

Abstract
Immune responses to HLA and development of anti-donor HLA (DSA) have been shown to play a role in chronic rejection following transplantation. We hypothesized that Abs to MHC changes microRNAs (miRNAs) leading to chronic lung allograft rejection. Microarray analysis was performed in a murine model of anti-MHC induced obliterative airway disease (OAD), a correlate of obliterative bronchiolitis. A unique profile of dysregulated miRNAs was detected in OAD mice on day 7 and 15 after Ab administration compared to control. Sixty-seven miRNAs were increased and 42 miRNAs were decreased in OAD mice on day 7. In addition, 15 miRNAs were over expressed and 16 miRNAs were under expressed in OAD mice on day 15. The expression of miR-16 and miR-195 were significantly decreased in lungs of OAD mice by qPCR and in situ hybridization with increases of H-2 Aa and H-2 Dma mRNA levels. Significant reduction of miR-16 and miR-195 levels were also noted in lung transplant (LTx) patients with DSA compared to LTx without DSA. Bioinformatic TargetScan and Reporter Assays identified the binding of miR-16 and miR-195 to the 3’UTR of Regulatory Factor X 5. qPCR and immunohistochemistry indicated post-transcriptional increases of Regulatory Factor X 5 mRNA and protein expression not only in OAD mice and but also in LTxR with DSA which was associated with increased expression of HLA-DPA1, HLA-DQA1, and HLA-DRA mRNA. Therefore, our results demonstrated that miRNAs induced by alloimmunity may play important roles in chronic rejection after LTx.
Introduction
Lung transplantations (LTx) are effective treatments for many diseases unresponsive to other conventional therapies. However, long-term survival of LTx recipients (LTxR) is often limited by the development of obliterative bronchiolitis (OB), a fibro-proliferative condition affecting small airways (1, 2). OB is generally thought to be a response to injury and inflammation resulting from acute vascular rejection, lymphocytic bronchiolitis, viral infections and other causes of bronchial injury (3). However, even as treatment for acute rejection and infection continue to improve, the incidence and severity of OB have not changed, resulting in lower graft and patient survival rates than those observed in other solid organ transplants (1, 4).
Multiple immune and non-immune mechanisms have been proposed to contribute to the pathogenesis of chronic rejection resulting in a slow and progressive deterioration of allograft function over months to years (5, 6). Histopathologically, chronic rejection is an inflammatory process resulting in replacement of allograft parenchyma with fibroproliferative changes eventually resulting in occlusion of small airways in the allograft (7). Previous studies have demonstrated strong association between the development of Abs to mismatched donor HLA and the development of Bronchiolitis Obliterans Syndrome (BOS), chronic rejection following human LTx (6). The development of alloimmune responses often precedes the development of BOS (8, 9) suggesting a pathogenic role for Abs to HLA in the development of chronic rejection.
MicroRNAs (miRNAs), highly conserved in both structure and function across species, are small noncoding RNA molecules of about 22 nucleotides that regulate the post-transcriptional expression of target genes (10). miRNAs, as immune regulators, may govern expression of genes relevant to allograft rejection, tolerance induction and post-transplant infection in recipients of organ transplants (11). In one report following renal transplantation it was demonstrated that 20 miRNAs were differentially expressed in acute rejection biopsies (12). Another report profiling miRNA expression following renal transplantation demonstrated that miR-142-5p, miR-155 and miR-223 were highly predictive of AR (13). MiR-155 (or MiR-150) has been reported to be a key player in adaptive immunity and T-cell mediated Ab responses (11, 14). It has been reported that miR-182 is induced by IL-2 and STAT5 that inhibited FOXO1 leading to clonal expansion of Th cells (15). Therefore, understanding the global changes in miRNA expression following anti-MHC induction can potentially provide insights into the pathogenesis of chronic allograft rejection in LTxR. However, analysis of human samples is limited by heterogeneity among patients and an inability to obtain samples at specific stages in the development of OB. To address these limitations, we employed a murine model of obliterative airway disease (OAD) to determine the mechanism(s) by which Abs to donor MHC may lead to the development of chronic rejection (16). In this model, intra-bronchial administration of Abs specific to MHC class I resulted in OAD including cellular infiltration, luminal occlusion, and fibrosis of the small airways, the central events seen during chronic lung allograft rejection (16). Using this OAD model we tested the hypothesis that there will be a sequential, stereotypic expression and patterns of miRNAs dysregulation that reflect pathophysiologic events in Ab-mediated rejection and development of chronic rejection both in the animal model of OAD induced by anti-MHC and human LTxR with de novo development of Abs to donor HLA.
Materials and Methods
Murine model of anti-MHC class I induced OAD
Murine mAb to H-2Kb (IgG2a, endotoxin free, measured by LAL assay), was administered intrabronchially at a dose of 200 μg per administration into wild-type C57BL/6 mice as reported earlier (16). Abs (200 μg) were administered with a 20-gauge catheter into the lung on days 1, 2, 3, 6, and then weekly thereafter. C1.18.4, (IgG2a), was given as controls. LentimiRa-GFP-miR-16 virus and lentimiRa-GFP-miR-195 virus were obtained from Applied Biological Materials (ABM) Inc. (Richmond, BC, Canada). 1×106 infectious units/ml were administered intratracheally into the anesthetized animals 3 days before Ab administration, then administered on day 3, 6 and weekly thereafter. All experiments in this study were performed in compliance with the guidelines of the Washington University School of Medicine Institutional Laboratory Animal Care and Use Committee.
Histological analysis
Lungs were fixed in 10% formaldehyde and sections cut at 5 μm thickness and stained with H&E and Masson’s trichrome. Lesions that displayed cellular infiltration, epithelial abnormalities, and fibro-proliferation were analyzed by random sampling. Morphometric analysis for fibrosis and cellular infiltration was performed. Fibrosis was calculated using Optimas software version (Media Cybernetics, Rockville, MD), as a percentage of total area enclosed by basement membrane. Cellular infiltration and epithelial abnormalities were similarly calculated as a percentage of the total bronchiole and vessels visualized, respectively.
Lung transplant recipients
Patients who underwent LTx at the Washington University School of Medicine between January 2011 and March 2013 were eligible for the study which was approved by the institutional review board at the Washington University School of Medicine, and written informed consent was obtained from all study patients. Thirty LTxR were included in this study. Among them, 15 developed anti-donor HLA (DSA) after LTx (DSA group) and the other 15 didn’t develop DSA and BOS after transplantation. These 30 LTxR were bilateral lung transplants and were matched for age, time from transplantation, underlying diagnosis, and immunosuppressive treatment. Bronchoalveolar lavage (BAL) fluid collected during bronchoscopy was centrifuged at 300 g and 4°C for 10 min to isolate cells. BAL cells were then washed with 20 mL of Hank balanced salt solution (Mediatech Inc; Herndon, VA) by three successive centrifugations.
PBMC isolation and lung tissues collection
PBMCs were separated from blood samples of OAD mice and mice receiving isotype control on days 7 and 15 using the Ficoll method (Amersham, Uppsala, Sweden). Lungs were collected from both groups of mice on days 7 and 15 and placed in RNAlater (Ambion, Foster City, CA). Briefly, lungs were perfused with PBS, the lung tissue was minced and placed in RPMI-1640 containing collagenase (2 mg/mL), incubated at 37°C for 2 h and strained through a 70-μm nylon cell strainer.
