

ABSTRACT
Human cytomegalovirus (HCMV) deregulates the cell cycle by several means, including inactivation of the anaphase-promoting complex/cyclosome (APC/C) E3 ubiquitin ligase. Viral proteins UL97 and UL21a, respectively, affect the APC/C by phosphorylation of APC/C coactivator Cdh1 and by inducing the degradation of subunits APC4 and APC5, which along with APC1 form the APC/C platform subcomplex. The aim of this study was to further characterize the mechanism of APC/C inactivation and define the relative contributions of UL21a and UL97 to APC/C substrate accumulation and to viral growth. We show that in uninfected cells, UL21a but not UL97 can disrupt APC/C function, leading to the accumulation of substrates. We find that UL21a is necessary and sufficient to induce the degradation of APC1, in addition to the previously reported APC4 and APC5. We also demonstrate that there is a previously unreported cellular mechanism for a specific decrease in the levels of all three platform subunits, APC1, APC4, and APC5, upon the depletion of any one of these subunits or of subunit APC8. Finally, we show that at a low multiplicity of infection, either UL97 or UL21a can partially complement a growth-defective mutant virus lacking both UL21a and UL97, with significantly greater benefit afforded by the expression of both proteins. This double mutant also can be partially rescued by inactivation of the APC/C using small interfering RNAs against specific subunits. These results further our understanding of HCMV's interaction with the cell cycle machinery and reveal a new cellular pattern of APC/C subunit downmodulation.
IMPORTANCE HCMV lytic infection subverts the host cell cycle machinery in multiple ways. A major effect is inactivation of the APC/C, which plays a central role in the control of cell cycle progression. This study provides further insight into the mechanism of inactivation. We discovered that the APC1 subunit, which along with APC4 and APC5 form the platform subcomplex of the APC/C, is an additional target of the degradation induced by HCMV protein UL21a. This study also shows for the first time that there is a unique cellular process in uninfected cells whereby depletion of APC1, APC4, APC5, or APC8 recapitulates the pattern of HCMV-mediated APC/C subunit degradation.
INTRODUCTION
Human cytomegalovirus (HCMV) infects the majority of the human population, causing significant morbidity and mortality in immunocompromised individuals, such as transplant patients and those with HIV. HCMV is also the leading viral cause of birth defects. These manifest as neural developmental defects ranging from hearing loss to calcification of the developing brain and death at a rate of 1 or 2 per 1,000 newborns.
HCMV lytic infection both modulates and is influenced by the host cell cycle. The virus preferentially infects cells in G0 or G1. Infection in other phases of the cell cycle results in a delay of immediate early gene expression until completion of mitosis in the case of a G2 infection. Infection during S phase remains unproductive in a certain percentage of cells. Early in infection, the virus causes a stimulation of resting cells into the cell cycle and subsequent arrest at the G1/S border (1–3). The infection inhibits host DNA replication, affects cyclin levels (4), prevents host DNA replication licensing (5–7), and inhibits the anaphase-promoting complex/cyclosome (APC/C) (8–10).
The APC/C is a large, multisubunit E3 ubiquitin ligase that targets a growing list of proteins for degradation by the proteasome. The APC/C orchestrates progression through the cell cycle by targeting the cyclins and other cell cycle-associated proteins for degradation to allow cells to proceed though cell cycle checkpoints. Its own activity is cyclical, showing activity in G1 and at anaphase and inhibition from S phase until the chromosomes are properly aligned in metaphase and the spindle assembly checkpoint is released. The APC/C also plays an important role in noncycling cells and is required to maintain low levels of cyclins to prevent unscheduled entry into the cell cycle. In postmitotic neurons, the APC/C is required for proper axon growth and morphogenesis (11, 12), for neural cell survival (13), and for maintenance of low levels of PFKFB3, a regulator of the rate of glycolysis whose accumulation can lead to excitotoxicity in neurons (14).
A 3-dimensional reconstruction at a resolution of 7.4 Å has recently been determined for a ternary complex of recombinant human APC/C with the coactivator Cdh1 and a high-affinity substrate, Hsl1 (15). It consists of three major subcomplexes: the tetratricopeptide repeat (TPR) subcomplex (subunits 3, 6, 7, and 8) that interacts with APC/C coactivators Cdh1 or Cdc20 to mediate substrate specificity, the catalytic E3 subcomplex (subunits 2 and 11), and the base or platform subcomplex (subunits APC1, APC4, and APC5) that attaches the TPR subunits to the catalytic core. Other subunits are APC15, which bridges APC5 and APC8 and is required for APC/C-bound mitotic checkpoint complex-dependent Cdc20 autoubiquitylation and degradation (16), the TPR accessory subunits APC12, APC13, and APC16, and APC10, which assists the regulatory subunits in substrate recognition. The interaction of TPR subunit APC8 with the platform subcomplex of APC1, APC4, and APC5 was found to require all of these subunits to maintain the structure of the subcomplex (17).
The APC is targeted by several viruses (reviewed in reference 18), but all appear to do so by different mechanisms. We and others have found that, upon HCMV infection, the APC4 and APC5 subunits are degraded, the TPR subunits dissociate from the catalytic core and Cdh1, Cdh1 is hyperphosphorylated, and APC/C substrates accumulate (5, 8, 9). We also demonstrated that the viral kinase UL97 is capable of phosphorylating the APC/C coactivator Cdh1 on several sites, but in the context of infection, UL97 is not required for APC/C inactivation and disassembly (10). It has since been reported that the HCMV protein responsible for degradation of APC4 and APC5 is UL21a, a small unstable protein expressed with early kinetics (19–21). Other viruses inactivate the APC/C by a variety of mechanisms, including competitive mimicry of APC11 by Orf virus poxvirus anaphase-promoting complex regulator (PACR) (22, 23) and binding of APC/C coactivators by human papillomavirus (HPV) E2 protein (24).
In this study, we aim to further elucidate the mechanism by which the APC/C is inactivated and the relative contributions of UL21a and UL97 to APC/C inactivation and to viral growth, as both proteins have multiple functions during the infection. We show that in the absence of other viral proteins, UL21a but not UL97 can disrupt APC/C function and cause the accumulation of substrates. Our findings demonstrate that UL21a expression alone leads to the degradation of APC1, as well as of APC4 and APC5. Our investigation into this viral process has also led us to a novel finding regarding APC/C subunit interdependency that points to a possible mechanism of UL21a-mediated APC/C subunit degradation. We show that the destruction of APC1, APC4, and APC5 can be mediated by a cellular process in the absence of infection when APC1, APC4, APC5, or APC8 is depleted by a small interfering RNA (siRNA). We finally demonstrate that exogenous inhibition of APC/C activity via siRNA against APC5 or APC8 is able to partially rescue a mutant virus lacking both UL21a and UL97 (denoted as Δ21a/97 virus).
MATERIALS AND METHODS
Cell culture and infection with HCMV.
Human foreskin fibroblasts (HFs) were cultured in HF growth medium (minimal essential medium [Gibco] supplemented with 10% fetal bovine serum, 1.5 μg/ml amphotericin B, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). Cells were incubated at 37°C with 7% CO2. To prepare for infections, cells were allowed to come to confluence and maintained for 3 days prior to infection to allow exit from the cell cycle. Virus was diluted in HF growth medium at one of the multiplicities of infection (MOI) indicated below and added to cells. After 6 h, virus was removed, cells were washed with phosphate-buffered saline (PBS), and fresh HF growth medium was added. For siRNA experiments, infection took place 96 h after siRNA transfection. For proteasome inhibition experiments, cells were treated with Cre and 100 nM salinosporamide A (SalA; gift from Bradley Moore, Scripps Institute of Oceanography, University of California, San Diego, CA) or vehicle and harvested 18 h later.
Lentiviral constructs.
The UL21a and UL97 open reading frames (ORFs) were amplified from the pHB5 bacterial artificial chromosome (BAC) and inserted downstream from the second loxP sequence in pLV-EF1α-loxp-Neo-loxp and pLV-EF1α-loxp-Puro-loxp, respectively (25). To accomplish this, UL21a was amplified using the forward and reverse primers 5′-GATCGAGTCGACATGGGAGGTAGCCCTGTTCC-3′ and 5′-GATCGGATCCTTAAAACTGGTCCCAATGTTCTTC-3′, placing a SalI site at the 5′ end. The amplicon was digested with SalI and then phosphorylated and ligated into the pLV-EF1α-loxp-Neo-loxp vector, which had been digested with unique SalI and SmaI sites. UL97 was amplified using forward and reverse primers 5′-GGGATCCACCGGCCCACTATGTCCTCCGCACTTCGGT-3′ and 5-CTTTACTTGTACCCCGCCTTTCCCCTCAGCAACCG-3′ and inserted into the SmaI-linearized pLV-EF1α-loxp-Puro-loxp backbone using the InFusion ligation-independent cloning kit (CloneTech).
