

Abstract
When naïve or memory T cells encounter foreign antigen along with proper co-stimulation they undergo rapid and extensive clonal expansion. In mammals, this type of proliferation is fair1y unique to cells of the adaptive immune system and requires a considerable expenditure of energy and cellular resources. While research has often focused on the roles of cytokines, antigenic signals, and co-stimulation in guiding T cell responses, data indicate that, at a fundamental level, it is cellular metabolism that regulates T cell function and differentiation and therefore influences the final outcome of the adaptive immune response. This review will focus on some earlier fundamental observations regarding T cell bioenergetics and its role in regulating cellular function, as well as recent work that suggests that manipulating the immune response by targeting lymphocyte metabolism could prove useful in treatments against infection and cancer.
Introduction
Activated T cells have an anabolic metabolism
Unlike the case for unicellular organisms where dramatic changes in nutrient availability affect proliferation, mammalian cells reside in nutrient-rich environments where cellular proliferation is controlled by extrinsic signals, such as growth factors, which regulate nutrient utilization [1]. One of the most striking changes to affect T cells after initial antigenic stimulation is an increase in cell size accompanied by a metabolic switch to glycolysis, which is required to support their growth, proliferation, and effector functions [2–4] (Figure 1). During TCR stimulation, signals from growth factor cytokines like IL-2, and the ligation of co-stimulatory CD28, lead to an increase in glycolysis by inducing the Pl3K-dependent activation of Akt [5,6]. Activated Akt can promote the mTOR (mammalian target of rapamycin) pathway, a key regulator of translation [7], as well as stimulate glycolysis by increasing glycolytic enzyme activiry and enhancing the expression of nutrient transporters, enabling increased utilization of glucose and amino acids [8,9,10••,11–13]. Together these changes lead to the increase in nutrient utilization and glucose metabolism that facilitates activation and proliferation.
Figure 1.
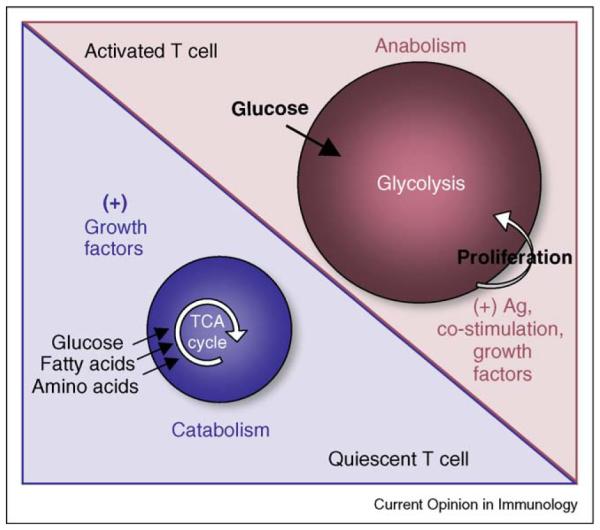
Activated and quiescent T cells have distinct metabolic phenotypes. Activated T cells (effector T cells) have an anabolic metabolism where they maintain a high rate of nutrient uptake and build biomass at the expense of ATP. In the presence of antigen and co stimulation, growth factor cytokines can stimulate glycolysis and support proliferation. Glycolysis provides ATP for proliferating T cells while fatty acids and amino acids are incorporated into cellular components. By contrast, quiescent T cells (naïve and memory T cells) have a catabolic metabolism where they use glucose, fatty acids, and amino acids for ATP generation through the TCA cycle and oxidative phosphorylation. Growth factor cytokines increase nutrient transporter expression and are important for cell survival, and in their absence, quiescent cells die of progressive atrophy.
