

ABSTRACT
Coronaviruses, the largest RNA viruses, have a complex program of RNA synthesis that entails genome replication and transcription of subgenomic mRNAs. RNA synthesis by the prototype coronavirus mouse hepatitis virus (MHV) is carried out by a replicase-transcriptase composed of 16 nonstructural protein (nsp) subunits. Among these, nsp3 is the largest and the first to be inserted into the endoplasmic reticulum. nsp3 comprises multiple structural domains, including two papain-like proteases (PLPs) and a highly conserved ADP-ribose-1″-phosphatase (ADRP) macrodomain. We have previously shown that the ubiquitin-like domain at the amino terminus of nsp3 is essential and participates in a critical interaction with the viral nucleocapsid protein early in infection. In the current study, we exploited atypical expression schemes to uncouple PLP1 from the processing of nsp1 and nsp2 in order to investigate the requirements of nsp3 domains for viral RNA synthesis. In the first strategy, a mutant was created in which replicase polyprotein translation initiated with nsp3, thereby establishing that complete elimination of nsp1 and nsp2 does not abolish MHV viability. In the second strategy, a picornavirus autoprocessing element was used to separate a truncated nsp1 from nsp3. This provided a platform for further dissection of amino-terminal domains of nsp3. From this, we found that catalytic mutation of PLP1 or complete deletion of PLP1 and the adjacent ADRP domain was tolerated by the virus. These results showed that neither the PLP1 domain nor the ADRP domain of nsp3 provides integral activities essential for coronavirus genomic or subgenomic RNA synthesis.
IMPORTANCE The largest component of the coronavirus replicase-transcriptase complex, nsp3, contains multiple modules, many of which do not have clearly defined functions in genome replication or transcription. These domains may play direct roles in RNA synthesis, or they may have evolved for other purposes, such as to combat host innate immunity. We initiated a dissection of MHV nsp3 aimed at identifying those activities or structures in this huge molecule that are essential to replicase activity. We found that both PLP1 and ADRP could be entirely deleted, provided that the requirement for proteolytic processing by PLP1 was offset by an alternative mechanism. This demonstrated that neither PLP1 nor ADRP plays an essential role in coronavirus RNA synthesis.
INTRODUCTION
Coronaviruses are enveloped, positive-strand RNA viruses that are broadly distributed among mammalian and avian species (1, 2). Members of this family are generally restricted to a narrow range of hosts. However, the emergence within 12 years of two major pathogens, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), has demonstrated that these viruses have the potential to cross species boundaries with grave consequences for human health (3, 4). Coronaviruses are sorted taxonomically into four genera: the alpha-, beta-, gamma-, and deltacoronaviruses (http://ictvonline.org/). SARS-CoV and MERS-CoV both fall into the Betacoronavirus genus, as does the extensively studied mouse hepatitis virus (MHV), which provides a valuable model for understanding the molecular biology, genetics, and biochemistry of these infectious agents.
Coronaviruses have the largest genomes of all RNA viruses and a correspondingly complex mechanism of RNA synthesis. During the course of infection, the genome is replicated by means of a negative-strand antigenome, and at least five subgenomic mRNAs are transcribed, also via negative-strand intermediates (5). The resulting 3′ nested set of subgenomic mRNAs, each comprising a 5′ genomic leader fused to a 3′ body sequence, is a defining characteristic of coronaviruses. Viral RNA synthesis is conducted by a very large replicase-transcriptase complex containing 15 or 16 subunits that are encoded by the 5′ two-thirds of the genome (Fig. 1). In one of the earliest events of infection, the genome is translated into two overlapping polyproteins, which are produced through a ribosomal frameshifting mechanism. These polyproteins are processed into their constituent nonstructural protein (nsp) subunits by one or two papain-like proteases (PLPs), contained within nsp3, and by a main protease contained within nsp5.
FIG 1.
