

Significance
Heat shock protein 70 (Hsp70) molecular chaperones help maintain protein homeostasis. Hsp70 functions require regulated promiscuous binding and release of a wide range of protein substrates. ATP binding to the Hsp70 nucleotide-binding domain (NBD) regulates the affinity and kinetics of substrate binding to their substrate-binding domain (SBD). Our work sought deeper understanding of the role of conformational dynamics for allosteric signaling in Hsp70s: The SBD undergoes a seesaw-like conformational change from a high substrate affinity state to one with lower substrate affinity, and we show that this conformational change results in drastic changes in conformational flexibility for the SBD that are essential for efficient substrate binding and release. These insights will help efforts to use Hsp70s as therapeutic targets.
Keywords: molecular chaperone, protein quality control, NMR chemical shift perturbations, conformational selection, entropically driven allostery
Abstract
Binding of ATP to the N-terminal nucleotide-binding domain (NBD) of heat shock protein 70 (Hsp70) molecular chaperones reduces the affinity of their C-terminal substrate-binding domain (SBD) for unfolded protein substrates. ATP binding to the NBD leads to docking between NBD and βSBD and releasing of the α-helical lid that covers the substrate-binding cleft in the SBD. However, these structural changes alone do not fully account for the allosteric mechanism of modulation of substrate affinity and binding kinetics. Through a multipronged study of the Escherichia coli Hsp70 DnaK, we found that changes in conformational dynamics within the βSBD play a central role in interdomain allosteric communication in the Hsp70 DnaK. ATP-mediated NBD conformational changes favor formation of NBD contacts with lynchpin sites on the βSBD and force disengagement of SBD strand β8 from strand β7, which leads to repacking of a βSBD hydrophobic cluster and disruption of the hydrophobic arch over the substrate-binding cleft. In turn, these structural rearrangements drastically enhance conformational dynamics throughout the entire βSBD and particularly around the substrate-binding site. This negative, entropically driven allostery between two functional sites of the βSBD–the NBD binding interface and the substrate-binding site–confers upon the SBD the plasticity needed to bind to a wide range of chaperone clients without compromising precise control of thermodynamics and kinetics of chaperone–client interactions.
Heat shock protein 70 (Hsp70) molecular chaperones are central players in protein quality control systems for organisms from bacteria to humans (1, 2). All Hsp70s consist of two highly conserved domains: an N-terminal nucleotide-binding domain (NBD), which regulates the affinity of substrate binding, and a C-terminal substrate-binding domain (SBD), which binds to exposed hydrophobic stretches of client proteins and is made up of a β-sandwich domain (the βSBD), and an α-helical lid (the αLid) (Fig. 1A). Hsp70 functions rely on interdomain allostery: ATP hydrolysis in the NBD controls thermodynamics and kinetics of substrate binding and release; in turn, substrate binding to the SBD stimulates ATPase activity in the NBD (3–5).
Fig. 1.
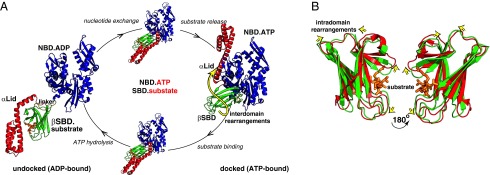
Structural changes in Hsp70 along its allosteric cycle. (A) Individual steps of the Hsp70 allosteric cycle: two end-point Hsp70 conformations (ADP-bound, domain-undocked and ATP-bound, domain-docked) and the ATP- and substrate-bound “allosterically active” intermediate require large rearrangements between the NBD (blue), βSBD (green), αLid (red), and interdomain linker (yellow). Schematic representations are shown for ADP-bound E. coli Hsp70 (DnaK) (PDB ID code 2KHO) and ATP-bound DnaK (PDB ID code 4B9Q, chain A); a previously built model (10) depicts the allosterically active intermediate. The yellow arrow highlights ATP-induced αLid dissociation from the βSBD. (B) Subtle structural perturbations in the βSBD between the two end-point Hsp70 states. Overlay of the structures of the βSBD from ADP-bound DnaK (green, PDB ID code 2KHO) and ATP-bound DnaK (red, PDB ID code B9Q). For A and B, the peptide substrate (orange) was modeled from the structure of the isolated SBD (PDB ID code 1DKZ). The yellow arrows highlight small, ATP-induced structural changes in the βSBD.
Mechanistic understanding of Hsp70 function has been greatly enhanced by recent breakthrough achievements in structural characterization of individual functional steps of the allosteric cycle for the Escherichia coli Hsp70 DnaK. New crystal structures and NMR analysis of DnaK have provided descriptions of three major functional states: ADP-bound (6, 7), ATP-bound (8, 9), and ATP/substrate-bound (10), which have distinct arrangements of the four structural units NBD, βSBD, αLid, and interdomain linker (Fig. 1A). In the ADP-bound (domain-undocked, linker-unbound) state, the two DnaK domains, the NBD and SBD, behave independently, with the interdomain linker exposed to the solvent and the αLid attached to the βSBD (6, 7). ATP binding results in linker interaction with the NBD (11); in turn, linker binding favors formation of an intimate interdomain NBD–βSBD interface and disfavors αLid interaction with the βSBD (8, 9). Taken together, ATP binding stabilizes a domain-docked, linker-bound conformation. Substrate binding to the ATP-bound, domain-docked conformation perturbs domain docking by favoring interaction between the βSBD and the αLid, while destabilizing the interaction of the NBD with the αLid and the βSBD, and at the same time maintaining NBD-linker binding, resulting in the domain-undocked, linker-bound conformation (10).
How do ligands (substrate or nucleotide) effect these domain rearrangements and conversely how do domain rearrangements modulate ATPase activity and substrate affinity? The nucleotide-binding site of the NBD is located remote from the interdomain interfaces, and thus ATP binding cannot directly (locally) affect domain communication. Instead, ATP binding leads to structurally subtle but functionally critical intradomain rearrangements among the four NBD subdomains (11, 12). ATP communicates with all four subdomains of the actin-like NBD, and the resulting arrangement of the subdomains favors binding of the hydrophobic interdomain linker to the NBD (12). Linker binding in turn enables further subdomain rearrangements and enhances ATPase activity (11). Moreover, linker binding brings the NBD and the SBD closer together, thus favoring formation of an NBD–SBD interface. In this way, linker binding allosterically links the nucleotide-binding site and interdomain interface (10).