Complementing cell lines.
Complementing cell lines were created by transducing HFs (passage 12-15) with lentivirus expressing the following bicistronic constructs. HF-21a cells (expressing UL21a) received pLV-EF1α-loxp-Neo-loxp-UL21a. HF-97 cells (expressing UL97) received pLV-EF1α-loxp-Puro-loxp-UL97. HF-21a/97 cells (expressing UL21a and UL97) first received pLV-EF1α-loxp-Neo-loxp-UL21a, followed by pLV-EF1α-loxp-Puro-loxp-UL97 several days later. The transductions were separated to avoid lentiviral recombination. Lentiviral particles were made by transfecting the lentiviral constructs into 293FT cells with ViraPower lentiviral packaging mix (Life Technologies), and transductions were performed as previously described (25). HF-97 cells were selected with 1 μg/ml puromycin, HF-21a cells were selected with 400 μg/ml G418, and HF-21a/97 cells were selected with both.
Construction of BAC recombinants.
pHB5-IE-Cre-1.2-flp has been described previously (25) and was used as the starting BAC for the mutant lacking UL21a (Δ21a) and the Δ21a/97 mutant. To construct deletions, the UL21a open reading frame was replaced by AmpR, and the UL97 ORF was replaced by TetR. AmpR and the promoter sequence were amplified from pcDNA3 using the following primer sequences: 5′-AGCCATGCAGCGTGCGGCGCCTCTCTCATGGATCCACTGTCACCGTCGCGGTGCGCGGAACCCCTATTTG-3′ and 5′-ATACAGACTTTTATATGATCCTTGTACAGATGTAAATAAAATGTTTTTATGGGGTCTGACGCTCAGTGG-3′. TetR was amplified from the pACYC184 vector using the following primer sequences: 5′-GTCGGTGTGGTAGCTAGTGCAGCCTTAGGAACAGGGAAGACTGTCGCCACGCCCCATACGATATAAGTTG-3′ and 5′-GTACCTTCTCTGTTGCCTTTCCCCTCAGCAACCGTCACGTTCCGCGTCCCGGACAGGAGCACGATCATG-3′. Recombination of the insert into the BAC was carried out in SW105, a modified strain of DH10B Escherichia coli, via heat-inducible recombinase. Clones were selected and confirmed by restriction endonuclease digestion and field inversion gel electrophoresis (FIGE) and by PCR. To grow deletion viruses, complementing cell lines were replated at low density. One day later, they were harvested and electroporated with the BAC along with pcDNA3-pp71 as described previously (25). Viral supernatants were harvested periodically, and their titers determined on the HF-21a/97 complementing cell lines.
Preparation of cell-permeable Cre.
Cell-permeable Cre recombinase was prepared as described in reference 26. Briefly, His-Tat-nuclear localization signal (NLS)-Cre protein was purified on Ni-NTA (Ni-nitrilotriacetic acid) Superflow resin and dialyzed against HEPES-buffered saline and HEPES-buffered glycerol, both tissue culture grade. The amount of Cre required to induce recombination in nearly all cells was determined by treating HF-97 cells with dilutions of Cre, followed by anti-UL97 immunofluorescence.
Depletion of APC/C subunits.
Cells were seeded at low density in antibiotic-free growth medium. The next day, depletion of a specific APC/C subunit was performed with 75 pmol siGENOME Smartpool siRNA (Dharmacon/Thermo Fisher) per well in 6-well dishes using Lipofectamine RNAiMax (Life Technologies). The transfection mixture was removed 4 to 6 h after transfection, and HF growth medium was added. The oligonucleotides targeting the sequences were as follows. For APC1, GGACAAGUGUGCACAAUUG, GGUUACAAUCCACGAGAAA, GAGUGGUUCGGGUGGGAAA, and GGCAUUGGCAGUUUAUAAA. For APC3, CAUGCAAGCUGAAAGAAUA, CAACACAAGUACCUAAUCA, GGAGAUGGAUCCUAUUUAC, and GGAAAUAGCCGAGAGGUAA. For APC4, GGACAUAGAUGAUGAAUGG, CAACACAGCUGGCGAGGUU, GCUCAAAUCUUCGUCAUGU, and GAGGAUGAAUCAAAUCUUC. For APC5, GAAGGCGAGUUGAAGGAUA, UGUCAGAGCUCAUCGAUAU, UAGGAAAGCGGUUGUAUUA, and GAACAGGCCUUAAGUCUUC. For APC6, GUAGAUGGCUUGCAAGAGA, GCUACAAGCUUACUUCUGU, UGGAAGAGCCCAUCAAUAA, and CUAUGGACCUGCAUGGAUA. For APC8, GCAGUUGCCUAUCACAAUA, UGAAACAGUUGAUAGCUUA, GUAGAAACGUGCUGUGUAA, and GAAAUUAAAUCCUCGGUAU. Finally, for nontargeting pool 1, UAGCGACUAAACACAUCAA, UAAGGCUAUGAAGAGAUAC, AGUAUUGGCCUGUAUUAG, and AUGAACGUGAAUUGCUCAA.
Western blot analysis.
Cells were harvested by trypsinization, washed with PBS, and pelleted. Pellets were lysed in Laemmli buffer with protease inhibitor cocktail (Sigma) and Halt phosphatase inhibitor cocktail (Thermo Scientific). Lysates were sonicated to shear DNA, and equal cell number equivalents of protein were run on Tris-glycine SDS gels and transferred to nitrocellulose. Membranes were probed with the following antibodies (Ab): rabbit (Rb) anti-geminin Ab (clone FL-209; Santa Cruz Biotechnology), mouse (Ms) anti-Cdc6 Ab (Neomarkers), Ms anti-Cdh1 Ab (Calbiochem), Ms anti-tubulin Ab (Sigma), Rb anti-APC1 Ab (A301-653A; Bethyl), Rb anti-APC3 Ab (Thermo Scientific), Rb anti-APC4 Ab (A301-176A; Bethyl), Rb anti-APC5 Ab (A301-026A; Bethyl), Rb anti-APC6 Ab (A301-165A; Bethyl), Rb anti-APC8 Ab (A301-181A Bethyl), and Rb anti-cyclin A Ab (Clone H-432, Santa Cruz Biotechnology). Antibodies to HCMV proteins included Ms anti-IE2 Ab (Chemicon), Ms anti-IE1/2 Ab (CH160; Virusys), Ms anti-pp28 Ab (Virusys), Ms anti UL44 Ab (Virusys), Rb anti-glutathione S-transferase (GST)-UL97 Ab (a kind gift from Don Coen), Rb anti-UL21a Ab (A kind gift from Dong Yu), Ms anti-UL57 Ab (Virusys), and Ms anti-pp65 Ab (Virusys).
Determination of viral titers.
Infected cell supernatants were centrifuged to remove cell debris, mixed with 1% dimethyl sulfoxide (DMSO), and stored at −80°C. Plaque assay was performed using the HF-21a/97 complementing cells to efficiently determine the titer of the UL21a/UL97 double mutant.
Immunofluorescence.
Cells grown on coverslips were fixed for 10 min at room temperature with 3.7% formaldehyde in PBS. Cells were permeabilized for 5 min with 0.2% Triton-X in PBS, washed twice, and blocked with 10% normal goat serum in PBS. Primary antibodies against UL21a or UL97 were diluted in 5% normal goat serum and incubated with the coverslips for 25 min at room temperature. After washing, the coverslips were incubated in Alexa Fluor 594 (Life Technologies)-labeled secondary antibodies, nuclei were stained with Hoechst in 5% normal goat serum, and cells were mounted on slides with Prolong gold antifade reagent (Life Technologies). Images were acquired on an epifluorescence microscope.
Real-time RT-PCR.