Modular composition of MHV nsp3. The upper schematic shows the MHV genome with numbered sections of the replicase-transcriptase polyprotein gene (rep 1a and rep 1b) representing the processed protein products nsp1 through nsp16. The expanded segment shows the domains of nsp3 as defined previously for MHV and other coronaviruses (12, 26, 66): Ubl1, ubiquitin-like domain 1 (14, 16); Ac, acidic region; PLP1, papain-like protease 1 (18, 19); ADRP, ADP-ribose-1″-phosphatase; Ubl2, ubiquitin-like domain 2 (70); PLP2, papain-like protease 2; NAB, nucleic acid-binding domain (13); G2M, coronavirus group 2 marker domain; TM1 and 2, transmembrane segments (10, 11); Y, coronavirus highly conserved domain (26). The region between ADRP and Ubl2-PLP2 corresponds to, but is highly diverged from, the SARS-unique domain (SUD) of SARS-CoV nsp3 (66). Residue numbers are those of mature MHV nsp3; the first residue of nsp3 equals residue 833 of the unprocessed replicase polyprotein. In the genome schematic, the cleavage sites for nsp3 PLP1 and PLP2 are denoted by open and closed circles, respectively; the cleavage sites for the nsp5 main protease are indicated by arrowheads.
The machinery of coronavirus RNA synthesis is built upon intracellular membranes. Three of the replicase subunits—nsp3, nsp4, and nsp6—become cotranslationally anchored in the endoplasmic reticulum (ER) by multiple transmembrane domains, and most of the remaining subunits are presumed to associate with these three or with each other. Positioning of the replicase on membranes, among other things, serves to concentrate all the components that must cooperate in the integrated reactions required to produce capped and polyadenylated mRNAs and progeny genomes. Such colocalization would be most critical in the earliest stages of infection. Assembly of the replicase-transcriptase complex also induces extensive remodeling of intracellular membranes, resulting in the formation of double-membrane vesicles (DMVs), convoluted membranes, and other structures (6, 7). This transformation appears to involve, or else to coopt, the cellular process of macroautophagy, and it is triggered by the membrane-bound cohort of the replicase subunits. Expression of just nsp3, nsp4, and nsp6 has been shown to be necessary and sufficient to generate DMVs and the other membrane structures seen during infection (8). However, it is not currently clear whether membrane rearrangements are brought about by the virus specifically for the purpose of RNA synthesis or if, instead, they are by-products of the developing struggle between the incoming virus and host cellular defenses (9).
Among the components of the replicase, the first to be inserted into the ER is nsp3, via membrane-spanning segments near its carboxy terminus (10, 11). nsp3 is also, by far, the largest of the replicase subunits, having multiple structural domains (12, 13), all of which are oriented on the cytoplasmic side of the ER. Some of these modules are present, and are often well conserved, across all genera of coronaviruses. At the amino terminus of nsp3 is a ubiquitin-like domain (Ubl1) (14) that is connected to a highly acidic (Ac) region of variable size (Fig. 1). In previous work, we discovered that MHV infection requires a critical interaction between Ubl1 and the viral nucleocapsid (N) protein (15). Genetic, biochemical, and biophysical evidence has mapped this interaction to the serine- and arginine-rich region that falls between the two RNA-binding domains of N protein (15, 16). Moreover, we have shown that there exists a complete correspondence between the Ubl1-N interaction and the requirement, common to all coronaviruses, for N protein to stimulate the infectivity of transfected genomic RNA (gRNA) (17). This correspondence led us to propose that the Ubl1-N interaction serves to tether the genome of the incoming virus to the nascent replicase, thereby coupling translation and assembly of the replicase to the formation of an initiation complex for RNA synthesis. This interaction is very likely to be essential, since deletion of MHV Ubl1 is lethal to MHV (17). In contrast, the Ac region can be deleted with little or no consequences for the virus.