As T cells undergo clonal expansion they preferentially ferment glucose to meet their energy demands, even though there is sufficient oxygen present to support mitochondrial oxidative phosphorylation [14–16]. This phenomenon is known as the Warburg effect [17], and is an unusual metabolic aspect of proliferating T cells and cancer cells. Since ATP production by aerobic glycolysis is much less efficient than by oxidative phosphorylation, a question remains as to why proliferating T cells favor this form of metabolism. One explanation, largely based on observations from Craig Thompson’s laboratory, is that glycolysis is an essentially anabolic form of metabolism that leaves cellular building blocks, such as amino acids and fatty acids untouched, as well as produces lactate, all of which can be incorporated into cellular components [18]. A cell that converts building blocks into biomass most efficiently will proliferate the fastest and in a host fighting an infection, rapid expansion of antigen-specific T cells could offer a decisive advantage [19].
Non-proliferating T cells have a catabolic metabolism
In contrast to proliferating T cells, quiescent T cells (i.e. naïve and memory cells), like most cells in normal tissues, interchangeably breakdown glucose, amino acids, and lipids to catabolically fuel ATP generation [2,18] (Figure 1). The posited effects of growth factors on resting T cell survival are related to their ability to modulate the surface expression of nutrient transporters [20]. Quiescent cells can also use autophagy (the break down of intracellular components) to supply the molecules to fuel oxidative phosphorylation [21]. There is growing evidence that quiescence is under active transcriptional control [22]. TOB1 (transducer of ERBB2 1) [23], LKLF (lung Krüppel-like factor) [24], and FOXO (Forkhead box class O) transcription factors all have been suggested to promote quiescence in lymphocytes by actively maintaining the expression of inhibitors of cellular activation [25,26]. Furthermore, FOXO transcription factors have been shown to modulate metabolic functions [27,28] and the family of Krüppel-like factors (KLFs) has been shown to regulate adipocyte differentiation and glucose homeostasis in mammals [29], which may suggest a degree of metabolic control in maintaining quiescence.
Implicit in the striking divergence of metabolic phenotypes between proliferating and quiescent T cells is the idea that the conversion, or switching, between differing metabolic states is required to effectively generate a given T cell fate [10••,18]. This not only applies to the switch from quiescence to glycolysis that accompanies naïve T cell activation, but also to the promotion of catabolism that appears to be important for the generation of quiescent memory T cells after infection [30••] (Figure 2). Each of these metabolic states represents a unique target of intervention for manipulating the T cell response and ameliorating disease.
Figure 2.
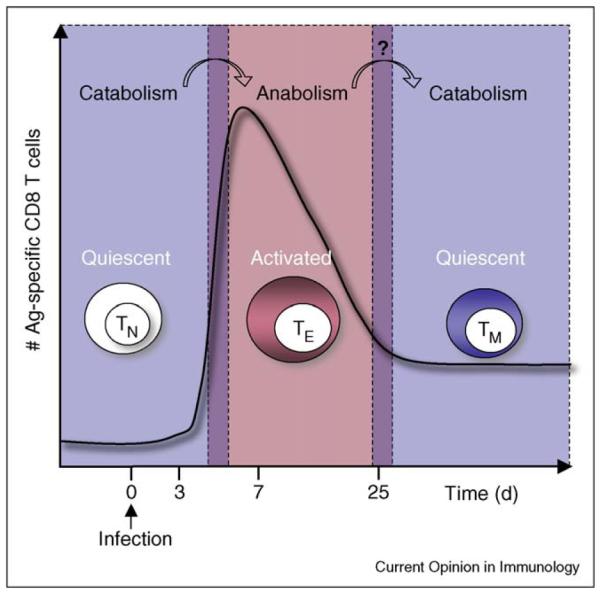
Metabolic transitions underpin T cell fate. In response to infection CD8 T cells undergo a developmental pattern characterized by first the expansion then contraction of antigen specific populations, followed by the persistence of long lived memory T cells. The striking divergence of metabolic phenotypes between activated and quiescent T cells suggests the idea that the conversion, or switching, between differing metabolic states is required for effective generation of a given T cell fate. This is true for the conversion from a resting metabolism to the highly glycolytic metabolism that is triggered by T cell activation. Recent data also suggest that the conversion to, or promotion of, catabolic processes (like fatty acid oxidation or autophagy) within antigen specific populations are important for the generation of memory T cells after infection. While cytokine receptor signaling supports a particular metabolic phenotype (Figure 1), the withdrawal of cytokines, or nutrients, can present a significant metabolic stress to a proliferating cell. In response to this stress, cells can promote catabolic pathways for survival. This appears to play a role in the generation of memory T cells following infection.