In turn, αLid dissociation from the βSBD in the domain-docked state modulates substrate affinity because αLid removal from the isolated SBD results in significant (∼10-fold) decrease in substrate affinity and enhanced substrate binding/release kinetics, increasing the magnitudes of the on- and off-rate constants by 12-fold and ∼100-fold (13)—a very similar effect to one observed in full-length DnaK upon ATP binding (domain docking). Strikingly, ATP-induced regulation of substrate affinity still occurs in DnaK lacking the αLid (14), suggesting that αLid detachment is not the only factor responsible for allosteric signal transduction in the SBD.
Comparing the SBD structure in ATP-bound (domain-docked) DnaK (8, 9) and earlier structures of isolated substrate-bound SBDs [which have been shown to be the same as the SBD structure in ADP-bound (domain-undocked) DnaK (6)] shows some small structural perturbations in the βSBD (Fig. 1B). In addition to these small structural changes, a growing pool of experimental evidence suggests that ATP binding (and resulting domain docking) significantly increases conformational dynamics in the βSBD. First, upon ATP binding, the SBD becomes more labile to trypsin digestion (15, 16) whereas isothermal titration calorimetry (ITC) results suggested a large (more than −18 kcal/mol) favorable entropic contribution upon ATP binding (17). In line with these observations, the enhanced conformational flexibility of ATP-bound DnaK makes its crystallization extremely challenging. The only two successful attempts to obtain X-ray structures of ATP-bound DnaK required significant modifications of the WT protein aiming at reduction of conformational flexibility—one by introduction of a disulphide (8) and the other by a loop modification (9)—and to enable crystallization. Nevertheless, even these relatively “rigid” DnaK variants displayed extremely high B-factors around the substrate-binding site (8, 9). Also, hydrogen–deuterium exchange data demonstrated that the substrate-binding loops undergo fast exchange with the solvent in the presence of ATP but not in the ADP-bound state (7, 18). In turn, solution NMR studies using the WT protein revealed enhanced mobility on the μs–ms timescale in the βSBD for the domain-docked conformation (7, 10). Does this ATP-induced conformational flexibility of the βSBD play an important role in allosteric signal transduction through the βSBD structure, especially taking into account the relatively modest structural changes for the βSBD between X-ray structures of the ATP- and ADP-bound forms? In this study, we have used solution NMR and computational modeling to elucidate the role of the βSBD conformational flexibility in mediating allosteric signaling in DnaK. Our results reveal that enhanced dynamics, triggered by conformational changes in key structural lynchpins in the βSBD, couples intradomain signal transduction with interdomain docking.
Results
Seesaw-Like “Rigid-Body” Subdomain Rearrangements in the βSBD Link the Substrate-Binding Site and an Interdomain Interface.
Functionally, the βSBD mediates two-way negative allostery: Substrate binding is disfavored upon domain docking, and domain docking is disfavored upon substrate binding. Recent X-ray structures of ATP-bound (domain-docked) DnaK (8, 9) provide detailed structural insights into changes of substrate affinity: Significant perturbations in the substrate-binding cleft in the docked conformation suggest that this state is incompatible with efficient substrate binding (Fig. 1B). However, overall similarity between domain-undocked and -docked βSBD structures (8, 9, 19) raises the question: How does domain-docking trigger conformational changes in the substrate-binding site, and, reciprocally, how does substrate–binding disfavor domain docking?
To elucidate the origins of coupling between substrate binding and domain docking, we compared the βSBD conformations extracted from domain-undocked DnaK (PDB ID code 2KHO) (6) and domain-docked DnaK (PDB ID code 4B9Q) (8) structures using the DynDom algorithm, which enables identification of subdomains and flexible linkers in protein conformational changes (20). This analysis revealed that the structural transition between these two βSBD states can be described in terms of a semirigid body seesaw-like rearrangement of two βSBD subdomains (or “dynamic domains” as defined by the DynDom program). Subdomain I comprises residues 420–437 and 456–485 (Fig. 2A, red), and subdomain II is defined as residues 400–419 and 440–455 (Fig. 2A, blue). ATP-binding results in an 18.6° rotation of subdomain I relative to subdomain II (Table S1). Three hinge/bending regions were identified by the DynDom analysis: residues 417–420, 438–440, and 454–457 (Fig. 2A, yellow). Intriguingly, all three hinge regions have been shown to play important roles in DnaK activity, supporting their role in the βSBD allosteric transition. (i) Residues 417–420 form direct contacts with the interdomain linker (8, 9) and have been shown to be crucial for Hsp70 allostery (9, 21). (ii) Even a subtle perturbation via mutation of the highly conserved Leu454 (located in the second hinge region) to isoleucine results in conformational redistribution in the DnaK conformational ensemble (10). (iii) Residues 438–440 directly interact with substrate through the highly conserved residue Ile438 (22). Importantly, elastic network analysis (23) of a single (domain-undocked) βSBD structure predicts a very similar subdomain rearrangement in the βSBD (Fig. S1), suggesting that βSBD subdomain rotation is not entirely induced by domain docking or an artifact of crystal packing, but rather it is an intrinsic feature of the βSBD structure.
Fig. 2.