Three other nsp3 domains are common to all coronaviruses. The PLPs harbored by nsp3 are responsible for the first three events of polyprotein processing (Fig. 1). In MHV nsp3, there are two of these modules: PLP1 performs the nsp1-nsp2 and nsp2-nsp3 cleavages, while PLP2 carries out the nsp3-nsp4 cleavage (18, 19). Many, but not all, PLPs also possess a deubiquitinase activity, which acts to counter host innate immunity (20). Additionally, it has been suggested that PLPs may have some role other than proteolysis in replicase function (21). Between PLP1 and PLP2 in MHV (or else upstream of the sole PLP of some coronaviruses) there is a macrodomain with ADP-ribose-1″-phosphatase (ADRP) and poly(ADP-ribose)-binding activity. The ADRP activity of this module has been shown to be dispensable in tissue culture (22, 23), but a role in viral RNA synthesis was proposed for the macrodomain, based on its nucleic acid-binding properties (24, 25). Finally, at the carboxy terminus of nsp3, there is a well-conserved region that has been dubbed the Y domain (26). Although nothing is known about its purpose, a particularly intriguing set of eight universally conserved cysteines and histidines occupies the amino-terminal 65 residues of this domain.
In order to better understand the functions of nsp3 in the replicase-transcriptase complex and how it contributes to the unique mechanism of coronavirus RNA synthesis, we began a dissection of MHV nsp3. Our effort was aimed at identifying those activities or structures in the huge nsp3 molecule that, like Ubl1, are essential to the replicase. We also wanted to test the possibility of uncovering unknown roles of the better-characterized modules, such as the PLPs and the ADRP domain. In the present study, we found that both PLP1 and ADRP could be completely deleted, provided that the requirement for the proteolytic processing activity of PLP1 was offset by an alternative mechanism. This indicated that neither PLP1 nor ADRP plays an essential role in RNA synthesis, per se. Moreover, in the course of bypassing the need for PLP1 activity, we showed that viable mutants of MHV could be generated in which nsp1 and nsp2 were simultaneously eliminated.
MATERIALS AND METHODS
Virus and cells.
Wild-type (WT), mutant, and revertant MHV strain A59 stocks were propagated in mouse 17 clone 1 (17Cl1) cells; plaque titrations and plaque purifications were carried out in mouse L2 cells. Monolayer cultures of both cell lines were maintained in Dulbecco's minimal essential medium (MEM) containing 10% fetal bovine serum. Spinner cultures of L2 cells, used for transfection of genomic RNA, were kept between densities of 1 × 105 and 2 × 106 cells/ml in Joklik's MEM containing 10% fetal bovine serum.
MHV mutant construction and analysis.
All MHV-A59 mutants in this study were created with an infectious cDNA system generously provided by Ralph Baric; the assembly of full-length cDNA and in vitro transcription of genomic RNA were carried out essentially as described previously (27). Viruses were generated by cotransfection of synthetic gRNA and MHV N mRNA through electroporation of suspension-grown L2 cells (17). Initial viral isolations were performed at 37°C for 1 to 5 days. The wild-type reference virus used throughout this work was previously obtained from isogenic wild-type full-length cDNA (17). To verify constructed viral mutants, RNA isolated from infected cell monolayers with Ultraspec reagent (Biotecx) was reverse transcribed with a random hexanucleotide primer and avian myeloblastosis virus reverse transcriptase (RT; Life Sciences). PCR amplification of cDNA was carried out with the Expand high-fidelity PCR system (Roche). For Sanger sequencing, RT-PCR products were purified with QIAquick spin columns (Qiagen). Whole-genome sequences (excluding 39 and 113 nucleotides [nt] at the 5′ and 3′ ends, respectively) were generated from 18 partly overlapping RT-PCR products spanning the genome. Equimolar amounts of amplicons were pooled, purified with an AMPure XP kit (Beckman Coulter), and quantitated on a Qubit fluorometer (Invitrogen). Tagged libraries of DNA fragments were prepared with a Nextera XT kit (Illumina), and 2- by 250-bp paired-end sequencing was performed on a MiSeq Sequencer (Illumina) using the MiSeq 500-cycle v2 kit. Viral genome sequences were fully assembled using SPAdes version 3.0.0 with the read correction mode active and K-mer sizes of 21, 33, 55, 77, 99, and 127.