Metabolic regulation of T cell responses
It is well documented that proliferating T cells require glucose to survive, and in the absence of glucose they cannot support their bioenergetic demands and undergo apoptosis [4,31,32]. Much less is known about how lipid metabolism regulates T cell responses. However, in a recent study of great interest, Bensinger et al. examined the influence of cellular lipid metabolism on the immune response and showed that LXR (Liver X receptor), a member of the nuclear receptor family of transcription factors that are important regulators of cholesterol, fatty acid, and glucose homeostasis, couples sterol metabolism to T cell proliferation [33••]. They found that sterol regulation during T cell activation was linked to transcriptional responses mediated by SREBP and LXR. LXR-β-deficient mice exhibited lymphoid hyperplasia and augmented responses to antigen, demonstrating that the proper regulation of sterol metabolism is essential for normal immune response development. A separate study showed that PPAR-γ, another member of the nuclear receptor family of transcription factors that regulates cellular differentiation and lipid metabolism and is closely related to LXR [34], selectively inhibits Th17 differentiation in a T cell intrinsic manner [35], further supporting the role of lipid metabolism in regulating T cell fate.
A recent study from our group showed that the CD8 T cell specific induction of fatty acid oxidation (FAO) following the peak of the primary T cell response to infection is important for the generation of CD8 T cell memory [30••]. We proposed that during a primary response to infection T cells experience a metabolic stress, perhaps due to nutrient and/or growth factor limitation (more on this point below) as infection is cleared and antigen concentrations decline and that during this period cells need to switch from glycolysis to other forms of catabolism, like FAO, in order to survive and develop into memory T cells. Our results also suggested that amino acid degradation and bile acid biosynthesis might be important for memory T cell development. This would be consistent with previous studies showing that antigen-presenting cells regulate amino acid availability to T cells, which alters their function and proliferation [36–38], as well as with Bensinger’s work showing the important role of sterol metabolism in T cell responses [33].
Lum et al. showed that the autophagic breakdown of cellular components is crucial for maintaining cell survival following growth factor withdrawal [39]. In light of these findings, an intriguing possibility is that during contraction and subsequent memory development T cells use autophagy to satisfy their bioenergetic needs. Consistent with this, Pua et al. have shown that autophagy is essential for T cell survival and proliferation [40]. They also suggested that it might have a physiologically significant role in the clearance of superfluous mitochondria in T lymphocytes as part of normal T cell homeostasis [41]. Future studies will need to determine what role this catabolic process serves in antigen-specific T cells after infection.
It remains unclear whether during the development of T cell memory there is an active switch to quiescence by cells that have adopted Warburg metabolism, or that those cells destined to become memory cells maintain a degree of ‘metabolic quiescence’ throughout development. Data from Prlic et al. showing that competition does not affect contraction kinetics suggest that the proposed growth factor/nutrient limitation at the peak of the T cell response is not due to constrained resources [42]. However, it is possible that cells are imparted with specific metabolic characteristics following their first encounter with antigen, and that this very early differentiation endows cells with their ability to use nutrients when they later experience a metabolic stress due to a perceived decline in and/or lack of signal from growth factor. This view is consistent with the model postulated by Chang et al. where the spectrum of cell fates is imparted to the clonal descendants of a single lymphocyte [43]. It will also be worth considering in future studies how different locations within the body, for example peripheral tissues versus lymphoid organs, could impose varying metabolic constraints on T cells to influence their development and subsequent longevity.
Harnessing immunity through metabolism
The fact that metabolism underlies the functional capacity of a T cell suggests that altering cellular metabolism may influence the final outcome of the adaptive response. In addition, lymphocytes may be particularly amenable to metabolic manipulation since their development is marked by striking changes in metabolism. Since clearance or control of pathogens or tumors usually requires T cell mediated immunity, the ability to deliberately manipulate T cell responses by regulating metabolism would be a powerful tool against cancers or infections. Several recent studies have shown that treating with pharmacological modulators of certain T cell intrinsic processes can effectively promote the formation of CD8 T cell memory [30,44••,45••].
