed by Southern blot analysis of DNA extracted from >100 individual colonies which arose after plating single cells on galactose-containing agar plates.
Figure 1. Ectopic Gene Conversion Assay.

(A) The experimental system to study ectopic gene conversion. A galactose inducible HO endonuclease generates a DSB within the 2 kb MATa sequence (marked by hygromycin resistance gene, HPH1) inserted at ARG5,6 on chromosome V. The homologous MATa-inc region on chromosome III is used as a donor for gene conversion. Both HML and HMR are deleted. Crossover and noncrossover products have different restriction fragment sizes and can be quantified on Southern blots.
(B) Southern blot analysis of the proportion of gene conversions with and without crossover in strains lacking Srs2 and/or Sgs1.
(C) Viability of srs2Δ, sgs1Δ, and rad59Δ cells after induction of a DSB.
We then tested the effect of deleting a number of genes implicated in DNA repair and recombination, including rad1Δ, mus81 Δ, mlh1Δ, msh2Δ, rad59Δ, sgs1Δ, and srs2Δ. The efficiency of DSB repair and viability of all tested mutants was comparable to wild-type, except sgs1Δ, srs2Δ, and rad59Δ (Figure 1C and data not shown). Repair efficiency and viability in sgs1Δ and rad59Δ was about 70%–80% of wild-type values. As we have shown previously, Srs2 is involved in recovery from the DNA damage checkpoint and although about 30% of the srs2Δ cells repair the break as assayed by Southern blots, only 2%–3% survive because of a failure to recover from checkpoint-mediated arrest (Vaze et al., 2002). The difference between the efficiency of repair and viability after DSB damage was seen only in srs2Δ. A severe inhibition of ectopic recombination by srs2Δ was also reported by Aylon et al. (2003).
Deletions of SGS1 and SRS2 caused significant increases in the proportion of gene conversions associated with crossing-over to 11.7 ± 2.4% or 16.6 ± 2.7%, respectively, compared to the wild-type 4.8 ± 1.0% among cells that repaired the break (Figure 2). Based on at least 10 independent experiments, the differences in crossover frequency are statistically significant. Single-colony analysis among cells that repaired the DSB revealed the same levels of crossover in the absence of helicases. Importantly, the results for srs2Δ were similar among the rare survivors, where 7 of 40 independent colonies contained crossovers (17.5%), compared to 17% as determined from the Southern blot in Figure 1B. All crossovers resulted in reciprocal translocations and do not arise from break-induced replication (BIR), since BIR would lead to death due to loss of essential genes distal to the DSB on chromosome V.
Figure 2. srs2Δ and sgs1Δ Cells Exhibit Increased Levels of Crossovers.

(A) Percentage of crossovers and noncrossovers among all cells that induced the DSB in the absence of DNA helicases Sgs1 and Srs2 or topoisomerase Top3.
(B) Level of crossovers among the cells that successfully repaired the DSB in the absence of Sgs1 and Srs2 or Top3 and in cells arrested in G2/M with nocodazole.
The increase in crossing-over in sgs1Δ cells occurs without a significant reduction in the overall efficiency of repair; but in the case of srs2Δ, the increase in crossovers among completed recombination events is ac- companied by a marked reduction in repair efficiency (Figures 2A–2B). This observation suggests that in the absence of SGS1, cells either change the pathway of repair or change the proportion of double HJ that are resolved as crossovers, whereas in the absence of SRS2, there is a failure to carry out ectopic recombination leading to noncrossovers.
To test if Srs2 is involved in crossover control in allelic recombination where the extent of homologous sequences is essentially unlimited, we used a diploid strain that has the MATa locus on one chromosome III, MATα-inc on the other, and is heterozygous for the distal markers THR4/thr4 (Figure 3). In addition, both HML and HMR donor sequences on the HO-cut chromosome were replaced by ADE1 (Malkova et al., 1996). We induced the HO break by plating cells on YEPGal plates and determined crossover frequency by scoring the number of Thr+/Thr− sectored colonies (Figure 3). The frequency of crossovers was 11.6% in wild-type cells and 26.8% in srs2Δ cells. Correcting for an equal number of mitoses where crossovers do not result in loss of heterozygosity total crossovers would be 23.2% and 53.6%, respectively. Our previous studies have shown that these sectored colonies arise almost exclusively from reciprocal recombination events and not from BIR (Malkova et al. 1996; A.M., M. Naylor, M. Yamaguchi, G.I., and J.E.H., unpublished data). Thus, srs2Δ increases crossing-over in allelic recombination about 2.5-fold, similar to its effect on ectopic recombination. Importantly, we did not observe any chromosome loss (Ade−Thr− colonies) in the absence of Srs2. This result suggests that Srs2 plays a crucial role in completing DSB repair only if the donor sequence shares a limited extent of homology with the recipient. One striking difference between allelic and ectopic recombination is in the kinetics of repair. Allelic recombination is much faster, being completed by 4 hr, whereas ectopic recombination requires about 7 hr (Figure 3).
Figure 3. Srs2 Suppresses Crossovers in Allelic Recombination.

(A) Allelic recombination assay, crossover frequency is measured as the frequency of sectored Thr4+/− colonies.