Global miRNA expression profiling
Total RNA was isolated using the mirVana miRNA isolation kit (Applied Biosystems, Foster City, CA). There were three individual RNA samples collected on day 7 and 15 from mice (n=3) receiving isotype control or anti-H-2Kb (Figure 1A). RNA quantity and quality was assessed spectrophotometrically with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) with Agilent RNA 6000 Nano Kit for total RNA. The latest version of Affymetrix platform for miRNA expression analysis (Genechip miRNA 3.0 array) based on mirBase version 17 (http://www.mirbase.org/) was used to obtain miRNA profiles. Normalization and statistical analysis were performed with Partek Genomic Suite 6.6 software by means of ANOVA test. Fold-change and p-values were applied to generate miRNA differentially expressed lists. Microarray data are available in the Gene Expression Omnibus at National Center for Biotechnology Information with the identification GSE58730.
Figure 1.

Histopathological analysis of murine model of anti-MHC class I induced OAD. (A) Workflow of miRNAs study on OAD model. Murine mAb to H-2Kb was given at a dose of 200 μg per administration into wild-type C57BL/6 mice. Abs (200 μg) were administered with a 20-gauge catheter into the lung on days 1, 2, 3, 6, and then weekly thereafter. C1.18.4, same isotype, was given on the days mentioned above as controls. Three individual RNA samples were collected on days 7 and 15 from animals receiving isotype or anti-H-2Kb Abs (n=3). The latest version of Affymetrix platform for miRNA expression analysis (miRNA 3.0 array) based on mirBase version 17 was used to obtain miRNA profiles. (B and C) The lungs were recovered on days 7 and 15. Formalin preserved tissues were sectioned and analyzed by H&E (upper panels) and Masson’s trichrome staining (lower panels). Images were captured and representative fields are depicted in the figure. Scale bar: 100μm.
TaqMan miRNA assay for individual miRNAs
Individual miRNA tests were performed on independent sets of PBMCs and lung tissues from additional groups (n=9) of OAD mice and mice receiving isotype control. First, total RNA isolated from each PBMC and lung tissues was subjected to RT using the miRNA specific primers from the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) as described previously. Thereafter, four miRNAs of interest (miR-16, miR-195, miR-200c, and miR-125b), differentially expressed from miRNA array, were further quantified by TaqMan miRNA assays (Applied Biosystems). RT- PCR was performed using Applied Biosystems 7900HT thermocycler. The 20 μl PCR reaction mixture included 8 μl of nuclease free water, 1 μl of RT products, 10 μl of 2x Taqman (AmpErase NO UNG) Universal PCR Master Mix and 1 μl of primer and probe mix of the TaqMan MicroRNA Assay kit (Applied Biosystems). Reaction was incubated in a 96-well optical plate at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. All qPCR reactions were performed in triplicate and the Ct values greater than 35 from the RT-PCR assays were treated as 35. The average expression levels of miRNAs were normalized using the reference gene snoRNA135 and subsequently the 2−Δct method was applied. The 2−ΔΔct method was used to express the levels of miRNAs.
miRNA in situ hybridization
In situ hybridizations were performed in 8-μm cryosections from the lung of mice with intra-bronchial administration Abs to H-2Kb or C1.18.4 isotype. In situ hybridizations of tissue sections were performed as previously described (17). After protease digestion, the digoxin-labeled locked nucleic acid (LNA)-scrambled control probe and LNA miR-16 and miR-195 antisense probe (Exiqon, Woburn, MA) were hybridized to the slides at 55°C for overnight. Following post hybridization washes with 0.2x SSC buffer at 55°C, 100 μl of rabbit anti-digoxin Ab (Sigma-Aldrich, Saint Louis, MO), diluted 1/500 in 10% sheep serum PBS buffer, was applied to the slides for 60 min at room temperature. Slides were counterstained with NBT-BCIP kit (Roche Lifescience, Indianapolis, IN), cover slipped, and mounted for viewing.
Immunofluorescence
The frozen cryosections of the lung of mice were fixed on cover slides in 4% formaldehyde for 20 min. After blocking, using PBS containing 5% goat serum for 45 min, cryosections were then incubated with FITC-conjugated anti-Maclura Pomifera Lectin Ab overnight at 4°C. The cryosections were washed 3 times using PBS. Then tissues were then mounted with DAPI-containing mounting solution. Fluorescent images were taken with a Zeiss confocal microscope (Carl Zeiss Microsystems).
RT-PCR
To determine the expression of mouse Regulatory factor X 5 (RFX5), mouse H-2 Aa and H-2 Dma, cDNA from both groups (n=9) of OAD and control animals was synthesized using SuperScript II RT with random hexamers (Life Technologies, Carlsbad, CA). To quantitate the expression of human RFX5, HLA-DPA1 HLA-DQA1, and HLA-DRA, cDNA from 30 LTx patients was reverse-transcribed by the same RT kit from Life Technologies. RT-PCR was performed using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). mRNA levels for candidate genes were measured by RT-PCR using gene-specific primers. (mouse Rfx5: F 5′-CAGCAGCATCTCATCTCTGC-3′, R 5′- ATAATGACCGTTCTCGAGGG -3′; mouse H2-Dma: F 5′- TGAGCAGAAGTCAGGAGCTG-3′, R 5′- GTGGGTCATCCCACAACACT-3′; mouse H2-Aa: F 5′- AGCCTCTGTGGAGGTGAAGA-3′, R 5′- TACTGGCCAATGTCTCCAGG-3′; mouse Gapdh: F 5′- CGTCCCGTAGACAAAATGGT-3′; Human RFX5: F 5′- GGCCGTGCAGAACAAAGTAG-3′, R 5′- TGAGGGGAGCTGAAGGTAGA-3′; Human HLA-DRA: F 5′- TGGAGTCCCTGTGCTAGGAT-3′, R 5′- ATAGAACTCGGCCTGGATGA-3′; Human HLA-DPA1: F 5′- CTTGGCTTTCCTGCTGAGTC-3′, R 5′- CCCTGTTGGTCTATGCGTCT-3′; Human HLA-DQA1: F 5′- CCCTGTGGAGGTGAAGACAT-3′, R 5′- CAAATTCATGGGTGTACTGGC-3′) Melting curve analysis was done at the end of the reaction to assess the quality of the final PCR products. The threshold cycle C(t) values were calculated by fixing the basal fluorescence at 0.05 units. Three replicates were used for each sample, and the average C(t) value was calculated. The ΔC(t) values were calculated as C(t) sample - C(t) GAPDH. The N-fold increase or decrease in expression was calculated by the ΔΔCt method using the C(t)GAPDH value as the reference point.
Immunohistochemical analysis
Frozen lung tissues were embedded in Freez Tissue matrix (OCT), and sections were cut at 8 μm thickness. The sections were fixed in cold alcohol for 2 min (−20°C) and air dried. The sections were treated with 3% H2O2 in EtOH for 10 min to block endogenous peroxidase activity. The sections were blocked with biotin/avidin system components for 15 min by blocking reagent (Avidin/Biotin Blocking Kit; BD Biosciences, San Jose, CA). Primary and secondary Abs were diluted with Ab dilution solution (BD Biosciences). The sections were incubated overnight (O/N) at 4°C with purified rat anti-rabbit polyclonal to RFX5 Abs (5.0 μg/ml; Abcam) followed by incubation for 30 min at room temperature with diluted biotin-conjugated goat anti-rat IgG (1:50; BD Pharmingen, Franklin Lakes, NJ). The sections were incubated with streptavidin-HRP and incubated for 30 min at room temperature (BD Pharmingen). The presence of positive cells was detected with the diaminobenzidine substrate kit (BD Pharmingen), counterstained with hematoxylin, and examined using a light microscope.