RNA was isolated from pellets using the DNA/RNA/protein extraction kit (Norgen) according to the manufacturer's instructions. Reverse transcription (RT) was performed on 1 μg of RNA per sample using ThermoScript reverse transcriptase (Life Technologies). APC1 (5′-TCATGGCTGGCTCAGGAAACCTAA-3′ and 5′-ACAGAGAAGAGCGGCAATGGAAGA-3′) and tubulin (sequences described previously [27]) were amplified using SYBR green (Life Technologies). APC5 (sequences described previously [10]) was amplified with TaqMan polymerase (Life Technologies). Real-time PCR was performed on an ABI Prism 7000 (Life Technologies), and relative quantifications were determined by comparison to standard curves for each cDNA. APC1 and APC5 transcript levels were normalized to the level of tubulin.
RESULTS
Creation of cell-permeable Cre-inducible fibroblast cell lines expressing UL21a and UL97 alone or together.
In order to study the individual and combined contributions of UL97 and UL21a to APC/C function both in the absence of other viral proteins and in the context of infection with a mutant lacking both genes, we used a Cre recombination-based system to conditionally express UL21a and UL97 in HF cells (Fig. 1A). HFs were transduced with bicistronic lentiviruses encoding a selectable marker flanked by loxP sites upstream from UL21a or UL97. Translation of this constitutively produced transcript yields the selectable marker gene product. In the presence of Cre recombinase, the selectable marker is recombined out, and translation of the viral protein commences. This allows the expression of each viral protein, alone or in combination, upon treatment with cell-permeable Cre (His-Tat-NLS-Cre) (26) or infection with the Cre-expressing wild-type (WT) or mutant HCMV. These cells are referred to as HF-21a, HF-97, and HF-21a/97 cells. The results in Fig. 1B show that treatment of HF-21a and HF-97 cells with cell-permeable Cre results in the expression of the corresponding viral proteins in a high percentage of the cells.
FIG 1.
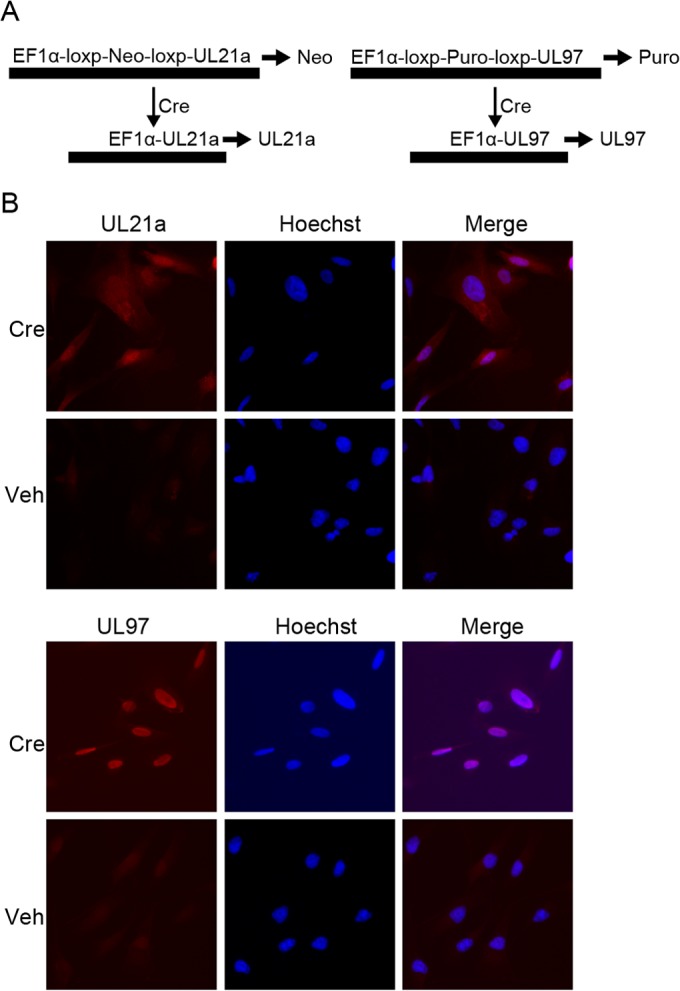
Creation of UL21a and UL97-expressing fibroblasts. (A) EF1α promoter drives constitutive expression of a loxP-flanked selectable marker (selected by neomycin for UL21a and puromycin for UL97). Cre recombinase induces the expression of viral proteins via removal of the selection marker. (B) Cell-permeable Cre induces the expression of UL21a and UL97. Complementing cell lines plated on coverslips were treated with Cre and harvested 72 h later. Coverslips were stained with UL97 and UL21a antibodies and counterstained with Hoechst to visualize nuclei. Veh, vehicle.
UL21a but not UL97 is sufficient to promote APC/C substrate accumulation in the absence of infection.
To study the effects of UL97 and UL21a, alone or together, on APC/C and substrate accumulation, we added cell-permeable Cre to HF-21a, HF-97, and HF-21a/97 cells after they were confluence synchronized in G0/G1. Cell-permeable Cre was also added to control confluence-synchronized HFs that did not express either protein. After 12 h of treatment, cells were washed twice with PBS, and growth medium was added. Cells were harvested 72 h after the addition of Cre and processed for Western blotting. For comparison, we infected HF with WT HCMV. We noted that Cre induction of the viral proteins in these cells, as well as infection with WT virus, results in a mobility shift of Cdh1 in cells expressing UL97, consistent with its phosphorylation by this viral kinase (Fig. 2). Upon the expression of UL21a, the APC/C substrates geminin and Cdc6 accumulated. This is consistent with previous reports (20) that UL21a causes substrate accumulation. However, UL97 was not sufficient to induce this accumulation. It appears that UL97 alone, despite its ability to phosphorylate Cdh1, is not sufficient to induce APC/C substrate accumulation in confluence-synchronized HFs. Additionally, there was no synergistic effect on geminin or Cdc6 levels from the expression of both proteins. We also show that the extent of APC/C substrate accumulation corresponds with the level of UL21a expressed. In an HF-21a/97 cell line expressing lower levels of UL21a (Fig. 2, left), the accumulation of substrates was less than that in a second line of HF-21a/97 cells expressing more UL21a (Fig. 2, right).
FIG 2.

UL21a but not UL97 expressed in uninfected cells promotes the accumulation of APC/C substrates. HF, HF-21a, HF-97, and HF-21a/97 cells were treated either with cell-permeable Cre to induce viral protein expression or with vehicle control for 12 h. Cells were then washed several times with PBS and maintained in HF medium. Cells were harvested 72 h after the beginning of Cre treatment and processed for Western blotting with antibodies to UL97, UL21a, geminin, Cdc6, Cdh1, APC1, APC4, APC5, APC6, and APC8. Tubulin served as a loading control. HF cells were infected at an MOI of 3 with WT HCMV (+) and harvested 24 hpi for comparison. Results for two different HF-21a/97 cell lines are shown; the cells whose blots are shown on the left had lower levels of expression of UL21a than those whose blots are shown on the right.
UL21a is necessary and sufficient for the degradation of APC4, APC5, and APC1.
To confirm that the Cre-induced expression of UL21a in the absence of other viral proteins also led to the degradation of APC4 and APC5 as previously reported (19), we assayed the lysates described above by Western blotting with antibodies to APC/C subunits. The results in Fig. 2 show that, as expected, the levels of APC4 and APC5 were significantly reduced during the infection with WT virus. The levels of APC4 and APC5 were also lower when UL21a was expressed alone or in the presence of UL97, and the magnitude of the decrease was dependent on the levels of UL21a. Surprisingly, we also found that in the infected cells or cells expressing UL21a, there was a loss of APC1, in addition to the lower levels of APC4 and APC5. There was not a global degradation of APC/C subunits, as APC6 and APC8 levels did not change.