(B) Viability and crossover frequency in WT and srs2Δ cells.
(C) Comparison of allelic and ectopic recombinational repair kinetics.
We also surveyed other genes that might affect crossing-over. Crossing-over was not affected by deleting RAD59 (data not shown), which plays roles in both RAD51-dependent and -independent recombination (Bai and Symington, 1996; Ira and Haber, 2002; Sugawara et al., 2000). Among genes whose absence causes a marked decrease in meiotic crossing-over, but not in total gene conversion, are the mismatch repair gene, MLH1 and the exonuclease EXO1 (Hunter and Borts, 1997; Khazanehdari and Borts, 2000; Tsubouchi and Ogawa, 2000), which are also expressed in mitotic cells. Deletion of either MLH1 or EXO1 does not change the level of crossover in our assay. The deletion of another mismatch repair gene, MSH2, also had no effect (data not shown).
A family of RAD1-related endonucleases, including Mus81, has recently been implicated in the cleavage of branched DNA structures, possibly including Holliday junctions (Boddy et al., 2001; Kaliraman et al., 2001). Neither rad1Δ nor mus81Δ mutants, alone or in double-mutant combination, were different with respect to crossover frequency from wild-type cells (data not shown).
Two Pathways of DSB Repair in Mitotic Cells Have Different Kinetics and Different Outcomes
Recently Allers and Lichten (2001) have shown that there are two pathways of ectopic DSB repair in meiosis. Gene conversions without crossing-over appear about 1 hr before crossovers; moreover, the appearance of crossovers depended on meiosis-specific genes controlled by NDT80. Here, studying ectopic mitotic DSB repair, we also find evidence for two kinetically distinct pathways of repair, producing noncrossovers and crossovers in the absence of meiosis-specific genes. As shown in Figure 4, the appearance of crossover products occurs about 30 min after gene conversions without exchange. We substantiated this finding by analyzing ectopic gene conversion in a strain in which the donor sequence was MATα-inc rather than MATa-inc, so that gene conversion restriction fragments with and without exchange have different sizes from the MATa locus that is cleaved by HO. Again, noncrossover products appear before crossovers (data not shown).
Figure 4. Kinetics of Ectopic DSB Repair.

Crossovers and noncrossovers appear with different kinetics in wild-type and sgs1Δ but not in srs2Δ. The total amount of product at 8 hr was 31 ± 5% in srs2Δ strain and 77 ± 18% in sgs1Δ relative to wild-type cells.
The kinetics of appearance of crossovers and non-crossovers in the absence of SGS1 was similar to that seen in wild-type cells (Figure 4). However, in the srs2Δ strain there was no apparent difference in the kinetics of gene conversions with and without crossing-over, with all events occurring at the time that crossovers arise in wild-type cells (Figure 4). This result, coupled with the reduced efficiency of DSB repair, suggests that srs2Δ has a much greater effect on the noncrossover pathway. It appears that the increase in crossovers from 5% to 17% in srs2Δ reflects a failure to complete non-crossover gene conversion whereas the pathway leading to crossovers remains unaffected (Figure 4). In four different experiments, we calculated the level of cross-overs at the beginning of repair (3–4 hr) and at the end of repair (8 hr). We observed a 1.79 ± 0.03-fold increase in crossover frequency relative to noncrossovers for wild-type cells and a 1.65 ±0.05-fold increase for sgs1Δ; however, in srs2Δ the frequency of crossover was unchanged (the ratio of crossover frequency at both the beginning and the end of repair was 1.02 ± 0.08).
Overexpression of RAD51 in srs2Δ Cells Nearly Eliminates the Noncrossover Pathway without Affecting Crossovers
The double mutant sgs1Δ srs2 Δ is severely impaired for growth (Gangloff et al., 2000; Lee et al., 1999) making it difficult to test directly if the effects of the deletions were epistatic, additive, or synergistic. However, the near-lethality of the double mutant is suppressed by a rad51Δ (Gangloff et al., 2000). Consequently, we were able to test DSB repair and crossing-over in the triple mutant sgs1Δ srs2Δ rad51Δ but under conditions when RAD51, essential for recombination, was under the control of a galactose-inducible promoter. We note that cell death due to expression of RAD51 (in the absence of expressing HO) does not appear for several cell generations. Cells form microcolonies, often with >20 cells, and most often arrest in the G2/M phase of the cell cycle (data not shown).
First, we tested repair efficiency and crossover level in the double mutant sgs1Δ rad51Δ GAL::RAD51 and srs2Δ rad51Δ GAL::RAD51, and the single mutant rad51Δ GAL::RAD51 strains. Overexpression of RAD51 slightly decreased the level of crossover in sgs1Δ and wild-type cells, with no reduction in repair efficiency. In srs2Δ cells, there was a further decrease in product formation from 31% with wild-type levels of Rad51 protein to 12 ± 4% when Rad51 was overexpressed and a dramatic increase in crossover frequency—from 16.6 ± 2.7% to 32 ± 5%—among the completed repair events (Figures 2A–2B). As all cells experience a DSB, the level of crossover was still 5% among all cells, similar to what is seen in wild-type cells. This observation again suggests that the absence of Srs2 predominantly affects the noncrossover pathway, particularly in the presence of excess Rad51. Previously it was reported that over-expression of Rad51 or Rad52 increases the MMS sensitivity of srs2Δ cells (Milne et al., 1995).