To isolate revertants of the N3S mutant, 12 independent plaques obtained at 37°C were each used to start cultures that were grown at 37°C and thereafter serially passaged at 39°C in L2 cells at a low multiplicity of infection. When an increase in growth rate was noted, following 10 to 13 passages, titers of viral supernatants were determined on L2 cells at 39°C, and a single larger plaque was chosen from each independent sample and purified for further analysis.
Genetic constructions were all made via manipulation of pBar-A3 (17), a modified version of MHV cDNA clone A (27) that is here referred to as pA-WT. For pA-N3S, clone A of the N3S mutant, the BsiWI-BstBI fragment of pA-WT (running from the leader through nsp3 Ubl1) was replaced with a fragment that was synthesized by PCR from overlapping oligonucleotides. The replacement fragment removed half of nsp1 and all of nsp2 and created 30 point mutations in 5′ gRNA secondary structures, as described in detail in Results. For pA-SSN3, clone A of the SSN3 mutant, the BamHI-BstBI fragment of pA-WT (running from nsp1 codon 86 through nsp3 Ubl1) was replaced with a synthetic fragment encoding residues 86 to 127 of nsp1, His7, and 58 residues of foot-and-mouth disease virus (FMDV) VP1-2A (from GenBank accession number X00871). Plasmid pA-SSN3x was constructed identically to pA-SSN3, except that the NPGP consensus autoprocessing sequence was changed to NLAP (28). An epitope-tagged version of pA-SSN3, designated pA-SSN3HA, was built in two steps. First, a synthetic fragment encoding the nsp3 Ac region, followed by FLAG and hemagglutinin (HA) tags, was inserted between the SalI and SacI sites bounding the deleted Ac region of the previously described pA3Ac2, which had been used to generate the ΔAc2 mutant (17). Then the BstBI-SpeI fragment of this intermediate (running from nsp3 Ubl1 to PLP1) was used to replace the corresponding fragment of pA-SSN3. Four derivative plasmids were obtained from pA-SSN3HA. One, pA-SSN3HAmutP, was generated by a two-step PCR that replaced the SacI-SpeI fragment (running from the start to the middle of nsp3 PLP1) to create the PLP1 catalytic mutation C289A. For the initial PLP1 deletion construct, pA-SSN3HAΔP1, the SacI-BstXI fragment encompassing PLP1 was removed from pA-SSN3HA, and following the creation of blunt ends, the vector was reclosed. In a second deletion mutant, pA-SSN3HAΔP2, the SacI-BstXI fragment of pA-SSN3HA was again removed, and this time it was replaced with a synthetic fragment that moved the deletion boundaries inward, as specified in Results. For the third deletion construct, pA-SSN3HAΔP3, the SacI-Acc65I fragment encompassing both PLP1 and ADRP was removed from pA-SSN3HA, and following the creation of blunt ends, the vector was reclosed. Oligonucleotides for PCR and DNA sequencing were obtained from Integrated DNA Technologies. The overall compositions of constructed plasmids were confirmed by restriction analysis, and all ligation junctions and regions resulting from PCR amplification were verified by DNA sequencing.
Viral growth kinetics.
To measure growth kinetics, confluent monolayers of 17Cl1 cells (75 cm2) were inoculated at a multiplicity of 1.0 PFU per cell for 2 h at 37°C, with rocking every 15 min. Following the removal of inocula, monolayers were washed three times with phosphate-buffered saline (PBS), and incubation was continued in fresh medium at 37°C. Sample aliquots of medium were withdrawn at various times from 2 to 48 h postinfection, and infectious titers were subsequently determined in L2 cells.