Since this result differed from that obtained previously, in which it was shown with a different antibody directed against APC1 (28, 29) that this subunit appeared to be stable and localized to the nucleus during infection (9), we confirmed the specificity of the antibody used here with siRNAs directed against the APC1 transcript and a nontargeting control siRNA. As shown by the results in Fig. 3A (right), the detected band disappears in the lane containing lysate from mock-infected cells in which APC1 was depleted by siRNA. For comparison, we show that in cells infected with WT HCMV, the band was not present when the cells were treated with either APC1 siRNA or nontargeting control siRNA. To document that the APC1 antibody used in this study detects a band of the correct molecular weight, we also included a lane that contained lysate from uninfected cells that had been immunoprecipitated with APC3 antibody (Fig. 3A, left, IP). We suspect that the previously used antibody, which was an affinity-purified rabbit antibody directed against a peptide of APC1 (aa 326 to 342) (28, 29), detects an alternative stable form, but we cannot confirm this as the antibody is no longer available. However, in support of the results shown here, a recent study showed by quantitative mass spectrometry that there is a 4- to 5-fold reduction in APC1 levels in infected cells (30). As further confirmation that UL21a is needed for the degradation of APC1, we infected HF cells with WT HCMV or a mutant virus lacking UL21a (Δ21a virus) and harvested the cells at 24 h postinfection (hpi) for Western blot analysis. The results in Fig. 3B show that, as was the case for APC4 and APC5, UL21a is necessary for degradation of APC1. To test whether APC1 degradation was proteasome dependent as has been shown for APC4 and APC5 (10), we induced UL21a expression in the HF-21a cells with Cre treatment in the presence or absence of the proteasome inhibitor salinosporamide A (SalA). We harvested the cells 18 h posttreatment for Western blot analysis. The results in Fig. 3C show that UL21a levels increased with proteasome inhibition, as expected, and that APC1 and APC5 degradation was prevented.
FIG 3.
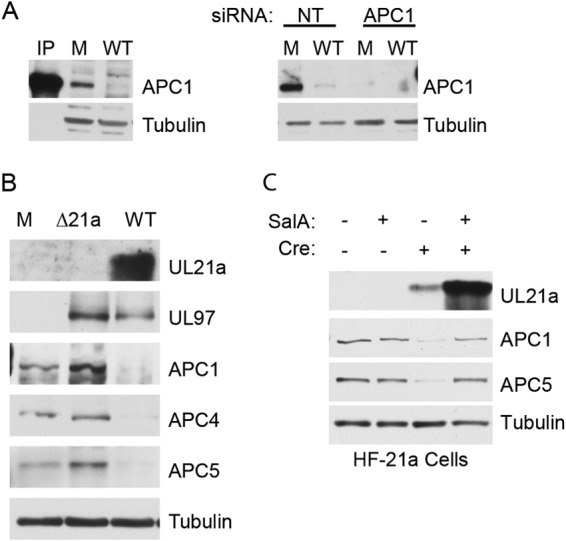
UL21a is necessary for APC1 degradation during infection. (A) Confirmation of anti-APC1 antibody specificity. (Left) Cells were mock infected (M) or infected with WT at high MOI and harvested 24 hpi for Western blotting with Ab to APC1. For molecular weight comparison, untreated HF cell lysate was immunoprecipitated with Ab to APC3 (IP) and run on the same gel. (Right) Cells were transfected with APC1 or nontargeting control (NT) siRNA. Four days posttransfection, cells were harvested and processed for Western blotting. (B) HF cells were confluence synchronized and mock infected or infected at high MOI with WT or Δ21a virus. Cells were harvested 24 hpi and processed for Western blotting with antibodies to UL21a, UL97, APC1, APC4, and APC5. Tubulin served as a loading control. (C) HF-21a cells were confluence synchronized and treated with cell-permeable Cre and/or 100 nM salinosporamide A (SalA) proteasome inhibitor as indicated. Cells were harvested 18 h posttreatment and processed for Western blotting with Ab to UL21a, APC1, APC5, and tubulin.
The APC/C degradation phenotype induced by HCMV can be mimicked by disruption of the APC8-APC1-APC4-APC5 interaction via siRNA-mediated depletion of individual APC/C subunits.
The specific APC/C subunit(s) that UL21a interacts with to mediate the degradation of the platform subunits is unknown. To further investigate the mechanism of APC/C subunit degradation by UL21a, we proceeded to test the hypothesis that UL21a might first target one of the platform subunits and that depletion of this subunit might prevent the degradation of the other two subunits. To address this, we first transfected HFs with siRNA against APC1 or APC5. Unexpectedly, we found that knocking down either of these subunits was sufficient to cause the degradation of APC1, APC4, and APC5 in uninfected cells. These experiments were repeated multiple times, and representative results are shown in Fig. 4. There was not global degradation of subunits, as the levels of two other APC/C subunits in the TPR subcomplex, APC6 and APC8, were not affected by the depletion of APC1 or APC5. To rule out off-target effects of the siRNA, we performed real-time RT-PCR on these samples and found that the siRNAs against APC1 and APC5 did not affect the RNA levels of the other subunit (Fig. 4B).
FIG 4.

Knockdown of APC1, APC4, APC5, or APC8 causes the degradation of APC1, APC4, and APC5 in uninfected cells. (A) HF cells were transfected with APC1 (1), APC5 (5), or nontargeting control (NT) siRNA or left untransfected (−). Cells were harvested 96 h posttransfection and processed for Western blotting with antibodies to APC1, APC4, APC5, APC6, and APC8. (B) RNA was extracted from the samples used for the experiment whose results are shown in panel A, and real-time RT-PCR was performed to detect the levels of APC1 and APC5 transcripts. Transcript levels were normalized to the level of the tubulin transcript. Error bars indicate standard deviations of the results from three replicates of a single experiment. (C) HF cells were transfected with APC3 (3), APC4 (4), APC6 (6), APC8 (8), or nontargeting (NT) control siRNA or left untransfected (−). After 96 h, the cells were harvested and processed for Western blotting as described for panel A.
We extended this experiment to include knockdown of the other APC/C subunit in the platform subcomplex, APC4, and three APC/C subunits in the TPR subcomplex, APC3, APC6, and APC8 (Fig. 4C). We were surprised to find that knockdown of APC8 is sufficient to cause a reduction of APC1, APC4, and APC5, and knockdown of any one of the group of APC1, APC4, and APC5 causes reduction of the other three but not of APC8. This was of interest since the recently solved structure of the human APC/C places APC1 next to APC8 (15). The levels of APC6 were not affected by depletion of these subunits, and the depletion of APC6, albeit incomplete, did not significantly affect the levels of the other subunits. In the case of APC3, we sometimes observed that its depletion modestly affected the levels of some of the other subunits.
This unexpected result may underlie the cellular mechanism by which the APC/C subunits are degraded during infection and demonstrates that UL21a may need only to disrupt the complex in order to effect APC1, APC4, and APC5 degradation.
A double mutant, the Δ21a/97 virus, is growth compromised compared to Δ21a and WT virus.
Previously, it was shown that a mutant virus that contained point mutations in UL21a that still allowed degradation of APC4 and APC5 seemed to have no growth defect but was severely growth impaired if a second mutation that eliminated the expression of UL97 was introduced (19). A second UL21a mutant virus that contained point mutations in UL21a that abrogated its ability to degrade APC4 and APC5 also replicated like the WT but was even more severely growth impaired than the first mutant when the UL97 mutation was introduced. One difficulty in interpreting these experiments was that the mutant viruses were not propagated on complementing cell lines and replication of the mutant viruses was analyzed in a multistep growth assay over 21 days. We therefore proceeded to adapt the method we previously used for complementation of IE2 mutant virus growth (25). We constructed a mutant virus that contained deletions of both UL21a and UL97, denoted as Δ21a/97 virus (Fig. 5A), and propagated it on the Cre-inducible HF-21a/97 cell line to facilitate growth and avoid compensatory mutations. BAC recombinant clones were monitored by PCR, restriction digest, and FIGE (Fig. 5B). In comparison to the results for the WT, the appearance of a 1.4-kbp fragment was diagnostic for deletion of UL21a, and the disappearance of the 13.5-kbp fragment indicated deletion of UL97 (Fig. 5B). Since the WT, Δ21a, and double mutant viruses express Cre under the direction of the IE promoter, there is induction of both UL21a and UL97 in the HF-21a/97 cells upon infection. The virus stock and viral supernatants produced in experiments also had their titers determined on HF-21a/97 cells in order to accurately determine the amounts of infectious virus produced.
FIG 5.
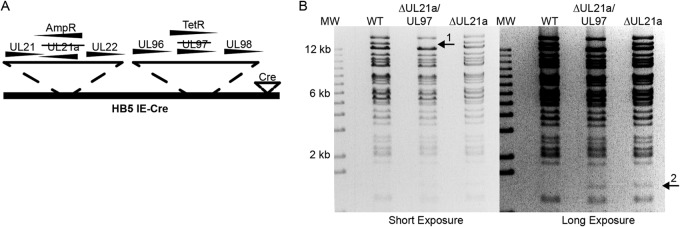
Creation of HCMV UL21a/UL97 double mutant. (A) Diagram of BAC recombinants. UL21a alone or UL21a and UL97 open reading frames in the wild-type (WT) Ad169 BAC HB5-IE-Cre/1.2-Flp (24) (referred to in all figures as WT) were replaced with ampicillin (UL21a) and tetracycline (UL97) resistance genes, respectively, and propagated on the HF-21a/97 complementing cells. (B) BamHI restriction digest of BAC recombinants and the molecular weight marker (MW) were resolved on an agarose gel. Arrows 1 and 2 indicate band changes predicted for mutation of UL97 and UL21a, respectively.