Coexpression of HO and RAD51 allowed recombination to be completed in the sgs1Δ srs2Δ rad51Δ (GAL::RAD51) triple mutant. Both the efficiency and kinetics of repair were comparable to srs2Δ (GAL::RAD51) alone, indicating that these helicases are not absolutely necessary for DSB repair. The level of crossing-over in the triple mutant was slightly higher (36 ± 4%) than in srs2Δ cells (GAL::RAD51) (32 ± 5%), but the difference was not statistically significant (Figure 2B). We propose that the lack of increase in crossovers in srs2Δ sgs1Δ compared to srs2Δ is due to the negative effect of RAD51 overexpression on crossovers observed in WT and sgs1Δ cells.
Recombination Defects of srs2Δ or sgs1Δ Mutants Are Suppressed by Overexpression of Sgs1 or Srs2, Respectively
Sgs1 and Srs2 are both 3′–5′ helicases and their absence results in a severe synthetic growth defect, suggesting that some of their functions can be redundant. Consistent with this idea, Mankouri et al. (2002) recently found that overexpression of SGS1 suppressed the sensitivity of srs2Δ to DNA damage. Here, we show that overexpressing SGS1 suppresses the high level of crossover in srs2Δ cells (Figure 5), increases recombination efficiency from 30% to 60%, and increases viability from 2%–3% to 32%. In one respect, overexpressing SGS1 did not suppress srs2Δ: the appearance of crossovers and noncrossovers continued to be coincident as shown for srs2Δ in Figure 4 (data not shown). Over-expressing SGS1 had no effect on the early appearance of noncrossovers in wild-type or sgs1Δ cells (data not shown).
Figure 5. Suppression of the Elevated Level of Crossing-Over in sgs1Δ and srs2Δ Strains by Overexpression of SRS2 or SGS1.
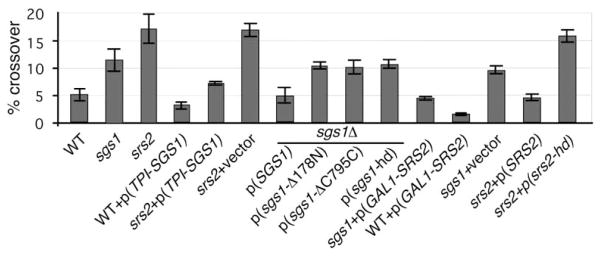
Genes were transcribed from their normal promoters unless otherwise specified.
We also tested whether overexpressing SRS2 could suppress the high crossover phenotype of sgs1Δ cells. Indeed, using a galactose-inducible SRS2 gene carried on a multicopy plasmid, overexpression of SRS2 decreased crossovers by half in sgs1Δ (Figure 5). Over-expressing Srs2 also decreased the level of product by half in wild-type cells and almost completely eliminated crossover products (<2%). As determined from Western blotting, Srs2 protein level was 20-fold higher than wild-type under these conditions (data not shown).
G2/M-Arrested Cells Have a High Level of Crossovers
When HO was induced in the cells arrested in G2/M with nocodazole total product formation was comparable to that found in logarithmically growing cells, but the frequency of crossing-over more than doubled to 12% (Figure 2B). Furthermore, srs2Δ cells, when arrested in G2/M, showed the same repair efficiency as randomly cycling cells but exhibited very high levels of crossing-over, approaching 25%. In contrast, sgs1Δ cells had the same level of exchange (about 10%) as in a random population of cells. The additive effect of G2/M arrest on crossing-over in srs2Δ but not in sgs1Δ cells is possibly due to lower Sgs1 abundance in G2/M (Frei and Gasser, 2000). To support this possibility, we showed that overexpressing SGS1 in wild-type G2/M-arrested cells reduced exchanges (data not shown). Overexpression of SGS1 in growing cells decreased crossover frequency by nearly 2-fold to about 2.5%–3% (Figure 5). It should be noted that sgs1Δ cells, and to a lesser extent srs2Δ cells, are slow-growing and accumulate cells in the G2 stage of the cell cycle; but this is unlikely to account for the increases in crossing-over that we see in these mutants in exponentially growing cultures. First, the phenotypes of the two helicase deletion mutants are distinctly different and therefore do not stem from holding cells in one stage of the cell cycle. Also, the top3 sgs1 double mutant that does not exhibit a significant growth defect shows the same higher frequency of crossing-over.
How Does the Sgs1 Helicase Suppress Crossing-Over?