Construction of plasmids
The primary miR-16 and miR-195 sequence was amplified from mouse genomic DNA and then the cloned DNA fragment was inserted into pcDNA3.1 vector (termed pcDNA3.1-miR-16 and pcDNA3.1-miR-195). The 3′ UTR fragment of mouse RFX5 containing a potential miR-16 and miR-195 binding site was cloned from mouse genomic DNA. The primers were as follows: Forward: 5′>CCTCTAGAATACCACCCATTTGTCCATT <3′ Reverse: 5′>CCCGGCCGGTCGAGCCATGTGAGCAAAAGG<3′ The PCR products were inserted into the luciferase reporter plasmid phRL-TK and the resulting plasmid was termed phRL-TK-RFX5. To further analyze the combination of RFX5 and miR-16 or miR-195, the sequence of the binding site 5′>TGCTGCTA<3′ was mutated to 5′>GAAGAAGG<3′ by PCR. The primers for mutation were as follows: Forward: 5′>GTTATTATACACAATGCTGCTATGAACATTCTTGTA<3′ Reverse: 5′>TACAAGAATGTTCATAGCAGCATTGTGTATAATAAC<3′ The resulting plasmid was termed phRL-TK-mutRFX5.
Cell Culture
The CHO and 3T3 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM medium (Gibco BRL, Grand Island, NY) supplemented with FBS (15%; Biocell Laboratories, Rancho Dominguez, CA), L-glutamine (2 mM), nonessential aminoacids (100 μM), HEPES (25 mM), sodium pyruvate (1 mM), penicillin (100 U/ml), and streptomycin (0.1 mg/ml).
miRNA mimics and Transfection
MiR-16 and miR-195 mimics were purchased from Ambion. 3T3 fibroblast cells were grown in 10% FBS in DMEM and transfected at 70-80% confluency in six well plates using Lipofectamin RNAi MAX™ (Life Technologies) with miRNA mimics at a final concentration of 10 nM unless indicated.
Luciferase assays
CHO cells were seeded at 105 cells per well 24 hr before transfection. Cells were transfected using Lipofectamine2000 transfection reagent (Life Technologies) with 495 ng of pcDNA3.1-miR-16 or pcDNA3.1-miR-195, 1 ng of phRL-TK-RFX5 or phRL-TK-mutRFX5 and 5 ng of firefly luciferase reporter plasmid (pGL3-control) was to normalize transfection efficiency. Luciferase activity was measured 36 hr after transfection by the Dual-Luciferase Reporter Assay System (Promega, Madison, WI).
Western blotting
Protein samples (25 μg/lane) were subjected to SDS-PAGE and then transferred to polyvinylidene difluoride membranes (Life technologies). The resultant membranes were blocked with 5% milk-TBST for 1 h at room temperature and then incubated with anti-RFX5 Ab (Abcam) overnight at 4°C, washed with TBST, incubated with appropriate secondary Ab (1:5000; Jackson ImmunoResearch) conjugated to horseradish peroxidase, washed, and visualized with ECL Western Blotting Detection Reagents (Amersham Biosciences). After stripping with Restore Western Blot Stripping Buffer (Pierce) for 20 min at room temperature, membranes were processed similarly with anti-GAPDH Ab (1:10,000 dilution, Abcam) as a loading control.
Statistical Analysis
qPCR and qualitative clinical variables were compared by the Mann Whitney U-test using GraphPad Prism program (Lo Jolla, CA). Independent and unpaired t-test corrections were performed to determine the significant difference between luciferase assays. The Pearson’s χ2 test was used to evaluate the correlation between miRNAs of miR-16, miR-195 and mRNAs of HLA-DPA1, HLA-DQA1, and HLA-DRA in LTxR. The correlation of relative miRNAs of miR-16, miR-195 and mRNAs of HLA-DPA1, HLA-DQA1, and HLA-DRA in LTxR was analyzed using linear correlation and regression. All data were expressed as mean ± SEM, unless otherwise specified. A p value <0.05 was regarded as statistically significant.
Results
Dysregulated miRNAs in murine OAD induced by Abs to MHC class I (anti-H-2Kb)
We developed the murine model in which OAD, correlate of BOS, following ligation of MHC molecules in the native lung by mAb to H-2Kb (16). Seven and 15 days after administration of Abs, the lung tissues were harvested and histopathological analysis was performed. Lungs from animals administered with anti-MHC class I Ab after 15 days demonstrated significant inflammatory cells around the vessels and bronchiole (Figure 1C). Further, there was epithelial hyperplasia and an increase in fibrosis (Figure 1C). However, there were no significant lesions on day 7 (Figure 1B). In addition, lungs from animals treated with control C1.18.4 Ab did not demonstrate any of the above pathology (Figures 1B and C).
Since we aimed at examination of the molecular mechanism(s) of the anti-MHC induced OAD, we chose early responses after Ab administration during the development of OAD. Differential expression of miRNAs after anti-MHC administration was determined on days 7 and 15. Out of 1000 mature mouse miRNAs examined, 367 miRNAs in PBMCs from both OAD mice and mice receiving isotype control were detectable and further analyzed (GEO accession #GSE58730). Clustering of miRNA expression patterns classified the OAD mice samples distinctly from those of mice receiving isotype control on days 7 and 15 (Figure 2A). Sixty-seven miRNAs were significantly increased and 42 miRNAs were significantly decreased in OAD mice as compared to those of mice receiving isotype control on day 7 (Table S1, p<0.05). In addition, 15 miRNAs were significantly over expressed and 16 miRNAs were under expressed in OAD mice as compared to those of mice receiving isotype control on day 15 (Table S2, p<0.05). Further, some clustered differential miRNAs showed similar expression patterns in OAD mice as compared to those of mice receiving isotype control. Three miRNAs (miR-1966, miR-3470b, and miR-3069) showed highly significant over expression (p<0.05) and three miRNAs (miR-467a-8, miR-195 and miR-16) were down regulated (p<0.05) after both days 7 and 15 of Ab administration (Figure 2B), suggesting that these dysregulated miRNAs may be important in the alloimmune response after LTx.
Figure 2.

Dysregulated miRNAs in murine OAD induced by Abs to MHC class I (anti-H-2Kb). (A) Heatmap of differential miRNAs between H-2Kb_7day vs Isotype_7day and H-2Kb_15day vs. Isotype_15day. Each column corresponds to the expression profile of three OAD mice and mice receiving isotype Abs on days 7 and 15. The color of each cell reflects the fold-change of expression level of the corresponding miRNA in the corresponding sample relative to the average expression level across all samples. Red represents a decrease in expression level, whereas yellow represents an increase. The scale (bottom) reflects miRNA abundance ratio in a given sample relative to the mean level for all samples. Hierarchical clustering of differential miRNAs expression was performed from OAD mice and mice receiving isotype Abs. (B) Venn diagram of differential miRNAs between H-2Kb_7day vs Isotype_7day and H-2Kb_15day vs. Isotype_15day. The Venn diagram shows the number and the specific miRNAs differentially expressed, and the 6 miRNAs shared (3 miRNAs up regulated and 3 miRNAs down regulated), in PBMC between OAD mice and mice receiving isotype Abs.