The transition between domain-undocked and domain-docked βSBD conformations involves seesaw-like “rigid-body” subdomain rearrangements. (A) The dynamic lobes as defined by a DynDom analysis of the βSBD from the ADP-bound (PDB ID code 2KHO) and ATP-bound (PDB ID code 4B9Q, chain A) states are colored in red (lobe I) and blue (lobe II); the bending/hinge regions are shown in yellow; residues from the strands β7 β8 and loop that are part of neither dynamic subdomains nor bending/hinge regions are shown in gray; substrate-binding loops and , as well as loops and , are labeled on the undocked structure (see also Fig. S1). The NBD-binding residues are shown as spheres (CA atoms only) and annotated; the peptide substrate (orange) was modeled from the isolated SBD X-ray structure (PDB ID code 1DKZ). (B) The βSBD/NBD domain-docking interface in domain-undocked and -docked βSBD conformations (top view); NBD-contacting residues are shown as orange sticks; βSBD residues that contact the NBD are colored in blue (Lys414/Asn415 and Gln442) and red (D481); the part of the αLid (residues I501–L507) that participates in the interdomain interface is colored green.
The fact that the substrate-binding site and NBD–βSBD contacts are located at interfaces between βSBD subdomains identified by DynDom and elastic network analyses suggests that βSBD subdomain rearrangement plays a direct role in βSBD-mediated allostery. First, the rotation of the subdomains significantly affects the arrangement of the substrate-binding loops and and thus should affect substrate-binding affinity and reciprocally could be regulated by substrate binding (Fig. 2A). Indeed, upon ATP binding, subdomain rotation opens the “hydrophobic arch” formed by (residue Ala429 of the subdomain I) and (residue Met404 of the subdomain II); the same arch has been shown to play a major role in substrate recognition and kinetics of substrate entry to the binding pocket (22, 24). Additionally, subdomain opening also limits substrate contacts with substrate-binding residues from and strands β3/β4 (specifically, Phe426, Ser427, Gln433, Ala435, Val436, and Ile438) and thus perturbs the substrate-binding cavity (8, 9).
Second, subdomain rotation results in rearrangements of three contact sites between the βSBD and the NBD and thus couples intradomain conformational changes in the βSBD with interdomain docking (Fig. 2A). The first NBD–βSBD binding site involves Lys414 and Asn415 from βSBD subdomain II and Asp326 from NBD subdomain IIA; the second contact involves Gln442 from the βSBD subdomain II and Asp148 from the NBD subdomain IA; finally, the third interaction site is formed by Asp481 from βSBD subdomain I and Ile168 from NBD subdomain IA (8, 9). Two of these sites are located in βSBD subdomain II, and the third site is in subdomain I. Consequently, subdomain rotation significantly changes the distance between the NBD-binding sites on the SBD: Lys414/Asn415, Gln442, and Asp481.
Interpreting the role of the βSBD in terms of a simple conformational selection model between the two states represented in the docked (ATP-bound) and undocked (isolated domain) crystal structures is confounded by observations in one region: Large structural rearrangements seen in strands β7 and β8 and loop upon ATP binding to DnaK (Fig. 2A, colored in gray) cannot be described in terms of rigid-body motions. Indeed, in the domain-undocked (ADP-bound) state, strand β8 (residues 494–502) forms an antiparallel β-sheet with strand β7. Upon domain docking, β8 dissociates from β7 and interacts with β5 (8, 9). As a result, only three β8 residues (Thr-500, Ile-501, and Lys-502) retain a stable β-strand conformation whereas the rest of β8 becomes largely unstructured, resulting in significant perturbations in a hydrophobic core that comprises highly conserved residues Leu454 (strand β5), Leu484 (strand β7), and Ile501 (strand β8). Remarkably, the β5/β7/β8 hydrophobic core does not directly interact with the NBD, but the rearrangement of strand β8 sterically prevents contacts between βSBD residue Gln442 and NBD residue Asp148 (Fig. 2B). Thus, β8 dissociation and disruption of the β5/β7/β8 hydrophobic core upon domain docking is linked to formation of a stable interdomain interface. Thus, the β5/β7/β8 hydrophobic core may act as a switch that triggers βSBD subdomain rotation.
The β5/β7/β8 Hydrophobic Core Is a Central Hub for the βSBD Allosteric Network.
We used NMR chemical shift perturbation (CSP) analysis to elucidate the role of the β5/β7/β8 core in triggering conformational transitions in the βSBD and to deduce how an allosteric signal is transduced through the βSBD. For our analysis, we examined the effect of mild (structurally nonperturbing) amino acid substitutions in apo (domain-undocked) DnaK. The mutations were made in putative allosteric “hotspots” deduced from the analysis above near the interdomain interface sites identified based on DynDom analysis: D481N, K414R, L454I, and I501A; in the interdomain linker L390V; and near the βSBD-αLid interface S505T and D511E.
CSPs from the allosteric substitutions near the βSBD–αLid interface and the β5/β7/β8 hydrophobic core revealed the allosteric network in the βSBD: Perturbations upon the K414R, S505T, and D511E substitutions (located on the βSBD–αLid interface) suggest allosteric coupling between the NBD-docking interface (the Lys-414 site), the αLid interface, and the β5/β7/β8 core (Fig. 3A, i). Perturbations in the β5/β7/β8 hydrophobic core resulted in widespread CSPs affecting almost the entire βSBD (Fig. 3A, ii), including subdomain II, the βSBD-αLid interface, the interface between subdomain I and subdomain II, and contacts between strands β5, β7, and β8 (Fig. 3A, ii). Intriguingly, a more subtle perturbation at the Ile-501 site upon the I501V substitution results in a similar CSP pattern to that observed upon the I501A substitution (Fig. 3B). Moreover, linear peak walking patterns between the WT protein and its I501V and I501A variants were observed for the majority of peaks arising from residues located distant from the I501 mutation site, suggesting that perturbations at Ile-501, the lynchpin residue of the β5/β7/β8 core, induce a conformational shift between two energetically similar βSBD conformations without significantly changing the structure, integrity, and stability of the βSBD. These results suggest that the majority of βSBD residues participate in a functional allosteric network that links the substrate-binding site and the interdomain interface and agree remarkably well with our previous statistical coupling analysis based on a large sequence alignment covering 926 Hsp70/110s (21), which suggested that more than 50% of the βSBD residues were evolutionary coupled.
Fig. 3.