The Sgs1 helicase interacts genetically and physically with topoisomerase III (Top3) through its N-terminal do- main (Fricke et al., 2001; Gangloff et al., 1994). Sgs1p also interacts with Rad51p at its C terminus (Wu et al., 2001). We introduced a set of centromeric plasmids carrying whole or partially deleted SGS1 genes (Fricke et al., 2001) into the sgs1Δ strain. The level of mutant proteins was the same for all constructs as determined by Western blotting (Fricke et al., 2001). As shown in Figure 5, only the plasmid carrying the intact SGS1 gene was able to fully suppress the high level of crossover observed in sgs1Δ. In contrast, plasmids carrying sgs1 mutants with a deletion or single amino acid mutation of the helicase domain (sgs1-ΔC795 or sgs1-hd) or a deletion of the N terminus (sgs1-ΔN178) that removes interaction with Top3 did not suppress the high frequency of crossing-over. These results show that both the helicase domain and the Top3 interaction domain are important for suppression of crossover frequency. To confirm that Sgs1’s effect on the level of crossover is Top3-dependent, we showed that both top3Δ and sgs1Δ top3Δ mutants have a similar phenotype to sgs1Δ (Figures 2A–2B). These results confirm that the Sgs1 and Top3 proteins are very potent in suppressing crossovers since they eliminate half of them. We also show that the helicase activity of Srs2 is required for the suppression of crossovers (Figure 5).
Sgs1p Prevents Rad51-Dependent Gene Conversion between Short Regions of Homology
Our previous study showed that there are two pathways of DSB repair in plasmids carrying inverted repeats, one of which is cleaved by HO: RAD51-dependent gene conversion and RAD51-independent break-induced replication coupled with single-strand annealing (BIR-SSA) (Ira and Haber, 2002; Kang and Symington, 2000). When there are only 33 bp of homology flanking the DSB, recombination is still 27% as efficient compared to long repeats. Nearly all of this recombination is RAD59-dependent (Figure 6). RAD51-dependent gene conversion requires a minimum of about 100 bp of homology on each DSB end (Ira and Haber, 2002). With short homology, Rad51p can apparently bind to ssDNA but cannot engage in productive recombination; thus, deleting RAD51 causes a surprising increase from 27% to 50% in recombinational repair (Ira and Haber, 2002). We now report that an sgs1Δ mutant has the same effect, increasing the DSB repair within short repeats to 55% (Figure 6). Deleting SGS1 did not have any impact on recombination when homology was ≥300 bp (data not shown). Apparently Sgs1p works in the RAD51-dependent pathway, since a rad59Δ sgs1Δ double mutant shows a higher level of repair (10%) than a rad59Δ single mutant (2%), whereas the rad51Δ sgs1Δ double mutant has the same level of repair (50%) as either single mutant. We suggest that Sgs1-Top3 is able to dismantle short Rad51-mediated strand invasion intermediates.
Figure 6. Sgs1p Prevents and Srs2 Facilitates Recombination between Short Homologous Sequences.
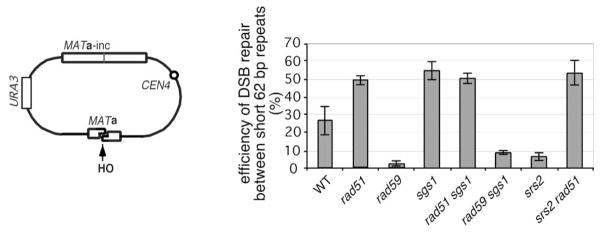
(A) In a plasmid used to study recombination, HO generates two 33 bp-long homologous DSB ends that can recombine with MATa-inc donor sequences situated in the opposite orientation.
(B) Effect of different mutations on the efficiency of DSB repair in the plasmid shown in (A).
Srs2 Facilitates Rad51-Independent Recombination Probably by Removal of Rad51
In contrast to sgs1Δ, the absence of the Srs2 helicase dramatically reduces DSB repair when the extent of homology is short (Ira and Haber, 2002). Recently, it was shown that Srs2 is able to remove Rad51 from ssDNA in vitro (Krejci et al., 2003; Veaute et al., 2003). One way in which srs2Δ cells might impair the RAD51-independent pathway would be by failing to remove Rad51 from ssDNA, which has an inhibitory effect on the alternative pathway. We therefore tested the efficiency of repair in rad51Δ srs2Δ cells. The recombination frequency was increased from 6% (srs2Δ) to over 50% (rad51Δ srs2Δ) (Figure 6). This result shows that the defect in srs2Δ cells is dependent on the presence of Rad51. This is consistent with the proposal that a helicase is able to remove Rad51 from ssDNA ends, allowing an alternative process to repair the DSB.
Discussion
Sgs1 and Srs2 DNA Helicases Suppress Crossovers in Mitotic Cells
We have shown that both Sgs1 and Srs2 proteins suppress crossing-over arising from DSB-induced homologous recombination. We suggest that Srs2 influences how DSBs are channeled into alternative recombination pathways, one of which yields a high proportion of crossovers whereas the other produces gene conversions with few if any crossovers. The two likely competing mechanisms are the double Holliday junction model (Szostak et al., 1983) and some variant of SDSA (reviewed in Pâques and Haber, 1999).
The strong evidence for the existence of two distinct mechanisms is that noncrossovers and crossovers do not appear with the same kinetics, as would be expected if they arose from alternative resolution of a common intermediate. Moreover, the balance between these two pathways can be altered by deletion or overexpression of SRS2. srs2Δ reduces the noncrossover pathway of ectopic recombination by 3-fold in RAD51 strains and by 5-fold when RAD51 is overexpressed. However, the absolute level of crossing-over, normalized to the number of cells that induced the DSB, remains constant. Also srs2Δ eliminates the kinetic difference in the appearance of crossovers and noncrossovers. Conversely, overexpressing SRS2 almost completely eliminates the crossover product. Together these results suggest that Srs2 is important in completing the noncrossover (SDSA) pathway in a way that is different from what occurs in the crossover pathway. In SDSA, the invading strand has to be displaced from the donor in order to repair the break, whereas in the double-HJ model, displacement of newly synthesized DNA is not necessary. We suggest that Srs2 facilitates strand displacement in the noncrossover SDSA pathway (Figure 7).