We employed TaqMan RT-PCR to validate the expression level of differentially expressed miRNAs. Four miRNAs (miR-16, miR-195, miR-200c, and miR-125b) were selected for validation based on target prediction analysis and published reports associated with MHC gene expression, Ag presentation and immune cell degranulation (18-20). In agreement with the microarray data, miR-16, miR-195, and miR-200c were significantly decreased (p<0.05), while miR-125b significantly increased, on both days 7 and 15 in PBMCs from OAD mice (n=9) compared to those of mice receiving isotype control (Figure 3). Taken together, these results identified a profile of miRNAs in the lungs with OAD following anti-MHC class I administration as well as in the PBMC from these animals suggesting a functional role for these miRNAs in the immune response leading to anti-MHC class I induced OAD.
Figure 3.

Validation of differential miRNAs from miRNA array. Four miRNAs from miRNA array were quantified by TaqMan miRNA assays using nine mice for each group (n=9). Expression fold changes of miRNAs were calculated by 2−ΔΔCt method, normalized to the expression of housekeeping gene snoRNA135. Expression fold changes of miRNAs in OAD mice were compared to those of mice receiving isotype Abs on days 7 and 15 after Ab administration. Statistical significance was evaluated and P-values were calculated with the Student’s t-test; error bars represent SEM.
MiR-16 and miR-195 expression was decreased in lungs from OAD mice and LTxR with Abs to donor HLA
To further determine the importance of these miRNAs in the alloimmune response, expression of miR-16 and miR-195 was analyzed in the lungs from anti-MHC induced OAD animals and the results presented in Figure 4A and 4B clearly demonstrate that they are significantly decreased in the lung tissues of OAD mice. The expression of the housekeeping gene snoRNA135 remained constant. To determine the cell type-specific localization of miR-16 and miR-195, in situ hybridization was conducted on cryo-sections of anti-H-2Kb or isotype control administered lungs using LNA anti-miR-16 and LNA anti-miR-195. MiR-16 and miR-195 was primarily detected in lung alveolus. As shown in Figure 4C, miR-16 and miR-195 expression was significantly down-regulated in the lung alveolus of OAD mice both days 7 and 15 compared to those of mice receiving isotype Abs (Figure 4C). Immunofluorescence staining using Ab against Maclura Pomifera Lectin, a specific marker for alveolar epithelium (21), demonstrated that miR-16 and miR-195 were expressed in the lung alveolus (Figure 4C and 4D). To determine whether similar reduction can be seen in human LTxR with DSA, we obtained BAL fluid following LTx. Significant reduction of miR-16 and miR-195 were also seen in the cells isolated from BAL fluid from LTxR with de novo development of DSA compared to LTxR without DSA (Figure 5). These results support the conclusion that miR-16 and miR-195 expression are down regulated following development of Abs to MHC.
Figure 4.

Validation of miRNA-16 and miRNA-195 in lung tissues from OAD mice. (A and B) qRT-PCR was used to detect expression of miR-16 (day 7: p=0.013 and day 15: p=0.009) and miR-195 (day 7: p=0.016 and day 15: p=0.004) in the local site in the lung tissues of OAD mice (n=9) and mice (n=9) receiving isotype Abs on days 7 and 15 after Ab administration. P-values were calculated with the Student’s t-test; error bars represent SEM. (C and D) In situ hybridization was conducted on cryosections of anti-H-2Kb or isotype Abs-challenged lungs using LNA anti-miR-16 and LNA anti-miR-195. Immunofluorescence staining using Ab against MPL was performed for lung alveolus localization using the same series of cryosections from the lung of OAD mice and mice receiving isotype Abs (n=3 each group). Images depict representative areas from the cryosections. Scale bar: 100μm.
Figure 5.
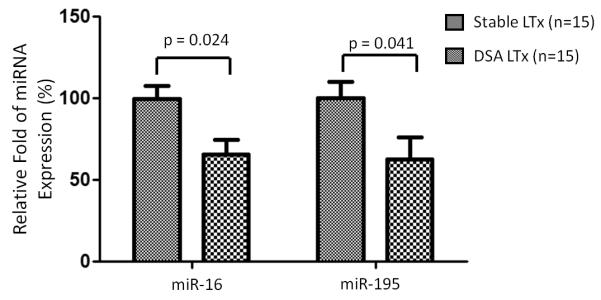
Validation of miRNA-16 and miRNA-195 in LTxR with de novo development of DSA (n=15) compared to LTxR without DSA (n=15). Expression fold changes of miRNAs were calculated by 2−ΔΔCt method, normalized to the expression of housekeeping gene RNU44. Expression fold changes of miRNAs in DSA LTxR were compared to those of stable LTxR. P-values were calculated with the Mann-Whitney test; error bars represent SEM.
RFX5 is a target of miR-16 and miRNA-195 in the mouse
Integration of miRNA target predictions from multiple algorithms has been reported to substantially increase the functional correlations and decrease the false-positive rate compared with single algorithms. Therefore, we determined common predicted targets of miR-16 and miR-195 using miRanda and Targetscan algorithms and both identified RFX5, which has been reported to be over expressed in transplanted organs (22). In our study the bioinformatics analysis clearly predicted the binding of miR-16 and miR-195 to the 3’UTR of RFX5 (data not shown). None of the other dysregulated miRNAs contained the predicted binding site in the 3’UTR of RFX5.
To determine if RFX5 is a target of miR-16 and miR-195 in the mouse, we constructed the plasmids that over expressed mmu-miR-16 and mmu-miR-195 in CHO cells (Figure 6A). We then examined binding of mouse miR-16 and miR-195 to the 3’UTR of mouse RFX5 mRNA using a luciferase assay. As the 3’UTR of RFX5 is inserted downstream of the luciferase open reading frame, specific binding to miR-16 and miR-195 prevents luciferase reporter gene expression (Figure 6B). In addition, mutation of the RFX5 binding site decreased specific binding to miR-16 and miR-195 and restores luciferase activity (Figure 6B) indicating that mouse RFX5 is indeed a target of miR-16 and miR-195.
Figure 6.

Rfx5 is a target of miRNA-16 and miRNA-195 in the mouse. (A) CHO cells were transfected with the pcDNA3.1-miR-16 and -miR-195 plasmid that overexpresses the mature mmu-miR-16 and miR-195. (B) The 3’UTR of mouse Rfx5, including the putative miRNA target site, was inserted downstream of a luciferase open reading frame in the phRL-TK-Rfx5 plasmid. A construct containing a mutated sequence of the miRNA binding site, phRL-TK-mutRfx5, was produced as a control. The different luciferase constructs were transfected into CHO cells with the construct pcDNA3.1-miR-16 and miR-195 that overexpresses the mature mmu-miR-16 and miR-195 or with the blank vector pcDNA-3.1. Luciferase activity was measured 36 hr after transfection by the Dual-Luciferase Reporter Assay System. All experiments (A and B) were repeated three times independently with consistent results. (C) Western blot assay demonstrated that Rfx5 protein level was down regulated when transfection of miR-16 and miR-195 in 3T3 cells. GAPDH was used as loading control.