We characterized the growth kinetics of Δ21a/97 virus by infecting confluence-synchronized HFs with Δ21a/97, Δ21a, or WT virus at a high MOI (MOI of 4) and collected cell pellets and cell supernatants at several times postinfection. Viral titers were determined by plaque assay on HF-21a/97 cells. We found the double Δ21a/97 mutant to be severely growth defective, releasing 2 to 3 log less virus than the WT and 10- to 50-fold less than Δ21a virus (Fig. 6A). The titer of the single mutant Δ21a virus was approximately 5-fold lower than that of the WT. Western blot analysis of viral proteins demonstrated that the double mutant was deficient in the production of late proteins (Fig. 6B) but not of early or IE proteins. While there was little difference in the levels of IE2 86 protein and early protein UL57, there were significant decreases in the levels of pp28 and the late IE2 protein p40 in the double mutant-infected cells and only modest decreases in the late protein levels in the Δ21 mutant-infected cells.
FIG 6.
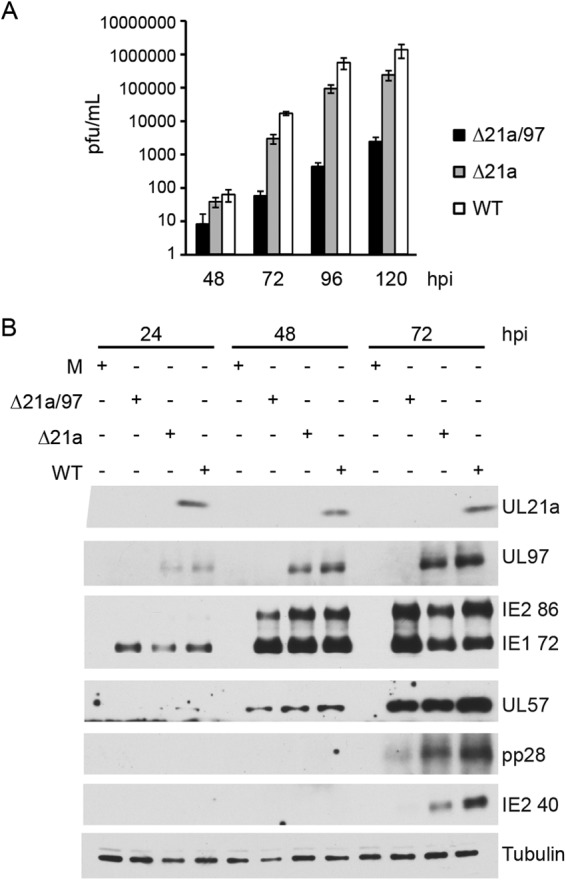
Δ21a/97 virus is defective for growth and deficient in late protein production. HF cells were confluence synchronized and mock infected (M) or infected with Δ21a/97, Δ21a, or WT virus at high MOI. (A) Supernatants were harvested 48, 72, 96, and 120 hpi and had their titers determined on HF-21a/97 cells. Shown are representative results of at least 2 independent experiments. Error bars indicate standard deviations. (B) Cells were harvested 24, 48, and 72 hpi and processed for Western blotting with antibodies to UL21a, UL97, IE proteins (IE2 86 and IE1 72), early protein (UL57), and late proteins (pp28 and IE2 40).
UL21a and UL97 contribute to but are not essential for the accumulation of APC/C substrates in infected cells.
We next tested the ability of Δ21a/97 virus to promote the accumulation of APC/C substrates. Cells were infected at high MOI with Δ21a/97, Δ21a, or WT virus and harvested at 24, 48, and 72 hpi for Western blot analysis. Lysates were probed for Cdh1 and several APC/C substrates. We found that at 24 hpi, there was a significant defect in the accumulation of APC/C substrates in cells infected with either mutant. At later times, however, APC/C substrates accumulated in cells infected with both mutants but with reduced efficiency in cells infected with the Δ21a/97 double mutant compared to their levels in cells infected with either Δ21a or WT virus (Fig. 7). These data indicate that other viral proteins or cellular proteins induced during the infection can contribute to blocking APC/C function in the absence of UL21a and UL97. As expected, in cells infected with the double mutant, Cdh1 failed to exhibit the change in migration that results from phosphorylation by UL97.
FIG 7.
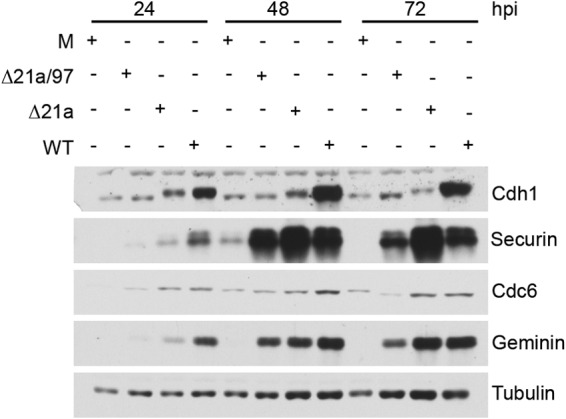
Cells infected with Δ21a/97 virus show a delay in the accumulation of APC/C substrates compared to their accumulation in cells infected with WT and Δ21a virus. HF cells were confluence synchronized and mock infected (M) or infected with Δ21a/97, Δ21a, or WT virus at an MOI of 4. Cells were harvested 24, 48, and 72 hpi and processed for Western blotting with antibodies to Cdh1, securin, geminin, and Cdc6.
The relative contributions of exogenously expressed UL97 and UL21a to complementation of the growth of double mutant Δ21a/97 virus are MOI dependent.
Having compared the influence of UL21a and UL97 on APC/C substrate levels in the context of the infection, we sought to characterize the relative contribution of each protein in the growth of the double mutant Δ21a/97 virus during a single-cycle HCMV infection at high and at low MOI. This was accomplished by comparing the results for Δ21a/97 mutant infection on HF cells to those on HF-21a, HF-97, and HF-21a/97 complementing cells, where UL21a and/or UL97 was provided exogenously upon the expression of Cre by the mutant virus. All cells were confluence synchronized in G0/G1 and then infected at a low MOI of 0.1 or a high MOI of 3. At 96 hpi, cell pellets were harvested for Western blot analysis. Supernatants were collected at various times postinfection to determine the titers of infectious virus produced.
Virion production was monitored by titration of supernatants on HF-21a/97 cells to provide complementing function. At both high and low MOI (Fig. 8A and B), infection of HF-21a cells resulted in higher titers of virus than infection of HF cells, with a greater increase in the titer when mutant virus was grown on HF-97 cells at high MOI. At low MOI, there was a delay in the production of virus on HF-21a cells, but at the last time point when supernatants were collected, the amount of virus produced on HF-21a cells was comparable to that produced on HF-97 cells. Infection of cells expressing both proteins (HF-21a/97 cells) with the double mutant at the low MOI resulted in still higher titers than infection of HF-97 cells, but the difference was less at high MOI. Cell pellets from these experiments harvested at 96 hpi were processed for Western blotting and probed for pp28, IE2 86, IE2 60, and IE2 40. Representative blots are shown in Fig. 8C and D. We observed that at high MOI, infection of HF-97 cells with the double mutant led to higher levels of the late proteins pp28, IE2 60, and IE2 40 than did infection of HF-21a cells, consistent with the titration results. (Fig. 8C). Likewise, the results of the Western blot analysis were consistent with the titers at low MOI, with infection on UL21a-expressing and UL97-expressing cells showing similar increases in late protein levels (Fig. 8D). At both high and low MOI, infection of HF-21a/97 cells led to the greatest increase in late protein expression compared to the results for double mutant infection of HFs.
FIG 8.