Figure 7. Model of Sgs1- and Srs2-Dependent Crossover Suppression.

(A) Srs2 promotes the noncrossover SDSA pathway.
(B) A HJ resolvase cuts double Holliday junctions to give crossovers and noncrossovers.
(C) Sgs1 acts together with Top3 to remove double Holliday junctions so that gene conversions will be recovered as noncrossovers.
In allelic recombination, srs2Δ causes the same increase in crossing-over as we see in ectopic recombination, but there isn’t any reduction in repair efficiency as we observed in ectopic recombination. Recently, Prado and Aguilera (2003) have suggested that limited regions of homology would decrease the possibility of forming a double HJ structure. In allelic recombination, homology is essentially unlimited and even if the invading strand cannot be displaced, new DNA synthesis would lead to formation of a double HJ intermediate (when both 3′ ends invade). Thus, in allelic recombination, strand invasion intermediates in srs2Δ cells would not be lost, but could be channeled into the crossover pathway, leading to the observed increase in crossovers.
There is mounting evidence that Srs2p may act by removing proteins from DNA undergoing recombination. Previously, we invoked such a mechanism to explain why Srs2p was required to resume cell cycle progression after DNA damage-induced checkpoint arrest and suggested that Srs2p’s role depended on Rad51p (Vaze et al., 2002). Recently, this idea has been supported by two in vitro studies demonstrating that Srs2p can remove Rad51p from ssDNA (Krejci et al., 2003; Veaute et al., 2003). We add further weight to this argument from in vivo experiments in which we conclude that the role of Srs2 is to remove Rad51 from ssDNA ends in circumstances where the extent of homology is very limited and the presence of Rad51 strongly inhibits intraplasmid DSB repair by a RAD59-, RAD50-dependent process (Ira and Haber, 2002).
Removal of Rad51 from ssDNA could inhibit recombination at the beginning by discouraging Rad51 filament formation, as seems to be the case in the UV sensitivity of srs2Δ(Aboussekhra et al., 1992), or it could be needed at later stages of recombination to promote recombination. For example, Srs2 could remove Rad51 during the strand exchange process in order to facilitate displacement of the base-paired invaded strand and the newly synthesized DNA from the template, in order to promote SDSA. Srs2 might also act to remove Rad51 from the second end of the DSB, preventing the formation of a double Holliday Junction intermediate and again facilitating SDSA (H. Klein, personal communication). Whatever the precise step, it seems to be distinct from steps shared in common by the noncrossover and crossover recombination pathways, as srs2Δ leaves crossovers largely unaffected.
Unlike Srs2, the absence of Sgs1 does not significantly affect the efficiency of DSB repair or the kinetics of product formation. However the absolute amount of crossing-over is twice higher in sgs1Δ cells. We note that sgs1Δ, top3Δ, and a double mutant all have the same phenotype. We suggest that Sgs1-Top3 act to remove double HJ structures, producing noncrossover outcomes (Figure 7). Such helicase and topoisomerase-dependent unwinding of double HJs has been suggested previously (Hastings, 1988; McGill et al., 1989; Nasmyth, 1982; Thaler and Stahl, 1988). Specifically, Top3 has been implicated in resolving recombination intermediates (Gangloff et al., 1999; Kwan et al., 2003). Moreover, Sgs1 and its human homologs BLM and WRN promote branch migration of Holliday junctions in vitro (Bennett et al., 1999; Constantinou et al., 2000; Karow et al., 2000). Consistent with our findings, Rockmill et al. (in press) have recently found that an sgs1 mutant (Δ795C) increases meiotic crossing-over in budding yeast.
In many assays of DNA metabolism, srs2Δ and sgs1Δ seem to have distinct phenotypes; yet our study has shown that overexpressed SGS1 substitutes for srs2Δ and vice versa. SGS1 overexpression suppresses three distinct aspects of recombination: the reduced efficiency of DSB repair, the increased crossovers, and the defect in recovery from the DNA damage checkpoint-imposed arrest. Conversely, overexpressing SRS2 suppresses the high level of crossover of sgs1Δ cells. It seems unlikely that Srs2 can work together with Top3; it is more likely that high levels of Srs2 suppresses formation of double HJs, as we observe almost no crossovers in wild-type cells overexpressing SRS2.
Regulation of Crossing-Over in Mitotic Recombination
We have shown that there are two kinetically distinct pathways of DSB repair in mitotic cells. This result is different in an important respect from the previous finding that noncrossovers preceded crossovers in meiotic recombination (Allers and Lichten 2001). In meiosis, crossovers are dependent on expression of the meiosis-specific transcription factor, NDT80, whereas noncross-overs were unperturbed in an ndt80Δ mutant. But in mitotic cells, where NDT80 is not expressed, there are still two distinct gene conversion pathways, one leading primarily to crossovers and one to noncrossovers. Another important difference is that the kinetically slower pathway, probably involving HJ intermediates, apparently leads to both crossovers and noncrossovers in mitotic cells, whereas in meiotic cells HJ intermediates lead only to crossovers.