To validate that the correlation between expression of miR-16 and miR-195 with RFX5 expression, we introduced miR-16 and miR-195 mimics into 3T3 mouse fibroblast cells. Western blot analysis demonstrated that RFX5 protein expression was down-regulated in 3T3 cells transfected with miR-16 or miR-195 as compared with control miR-transfected 3T3 fibroblast cells (Figure 6C) indicating that there is a miR-16 and miR-195 mediated post-transcriptional regulation leading to decrease in RFX5 protein expression.
RFX5 was up regulated in OAD mice and LTxR with de novo development of DSA
We analyzed RFX5 expression at the mRNA levels in OAD model. There was a significant increase in RFX5 mRNA levels at both time-points, days 7 and 15 following anti-MHC class I administration (Figure 7A, day 7: p=0.022 and day 15: p=0.013). The housekeeping gene, β-Actin demonstrated constant expression levels on both H-2Kb mice and mice receiving isotype control. In addition, western blot analysis demonstrated that RFX5 protein level was up regulated in OAD mice compared to those in mice receiving isotype control (Figure 7B). Furthermore, immunohistochemical staining of RFX5 in lung tissues demonstrated that RFX5 was up-regulated in OAD mice compared to those in mice receiving isotype control (Figure 7C). These data demonstrate that there is a post-transcriptional regulation leading to decrease in RFX5 expression during anti-MHC induced OAD.
Figure 7.

Rfx5 was up regulated in OAD mice and LTxR who developed DSA compared to control. RNA from OAD mice and isotype mice on days 7 and 15 (n=9). (A) RT-PCR of Rfx5. Data was normalized to GAPDH expression. P-values were calculated with the Student’s t-test. (B) Western blot assay of Rfx5 protein expression. GAPDH was used as loading control. (C) Immunohistochemical staining of Rfx5 in lung tissues from OAD mice and mice receiving isotype Abs. Images were captured and representative areas depicted in the figure. Frozen cryosections from at least three different mice of each group were chosen for experiments of immunohistochemical staining. Scale bar: 100μm. (D) RT-PCR of Rfx5 mRNA levels in LTxR with de novo development of DSA (n=15) compared to LTxR without DSA (n=15). P-values were calculated with the Mann-Whitney test; error bars represent SEM.
To determine whether RFX5 is up regulated following DSA development in human LTxR, we analyzed 15 LTxR with DSA and 15 stable LTxR with no detectable DSA. Significant increases in RFX5 mRNA level in LTxR who developed DSA was noted when compared to stable LTxR (n=15, p=0.029, Figure 7D). This was also associated with a concomitant decrease of miR-16 and miR-195 expression. Therefore, the noted post-transcriptional increase in RFX5 mRNA level in both OAD animals and LTxR with de novo development of donor specific Ab to HLA strongly suggest an important role for the miRNA-16 and miRNA-195 in regulating the immune responses leading to chronic rejection.
H-2 Aa and H-2 Dma and HLA-DPA1, HLA-DQA1, and HLA-DRA were up-regulated in anti-MHC class I induced OAD in mice and LTxR with de novo development of DSA
RFX5 has been reported to be essential for the control of MHC class II gene expression (23, 24). To investigate the relationship between miR-16, miR-195 levels and MHC class II gene expression, qRT-PCR was performed. As shown in Figure 8A and 8B, there was a significant increase of mouse H-2 Aa (day 7: p=0.025 and day 15: p=0.011) and H-2 Dma (day 7: p=0.016 and day 15: p=0.005) mRNA levels in the lung tissues of anti-MHC class I induced OAD mice compared to those in mice receiving isotype control. More important is our finding that an inverse relationship between miR-16, miR-195 and HLA-DPA1, HLA-DQA1, and HLA-DRA mRNA transcript levels was also demonstrable in human LTxR with de novo development of DSA (Figure 8C-8H). These data clearly demonstrate that Ab mediated miRNA dysregulation results in increased RFX5 expression, which will lead to activation of MHC class II gene expression.
Figure 8.

Human MHC II molecules and mouse H-2 molecules are inversely expressed with miR-16 and miR-195 in OAD mice and DSA LTxR. (A-B) RT-PCR of mouse H-2 Aa and H-2 Dma mRNA levels in the lung tissues of OAD mice groups compared to those in isotype groups (n=9). P-values were calculated with the Student’s t-test. (C-H) RT-PCR of miR-16, miR-195 and HLA-DPA1, HLA-DQA1, and HLA-DRA mRNA transcript levels (n=15). The Pearson’s χ2 test was used to evaluate the correlation between miRNAs of miR-16, miR-195 and mRNAs of HLA-DPA1, HLA-DQA1, and HLA-DRA in LTxR. The correlation of relative miRNAs of miR-16, miR-195 and mRNAs of HLA-DPA1, HLA-DQA1, and HLA-DRA in LTxR was analyzed using linear correlation and regression.
Overexpression of miR-16 and miR-195 significantly reduces induction of RFX5, H-2Aa and H-2 Dma expression after anti-MHC class I Abs administration
To further demonstrate the in vivo relevance, we induced miR-16 and miR-195 over expression in the lungs to determine their effects in the lung of OAD mice using lentivirus based gene delivery. qRT-PCR was performed to assess miR-16 and miR-195 expression after virus infection and anti-H-2Kb administration. MiR-16 and miR-195 were compensatoraly expressed on day 7 or day 15 after anti-H-2Kb administration when infected with miR-16 and miR-195 lentivirus compared to those infected with lenti-GFP control (Figure 9A and 9B). Further qRT-PCR demonstrated that there were no significant up-regulation of RFX5, H-2Aa and H-2 Dma mRNA expression levels on days 7 and 15 in OAD mice infected with lenti-miR-16 and miR-195 virus (Figure 9C, 9D and 9E). These data demonstrated that enforced expression of miRNA-16 and miRNA-195 diminishes induction of RFX5 expression and consequently decreases MHC expression after anti-MHC class I administration.
Figure 9.

Overexpression of miR-16 and miR-195 diminishes up regulation of Rfx5, H-2Aa and H-2 Dma expression. (A and B) qRT-PCR was used to detect expression of miR-16 and miR-195 in the lung tissues of OAD mice (n=3~5) on days 7 and 15 after lentivirus (miR-16 and miR-195) infection and Ab administration. P-values were calculated with the Student’s t-test. (C) RT-PCR of Rfx5 in the lung tissues of OAD mice (n=3~5) on day 7 and 15 after lentivirus (miR-16 and miR-195) infection and Ab administration. Data was normalized to GAPDH expression. P-values were calculated with the Student’s t-test. (D and E) RT-PCR of mouse H-2 Aa and H-2 Dma mRNA levels in the lung tissues of OAD mice (n=3~5) on days 7 and 15 after lentivirus (miR-16 and miR-195) infection and Ab administration. P-values were calculated with the Student’s t-test.