Allosteric networks in the βSBD. (A) Three types of CSP response were observed from the soft mutations: (i) propagated through the βSBD- αLid interface, (ii) widespread through the entire βSBD, and (iii) local perturbations around the mutation sites. CSPs for individual residues were calculated using differences in chemical shifts for backbone amide 1H (ΔωH) and 15N (ΔωN) using the following equation: CSPs are mapped onto the domain-undocked structure of the SBD. CSPs are shown in yellow when ΔωH > 0.03 ppm and/or ΔωN > 0.3 ppm and red if CSPs larger than 0.1 (see Fig. S2); the mutation sites are shown in blue; residues without experimental data are shown in black. (B) Several representative regions from amide transverse relaxation optimized spectroscopy (TROSY) spectra of apo DnaK (black) and its I501V (green) and I501A (red) variants showing a “peak walking” pattern upon perturbations in the Ile-501 site.
In contrast, the D481N and L390V substitutions resulted in CSPs that were local to the mutated residue (Fig. 3A, iii). Interestingly, in contrast to another NBD contact residue, Gln442, Asp481 is positioned such that productive domain docking does not require large intradomain rearrangements for it to be accessible at the interface. Similarly, the linker residue Leu-390 is exposed to the solvent in the ADP-bound states and thus accessible for docking without conformational change. Interestingly, residues D481 and L390 are not only crucial for interdomain allostery (8, 9, 25), but even subtle perturbations at these sites (e.g., the D481N and L390V substitution) result in significant destabilization of domain docking in the presence of ATP (10). These facts, together with the absence of long-range CSPs in the βSBD upon the D481N substitution in the apo (domain-undocked) state, suggest that Asp481 and Leu390 are important for formation of the interdomain interface upon domain docking rather than for allosteric control of the βSBD conformational ensemble.
All together, our results experimentally demonstrated that perturbations at the interdomain interface and in the β5/β7/β8 core are propagated through the βSBD structure, suggesting allosteric networks of energetically coupled βSBD residues that link the βSBD–NBD and βSBD–αLid interfaces, the β5/β7/β8 core, and the substrate-binding site. Importantly, communication within these networks is possible even in the absence of significant changes in βSBD structure: Perturbations in the allosteric hotspots (i.e., the β5/β7/β8 core) result from redistributions in the functional ensemble of βSBD conformations that have very similar structure (Figs. 1B and 2A) but significantly different ability to bind substrate and to dock with the NBD.
Perturbations at the Substrate-Binding Site Enable Modulation of the βSBD Conformation.
Next, we asked how the allosteric signal propagates in the opposite direction: from the substrate-binding site to the interdomain interface. Our previous CSP analysis on the isolated (domain-undocked) βSBD revealed that substrate binding results in small, but significant, long-range CSPs for residues 416–420 (14), despite the fact that no significant changes were observed between the X-ray structures of substrate-unbound (PDB ID code 4R5L) (26) and substrate-bound SBD (PDB ID code 1DKZ) (19). Interestingly, our DynDom results identified this part of the protein as a hinge/bending region between two βSBD subdomains, providing a plausible explanation for these NMR perturbations: Substrate binding to domain-docked DnaK results in a small redistribution in subdomain arrangements that predominantly should affect subdomain interfaces and hinge/bending regions. To validate this model, we analyzed the effect of perturbations in loop at the substrate-binding site on the βSBD conformational ensemble. To monitor long-range changes upon perturbation of , we performed CSP analysis on a previously characterized DnaK construct deficient for peptide binding (9). For this construct, called DnaK-, the entire (TAEDNQS) was replaced by a shorter sequence (MGG) from a DnaK homologous protein, Hsp110 (9, 27). In the domain-undocked conformation, this construct has low substrate affinity–similar to domain-docked (ATP-bound) WT DnaK (27). Intriguingly, the X-ray structure of this loop variant showed no significant changes from the domain-undocked SBD structure (rmsd ≤ 0.4 Å) apart from local perturbations in itself, which result in a slightly more open arrangement of and relative to (9). By contrast, our CSP analysis, which reports on the solution conformational ensemble, revealed widespread long-range perturbations in the βSBD upon the substitution (Fig. 4), suggesting that the substitutions alter the βSBD conformational distribution in functionally significant ways, experimentally validating two-way allosteric coupling between and β8, as predicted by our DynDom analysis. Our results fully agree with the fact that the substitution not only perturbs substrate binding locally but also offers an explanation for its facilitation of crystallization of the otherwise transient ATP-bound DnaK conformation by shifting the population of this domain to this state (9). Moreover, in the Hsp110 protein (where this substitution occurs naturally), the domain-docked conformation seems to be much more stable than it is in DnaK (27, 28).
Fig. 4.

Loop enables control of the βSBD conformation. Histogram of CSPs between DnaK and DnaK- (Left). CSPs were calculated as in Fig. 3. Residues with significant CSPs (more than 0.05 ppm) are colored red. Red dots highlight residues for those peaks that were present in the NMR spectra of DnaK but could not be found in the spectrum of DnaK- (i.e., these residues either have CSPs larger than ∼0.2 ppm or become very dynamic on the μs–ms timescale). The bars across the top show β strands in the protein. Only data for residues 393–501 are shown. On the Right, the CSPs are mapped onto the undocked βSBD structure (taken from PDB ID code 2KHO).
Perturbations at the Substrate-Binding Site Enable Modulation of βSBD Conformational Flexibility.