Roles of BLM/WRN Helicases in Mammalian Recombination and Genome Integrity
The BLM/WRN helicases in human cells may act similarly to Sgs1 and Srs2 in Saccharomyces, to resolve recombination intermediates and to suppress loss of heterozygosity. BLM syndrome cells have very high levels of sister chromatid exchange (SCE) and the putative Top3α interaction domain of BLM is necessary to suppress elevated SCE (Hu et al., 2001; Wu et al., 2000). It is possible that BLM mutant cells have a higher incidence of spontaneous DNA damage, resulting in increased recombination, but our results with sgs1 mutants suggest that increased SCE could result from an increased proportion of crossovers arising from the same number of lesions.
Both BLM-deficient ES cells and yeast sgs1Δ cells show an elevated rate of spontaneous recombination, which leads to an increase in LOH (Ajima et al., 2002; Luo et al., 2000). It is unclear if the higher level of LOH is attributable to increased DNA damage, to an increased ability to recombine between inherently divergent sequences of homologous chromosomes or to an increased resolution of recombination events as crossovers (Shao et al., 2001; Myung et al., 2001).
Recently, the role of D. melanogaster BLM has also been studied in its repair of a DSB created by excision of a P element (Adams et al., 2003). A defect in DmBLM reduced the efficiency of SDSA, but apparently at a stage after strand invasion, as complete repair events were lost in favor of outcomes that appear to have arisen by a lack of processivity of DNA replication at the two ends of the DSB, resulting in single-strand annealing between fortuitous direct-repeated sequences within the region being copied. It is possible that the effect we see in srs2Δ on SDSA could arise from a similar defect, but our substrates do not have the internally repeated sequences that would be needed to detect aborted SDSA.
WRN mutations also affect homologous recombination, both reducing the efficiency of recombination and increasing the proportion of recombination events that are crossover-associated (Prince et al., 2001). Expression of bacterial Holliday junction resolvase RusA rescues the WRN recombination defect, which also suggests a role of WRN in resolution of HJ (Saintigny et al., 2002). Since no interaction has been found between human Top3 and WRN, the endonuclease activity necessary to resolve recombination intermediates needs to be determined. WRN cells do not show an increase in SCE, so the roles of BLM and WRN are likely to be different, however both have implications in the resolution of recombination intermediates. In one respect, WRN defects more closely resemble those of srs2 than sgs1. In both yeast and mammals, it appears that srs2 and WRN mutant cells can initiate and complete some types of recombination, but are defective in recovery (Saintigny et al., 2002; Vaze et al., 2002).
In summary, we find that both Sgs1 and Srs2 helicases suppress crossing-over in mitotic cells, but by different mechanisms. Both the choice of repair pathway and the resolution of Holliday junctions appear to be affected. Further studies are needed to determine how many different steps in homologous recombination require one or both of these helicases.
Experimental Procedures
Strains and Plasmids
Strains used in the study are presented in Supplemental Table S1 available at http://www.cell.com/cgi/content/full/115/4/401/DC1. Overexpression plasmids used in the study: multicopy pYES containing SRS2 gene under the control of GAL1 promoter; centromeric plasmid pRS316 containing SGS1 gene under TPI1 promoter; and centromeric plasmid YCplac22 containing either wild-type SRS2 or the srs2-K41A helicase mutant. Strains used to study the frequency of crossover carry MATa and MATa-inc sequences on chromosome III and chromosome V (Figure 1). Plasmid pGI365 used in the study has two inverted MATa perfectly homologous repeats of 62 bp and was described in detail previously (Ira and Haber, 2002).
Measurement of Viability, Product Formation, Crossover Frequency, and Plasmid Retention
To determine viability cells were grown o/n in YEP-lactate and the dilutions were plated onto YEP-Gal and YEPD plates. Plates were incubated at 30°C for 2–4 days. The number of colonies was counted and the viable proportion was derived by dividing colony-forming units on YEP-galactose by that on YEPD. Physical analysis of crossover frequencies in the ectopic recombination assay described in Figure 1A was done with DNA samples isolated from cells 8 hr after induction of HO endonuclease. Recombination between the repeats was induced by HO induction with 2% galactose in YEP lactate media when cell density was 1 ×107. HO endonuclease cuts the MAT sequence in all cells within one hour. Isolated DNA was digested with EcoRI enzyme and separated on 0.8% agarose gel. Southern blotting was done as described (Church and Gilbert, 1985), and the blots were probed with an 800 bp MATa fragment. Density of the gene conversion and crossover bands was done using Bio-Rad Quantity One software. To determine the frequency of crossovers among cells that repaired the break, we divided the intensity of the crossover band by the intensities of the noncrossover and the crossover bands. To determine the frequency of crossing-over among the all cells (whether they repaired the break or not), we divided the normalized intensity of the crossover band by the normalized intensity of the MATa uncut band at time = 0 hr.