Discussion
In this study, we determined the profile of miRNA expression in a murine model of OAD induced by Abs to MHC class I molecule and translated the findings in human LTxR with de novo development of Abs to mismatched donor HLA. We demonstrated that miRNAs were differentially expressed in OAD mice in comparison with mice receiving isotype control indicating that miRNAs may play important regulatory functions leading to chronic rejection following alloimmune responses. We further focused on two miRNAs, miR-16 and miR-195 which are significantly decreased since they are predicted to post-transcriptionally inhibit RFX5 which can up regulate MHC class II expression. We demonstrated that both of these miRNAs are down regulated in OAD mice as well as in LTxRs with de novo development of DSA. Further, in the murine model of OAD induced by anti-MHC I and LTxR with de novo development of DSA we demonstrated that it can lead to up regulation of murine MHC class II (H-2 Aa and H-2 Dma) and members of HLA class II molecules (HLA-DPA1, HLA-DQA1, and HLA-DRA) respectively which we propose will result in augmented immune responses against both allo and self-Ags leading to the development of BOS following human LTx.
There is increasing evidence that de novo development of Abs to mismatched donor MHC is a critical component in the immunopathogenesis of chronic rejection following organ transplantation (25, 26). Studies from our lab (27, 28) and others (29) have demonstrated that development of Abs to MHC precede the development of Abs to tissue restricted self-Ags and chronic rejection. The ligation of MHC class I molecules by its specific Abs on epithelial and endothelial cells have also been shown to induce the activation of signaling cascades leading to up regulation of MHC class II molecules, secretion of inflammatory cytokines, chemokines, fibrogenic growth factors, and adhesion molecules, all proposed to play an important role in the pathogenesis of chronic rejection (8, 30). With the goal to specifically address the role of Abs to mismatched donor HLA in chronic lung allograft rejection, we developed a murine model of OAD in which MHC class I Abs were intrabronchially administered into mice (16). The animals following administration of Abs developed cellular immune responses including Th17 cells to self-Ags with marked IL17 induction leading to immune responses to tissue restricted self-Ags (Collagen V and K-alpha 1 tubulin) resulting in classic OAD lesions as manifested by epithelial hyperplasia, cellular infiltration around the bronchiole, luminal occlusion and fibrosis around the smaller bronchioles (16). Analysis of the miRNA profile in the context of LTx or OAD mouse offers new insights into the pathogenesis of lung inflammation. The dynamics of miRNA expression followed two clear patterns: a) miRNAs (7, 15, 19, 301, 467 and 1954) that targeting proinflammatory genes were turned down, and b) miRNAs (375, 431 and 433) that promote cell survival and proliferation were markedly up-regulated at day 7 post ligation. Since the up regulated miRNAs were not lung specific and have been known to be expressed in islets of Langerhans, brain, heart and B cell lymphomas (31-33), we decided to analyze the effect of down-regulated miRNAs. Our observation of a reduced miR148b expression in the OAD mouse is suggestive of an immunomodulatory roles for MHC I ligation with Abs. Liu et al. (34) have previously demonstrated a regulatory role for miR-148 that impairs production of inflammatory cytokines, expression of MHC class II and proliferation of Ag specific T cells. Therefore down-modulation of miR-148 in OAD mouse is likely to have contributed to the proinflammatory spread of the immune response. By another example of the miRNA dependent modulation of the immune response is miR-301 mediated inflammation where miR-301 regulated the development of Th17 through IL-6/IL-23-STAT3 as observed in experimental autoimmune encephalomyelitis model (35).
There are many reports demonstrating that over expression and induction of MHC class II Ags correlate significantly with organ-specific autoimmunity and rejection of transplanted organs (36-41). MHC class II is not usually expressed on mouse bronchial epithelium and is not targeted by miRNAs. However, induction of MHC class II Ags de novo have been described during acute unmodified pulmonary allograft rejection (42). It has been proposed that it is due to the consequence of increased inflammation at the local site ongoing during rejection. In this report we demonstrate dysregulation of miRNAs as a contributing factor influencing expression of MHC molecules in the endothelium and epithelium which can not only augment the immune responses but also present self-Ags to the immune system resulting in the development of chronic rejection.
Recently two regulatory factors (viz. RFX5 and CIITA) have been shown to control expression of MHC class II and HLA-DM. RFX5 is a transcription factor which specifically binds to the X box of MHC class II promoters which is essential for the expression and function of MHC class II molecules (43, 44) while CIITA serves as the transcriptional co-activator of the MHC class II genes. It is well established that RFX5 is essential for activation of MHC II promoters (24). Mutations of RFX5 that abrogate MHC II expression have been identified in at least five unrelated MHC II deficiency patients (45-50). In addition, RFX5 or CIITA deficiency in knockout mice is characterized by a severe deficiency in MHC-II expression (24). RFX5 is known to form a hetero dimer by combining with two other unrelated proteins (i.e. RfxAP and RfxANK) that is responsible for binding to the cis-acting X box element of MHC-II promoters (51-53). However, very little is known about the post-transcriptional regulation of RFX5 expression. Herein, we show that miR-16 and miR-195 specifically bind to the 3’UTR of RFX5 mRNA in the mouse. Additionally, a significant increase in RFX5 mRNA level was evident in PBMC and lung tissues of OAD mice. We also demonstrated that miR-16 and miR-195 expression was decreased not only in the lungs of OAD mice but also in the lung tissues from LTxR with de novo development of Abs against donor HLA (Figure 4A, 4B, and Figure 5). Further, in situ hybridization demonstrated that miR-16 and miR-195 expression was significantly down-regulated in the lung alveolus of OAD mice on days 7 and 15 compared to those of mice receiving isotype controls (Figure 4C and 4D). Based on this, we propose that regulation of miR-16 and miR-195 targeting RFX5 mRNA may lead to up regulation of MHC class II molecules. Our observation suggests and provides an instance of miRNA down modulation leading to up regulation of the target genes in lung pathology. While this concept necessitates further studies, up regulation of MHC class II as documented here can augment Ag presentation not only of donor allo-Ags but also self-Ags which may play an important role in the immunopathogenesis of BOS development following human LTx.
One of the perceived limitations of our model is that we delivered Abs to MHC by the intrabronchial route which is not physiologic. Another limitation is that the analysis was largely based upon Targetscan that predicts many other potential targets for miRNAs. Therefore, we cannot exclude the possibility of other potential targets for miR-16 and miR-195 that may have impact on immune pathways and regulate innate and adoptive immunity. However, based upon similar findings in LTxR with development of DSA, we are confident that these miRNAs mediated regulation of MHC molecules are playing an important role in the pathogenesis of chronic rejection.
In summary, we, for the first time, demonstrated specific dysregulation of miRNAs during lung allograft rejection both in a murine model of OAD induced by Abs to MHC class I molecules as well as in human LTxR with de novo development of DSA which is generally accepted as an important risk factor for development of chronic rejection following human LTx. We also demonstrated that miRNA-16 and miRNA-195 which are selectively down regulated can indeed post-transcriptionally regulate RFX5 mRNA leading to increased expression of MHC molecules and thereby will lead to efficient allo as well as self-Ag presentation which can lead to chronic rejection following organ transplantation.
Supplementary Material
Acknowledgements
We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for technical support with microRNA array. We also thank Dr. Yongjun Yin and Dr. David Ornitz from Departments of Developmental Biology, Radiology, and Medicine, Washington University School of Medicine for their help in miRNA experiments of in situ hybridization. We thank Billie Glasscock for her assistance in preparing this manuscript.