Northern blotting.
RNA was extracted from infected cell monolayers with TRI-Reagent (Zymo) according to the manufacturer's instructions. Northern blotting of intracellular RNA was performed as previously described in detail (29) by using a PCR-amplified probe corresponding to the 3′-most 539 nucleotides of the N gene and the entire 3′ untranslated region (UTR) of the MHV genome. The probe was labeled with an AlkPhos Direct kit, and blots were visualized using CDP-Star detection reagent (GE Healthcare).
In vitro translation.
Capped mRNAs were produced with T7 RNA polymerase (mMessage mMachine; Ambion) by runoff transcription of BstZ17I-truncated pA-WT or SpeI-truncated pA-N3S, pA-N3SΔ1, pA-N3SΔ2, pA-SSN3, or pA-SSN3x. Synthetic transcripts (0.46 μg) were translated in 17 μl of rabbit reticulocyte lysate (Ambion) labeled with [35S]methionine (MP Biomedicals); protein products were analyzed in 10% polyacrylamide gels by SDS-PAGE, followed by fluorography. Vector pA-N3SΔ1 was derived from pA-N3S by removal of the BsiWI-NcoI fragment and religation of the plasmid following the creation of blunt ends. The same strategy was used to make pA-N3SΔ2 by removal of the ApaI-NcoI fragment.
Western blotting.
Sets of lysates were prepared from 75-cm2 confluent monolayers of L2 cells that were mock infected or infected with wild-type and mutant MHV at either a high or a low multiplicity of infection at 37°C. At 8 to 10 h postinfection (for infections begun at a multiplicity of 1 PFU per cell) or 17 to 27 h postinfection (for infections begun at a multiplicity of 0.01 PFU per cell), monolayers were washed twice with PBS and then lysed by the addition of 500 μl of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1.0% Nonidet P-40, 0.7 μg/ml pepstatin, 1.0 μg/ml leupeptin, 1.0 μg/ml aprotinin, and 0.5 mg/ml Pefabloc SC (Roche). Lysed cells were held on ice for 15 to 30 min and then clarified by centrifugation. For immunoprecipitations, lysates were cleared with preimmune antiserum and were then incubated for 1 h at 4°C with anti-nsp3 antibody D3 (30) or VU164 (31), generously provided by Susan Baker (Loyola University Chicago, Maywood, IL) or by Mark Denison (Vanderbilt University, Nashville, TN), respectively. Samples were incubated for an additional 1 h at 4°C with a 75% slurry of nProtein A Sepharose (GE Healthcare) in lysis buffer. Sepharose beads were collected by centrifugation, washed three times with lysis buffer, and used directly for SDS-PAGE sample preparation. Lysate samples or immunoprecipitates were separated by SDS-PAGE through 7.5% or 4-to-15% gradient polyacrylamide gels, with prestained protein markers (NEB) in flanking lanes, and were transferred to polyvinylidene difluoride membranes. Blots were probed with one of the following: anti-HA monoclonal antibody (MAb 12CA5; Roche), anti-nsp1 antibody VU221 (32), or anti-nsp8 antibody VU124 (33); the last two antibodies were also gifts from Mark Denison. Bound primary antibodies were visualized using a chemiluminescent detection system (ECL or West Dura; Pierce).
RESULTS
Construction of a mutant containing nsp3 as the first subunit of the replicase-transcriptase complex.