The kinetics of product formation (noncrossovers and crossovers) at given time point were determined by dividing the normalized intensity of band corresponding to product by normalized intensity of product band 8 hr after induction (maximum value). Overall product formation compared to wild-type was determined by dividing the normalized intensity of band corresponding to products at 8 hr time point by normalized intensity of product bands in wild-type 8 hr after HO induction.
Crossing-over in HO-induced allelic recombination was determined as described previously (Malkova et al. 1996). Intraplasmid recombination assays were performed as described previously (Ira and Haber, 2002).
Acknowledgments
We thank Francis Fabre for plasmids carrying GAL::RAD51, and Iian Hickson, Steve Brill, Kelvin Kwan, James Wang, and Hannah Klein for various strains and plasmids. We thank Eric Coïc and Moru Vaze for helpful suggestions. This work was supported by NIH grants GM20056 and GM61766.
References
- Aboussekhra A, Chanet R, Adjiri A, Fabre F. Semi-dominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol. 1992;12:3224–3234. doi: 10.1128/mcb.12.7.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–267. doi: 10.1126/science.1077198. [DOI] [PubMed] [Google Scholar]
- Aguilera A, Klein HL. Genetic control of intrachromosomal recombination in Saccharomyces cerevisiae. I Isolation and genetic characterization of hyper-recombination mutations. Genetics. 1988;119:779–790. doi: 10.1093/genetics/119.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajima J, Umezu K, Maki H. Elevated incidence of loss of heterozygosity (LOH) in an sgs1 mutant of Saccharomyces cerevisiae: roles of yeast RecQ helicase in suppression of aneuploidy, interchromosomal rearrangement, and the simultaneous incidence of both events during mitotic growth. Mutat Res. 2002;504:157–172. doi: 10.1016/s0027-5107(02)00089-1. [DOI] [PubMed] [Google Scholar]
- Allers T, Lichten M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell. 2001;106:47–57. doi: 10.1016/s0092-8674(01)00416-0. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Bitan-Banin G, Kupiec M. Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:1403–1417. doi: 10.1128/MCB.23.4.1403-1417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Symington LS. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996;10:2025–2037. doi: 10.1101/gad.10.16.2025. [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Keck JL, Wang JC. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J Mol Biol. 1999;289:235–248. doi: 10.1006/jmbi.1999.2739. [DOI] [PubMed] [Google Scholar]
- Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR, 3rd, Russell P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001;107:537–548. doi: 10.1016/s0092-8674(01)00536-0. [DOI] [PubMed] [Google Scholar]
- Church GM, Gilbert W. The genomic sequencing technique. Prog Clin Biol Res. 1985;177:17–21. [PubMed] [Google Scholar]
- Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Esposito MS. Evidence that spontaneous mitotic recombination occurs at the two-strand stage. Proc Natl Acad Sci USA. 1978;75:4436–4440. doi: 10.1073/pnas.75.9.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA. 2002;99:16887–16892. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Kaliraman V, Brill SJ. Mapping the DNA topoisomerase III binding domain of the Sgs1 DNA helicase. J Biol Chem. 2001;276:8848–8855. doi: 10.1074/jbc.M009719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, de Massy B, Arthur L, Rothstein R, Fabre F. The essential role of yeast topoisomerase III in meiosis depends on recombination. EMBO J. 1999;18:1701–1711. doi: 10.1093/emboj/18.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–2053. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- Hastings PJ. Recombination in the eukaryotic nucleus. Bio-essays. 1988;9:61–64. doi: 10.1002/bies.950090206. [DOI] [PubMed] [Google Scholar]
- Hickson ID, Davies SL, Li JL, Levitt NC, Mohaghegh P, North PS, Wu L. Role of the Bloom’s syndrome helicase in maintenance of genome stability. Biochem Soc Trans. 2001;29:201–204. doi: 10.1042/0300-5127:0290201. [DOI] [PubMed] [Google Scholar]
- Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, Johnson FB, Guarente L, Ellis NA. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum Mol Genet. 2001;10:1287–1298. doi: 10.1093/hmg/10.12.1287. [DOI] [PubMed] [Google Scholar]
- Hunter N, Borts RH. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev. 1997;11:1573–1582. doi: 10.1101/gad.11.12.1573. [DOI] [PubMed] [Google Scholar]
- Ira G, Haber JE. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol. 2002;22:6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V, Mullen JR, Fricke WM, Bastin-Shanower SA, Brill SJ. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001;15:2730–2740. doi: 10.1101/gad.932201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang LE, Symington LS. Aberrant double-strand break repair in rad51 mutants of Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:9162–9172. doi: 10.1128/mcb.20.24.9162-9172.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow JK, Constantinou A, Li JL, West SC, Hickson ID. The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc Natl Acad Sci USA. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- Khazanehdari KA, Borts RH. EXO1 and MSH4 differentially affect crossing-over and segregation. Chromosoma. 2000;109:94–102. doi: 10.1007/s004120050416. [DOI] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- Kwan KY, Moens PB, Wang JC. Infertility and aneuploidy in mice lacking a type IA DNA topoisomerase III beta. Proc Natl Acad Sci USA. 2003;100:2526–2531. doi: 10.1073/pnas.0437998100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Johnson RE, Yu SL, Prakash L, Prakash S. Requirement of yeast SGS1 and SRS2 genes for replication and transcription. Science. 1999;286:2339–2342. doi: 10.1126/science.286.5448.2339. [DOI] [PubMed] [Google Scholar]