Next, to further examine how perturbations around the substrate-binding site affect the βSBD conformation, we performed similar CSP analysis on another previously characterized mutant deficient for peptide binding (9). For this construct (called DnaK-PP), two highly conserved residues from loop , Gly461, and Gly468 were substituted by prolines (9). Similar to the DnaK- construct, the previous biochemical characterization of DnaK-PP revealed that it had very low substrate affinity even in the absence of ATP (9). Importantly, neither Gly461 nor Gly468 is directly involved in substrate binding, suggesting that these mutations act indirectly rather than directly perturbing interactions with substrate. As previously suggested (9), perturbations in loop somehow favor the βSBD conformation with low substrate-binding affinity even in the absence of domain docking, but they do not affect domain docking in the presence of ATP. Fully in line with this suggestion, our NMR analysis revealed that these two substitutions did not significantly affect the NBD in either the docked or undocked DnaK conformation: No significant changes were observed for NBD resonances in either the presence or the absence of ATP, suggesting that, similar to the WT protein, this variant adopts the same domain-undocked conformation in the presence of ADP and the domain-docked conformation upon ATP binding (Fig. 5A). In contrast, the substitutions drastically affect βSBD conformation (Fig. 5B): All βSBD peaks disappeared from the spectra of both domain-docked and domain-undocked states upon introduction of the substitutions, which can be explained only by enhanced μs–ms dynamics for the entire βSBD upon introduction of the mutations. Importantly, the fact that these enhanced dynamics are not associated with nonfunctional βSBD destabilization and/or unfolding—the construct enables highly specific and delicate NBD–βSBD contacts—suggests that the substitutions in the loop result in thermodynamic redistribution of the ensemble of functional βSBD conformations. Interestingly, a similar effect (enhanced μs–ms flexibility) was observed for all βSBD peaks in ATP-bound WT DnaK (7, 10). Moreover, the enhanced flexibility reflected in the NMR data are fully consistent with the long-time observation of enhanced proteolytic lability for the SBD in ATP-bound DnaK (15, 16), and other indications of dynamics, such as ITC results (17) (see also Introduction), suggesting that enhanced conformational flexibility is a unique and functionally important feature of the docked-like βSBD conformation.
Fig. 5.

Perturbations in loop affect βSBD conformational flexibility without affecting domain rearrangement. Representative regions from amide TROSY spectra of DnaK (black) and DnaK-PP (red) in their apo and ATP-bound states illustrating (A) NMR fingerprints upon ATP binding. (B) Representative region peaks corresponding to the NBD are labeled in blue (assignments from ref. 10); peaks corresponding to the βSBD are labeled in green labels (for apo protein; assignments from ref. 10) or indicated by green arrows (for ATP-bound DnaK: site-specific βSBD assignments are not available). Schematics illustrate that substitutions in loop had dramatic dynamic effects throughout the entire βSBD but did not affect the NBD in either the docked or undocked DnaK conformation.
NBD Docking, β8 Strand Rearrangement, and Substrate-Binding Control βSBD Flexibility.
To elucidate the link between ATP-induced domain docking, substrate binding, and intradomain flexibility of the βSBD, we used full-atom molecular dynamic (MD) simulations. As expected from previously reported attempts to monitor domain-docking/domain-undocking transitions in Hsp70s using full-atom MD simulations (29), no full transition between the βSBD end-point conformations (NBD-docked and -undocked) was observed in our MD simulations of the full-length DnaK. Thus, to monitor conformational transitions in the βSBD, we dissected key functional steps of the Hsp70 allosteric cycle—dissociation from the NBD, β8 rearrangement, and substrate binding—using truncations of functionally and structurally important regions that enable conformational transitions in the DnaK to occur on the MD timescale (Table S2).
First, to test whether MD can reproduce experimental results and demonstrate that substrate binding significantly reduces conformational flexibility of the substrate-binding site and favors arch closure (14, 30, 31), we performed MD simulations of the domain-undocked βSBD conformation in the presence and the absence of a short model substrate peptide NR (NRLLLTG) (32). In full agreement with experimental observations, our MD simulations demonstrated that loops and are highly dynamic in the absence of substrate but become rigid in the substrate-bound state, as indicated by the rms fluctuations (rmsf) of individual CA atoms calculated over MD trajectories (Fig. 6, Top).
Fig. 6.

Changes in βSBD conformational flexibility upon NBD and substrate binding (see also Fig. S3). The rmsf of individual CA atoms calculated over 100-ns MD trajectories of (Top) the domain-undocked βSBD in the presence (red) and absence (black) of the substrate peptide NR (U-βSBD and U-βSBD+NR; Table S2) and domain-docked βSBD (D-βSBD, Bottom) in the presence (+NBD, red) and in the absence of the NBD (see MD details in Table S2). Local changes in dynamics are annotated in red and long-range changes in blue.
Next, to monitor how NBD docking affects βSBD flexibility, we performed MD simulations on the domain-docked conformation of the βSBD in the presence and in the absence of the NBD (using as a starting structure the ATP-bound X-ray conformation, PDB ID code 4B9Q) (Table S2). NBD removal results in a very significant increase in βSBD dynamics even in the absence of significant changes in βSBD structure (Fig. 6, Bottom). The largest effects were observed for the NBD-binding loops containing residues D481 () and K414 (), suggesting that NBD binding stabilizes the K414/Q442/D481 arrangement required for NBD–βSBD docking. Notably, NBD docking results not only in local stabilization of the NBD-docking loops, but also in long-range dynamic effects: for example, suppressing flexibility of the substrate-binding loop (in blue in Fig. 6).
To further elucidate the mechanism of coupling between structural rearrangements in the βSBD and conformational flexibility of the substrate-binding loops, we monitored the distance between two arch residues, Met404 () and Ala429 (), as a function of simulation time for different MD trajectories (Fig. 7 and Fig. S4). In the domain-undocked, substrate-bound state, the βSBD spends the vast majority of time with stable contacts within the / arch [labeled “C” (closed) in Fig. 7, Left]. The fact that only very small distance variations were observed during the MD simulations suggests the presence of only very limited dynamics around the substrate-binding site. By contrast, in the domain-docked conformation, there are no stable arch contacts between loops and , and the βSBD samples two distinct / arrangements. One of these loop arrangements is close to the / conformation observed in the X-ray structure of domain-docked DnaK [labeled “O” (open) in Fig. 7, Right] whereas another arrangement represents a transient partially open conformation [labeled “I” (intermediate) in Fig. 7]. The very significant variation in distance between arch residues for both loop arrangements of domain-docked βSBD indicates that this βSBD conformation is highly dynamic on the sub-ns to ns timescale. Taken together, these MD results suggest that domain-docked and -undocked βSBD conformations differ not only in the arrangement of the substrate-binding loops but also in the dynamics around the substrate-binding site (Fig. 7). Thus, the computational simulations are in excellent agreement with the surprisingly large βSBD B-factors in X-ray structures of domain-docked DnaK (8, 9) and the enhanced hydrogen–deuterium exchange observed around the substrate-binding site (7, 18).