To begin to dissect the amino-terminal domain structure of nsp3, it was first necessary to disentangle nsp3 from its obligation to process upstream nsp subunits. The PLP1 module of MHV nsp3 carries out the nsp1-nsp2 and nsp2-nsp3 cleavage events of replicase-transcriptase polyprotein maturation (Fig. 1). Our first strategy to obviate these cleavages was to create a mutant, designated nsp3-start (N3S), in which polyprotein translation initiated with nsp3, while most of the preceding coding region was deleted. The major constraint on the construction of the N3S mutant was the need to preserve genomic elements that participate in viral RNA synthesis. The 5′ ends of coronavirus genomes, including the 5′ UTR and part of the nsp1 coding region, contain multiple cis-acting RNA structures that are critical for replication and transcription (34). In MHV and other betacoronaviruses, the genus in which these structures have been most extensively studied, the genomic 5′ end contains a series of at least eight stem-loops (SL) (35–39), as well as a four-way junction formed by a long-range interaction between the regions upstream of SL 5 (SL5) and downstream of SL7 (40) (Fig. 2A). Mutations or deletions in many of these elements have been found to debilitate or kill the virus or to abrogate defective interfering (DI) RNA replication.
FIG 2.

Strategy for construction of the nsp3-start (N3S) mutant. (A) Schematics of the genomic 5′ ends of wild-type MHV and the N3S mutant. Numbered RNA structures, stem-loops SL1 through SL7 (35, 36, 39) and SL8 (37), have been characterized previously. LR indicates a long-range interaction between the regions upstream of SL5 and downstream of SL7 (40). We have designated three structures predicted for the region downstream of SL8 as SLA, SLB, and SLC. Asterisks in the N3S genome show the positions of AUG start codons that were knocked out; open circles in the wild-type genome denote cleavage sites for PLP1. Note that the schematics do not accurately depict the relative lengths or extent of overlap of the 5′ RNA secondary structures and the nsp1 ORF. (B) Details of the changes engineered in the N3S mutant to disrupt 5 in-frame start codons and 6 out-of-frame start codons upstream of the nsp3 ORF. Only structures that contain start codons are shown. Residue numbering begins from the 5′ end of the wild-type genome; the N3S mutant has a deletion removing wild-type nucleotides 595 through 2706. Nucleotides of in-frame start codons are boxed; nucleotides of out-of-frame start codons are circled. In SL4, the start and stop codons of the uORF, denoted by stars, were unaltered in the N3S mutant. In SL5, the wild-type nsp1 start codon is labeled. N3S mutations, indicated by arrows, were made to knock out start codons, to preserve RNA secondary structure, or to optimize the context of the start codon that was juxtaposed to the start of the nsp3 ORF.
We consequently designed the 5′ UTR of the N3S mutant to contain a modified version of the 5′-most 594 nucleotides of the MHV genome, thereby retaining some margin beyond the 467-nt minimal segment previously shown to be sufficient to support DI RNA replication (41, 42). The AUG codon that occurs at nucleotides 592 to 594 was fused to the beginning of the nsp3 coding region (nt 2707 of the wild-type sequence) to serve as the start codon for the replicase-transcriptase polyprotein (Fig. 2B); at the −4 position relative to the AUG codon, nt 588 was mutated to optimize the context for translation initiation (43). To ensure utilization of the newly created start codon, the original start codon for nsp1 and the other four in-frame upstream AUG codons were each knocked out by mutations at 2 or 3 positions. Additionally, six out-of-frame upstream AUG codons were each mutated at 1 or 2 positions. Further point mutations were made to restore any base pairs in RNA secondary structures that would be disrupted by the start codon knockouts. The RNA secondary structure shown in Fig. 2 is the lowest free-energy mfold structure (44) for wild-type MHV nucleotides 79 to 594 (with no imposed constraints to generate the long-range interaction). The mfold-predicted structure for the N3S mutant was verified to be identical to that of the wild type. The only upstream start codon that was not eliminated was that for the small upstream open reading frame (uORF) in SL4 (Fig. 2B). Similarly situated uORFs are found in the majority of coronavirus genomes, and in MHV there exists selective pressure for the uORF to be maintained (45).