An important study by Araki et al. demonstrated that mTOR, a key regulator of translation, regulates CD8 T cell memory in a cell-intrinsic manner [45••]. The crucial role of mTOR signaling in regulating T cell fate is highlighted by another important study showing that it dictates decisions between effector and regulatory T cell lineage commitment [46]. The PI3K/Akt/mTOR pathway is used by cells to respond to growth factors and serves to augment many of the metabolic activities that support cellular biosynthesis [13]. Signals emanating from this pathway result in increased nutrient uptake, enhanced glycolytic enzyme activity, and promote the biosynthesis of macromolecules [10••]. Blocking the mTOR pathway can inhibit proliferation [47] while favoring the promotion of other forms of nutrient catabolism [48–53]. We, and Araki et al., demonstrated that the mTOR inhibitor rapamycin promotes CD8 T cell memory and it is tempting to speculate that this is due to the promotion of catabolic metabolism in CD8 T cells. However, it remains possible that the effects are due to blocking cell cycle progression through the inhibition of protein synthesis. Our group also showed that the anti-diabetic drug metformin, which activates AMPK (AMP-activated protein kinase) enhances CD8 T cell memory generation. Active AMPK can inhibit the mTOR pathway [54] as well as more directly promote fatty acid catabolism [55,56]. It is likely that rapamycin and metformin act in both of these ways to regulate metabolism and augment CD8 T cell memory development.
A recent study by Gattinoni et al. implicated the Wnt-β-catenin developmental pathway in regulating CD8 T cell memory [44••]. By inhibiting gsk3-β (glycogen synthase kinase 3-β), and thereby promoting Wnt signaling, they showed that effector CD8 T cell differentiation was arrested and the development of ‘stem cell like’ memory CD8 T cells was induced. Interestingly, gsk3-β inhibition also inhibits the mTOR pathway in an AMPK-dependent manner, demonstrating that Wnt and energy signals can integrate to regulate cell growth [57]. While more work needs to be done to establish the precise mechanisms of regulation, it is clear that using pharmacological modulators of certain CD8 T cell intrinsic processes, like developmental or metabolic pathways, can promote immunologic memory and may become a powerful tool for treating human disease [30••,44••,45••].
Previous work has shown that CD8 T cells from a chronic viral infection display marked defects in the expression of metabolic genes [58]. Rutishauser et al. recently demonstrated that the transcription factor Blimp-1 promotes CD8 T cell terminal differentiation and represses the acquisition of memory T cell properties [59], Kallies et al. showed that Blimp-1 is required for effector CD8 T cell differentiation [60], and Shin et al. identified Blimp-1 as a regulator of CD8 T cell exhaustion during chronic viral infection that acts by repressing key aspects of normal memory CD8 T cell differentiation [61]. It will be interesting to determine if the terminal differentiation conferred by Blimp-1, or by active gsk3-β, imparts an inappropriate metabolic phenotype that hinders T cell development, or if metabolic perturbations affect the expression of these key regulators.
Bmi-1, a crucial regulator of cellular self-renewal, has been shown to be a molecular determinant of the capacity of antigen-specific CD8 T cells to persist during chronic infection, and thus a possible regulator of replicative competence [62]. In conjunction with this, it has also been shown that Bmi-1 regulates CD4 T cell memory survival via repression of Noxa [63], a gene that controls susceptibility to apoptosis under glucose limitation in dividing T cells [32] while another study demonstrated that Bmi-1-deficient cells have impaired mitochondrial function, increased reactive oxygen species, and subsequent engagement of the DNA damage response pathway [64]. Together, these data may further suggest an underlying role for metabolism in regulating longevity and senescence in T cells.