Fig. 7.
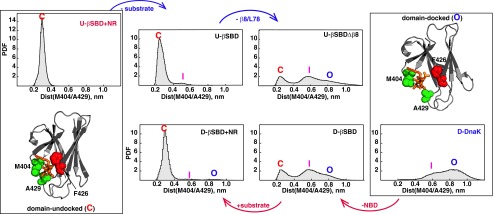
Conformational events enabling rearrangement of substrate-binding loops . Probability density functions (PDFs) for the distance distribution between arch residues Met404 and Phe429 were obtained from MD trajectories of different DnaK constructs (see also Table S2 and Fig. S4): (Top row, from left to right) PDFs for the domain-undocked conformation (PDB ID code 2KHO as a starting structures) of the isolated βSBD (U-βSBD) in the presence and the absence of substrate (the peptide NR, annotated “-substrate”) and upon removal of β8/ (U-βSBDΔβ8). (Bottom row, from right to left) PDFs for the domain-docked conformation (PDB ID code 4B9Q, chain A as a starting structure) of DnaK (D-DnaK); and the isolated βSBD (D-βSBD) in the absence and the presence of NR substrate. Three distinct / arrangements are labeled C for “closed” (red label), I for “intermediate” (magenta label), and O for “open” (blue label). The corresponding structures for the C and O states are shown in the boxes at Left and Right. The arch residues Met404 and Phe429 are shown in spacefill on the βSBD conformation of ADP-bound (PDB ID code 2KHO) and ATP-bound (PDB ID code 4B9Q, chain A) DnaK. The peptide substrate (orange) was modeled from the isolated SBD X-ray structure (PDB ID code 1DKZ); side chain atoms for substrate-binding residue Phe426 (located on the lobe I) are shown in red spacefill.
Next, we carried out MD simulations on truncated DnaK constructs that mimic different stages in the transition between domain-docked and domain-undocked conformations. (In all of these simulations, the αLid was removed to focus on intradomain contributions.) First, we removed the NBD domain from the βSBD, using the crystal structure of domain-docked, ATP-bound DnaK as a starting point (8), and then ran MD simulations for the isolated βSBD. Strikingly, removal of the NBD contacts is adequate to cause partial closure of the substrate-binding loops (Fig. 7, red arrow). This finding suggests that interactions with the NBD are required to stabilize the fully open / arrangement, and in turn, destabilization/removal of NBD–βSBD contacts favors partially open intermediate [labeled “I” (intermediate) in Fig. 7]. In turn, the addition of substrate to this conformation is sufficient for formation of a stable /arch (Fig. 7, red arrow).
For the domain-undocked βSBD, removal of substrate slightly enhances dynamics around the substrate-binding site (Fig. 7, blue arrow). Dissociation of strand β8 (as observed in the crystal structure of ATP-bound DnaK) and removal of loop were explored by virtual protein engineering: truncation to residue Gly494. Even in the domain-undocked βSBD conformation, removal of strand β8 and loop resulted in opening of the substrate-binding loops and drastic enhancement in loop flexibility around the substrate-binding site (Fig. 7, blue arrow), showing that β5/β7/β8 and / interactions are absolutely essential for the formation of a stable - arch. These results argue that formation of an interface with the NBD, binding of substrate, and specific interactions with strand β8 and loop are key trigger points that control dynamics around the substrate-binding site.
Discussion
Growing evidence suggests that many chaperone systems, including Hsp60 (33), the trigger factor chaperone (34), and small HSPs (35), rely on conformational dynamics of their substrate-binding domains to ensure promiscuous binding of a wide range of client proteins without compromising very specific processes promoting client folding. The dynamic nature of complexes between these chaperones and their client proteins has been suggested to play a key role in efficient folding: Binding that is too tight and rigid would protect aggregation-prone regions from aggregation but would also prevent client proteins from efficient folding. In contrast to Hsp70s, these other classes of chaperone contain multiple client-binding sites: While interacting transiently with client proteins through individual binding sites, these chaperones prevent protein aggregation without compromising efficient folding. Because the Hsp70 chaperones contain only a single substrate-binding site, their chaperone functions presumably rely on large structural changes, which in turn enable precise control of the thermodynamics and kinetics of substrate binding using energy of ATP (3, 4, 36). Nevertheless, mounting evidence suggests that functionally different Hsp70 conformations have drastically different conformational flexibility (see Introduction)—the role of this conformational dynamics for the Hsp70 functional cycle and substrate binding has been elusive. In this study we addressed this question and elucidated the relationship between structural features, conformational flexibility, and function of different Hsp70 conformations.
In line with previous results (see Introduction), our analysis revealed that the βSBD plays an active allosteric role enabling control of substrate binding and release. Despite the overall similarity of the domain-docked and domain-undocked βSBD structures (8, 9), the majority of βSBD residues participate in an allosteric network that links the substrate-binding site and the interdomain interface. We identified the two most critical allosteric hotspots in the βSBD, which are located in (i) loops and surrounding the substrate-binding site and (ii) the β5/β7/β8 hydrophobic core located near the interdomain interface. Even minor perturbations in these hotspots propagate through the entire βSBD, suggesting how substrate-binding and nucleotide-induced domain docking can regulate Hsp70 functions. Strikingly, the recent discovery of new allosteric inhibitors of Hsp70s (26, 37) demonstrates that, indeed, the region around the β5/β7/β8 hydrophobic core is a promising druggable site, fully in line with our results. Moreover, the exquisite sensitivity to subtle changes in these βSBD allosteric hotspots suggests how evolutionary tuning of Hsp70s and modulation by posttranslational modifications and cochaperone interactions can alter Hsp70 functional behavior.