The relative requirements for exogenous expression of UL97 and UL21a in complementing the growth of double mutant Δ21a/97 virus are MOI dependent. HF, HF-21a, HF-97, and HF-21a/97 cells were confluence synchronized and infected at an MOI of 3 (A, C) or an MOI of 0.1 (B, D). Supernatants were collected, their titers were determined on HF-21a/97 cells (A, B), and cells were harvested 96 hpi and processed for Western blotting with antibodies to UL21a, UL97, IE2 proteins (IE2 86, IE2 60, and IE2 40), and pp28 (C, D). Shown are representative results from 2 or 3 independent experiments. Error bars indicate standard deviations.
Exogenous inactivation of the APC/C during double mutant infection results in increases in late protein expression and viral titers.
As both UL97 and UL21a perform other functions during the infection apart from phosphorylating Cdh1 and mediating the degradation of APC/C subunits, respectively, we were interested in determining the degree of rescue of the double mutant's growth deficiency that results from inactivation of the APC/C. To decrease the activity of the APC/C, we used siRNAs to deplete APC5 and APC8. A nontargeting siRNA was used as a control. The growth of the double mutant on the various siRNA-treated cells was assessed by measuring late viral protein accumulation and viral titers. Efficient knockdown of both APC5 and APC8 took several days, resulting in the cells coming to confluence (and thus a G0/G1 state) prior to infection. Since one function of UL21a is to degrade cyclin A2, we wanted to ensure that this would not be a confounding variable in the experiment. To document that the siRNA-treated cells are not cycling and do not express significant levels of cyclin A2, we harvested the cells at 96 h posttransfection and probed for cyclin A2 along with APC/C subunits 5 and 8 (Fig. 9A). We show that under these conditions, confluent siRNA-treated cells have only low levels of cyclin A2, in contrast with the very high levels found in asynchronous cycling nontreated cells and in siRNA-treated cells that were reseeded at low density to allow the cells to enter G1 and then harvested 24 h later when they entered S phase of the cell cycle.
FIG 9.

Knockdown of APC5 or APC8 results in increased growth of Δ21a/97 virus. (A) HF cells were transfected with APC5 (5), APC8 (8), or nontargeting control (NT) siRNA or left untreated (−). At 72 h after transfection, some cells were replated at lower density to release them into G1 phase (Released), while other cells were maintained at confluence (Confluent). The untreated cells were maintained in a cycling state (Cyc) by passaging them several times prior to harvest. At 96 h posttransfection, cells were harvested and processed for Western blotting with antibodies against APC5, APC8, cyclin A2, and tubulin. (B, C) HF cells were transfected with siRNAs as described for panel A. At 96 h after transfection, cells were mock infected (B). (C) Cells were mock infected or infected at an MOI of 0.5 with Δ21a/97 or WT virus. Cells were harvested 96 hpi and processed for Western blotting with antibodies to APC5, APC8, geminin, and Cdc6 (B) and antibodies to IE2 proteins (IE2 86, IE2 60, and IE2 40) and pp28 (C). The asterisk indicates another possible form of IE observed with the current lot of antibody. (D) Cell supernatants were harvested, and their titers were determined on HF-21a/97 cells. Shown are the results of two independent experiments.
In the experiment whose results are shown in Fig. 9B and C, cells were mock infected (Fig. 9B) or infected (Fig. 9C) with Δ21a/97 or WT virus at an MOI of 0.5 4 days after transfection with siRNA. In the uninfected cells depleted of the APC/C subunits (Fig. 9B), there was accumulation of substrates, with slightly greater accumulation in cells treated with siRNA to APC8, probably due to the fact that depletion of APC8 also resulted in degradation of APC1, APC4, and APC5. It should be noted that, in accord with the results described above, depletion of APC8 also resulted in depletion of APC5. Supernatant was collected at various times postinfection, and the cells were harvested at 96 hpi for Western blot analysis. Membranes were probed for APC5 and APC8, to ensure maintenance of protein knockdown late in the infection, and for viral late proteins (Fig. 9C). Western blot analysis showed that the knockdown of both APC/C proteins was maintained until at least 96 hpi and that APC/C subunit knockdown resulted in a greater accumulation of APC/C substrates in cells infected with the double mutant. Knockdown of APC5 and, to a greater extent, knockdown of APC8 also resulted in increased production of late proteins. Supernatants were assayed for viral production by titration on the complementing HF-21a/97 cells. In accord with the levels of viral late proteins observed, viral titers were significantly increased by depletion of APC5, with a greater increase observed in cells depleted of APC8 (Fig. 9D). A comparison of the results in Fig. 8B with those in Fig. 9D shows that the increase in viral titer when mutant virus was grown on cells expressing UL21a relative to the titer when virus was grown on HF cells was comparable to the increase when mutant virus was grown on HF cells that were depleted of APC5 or APC8. While the mutant still showed debilitated growth relative to the growth of the WT, depletion of APC/C subunits was able to increase viral growth in a single-cycle infection. The growth of WT virus was not affected by knockdown of either protein.
DISCUSSION
In this study, we discovered that APC1 is an additional target of degradation by the HCMV protein UL21a. Particularly noteworthy is our finding that recapitulation of the pattern of APC/C subunit degradation induced by HCMV occurs in uninfected cells upon depletion of APC1, APC4, APC5, or APC8. Finally, we show that when complementing cells are infected at low MOI with a mutant virus that lacks both UL97 and UL21a (Δ21a/97 mutant), there is a delay in the production of infectious virus on cells expressing UL21a relative to the results for cells expressing UL97, but the final viral titers and levels of late proteins are comparable. The growth of the double mutant is further enhanced on cells expressing both proteins. In contrast, when complementing cells are infected at high MOI with the double mutant, UL97 provides greater increases in the accumulation of late viral proteins and production of infectious virus than UL21a, and there is only a modest further increase when the virus is grown on cells that express both proteins. These results are consistent with those of Prichard et al. (31), who showed that the complementation of a UL97 deletion virus by exogenous UL97 was more efficient at high MOI.
Our results also confirm the studies by Fehr et al. (19) that show that UL21a can disable the APC/C and is responsible for the degradation of APC4 and APC5. In addition, we find that UL21a expression leads to APC1 degradation. In support of our antibody-based observation, the reduction in APC1 levels during HCMV infection was recently confirmed by quantitative mass spectrometry (30). We also show that UL97 expression in uninfected cells does not cause the accumulation of APC/C substrates despite phosphorylation of Cdh1. The UL97 protein that was expressed was properly localized to the nucleus, but it is possible that the phosphorylation of Cdh1 by UL97 is incomplete. Phosphorylation of Cdh1 leaves the APC/C itself intact, so it may be that a relatively small amount of unphosphorylated Cdh1 would be sufficient to coactivate the APC/C.
In the course of experiments to determine the mechanism of APC/C subunit degradation, we tested the hypothesis that depletion of one of the platform subunits might prevent UL21a-mediated degradation of the other subunits. We were surprised to see that knocking down any one of the three platform subunits led to the degradation of others in the absence of infection. Moreover, depletion of APC8, a member of the TPR complex, also led to degradation of all of the platform subunits, but APC8 was not degraded either by UL21a or by depletion of a platform subunit. To our knowledge, this phenomenon has not been described previously. However, the result is not totally unexpected since it was previously shown that the binding of APC1, APC4, APC5, and APC8 is interdependent in vitro (17) and the recently solved structure of the human APC/C places APC1 next to APC8 (15). This process does confound the interpretation of RNA interference experiments intended to delineate the sufficiency of any of these individual subunits in this and other viral or cellular events.
It is currently unclear whether the subunits are degraded by some active process or whether they are degraded as part of normal APC/C turnover, with newly synthesized subunits being recognized as improperly folded proteins if they do not associate correctly. Our studies show that there is a clear cellular process in place for decreasing the levels of the APC/C subunits APC1, APC4, and APC5 but not those of APC3, APC6, and APC8 if the APC/C becomes destabilized. These results suggest that UL21a may need only to disrupt the association of the APC/C subunits, and a cellular process will then mediate the degradation of subunits APC1, APC4, and APC5. To date, our time course experiments have been unable to distinguish whether degradation of the subunits precedes or follows release from the rest of the complex, but it is possible that the use of new high-resolution imaging tools may help answer the question.