T cell life span: parallels with organismal life span
Immunological memory is the basis of vaccination, and promoting the generation of long-lived memory T cells is a focus of vaccine development. The greatest insights into the molecular pathways that control longevity have come from studies in worms, yeast, and fruit flies. This work has shown that insulin/IGF signaling, DAF-16/FOXO transcription factors, and TOR all play crucial roles in regulating metabolism as well as life span [65–70]. The cellular response to environmental or metabolic stress is functionally important in longevity and the genes that regulate longevity, may have actually evolved to protect animals from harsh environmental conditions [71]. Interestingly, the stress resistance of vertebrate cells in culture has been shown to positively correlate with the life spans of the species from which they originated [71,72].
Metabolic stress can be imposed by dietary restriction. Dietary restriction is defined as a reduction in nutrient availability without malnutrition, and has long been known to increase lifespan in species ranging from yeast to mammals [73]. Studies suggest that blocking mTOR activity may mimic dietary restriction, and consistent with this the AMPK–FOXO pathway, which inhibits TOR activity, is implicated in mediating longevity by dietary restriction in C. elegans [74]. In addition, the mTOR inhibitor rapamycin and AMPK activators like metformin have been shown to increase mammalian life span [75•,76], which is consistent with observations that these drugs promote T cell life span [30••,45••]. Furthermore, dietary restriction causes an increase in fat metabolism. A study from Wang et al. has linked fat metabolism with longevity in C. elegans [77], which is consistent with the idea that inducing the breakdown of fat in T cells can increase their life span [30••]. If we consider the possibility that rapamycin and metformin impose a ‘drug-enforced dietary restriction’ on T cells it may provide us with a more global understanding as to why these drugs promote the development of long-lived memory T cells. Since dietary restriction increases life span across many species, it is worth applying a model of the longevity pathway [78••] to T cells (Figure 3). Further work is needed to determine precisely how these molecular pathways could regulate T cell life span.
Figure 3.

Analogous model of T cell longevity (left panel). The mTOR inhibitor rapamycin and the AMPK activator metformin have both been shown to promote the development of long-lived memory CD8 T cells. Longevity pathway (right panel). A conserved signaling cascade controls life span in yeast, worms, flies, and mice (figure adapted from [78••]).
Metabolic contributions
The question as to precisely what fluctuations in metabolism provide to T cells during different phases of an immune response remains incompletely answered. One assumption is that switching to catabolic forms of metabolism in the face of metabolic stress, that is, after infection is cleared and associated signals dissipate, is a survival mechanism that provides energy in the form of ATP, and that this allows T cells to assume quiescence and become long-lived. However, it may be more complicated than this. For a T cell, inducing FAO after the peak of the effector response may not only provide energy, but also promote the breakdown of inflammatory mediators like eicosanoids and leukotrienes, or alter lipid raft formation and influence signaling events. It is also possible that changes in FAO alter T cell gene expression to promote longevity after infection. ATP citrate lyase, an important enzyme for fatty acid biosynthesis, has recently been shown to link growth factor-induced increases in nutrient metabolism to the regulation of histone acetylation and gene expression [79]. It is also important to note that glucose is used not only to fuel glycolysis, but also for glycosylation of protein. Differences in glycosylation can have enormous effects on T cell function, trafficking, and susceptibility to death [80,81]. Lastly, it should be considered that changes in metabolism, such as an increase in catabolic processes like autophagy, could alter the accumulation and degradation of damaged proteins and thus affect T cell survival. Together, all of these cellular processes are intertwined by the single common thread that metabolism influences each of them. Defining metabolic requirements of T cells during different phases of the immune response will be important for our future understanding of T cell development.
Conclusion
Successful disease prevention or therapy often depends on T cells performing to their expected capacity. The ability to alter T cell performance could prove useful in settings of prophylactic vaccination where the induction of a population of long-lived memory T cells is the goal, or in cases of therapeutic cancer vaccines, where effective tumor therapy depends on the reliable ex vivo expansion of a patient’s T cells. If we can manipulate T cell metabolism, and many drugs that do this are already approved for use in humans, we may be able to shape immune responses and open new ways in which we approach the treatment of human disease.