Why is the βSBD conformational ensemble so sensitive to even minor perturbations in these allosteric hotspots? The transition between the two energetically and structurally similar βSBD conformations seen in crystal structures of ADP- and ATP-bound states of DnaK can be described as a rigid body rotation of two βSBD subdomains, providing a plausible model for allosteric coupling between substrate binding and domain docking without major structural changes between the functional states: The βSBD structure allows (at least) two orthogonal subdomain orientations whereas both functional sites (namely, the substrate-binding cleft and the NBD interaction surface) are located on the interfaces between these two subdomains. Importantly, substrate binding and domain docking stabilize “orthogonal” subdomain rearrangements that are compatible with either domain docking or substrate binding, providing an explanation for the dual allosteric regulation of these two events in the absence of large structural changes. In addition, the β5/β7/β8 structural rearrangement upon domain docking is a triggering event for the βSBD subdomain rotations: Two orthogonal interstrand interactions, β7–β8 and β5–β8, stabilize βSBD states compatible with either domain docking or substrate binding. This mechanism is similar to the Monod–Wyman–Changeux (MWC) model (38), with substrate binding and domain docking playing the roles of allosteric effectors that modify a preexisting conformational equilibrium in the βSBSD.
In the domain-docked conformation, the lack of stable contacts between two βSBD subdomains around the substrate-binding site (particularly between loops and ) results in drastically enhanced conformational flexibility around the substrate-binding site. These conformational dynamics around the substrate-binding loops in the domain-docked conformation are absolutely essential for fast and efficient substrate binding to the Hsp70. Indeed neither “end-point” (rigid X-ray) βSBD conformation enables efficient substrate binding (Fig. 8): The domain-docked βSBD unlikely allows substrate to bind to the substrate-binding pocket due to large distortions in the pocket (binding is restricted thermodynamically) whereas, in the domain-undocked βSBD, the / arch limits substrate entry to the pocket (binding is restricted kinetically). Critically, flexibility around the substrate-binding site results in transient population of the βSBD conformation suitable for binding (both kinetically and thermodynamically) and thus can facilitate efficient interaction with substrates and consequent conformational changes in the βSBD and the entire protein. Moreover, we suggest that this conformational flexibility and adjustability of the substrate-binding pocket plays a key role for promiscuous substrate recognition of Hsp70s without compromising the robustness of the allosteric cycle, allowing different substrate proteins (with a wide range of sizes and shapes) to interact with the substrate-binding pocket without steric clashes.
Fig. 8.

The role of conformational flexibility in the Hsp70 functional cycle. (A) Diagrams schematically illustrate the Hsp70 allosteric cycle and highlight that conformational flexibility of the βSBD is absolutely essential for structural transitions between different Hsp70 conformations (listed in Fig. 1). (B) Schematics showing (Left) the rigid conformational landscape of the βSBD conformation with high substrate affinity populated in domain-undocked (ADP-bound) DnaK and (Right) the dynamic conformational landscape of the βSBD conformation with low substrate affinity populated in domain-docked (ATP-bound) DnaK.
As a result, this mechanism enables several unique functional features of Hsp70s: the ability to bind to a wide array of substrates (39), including fully unfolded proteins (40), misfolded species (41) and even aggregates (42), and, moreover, the capacity to precisely regulate domain docking and fine-tune the thermodynamics and kinetics of substrate binding using ATP binding and hydrolysis. Taking together, entropy and conformational flexibility play a crucial role for allosteric signal transduction in the βSBD: Interdomain docking not only results in intradomain structural rearrangements in the βSBD but also enhanced functionally important conformational flexibility in the substrate-binding site. Consequently, our study provides an example of entropically driven allostery in a multidomain protein and interplay of thermodynamics, kinetics, and binding in an allosteric mechanism.
Materials and Methods
Expression and purification of uniformly 2H-,13C-, and 15N-labeled protein samples of DnaK(1–552) T199A/L542Y/L543E (called DnaK in the text; see details in SI Text) and its variants were performed according to published methods (10). NMR samples contained ∼300 μM protein, 10 mM of potassium phosphate (pH 7.0), and 5 mM MgCl2. NMR spectra in this study were obtained at 26 °C on a 700-MHz Agilent NMR system equipped with a cryogenically cooled triple-resonance probe (for more details, see SI Text).
All MD simulations were performed using GROMACS (version 4.6) (43) and the CHARMM27 all-hydrogen force field with the TIP3P water model (Table S2; for details see SI Text); domain-undocked (PDB ID code 2KHO) and domain-docked (PDB ID code 4B9Q, chain A) DnaK structures were used as starting structures.
Supplementary Material
Acknowledgments
All MD simulations were carried out on the ARC2 server, which belongs to the High Performance Computing facilities at the University of Leeds. This work was supported by National Institutes of Health Grant GM027616 (to L.M.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1506692112/-/DCSupplemental.