This work is supported by NIH HL092514/HL056643 and the BJC foundation (TM)
Abbreviations
- BOS
Bronchiolitis Obliterans Syndrome
- BAL
bronchoalveolar lavage
- DSA
anti-donor HLA
- LNA
locked nucleic acid
- LTx
lung transplant
- LTxR
lung transplant recipient
- miRNAs
microRNAs
- OAD
obliterative airway disease
- OB
obliterative bronchiolitis
- RFX5
Regulatory Factor X 5
Footnotes
Disclosures The authors have no financial conflict of interest.
The microarray raw data presented in this article has been submitted to Gene Expression Omnibus (GEO), NCBI (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE58730.
References
- 1.Boehler A, Estenne M. Post-transplant bronchiolitis obliterans. Eur Respir J. 2003;22:1007–1018. doi: 10.1183/09031936.03.00039103. [DOI] [PubMed] [Google Scholar]
- 2.Hachem RR, Trulock EP. Bronchiolitis obliterans syndrome: pathogenesis and management. Semin Thorac Cardiovasc Surg. 2004;16:350–355. doi: 10.1053/j.semtcvs.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Sharples LD, McNeil K, Stewart S, Wallwork J. Risk factors for bronchiolitis obliterans: a systematic review of recent publications. J Heart Lung Transplant. 2002;21:271–281. doi: 10.1016/s1053-2498(01)00360-6. [DOI] [PubMed] [Google Scholar]
- 4.Estenne M, Hertz MI. Bronchiolitis obliterans after human lung transplantation. Am J Respir Crit Care Med. 2002;166:440–444. doi: 10.1164/rccm.200201-003pp. [DOI] [PubMed] [Google Scholar]
- 5.Smith MA, Sundaresan S, Mohanakumar T, Trulock EP, Lynch JP, Phelan DL, Cooper JD, Patterson GA. Effect of development of antibodies to HLA and cytomegalovirus mismatch on lung transplantation survival and development of bronchiolitis obliterans syndrome. J Thorac Cardiovasc Surg. 1998;116:812–820. doi: 10.1016/S0022-5223(98)00444-9. [DOI] [PubMed] [Google Scholar]
- 6.Tiriveedhi V, Weber J, Seetharam A, Mohanakumar T. Cross-talk of alloimmune response and autoimmunity: role in pathogenesis of chronic rejection. Discov Med. 2010;9:229–235. [PubMed] [Google Scholar]
- 7.Hachem RR. Lung allograft rejection: diagnosis and management. Curr Opin Organ Transplant. 2009;14:477–482. doi: 10.1097/MOT.0b013e32832fb981. [DOI] [PubMed] [Google Scholar]
- 8.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Hum Immunol. 2003;64:521–529. doi: 10.1016/s0198-8859(03)00038-7. [DOI] [PubMed] [Google Scholar]
- 9.Jaramillo A, Smith MA, Phelan D, Sundaresan S, Trulock E, Lynch J, Cooper J, Patterson GA, Mohanakumar T. Temporal relationship between the development of anti-HLA antibodies and the development of bronchiolitis obliterans syndrome after lung transplantation. Transplant Proc. 1999;31:185–186. doi: 10.1016/s0041-1345(98)01495-x. [DOI] [PubMed] [Google Scholar]
- 10.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 11.Harris A, Krams SM, Martinez OM. MicroRNAs as immune regulators: implications for transplantation. Am J Transplant. 2010;10:713–719. doi: 10.1111/j.1600-6143.2010.03032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sui W, Dai Y, Huang Y, Lan H, Yan Q, Huang H. Microarray analysis of MicroRNA expression in acute rejection after renal transplantation. Transpl Immunol. 2008;19:81–85. doi: 10.1016/j.trim.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Anglicheau D, Sharma VK, Ding R, Hummel A, Snopkowski C, Dadhania D, Seshan SV, Suthanthiran M. MicroRNA expression profiles predictive of human renal allograft status. Proc Natl Acad Sci U S A. 2009;106:5330–5335. doi: 10.1073/pnas.0813121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Stittrich AB, Haftmann C, Sgouroudis E, Kuhl AA, Hegazy AN, Panse I, Riedel R, Flossdorf M, Dong J, Fuhrmann F, Heinz GA, Fang Z, Li N, Bissels U, Hatam F, Jahn A, Hammoud B, Matz M, Schulze FM, Baumgrass R, Bosio A, Mollenkopf HJ, Grun J, Thiel A, Chen W, Hofer T, Loddenkemper C, Lohning M, Chang HD, Rajewsky N, Radbruch A, Mashreghi MF. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol. 2010;11:1057–1062. doi: 10.1038/ni.1945. [DOI] [PubMed] [Google Scholar]
- 16.Fukami N, Ramachandran S, Saini D, Walter M, Chapman W, Patterson GA, Mohanakumar T. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182:309–318. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colvin JS, Feldman B, Nadeau JH, Goldfarb M, Ornitz DM. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev Dyn. 1999;216:72–88. doi: 10.1002/(SICI)1097-0177(199909)216:1<72::AID-DVDY9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Chaudhuri AA, So AY, Sinha N, Gibson WS, Taganov KD, O’Connell RM, Baltimore D. MicroRNA-125b potentiates macrophage activation. J Immunol. 2011;187:5062–5068. doi: 10.4049/jimmunol.1102001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 20.Wendlandt EB, Graff JW, Gioannini TL, McCaffrey AP, Wilson ME. The role of microRNAs miR-200b and miR-200c in TLR4 signaling and NF-kappaB activation. Innate Immun. 2012;18:846–855. doi: 10.1177/1753425912443903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams MC, Dobbs LG. Expression of cell-specific markers for alveolar epithelium in fetal rat lung. Am J Respir Cell Mol Biol. 1990;2:533–542. doi: 10.1165/ajrcmb/2.6.533. [DOI] [PubMed] [Google Scholar]
- 22.Renella R, Picard C, Neven B, Ouachee-Chardin M, Casanova JL, Le Deist F, Cavazzana-Calvo M, Blanche S, Fischer A. Human leucocyte antigen-identical haematopoietic stem cell transplantation in major histocompatiblity complex class II immunodeficiency: reduced survival correlates with an increased incidence of acute graft-versus-host disease and pre-existing viral infections. Br J Haematol. 2006;134:510–516. doi: 10.1111/j.1365-2141.2006.06213.x. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty M, Bhattacharya D, Mukhopadhyay C, Chakrabarti A. Structure and conformational studies on dityrosine formation in the DNA binding domain of RFX5. Biophys Chem. 2010;149:92–101. doi: 10.1016/j.bpc.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Clausen BE, Waldburger JM, Schwenk F, Barras E, Mach B, Rajewsky K, Forster I, Reith W. Residual MHC class II expression on mature dendritic cells and activated B cells in RFX5-deficient mice. Immunity. 1998;8:143–155. doi: 10.1016/s1074-7613(00)80467-7. [DOI] [PubMed] [Google Scholar]
- 25.Sundaresan S, Mohanakumar T, Smith MA, Trulock EP, Lynch J, Phelan D, Cooper JD, Patterson GA. HLA-A locus mismatches and development of antibodies to HLA after lung transplantation correlate with the development of bronchiolitis obliterans syndrome. Transplantation. 1998;65:648–653. doi: 10.1097/00007890-199803150-00008. [DOI] [PubMed] [Google Scholar]
- 26.Jaramillo A, Fernandez FG, Kuo EY, Trulock EP, Patterson GA, Mohanakumar T. Immune mechanisms in the pathogenesis of bronchiolitis obliterans syndrome after lung transplantation. Pediatr Transplant. 2005;9:84–93. doi: 10.1111/j.1399-3046.2004.00270.x. [DOI] [PubMed] [Google Scholar]