Acknowledgements
I would like to thank Edward Pearce, Matt Walsh, Pedro Cejas, Rusty Jones, and Yongwon Choi who contributed to the ideas and discussions that are the basis of this review.
The author’s laboratory is supported by a grant from the Emerald Foundation.
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Raff MC. Size control: the regulation of cell numbers in animal development. Cell. 1996;861:173–175. doi: 10.1016/s0092-8674(00)80087-2. [DOI] [PubMed] [Google Scholar]
- 2.Krauss S, Brand MD, Buttgereit F. Signaling takes a breath—new quantitative perspectives on bioenergetics and signal transduction. Immunity. 2001;15:497–502. doi: 10.1016/s1074-7613(01)00205-9. [DOI] [PubMed] [Google Scholar]
- 3.Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res. 1973;77:127–135. doi: 10.1016/0014-4827(73)90561-2. [DOI] [PubMed] [Google Scholar]
- 4.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–692. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 5.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 6.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 8.Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med. 2004;200:659–669. doi: 10.1084/jem.20040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 10••.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–178. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- The authors provide a concise framework regarding what is known about T cell metabolism to date and draw attention to the crucial role of metabolism in T cell activation and function.
- 11.Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size. CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33:2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- 12.Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Wang T, Marquardt C, Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature. 1976;261:702–705. doi: 10.1038/261702a0. [DOI] [PubMed] [Google Scholar]
- 15.Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem. 1994;269:31484–31490. [PubMed] [Google Scholar]
- 16.Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997;11:388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- 17.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 18.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 19.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naïve T cells. J Immunol. 2001;167:6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 21.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 22.Tzachanis D, Boussiotis VA. Tob, a member of the APRO family, regulates immunological quiescence and tumor suppression. Cell Cycle. 2009;8:1019–1025. doi: 10.4161/cc.8.7.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzachanis D, Freeman GJ, Hirano N, van Puijenbroek AA, Delfs MW, Berezovskaya A, Nadler LM, Boussiotis VA. Tob is a negative regulator of activation that is expressed in anergic and quiescent T cells. Nat Immunol. 2001;2:1174–1182. doi: 10.1038/ni730. [DOI] [PubMed] [Google Scholar]
- 24.Kuo CT, Veselits ML, Leiden JM. LKLF: a transcriptional regulator of single-positive T cell quiescence and survival. Science. 1997;277:1986–1990. doi: 10.1126/science.277.5334.1986. [DOI] [PubMed] [Google Scholar]
- 25.Liu JO. The yins of T cell activation. Sci STKE. 2005;2005:re1. doi: 10.1126/stke.2652005re1. [DOI] [PubMed] [Google Scholar]
- 26.Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. 2003;24:380–386. doi: 10.1016/s1471-4906(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 27.Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Curr Diab Rep. 2009;9:208–214. doi: 10.1007/s11892-009-0034-5. [DOI] [PubMed] [Google Scholar]
- 28.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 29.Brey CW, Nelder MP, Hailemariam T, Gaugler R, Hashmi S. Kruppel-like family of transcription factors: an emerging new frontier in fat biology. Int J Biol Sci. 2009;5:622–636. doi: 10.7150/ijbs.5.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study showed that treating mice after infection with either rapaymcin or metformin, two drugs that augment fatty acid oxidation, enhanced the development of memory CD8 T cells. On the basis of their observations, the authors suggest that promoting catabolic pathways of metabolism, like fatty acid oxidation, is crucial to the development of long-lived memory T cells.
- 31.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factorssurvival through effects can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alves NL, Derks IA, Berk E, Spijker R, van Lier RA, Eldering E. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity. 2006;24:703–716. doi: 10.1016/j.immuni.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 33••.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The authors demonstrated that the transcriptional regulation of intracellular cholesterol homeostasis impacts T cell proliferation during acquired immune responses. Their findings implicate LXR signaling in a metabolic checkpoint that modulates cell proliferation and immunity.