Our studies also show that the growth of the Δ21a/97 double mutant is defective compared to that of both Δ21a and WT virus with respect to late protein expression and production of infectious virus. As is the case with some other HCMV mutants, the growth defect of the double mutant is MOI dependent and much greater at low MOI than high MOI. Interestingly, APC/C substrates accumulated, albeit with a delay, in cells infected with either Δ21a or Δ21a/97 virus. Thus, even in the absence of UL21a (in the presence or absence of UL97), APC/C substrates begin to accumulate later in the infection. This may be due to a yet-to-be-discovered viral or induced cellular protein that can disable APC/C function. Alternatively, the infection may activate some cellular mechanism that normally operates during S/G2 phase to inactivate the APC/C and allow substrate accumulation. Regardless of the mechanism, further inactivation of the APC/C by depletion of APC/C subunits APC5 and APC8 during Δ21a/97 virus infection leads to a growth benefit in a single cycle of infection.
Previously, a double mutant virus that contained a UL97 deletion and UL21a point mutations that rendered UL21a incapable of interacting with the APC/C was shown to be more growth impaired than a double mutant that contained a UL97 deletion and UL21a point mutations where UL21a was still able to degrade APC4 and APC5 (19), suggesting that inactivation of the APC/C was important in the absence of UL97. One difficulty in interpreting these experiments was that the mutant viruses were not propagated on complementing cell lines and the replication of the mutant viruses was analyzed in a multistep growth assay over 21 days where the cells continued to proceed through the cell cycle. They were also conducted in MRC5 fibroblasts, where the growth defect of an UL21a deletion virus is significantly greater than that in HF cells. In our studies, we constructed a Δ21a/97 double mutant virus that was unable to express any UL21a and UL97. By growing the double mutant virus on complementing HFs, we were able to avoid the complication of any second-site mutations that might change the viral growth. We were also able to show directly, in a single-cycle growth experiment in cells that were confluence synchronized and maintained in G0/G1, that the increase in viral titer in cells expressing UL21a was comparable to that in cells in which the APC/C was inactivated by depletion of APC/C subunits.
The benefit resulting from knocking down APC5 and APC8 for Δ21a/97 mutant replication is impressive considering that during the infection, both UL21a and UL97 possess other functions that are independent of the APC/C. UL97 in particular is multifunctional. It phosphorylates several viral proteins, as well as itself, and a number of cellular proteins, including Rb, lamins A and C, p32, and eukaryotic elongation factor 1 delta (32–43). Studies of UL97 deletion mutants indicate a role for this protein in viral DNA synthesis, DNA encapsidation, capsid maturation, nuclear egress, and cytoplasmic secondary envelopment (38, 41, 44–46), although the extent of the defects appears to depend on the culture conditions. Which of these functions are being complemented by UL21a or by inactivation of the APC/C remains to be determined.
It has been reported that UL21a, in addition to inactivating the APC/C, functions at a later time to promote viral DNA replication and late gene expression (21). One recently discovered function of UL21a is associated with proteasome-dependent degradation of cyclin A2, and the domain responsible for interaction with cyclin A2 is separate from that which interacts with the APC/C (47). It was reported that cyclin A2 degradation was necessary for efficient multistep replication in MRC5 cells, and siRNA-mediated depletion of cyclin A2 removed the growth defect seen in a UL21a deletion mutant. However, these experiments were done at low MOI in MRC5 cells progressing through the cell cycle, and viral titers were assayed after virus had undergone multiple cycles of replication. Additionally, cells were infected in S phase, when cyclin A2 levels are very high. Recent work by Eifler et al. examined UL21a-mediated cyclin A2 degradation in confluence-synchronized HEL cells (48). They found that degradation of cyclin A2 by UL21a was required to prevent mitotic catastrophe, as in the absence of UL21a's cyclin A2 degradation function, infected cells were not prevented from entering mitosis and mitotic aberrations and cell death followed. This appeared to be specific to the cyclin A2 degradation locus of UL21a, RXL2. While the UL21a null mutant also induced the accumulation of cyclin A2, a similar level of mitotic catastrophe and cell death did not take place. Correspondingly, the increase in cyclin A2 in cells infected with the UL21a RXL2 point mutant was much greater than that seen during infection with the UL21a null mutant. In agreement with this work, we also see small increases in cyclin A2 levels during infection with the UL21a null mutant and the UL21a/UL97 double mutant, but these increases are very modest in comparison to the levels observed in cycling HF cells.
Interestingly, cyclin A2 is a substrate of the APC/C, and inactivation of the APC/C should lead to accumulation of cyclin A2. However, we previously showed that cyclin A2 transcription is inhibited during the infection, and if the virus is grown in confluence-synchronized cells, which remain in G0/G1, there is very little cyclin A2 until later times in the infection (49). We show here that the cyclin A2 levels remain very low in our APC/C subunit depletion experiment when cells are maintained in a G0/G1 state (Fig. 9), and thus, the levels are negligible at the time of infection compared to the levels in asynchronous, cycling cells. It is only when cells are infected in S phase, where there are already high levels of cyclin A2, that it might be necessary for UL21a to degrade the cyclin A2 in order for the infection to initiate. The situation is likely more complicated, as it has been reported that the HCMV tegument protein pp150 also binds to cyclin A2, and this binding between pp150 on the incoming particle and cyclin A2 is responsible for the block in initiation of HCMV IE gene expression when cells are infected in S phase (50, 51). We cannot exclude that increases in cyclin A2 contribute to the growth deficiency of our double mutant. However, direct comparison to the above-mentioned studies is difficult due to the difference in cell types, as different cell types may respond differently to a given level of cyclin A2. Another possibility includes yet-to-be-discovered functions of UL21a that are required for efficient viral DNA synthesis and late gene expression, and these are likely cell type dependent (e.g., there is greater impairment of replication of UL21a deletion mutants in MRC5 cells than in HFs).
It is still to be determined what the functional relevance of APC/C inactivation is in HCMV infection in vivo. However, the fact that inactivation of the APC/C by HCMV is seen not only in fibroblasts but also in aortic endothelial cells and neural progenitors (unpublished results) suggests that APC/C inactivation serves some additional role in vivo that is undetectable in a tissue culture system. Experiments are in progress to determine more precisely the molecular mechanisms governing disassembly of the APC/C and degradation of specific subunits in the presence of UL21a.
ACKNOWLEDGMENTS
We thank Don Coen for the UL97 antibody, Dong Yu for the UL21a antibody, and Bradley Moore for salinosporamide A. We especially want to thank Jean-Philippe Belzile for valuable advice and other members of the Spector laboratory for helpful discussions.
This work was supported by NIH grants R01CA034729 and R21AI083991 to D.H.S. and training grant T32DK007541.