References
- 1.Mayer MP, Bukau B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62(6):670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: Brothers in arms. Mol Cell. 2010;40(2):238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Zuiderweg ERP, et al. Allostery in the Hsp70 chaperone proteins. Top Curr Chem. 2013;328:99–153. doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer MP. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci. 2013;38(10):507–514. doi: 10.1016/j.tibs.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Mayer MP, Brehmer D, Gässler CS, Bukau B. Hsp70 chaperone machines. Adv Protein Chem. 2001;59:1–44. doi: 10.1016/s0065-3233(01)59001-4. [DOI] [PubMed] [Google Scholar]
- 6.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ERP. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci USA. 2009;106(21):8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swain JF, et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26(1):27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48(6):863–874. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 9.Qi R, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol. 2013;20(7):900–907. doi: 10.1038/nsmb.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhuravleva A, Clerico EM, Gierasch LM. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell. 2012;151(6):1296–1307. doi: 10.1016/j.cell.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci USA. 2011;108(17):6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharya A, et al. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J Mol Biol. 2009;388(3):475–490. doi: 10.1016/j.jmb.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buczynski G, Slepenkov SV, Sehorn MG, Witt SN. Characterization of a lidless form of the molecular chaperone DnaK: Deletion of the lid increases peptide on- and off-rate constants. J Biol Chem. 2001;276(29):27231–27236. doi: 10.1074/jbc.M100237200. [DOI] [PubMed] [Google Scholar]
- 14.Pellecchia M, et al. Structural insights into substrate binding by the molecular chaperone DnaK. Nat Struct Biol. 2000;7(4):298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- 15.Liberek K, Skowyra D, Zylicz M, Johnson C, Georgopoulos C. The Escherichia coli DnaK chaperone, the 70-kDa heat shock protein eukaryotic equivalent, changes conformation upon ATP hydrolysis, thus triggering its dissociation from a bound target protein. J Biol Chem. 1991;266(22):14491–14496. [PubMed] [Google Scholar]
- 16.Kamath-Loeb AS, Lu CZ, Suh WC, Lonetto MA, Gross CA. Analysis of three DnaK mutant proteins suggests that progression through the ATPase cycle requires conformational changes. J Biol Chem. 1995;270(50):30051–30059. doi: 10.1074/jbc.270.50.30051. [DOI] [PubMed] [Google Scholar]
- 17.Taneva SG, Moro F, Velázquez-Campoy A, Muga A. Energetics of nucleotide-induced DnaK conformational states. Biochemistry. 2010;49(6):1338–1345. doi: 10.1021/bi901847q. [DOI] [PubMed] [Google Scholar]
- 18.Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281(24):16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- 19.Zhu X, et al. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272(5268):1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayward S, Lee RA. Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J Mol Graph Model. 2002;21(3):181–183. doi: 10.1016/s1093-3263(02)00140-7. [DOI] [PubMed] [Google Scholar]
- 21.Smock RG, et al. An interdomain sector mediating allostery in Hsp70 molecular chaperones. Mol Syst Biol. 2010;6:414. doi: 10.1038/msb.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rüdiger S, Buchberger A, Bukau B. Interaction of Hsp70 chaperones with substrates. Nat Struct Biol. 1997;4(5):342–349. doi: 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- 23.Suhre K, Sanejouand YH. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32(Web Server issue):W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rüdiger S, Mayer MP, Schneider-Mergener J, Bukau B. Modulation of substrate specificity of the DnaK chaperone by alteration of a hydrophobic arch. J Mol Biol. 2000;304(3):245–251. doi: 10.1006/jmbi.2000.4193. [DOI] [PubMed] [Google Scholar]
- 25.Kumar DP, et al. The four hydrophobic residues on the Hsp70 inter-domain linker have two distinct roles. J Mol Biol. 2011;411(5):1099–1113. doi: 10.1016/j.jmb.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leu JI, Zhang P, Murphy ME, Marmorstein R, George DL. Structural basis for the inhibition of HSP70 and DnaK chaperones by small-molecule targeting of a C-terminal allosteric pocket. ACS Chem Biol. 2014;9(11):2508–2516. doi: 10.1021/cb500236y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, et al. Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. J Biol Chem. 2012;287(8):5661–5672. doi: 10.1074/jbc.M111.275057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131(1):106–120. doi: 10.1016/j.cell.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicolaï A, Delarue P, Senet P. Decipher the mechanisms of protein conformational changes induced by nucleotide binding through free-energy landscape analysis: ATP binding to Hsp70. PLOS Comput Biol. 2013;9(12):e1003379. doi: 10.1371/journal.pcbi.1003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens SY, Cai S, Pellecchia M, Zuiderweg ERP. The solution structure of the bacterial HSP70 chaperone protein domain DnaK(393-507) in complex with the peptide NRLLLTG. Protein Sci. 2003;12(11):2588–2596. doi: 10.1110/ps.03269103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swain JF, Schulz EG, Gierasch LM. Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J Biol Chem. 2006;281(3):1605–1611. doi: 10.1074/jbc.M509356200. [DOI] [PubMed] [Google Scholar]
- 32.Pierpaoli EV, Gisler SM, Christen P. Sequence-specific rates of interaction of target peptides with the molecular chaperones DnaK and DnaJ. Biochemistry. 1998;37(47):16741–16748. doi: 10.1021/bi981762y. [DOI] [PubMed] [Google Scholar]
- 33.Joachimiak LA, Walzthoeni T, Liu CW, Aebersold R, Frydman J. The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell. 2014;159(5):1042–1055. doi: 10.1016/j.cell.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saio T, Guan X, Rossi P, Economou A, Kalodimos CG. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science. 2014;344(6184):1250494. doi: 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stengel F, et al. Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc Natl Acad Sci USA. 2010;107(5):2007–2012. doi: 10.1073/pnas.0910126107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayer MP, Bukau B. Hsp70 chaperone systems: Diversity of cellular functions and mechanism of action. Biol Chem. 1998;379(3):261–268. [PubMed] [Google Scholar]
- 37.Hassan AQ, et al. The novolactone natural product disrupts the allosteric regulation of Hsp70. Chem Biol. 2015;22(1):87–97. doi: 10.1016/j.chembiol.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Changeux J-P. Allostery and the Monod-Wyman-Changeux model after 50 years. Annu Rev Biophys. 2012;41:103–133. doi: 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]
- 39.Clerico EM, Tilitsky JM, Meng W, Gierasch LM. How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol. 2015;427(7):1575–1588. doi: 10.1016/j.jmb.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science. 2002;295(5561):1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 41.Sharma SK, De los Rios P, Christen P, Lustig A, Goloubinoff P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat Chem Biol. 2010;6(12):914–920. doi: 10.1038/nchembio.455. [DOI] [PubMed] [Google Scholar]
- 42.Diamant S, Ben-Zvi AP, Bukau B, Goloubinoff P. Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery. J Biol Chem. 2000;275(28):21107–21113. doi: 10.1074/jbc.M001293200. [DOI] [PubMed] [Google Scholar]
- 43.Pronk S, et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.