- Liberi G, Chiolo I, Pellicioli A, Lopes M, Plevani P, Muzi-Falconi M, Foiani M. Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J. 2000;19:5027–5038. doi: 10.1093/emboj/19.18.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- Malkova A, Ivanov EL, Haber JE. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Klein F, Leung WY, Haber JE. HO endo-nuclease-induced recombination in yeast meiosis resembles Spo11-induced events. Proc Natl Acad Sci USA. 2000;97:14500–14505. doi: 10.1073/pnas.97.26.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Craig TJ, Morgan A. SGS1 is a multicopy suppressor of srs2: functional overlap between DNA helicases. Nucleic Acids Res. 2002;30:1103–1113. doi: 10.1093/nar/30.5.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill C, Shafer B, Strathern J. Coconversion of flanking sequences with homothallic switching. Cell. 1989;57:459–467. doi: 10.1016/0092-8674(89)90921-5. [DOI] [PubMed] [Google Scholar]
- Milne GT, Ho T, Weaver DT. Modulation of Saccharomyces cerevisiae DNA double-strand break repair by SRS2 and RAD51. Genetics. 1995;139:1189–1199. doi: 10.1093/genetics/139.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Chen C, Kolodner RD. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet. 2001;27:113–116. doi: 10.1038/83673. [DOI] [PubMed] [Google Scholar]
- Nasmyth KA. Molecular genetics of yeast mating type. Annu Rev Genet. 1982;16:439–500. doi: 10.1146/annurev.ge.16.120182.002255. [DOI] [PubMed] [Google Scholar]
- Nassif N, Penney J, Pal S, Engels WR, Gloor GB. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol. 1994;14:1613–1625. doi: 10.1128/mcb.14.3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâques F, Haber JE. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:6765–6771. doi: 10.1128/mcb.17.11.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A. Control of cross-over by single-strand DNA resection. Trends Genet. 2003;19:428–431. doi: 10.1016/S0168-9525(03)00173-2. [DOI] [PubMed] [Google Scholar]
- Prince PR, Emond MJ, Monnat RJ., Jr Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001;15:933–938. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockmill B, Fung JC, Branda SS, Roeder GS. The Sgs1 helicase regulates chromosome synapsis and meiotic crossing over. Curr Biol. 2003;22:1954–1962. doi: 10.1016/j.cub.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Rong L, Palladino F, Aguilera A, Klein HL. The hyper-gene conversion hpr5–1 mutation of Saccharomyces cerevisiae is an allele of the SRS2/RADH gene. Genetics. 1991;127:75–85. doi: 10.1093/genetics/127.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in Werner syndrome. Mol Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacha A, Kleckner N. Identification of double Holliday junctions as intermediates in meiotic recombination. Cell. 1995;83:783–791. doi: 10.1016/0092-8674(95)90191-4. [DOI] [PubMed] [Google Scholar]
- Shao C, Stambrook PJ, Tischfield JA. Mitotic recombination is suppressed by chromosomal divergence in hybrids of distantly related mouse strains. Nat Genet. 2001;28:169–172. doi: 10.1038/88897. [DOI] [PubMed] [Google Scholar]
- Shen J, Loeb LA. Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev. 2001;122:921–944. doi: 10.1016/s0047-6374(01)00248-2. [DOI] [PubMed] [Google Scholar]
- Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R. Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics. 2002;162:647–662. doi: 10.1093/genetics/162.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles–a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Stark JM, Jasin M. Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol. 2003;23:733–743. doi: 10.1128/MCB.23.2.733-743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE. DNA length dependence of the single-strand annealing pathway and the role of Sac-charomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol. 2000;20:5300–5309. doi: 10.1128/mcb.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- Thaler DS, Stahl FW. DNA double-chain breaks in recombination of phage lambda and of yeast. Annu Rev Genet. 1988;22:169–197. doi: 10.1146/annurev.ge.22.120188.001125. [DOI] [PubMed] [Google Scholar]
- Tsubouchi H, Ogawa H. Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol Biol Cell. 2000;11:2221–2233. doi: 10.1091/mbc.11.7.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaze M, Pellicioli A, Lee S, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber J. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires srs2 helicase. Mol Cell. 2002;10:373–385. doi: 10.1016/s1097-2765(02)00593-2. [DOI] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- Virgin JB, Bailey JP, Hasteh F, Neville J, Cole A, Tromp G. Crossing over is rarely associated with mitotic intragenic recombination in Schizosaccharomyces pombe. Genetics. 2001;157:63–77. doi: 10.1093/genetics/157.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ. SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID. The Bloom’s syndrome gene product interacts with topoisomerase III. J Biol Chem. 2000;275:9636–9644. doi: 10.1074/jbc.275.13.9636. [DOI] [PubMed] [Google Scholar]
- Wu L, Davies SL, Levitt NC, Hickson ID. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001;276:19375–19381. doi: 10.1074/jbc.M009471200. [DOI] [PubMed] [Google Scholar]