REFERENCES
- 1.Lu M, Shenk T. 1996. Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J Virol 70:8850–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bresnahan WA, Boldogh I, Thompson EA, Albrecht T. 1996. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 224:150–160. doi: 10.1006/viro.1996.0516. [DOI] [PubMed] [Google Scholar]
- 3.Dittmer D, Mocarski ES. 1997. Human cytomegalovirus infection inhibits G1/S transition. J Virol 71:1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jault FM, Jault JM, Ruchti F, Fortunato EA, Clark C, Corbeil J, Richman DD, Spector DH. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol 69:6697–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biswas N, Sanchez V, Spector DH. 2003. Human cytomegalovirus infection leads to accumulation of geminin and inhibition of the licensing of cellular DNA replication. J Virol 77:2369–2376. doi: 10.1128/JVI.77.4.2369-2376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian Z, Xuan B, Hong TT, Yu D. 2008. The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments. J Virol 82:3452–3465. doi: 10.1128/JVI.01964-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiebusch L, Uecker R, Hagemeier C. 2003. Human cytomegalovirus prevents replication licensing by inhibiting MCM loading onto chromatin. EMBO Rep 4:42–46. doi: 10.1038/sj.embor.embor707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiebusch L, Bach M, Uecker R, Hagemeier C. 2005. Human cytomegalovirus inactivates the G0/G1-APC/C ubiquitin ligase by Cdh1 dissociation. Cell Cycle 4:1435–1439. doi: 10.4161/cc.4.10.2077. [DOI] [PubMed] [Google Scholar]
- 9.Tran K, Mahr JA, Choi J, Teodoro JG, Green MR, Spector DH. 2008. Accumulation of substrates of the anaphase-promoting complex (APC) during human cytomegalovirus infection is associated with the phosphorylation of Cdh1 and the dissociation and relocalization of APC subunits. J Virol 82:529–537. doi: 10.1128/JVI.02010-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran K, Kamil JP, Coen DM, Spector DH. 2010. Inactivation and disassembly of the anaphase-promoting complex during human cytomegalovirus infection is associated with degradation of the APC5 and APC4 subunits and does not require UL97-mediated phosphorylation of Cdh1. J Virol 84:10832–10843. doi: 10.1128/JVI.01260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A. 2004. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- 12.Stegmuller J, Huynh MA, Yuan Z, Konishi Y, Bonni A. 2008. TGFbeta-Smad2 signaling regulates the Cdh1-APC/SnoN pathway of axonal morphogenesis. J Neurosci 28:1961–1969. doi: 10.1523/JNEUROSCI.3061-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almeida A, Bolaños JP, Moreno S. 2005. Cdh1/Hct1-APC is essential for the survival of postmitotic neurons. J Neurosci 25:8115–8121. doi: 10.1523/JNEUROSCI.1143-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. 2009. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- 15.Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D. 2014. Molecular architecture and mechanism of the anaphase-promoting complex. Nature 513:388–393. doi: 10.1038/nature13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uzunova K, Dye BT, Schutz H, Ladurner R, Petzold G, Toyoda Y, Jarvis MA, Brown NG, Poser I, Novatchkova M, Mechtler K, Hyman AA, Stark H, Schulman BA, Peters JM. 2012. APC15 mediates CDC20 autoubiquitylation by APC/C(MCC) and disassembly of the mitotic checkpoint complex. Nat Struct Mol Biol 19:1116–1123. doi: 10.1038/nsmb.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thornton BR, Ng TM, Matyskiela ME, Carroll CW, Morgan DO, Toczyski DP. 2006. An architectural map of the anaphase-promoting complex. Genes Dev 20:449–460. doi: 10.1101/gad.1396906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fehr AR, Yu D. 2013. Control the host cell cycle: viral regulation of the anaphase-promoting complex. J Virol 87:8818–8825. doi: 10.1128/JVI.00088-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fehr AR, Gualberto NC, Savaryn JP, Terhune SS, Yu D. 2012. Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a. PLoS Pathog 8:e1002789. doi: 10.1371/journal.ppat.1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fehr AR, Yu D. 2010. Human cytomegalovirus gene UL21a encodes a short-lived cytoplasmic protein and facilitates virus replication in fibroblasts. J Virol 84:291–302. doi: 10.1128/JVI.01116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fehr AR, Yu D. 2011. Human cytomegalovirus early protein pUL21a promotes efficient viral DNA synthesis and the late accumulation of immediate-early transcripts. J Virol 85:663–674. doi: 10.1128/JVI.01599-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mo M, Fleming SB, Mercer AA. 2009. Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11. Proc Natl Acad Sci U S A 106:19527–19532. doi: 10.1073/pnas.0905893106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mo M, Fleming SB, Mercer AA. 2010. Orf virus cell cycle regulator, PACR, competes with subunit 11 of the anaphase promoting complex for incorporation into the complex. J Gen Virol 91:3010–3015. doi: 10.1099/vir.0.026054-0. [DOI] [PubMed] [Google Scholar]
- 24.Bellanger S, Blachon S, Mechali F, Bonne-Andrea C, Thierry F. 2005. High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 4:1608–1615. doi: 10.4161/cc.4.11.2123. [DOI] [PubMed] [Google Scholar]
- 25.Sanders RL, Clark CL, Morello CS, Spector DH. 2008. Development of cell lines that provide tightly controlled temporal translation of the human cytomegalovirus IE2 proteins for complementation and functional analyses of growth-impaired and nonviable IE2 mutant viruses. J Virol 82:7059–7077. doi: 10.1128/JVI.00675-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nolden L, Edenhofer F, Peitz M, Brustle O. 2007. Stem cell engineering using transducible Cre recombinase. Methods Mol Med 140:17–32. doi: 10.1007/978-1-59745-443-8_2. [DOI] [PubMed] [Google Scholar]
- 27.DuRose JB, Li J, Chien S, Spector DH. 2012. Infection of vascular endothelial cells with human cytomegalovirus under fluid shear stress reveals preferential entry and spread of virus in flow conditions simulating atheroprone regions of the artery. J Virol 86:13745–13755. doi: 10.1128/JVI.02244-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heilman DW, Teodoro JG, Green MR. 2006. Apoptin nucleocytoplasmic shuttling is required for cell type-specific localization, apoptosis, and recruitment of the anaphase-promoting complex/cyclosome to PML bodies. J Virol 80:7535–7545. doi: 10.1128/JVI.02741-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teodoro JG, Heilman DW, Parker AE, Green MR. 2004. The viral protein apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev 18:1952–1957. doi: 10.1101/gad.1198404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. 1999. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J Virol 73:5663–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baek M-C, Krosky PM, Coen DM. 2002. Relationship between autophosphorylation and phosphorylation of exogenous substrates by the human cytomegalovirus UL97 protein kinase. J Virol 76:11943–11952. doi: 10.1128/JVI.76.23.11943-11952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamil JP, Coen DM. 2007. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J Virol 81:10659–10668. doi: 10.1128/JVI.00497-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krosky PM, Baek M-C, Jahng WJ, Barrera I, Harvey RJ, Biron KK, Coen DM, Sethna PB. 2003. The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase. J Virol 77:7720–7727. doi: 10.1128/JVI.77.14.7720-7727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marschall M, Freitag M, Suchy P, Romaker D, Kupfer R, Hanke M, Stamminger T. 2003. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 311:60–71. doi: 10.1016/S0042-6822(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 36.Thomas M, Rechter S, Milbradt J, Auerochs S, Muller R, Stamminger T, Marschall M. 2009. Cytomegaloviral protein kinase pUL97 interacts with the nuclear mRNA export factor pUL69 to modulate its intranuclear localization and activity. J Gen Virol 90:567–578. doi: 10.1099/vir.0.005827-0. [DOI] [PubMed] [Google Scholar]
- 37.Baek M-C, Krosky PM, Pearson A, Coen DM. 2004. Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 324:184–193. doi: 10.1016/j.virol.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 38.Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog 5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- 40.Kawaguchi Y, Matsumura T, Roizman B, Hirai K. 1999. Cellular elongation factor 1delta is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J Virol 73:4456–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marschall M, Marzi A, Aus Dem Siepen P, Jochmann R, Kalmer M, Auerochs S, Lischka P, Leis M, Stamminger T. 2005. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem 280:33357–33367. doi: 10.1074/jbc.M502672200. [DOI] [PubMed] [Google Scholar]
- 42.Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol 82:5054–5067. doi: 10.1128/JVI.02174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, Coen DM. 2015. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J Virol 89:523–534. doi: 10.1128/JVI.02426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krosky PM, Baek MC, Coen DM. 2003. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J Virol 77:905–914. doi: 10.1128/JVI.77.2.905-914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milbradt J, Auerochs S, Sticht H, Marschall M. 2009. Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J Gen Virol 90:579–590. doi: 10.1099/vir.0.005231-0. [DOI] [PubMed] [Google Scholar]
- 46.Wolf DG, Courcelle CT, Prichard MN, Mocarski ES. 2001. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc Natl Acad Sci U S A 98:1895–1900. doi: 10.1073/pnas.98.4.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caffarelli N, Fehr AR, Yu D. 2013. Cyclin A degradation by primate cytomegalovirus protein pUL21a counters its innate restriction of virus replication. PLoS Pathog 9:e1003825. doi: 10.1371/journal.ppat.1003825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eifler M, Uecker R, Weisbach H, Bogdanow B, Richter E, Konig L, Vetter B, Lenac-Rovis T, Jonjic S, Neitzel H, Hagemeier C, Wiebusch L. 2014. PUL21a-cyclin A2 interaction is required to protect human cytomegalovirus-infected cells from the deleterious consequences of mitotic entry. PLoS Pathog 10:e1004514. doi: 10.1371/journal.ppat.1004514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salvant BS, Fortunato EA, Spector DH. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72:3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bogdanow B, Weisbach H, von Einem J, Straschewski S, Voigt S, Winkler M, Hagemeier C, Wiebusch L. 2013. Human cytomegalovirus tegument protein pp150 acts as a cyclin A2-CDK-dependent sensor of the host cell cycle and differentiation state. Proc Natl Acad Sci U S A 110:17510–17515. doi: 10.1073/pnas.1312235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oduro JD, Uecker R, Hagemeier C, Wiebusch L. 2012. Inhibition of human cytomegalovirus immediate-early gene expression by cyclin A2-dependent kinase activity. J Virol 86:9369–9383. doi: 10.1128/JVI.07181-11. [DOI] [PMC free article] [PubMed] [Google Scholar]