- 34.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 35.Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, Alferink J, Novak N, Beyer M, Mayer G, et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206:2079–2089. doi: 10.1084/jem.20082771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mellor AL, Munn DH. IDO: expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 37.Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, Clarke F, Sitia R, Rubartelli A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A. 2002;99:1491–1496. doi: 10.1073/pnas.022630299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edinger AL, Thompson CB. Antigen-presenting cells control T cell proliferation by regulating amino acid availability. Proc Natl Acad Sci U S A. 2002;99:1107–1109. doi: 10.1073/pnas.042707999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 40.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 42.Prlic M, Bevan MJ. Exploring regulatory mechanisms of CD8+ T cell contraction. Proc Natl Acad Sci U S A. 2008;105:16689–16694. doi: 10.1073/pnas.0808997105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 44••.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study showed that the pharmacolgical manipulation of the developmental pathway Wnt can promote the development of CD8 T memory stem cells.
- 45••.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP. Ahmed R: mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kay JE, Kromwel L, Doe SE, Denyer M. Inhibition of T and B lymphocyte proliferation by rapamycin. Immunology. 1991;72:544–549. [PMC free article] [PubMed] [Google Scholar]
- 48.Brown NF, Stefanovic-Racic M, Sipula IJ, Perdomo G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism. 2007;56:1500–1507. doi: 10.1016/j.metabol.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Noda T, Ohsumi Y. Tor: a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 50.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–2326. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 51.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 52.Sipula IJ, Brown NF, Perdomo G. Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism. 2006;55:1637–1644. doi: 10.1016/j.metabol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Peng T, Golub TR, Sabatini DM. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol. 2002;22:5575–5584. doi: 10.1128/MCB.22.15.5575-5584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 55.Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- 56.Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem. 1994;269:22162–22168. [PubMed] [Google Scholar]
- 57.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 58.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 59.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31:283–295. doi: 10.1016/j.immuni.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 61.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, Wherry EJ. A role for the transcriptional repressor Blimp 1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–320. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heffner M, Fearon DT. Loss of T cell receptor-induced Bmi-1 in the KLRG1(+) senescent CD8(+) T lymphocyte. Proc Natl Acad Sci U S A. 2007;104:13414–13419. doi: 10.1073/pnas.0706040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamashita M, Kuwahara M, Suzuki A, Hirahara K, Shinnaksu R, Hosokawa H, Hasegawa A, Motohashi S, Iwama A, Nakayama T. Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J Exp Med. 2008;205:1109–1120. doi: 10.1084/jem.20072000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kapahi P, Zid B. TOR pathway: linking nutrient sensing to life span. Sci Aging Knowledge Environ. 2004;2004:PE34. doi: 10.1126/sageke.2004.36.pe34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- 68.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 69.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 70.Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 71.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Kapahi P, Boulton ME, Kirkwood TB. Positive correlation between mammalian life span and cellular resistance to stress. Free Radic Biol Med. 1999;26:495–500. doi: 10.1016/s0891-5849(98)00323-2. [DOI] [PubMed] [Google Scholar]
- 73.Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–1074. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greer EL, Banko MR, Brunet A. AMP-activated protein kinase and FoxO transcription factors in dietary restriction-induced longevity. Ann N Y Acad Sci. 2009;1170:688–692. doi: 10.1111/j.1749-6632.2009.04019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75•.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study showed that inhibiting the mammalian target of rapamycin (mTOR) increases life span in mice.
- 76.Anisimov VN, Semenchenko AV, Yashin AI. Insulin and longevity: antidiabetic biguanides as geroprotectors. Biogerontology. 2003;4:297–307. doi: 10.1023/a:1026299318315. [DOI] [PubMed] [Google Scholar]
- 77.Wang MC, O’Rourke EJ, Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 2008;322:957–960. doi: 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78••.Kaeberlein M, Kapahi P. Cell signaling. Aging is RSKy business. Science. 2009;326:55–56. doi: 10.1126/science.1181034. [DOI] [PubMed] [Google Scholar]
- The authors discuss recent results concerning lifespan and rapamycin treatment in mice. They provide a clear explanation on how these observations fit with the highly conserved longevity pathway that links nutrient availability to aging in organisms as diverse as yeast and mice.
- 79.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 81.Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. 2008;8:874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]