- 27.Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, Steward N, Aloush A, Hachem R, Trulock E, Meyers B, Patterson GA, Mohanakumar T. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant. 2011;30:624–631. doi: 10.1016/j.healun.2011.01.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takenaka M, Tiriveedhi V, Subramanian V, Hoshinaga K, Patterson AG, Mohanakumar T. Antibodies to MHC class II molecules induce autoimmunity: critical role for macrophages in the immunopathogenesis of obliterative airway disease. PLoS One. 2012;7:e42370. doi: 10.1371/journal.pone.0042370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haque MA, Mizobuchi T, Yasufuku K, Fujisawa T, Brutkiewicz RR, Zheng Y, Woods K, Smith GN, Cummings OW, Heidler KM, Blum JS, Wilkes DS. Evidence for immune responses to a self-antigen in lung transplantation: role of type V collagen-specific T cells in the pathogenesis of lung allograft rejection. J Immunol. 2002;169:1542–1549. doi: 10.4049/jimmunol.169.3.1542. [DOI] [PubMed] [Google Scholar]
- 30.Harris PE, Bian H, Reed EF. Induction of high affinity fibroblast growth factor receptor expression and proliferation in human endothelial cells by anti-HLA antibodies: a possible mechanism for transplant atherosclerosis. J Immunol. 1997;159:5697–5704. [PubMed] [Google Scholar]
- 31.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 32.Wu D, Murashov AK. MicroRNA-431 regulates axon regeneration in mature sensory neurons by targeting the Wnt antagonist Kremen1. Front Mol Neurosci. 2013;6:35. doi: 10.3389/fnmol.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gotanda K, Hirota T, Matsumoto N, Ieiri I. MicroRNA-433 negatively regulates the expression of thymidylate synthase (TYMS) responsible for 5-fluorouracil sensitivity in HeLa cells. BMC Cancer. 2013;13:369. doi: 10.1186/1471-2407-13-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Zhan Z, Xu L, Ma F, Li D, Guo Z, Li N, Cao X. MicroRNA-148/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKIIalpha. J Immunol. 2010;185:7244–7251. doi: 10.4049/jimmunol.1001573. [DOI] [PubMed] [Google Scholar]
- 35.Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci U S A. 2012;109:E1248–1257. doi: 10.1073/pnas.1114325109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bottazzo GF, Pujol-Borrell R, Hanafusa T, Feldmann M. Role of aberrant HLA-DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet. 1983;2:1115–1119. doi: 10.1016/s0140-6736(83)90629-3. [DOI] [PubMed] [Google Scholar]
- 37.He J, Li C, Yuan XN, Zhang JL, Li Y, Wei XD, Hou JQ. Anti-human leukocyte antigens and anti-major histocompatibility complex class I-related chain A antibody expression in kidney transplantation during a four-year follow-up. Chin Med J (Engl) 2013;126:2815–2820. [PubMed] [Google Scholar]
- 38.Ayala Garcia MA, Gonzalez Yebra B, Lopez Flores AL, Guani Guerra E. The major histocompatibility complex in transplantation. J Transplant. 2012;2012:842141. doi: 10.1155/2012/842141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox ST, Stephens HA, Fernando R, Karasu A, Harber M, Howie AJ, Powis S, Zou Y, Stastny P, Madrigal JA, Little AM. Major histocompatibility complex class I-related chain A allele mismatching, antibodies, and rejection in renal transplantation. Hum Immunol. 2011;72:827–834. doi: 10.1016/j.humimm.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 40.Morales-Buenrostro LE, Alberu J. Anti-major histocompatibility complex class I-related chain A antibodies in organ transplantation. Transplant Rev (Orlando) 2008;22:27–38. doi: 10.1016/j.trre.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 41.Bach FH. The major histocompatibility complex in transplantation immunology. Transplant Proc. 1973;5:23–29. [PubMed] [Google Scholar]
- 42.Romaniuk A, Prop J, Petersen AH, Wildevuur CR, Nieuwenhuis P. Expression of class II major histocompatibility complex antigens by bronchial epithelium in rat lung allografts. Transplantation. 1987;44:209–214. doi: 10.1097/00007890-198708000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Villard J, Peretti M, Masternak K, Barras E, Caretti G, Mantovani R, Reith W. A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol Cell Biol. 2000;20:3364–3376. doi: 10.1128/mcb.20.10.3364-3376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stavride P, Arampatzi P, Papamatheakis J. Differential regulation of MHCII genes by PRMT6, via an AT-hook motif of RFX5. Mol Immunol. 2013;56:390–398. doi: 10.1016/j.molimm.2013.05.235. [DOI] [PubMed] [Google Scholar]
- 45.Brickey WJ, Wright KL, Zhu XS, Ting JP. Analysis of the defect in IFN-gamma induction of MHC class II genes in G1B cells: identification of a novel and functionally critical leucine-rich motif (62-LYLYLQL-68) in the regulatory factor X 5 transcription factor. J Immunol. 1999;163:6622–6630. [PubMed] [Google Scholar]
- 46.DeSandro A, Nagarajan UM, Boss JM. The bare lymphocyte syndrome: molecular clues to the transcriptional regulation of major histocompatibility complex class II genes. Am J Hum Genet. 1999;65:279–286. doi: 10.1086/302519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peijnenburg A, Van Eggermond MJ, Gobin SJ, Van den Berg R, Godthelp BC, Vossen JM, Van den Elsen PJ. Discoordinate expression of invariant chain and MHC class II genes in class II transactivator-transfected fibroblasts defective for RFX5. J Immunol. 1999;163:794–801. [PubMed] [Google Scholar]
- 48.Peijnenburg A, Van Eggermond MC, Van den Berg R, Sanal O, Vossen JM, Van den Elsen PJ. Molecular analysis of an MHC class II deficiency patient reveals a novel mutation in the RFX5 gene. Immunogenetics. 1999;49:338–345. doi: 10.1007/s002510050501. [DOI] [PubMed] [Google Scholar]
- 49.Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, Reith W. A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome) Genes Dev. 1995;9:1021–1032. doi: 10.1101/gad.9.9.1021. [DOI] [PubMed] [Google Scholar]
- 50.Villard J, Reith W, Barras E, Gos A, Morris MA, Antonarakis SE, Van den Elsen PJ, Mach B. Analysis of mutations and chromosomal localisation of the gene encoding RFX5, a novel transcription factor affected in major histocompatibility complex class II deficiency. Hum Mutat. 1997;10:430–435. doi: 10.1002/(SICI)1098-1004(1997)10:6<430::AID-HUMU3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 51.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. Embo J. 1997;16:1045–1055. doi: 10.1093/emboj/16.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, Sanchez JC, Hochstrasser DF, Mach B, Reith W. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet. 1998;20:273–277. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- 53.Reith W, Satola S, Sanchez CH, Amaldi I, Lisowska-Grospierre B, Griscelli C, Hadam MR, Mach B. Congenital immunodeficiency with a regulatory defect in MHC class II gene expression lacks a specific HLA-DR promoter binding protein, RF-X. Cell. 1988;53:897–906. doi: 10.1016/s0092-8674(88)90389-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.