Following multiple trials, a single isolate of the N3S mutant was obtained. Initially, the sequence of the 5′ genomic end of this recombinant was determined, confirming that it contained exactly the engineered 30 point mutations and 2,112-nt deletion (Fig. 2B) and, in particular, that the 5′-most AUG codon occurred at nt 592 to 594 (Fig. 3A). To verify the functionality of this relocated start codon, we translated synthetic mRNAs that were identical to the 5′ ends of the wild-type or N3S mutant genomes. These mRNAs were generated by runoff in vitro transcription of truncated pA-WT or pA-N3S, the first of the seven plasmids used for assembly of full-length wild-type or N3S mutant cDNAs, respectively (Fig. 3B). Additionally, we produced two N3S-related mRNAs from plasmids pA-N3SΔ1 and pA-N3SΔ2, which contained deletions in the 5′ UTR of pA-N3S. The mRNAs were translated in a reticulocyte lysate system, and [35S]methionine-labeled protein products were analyzed by SDS-PAGE. For pA-WT mRNA, a product corresponding to the entire nsp1 fused to a small fragment of nsp2 had a size consistent with its expected molecular mass of 40.2 kDa (Fig. 3C). Translation of pA-N3S mRNA yielded a partial nsp3 fragment with an apparent molecular mass of 46 kDa, somewhat larger than its calculated size of 35.9 kDa. However, since a product of identical mobility was obtained with pA-N3SΔ1 and pA-N3SΔ2 mRNAs, the first of which has a 5′ UTR of only 25 nt, this showed that translation of pA-N3S mRNA had started at the expected location. As anticipated for cap-dependent protein synthesis, the efficiency of translation of the N3S-related mRNAs increased as the size of the 5′ UTR decreased. The aberrantly slow migration of the partial nsp3 product was likely due to the highly acidic (Ac) region near the amino terminus of nsp3 (Fig. 1); the same mobility was seen for an almost identical nsp3 fragment that was initiated by a completely different mechanism (see below). A faint, slower-migrating band was observed for translated pA-N3S mRNA (Fig. 3C, open circle), but this same band was present for the 5′-UTR-deleted N3S mRNAs and it therefore could not have resulted from upstream initiation at a noncanonical start codon.
FIG 3.

Confirmation of the N3S mutant. (A) Segment of sequence of an RT-PCR product from total RNA purified from N3S-infected cells. The newly created nsp3 start codon is boxed. (B) Schematics of mRNAs generated by runoff in vitro transcription of truncated T7 vectors. Plasmids pA-WT and pA-N3S were those used to produce the 5′-most fragment of the full-length genomic cDNA for wild-type virus and N3S mutant virus, respectively. The length of the 5′ UTR is given above each mRNA. Deleted derivatives of pA-N3S reduced the size of the 5′ UTR of the encoded mRNA from 591 nt to either 25 nt (pA-N3SΔ1) or 249 nt (pA-N3SΔ2). The three pA-N3S-related plasmids were truncated with SpeI; pA-WT was truncated with BstZ17I. In the 5′ UTRs of mRNAs, the uORF is denoted by an open rectangle; asterisks represent start codon knockouts. The predicted molecular masses of translation products are indicated above the mRNAs. (C) In vitro-transcribed mRNAs were translated in a reticulocyte lysate system, and [35S]methionine-labeled protein products were analyzed by SDS-PAGE, followed by fluorography. Mock, control without added mRNA; [nsp3], nsp3 partial product from pA-N3S-related runoff mRNAs; nsp1-[nsp2], nsp1-partial nsp2 product from pA-WT runoff mRNA. The open circle denotes an artifactual band obtained with pA-N3S-related runoff mRNAs. The [nsp3] and nsp1-[nsp2] proteins contain 4 and 10 methionine residues, respectively.
Together, these results demonstrated that the complete elimination of nsp1 expression coupled with deletion of the entirety of nsp2 does not abolish the viability of MHV in tissue culture. This represents the largest truncation of the replicase-transcriptase that has yet been made, although it must be noted that successful individual deletion of nsp2 in both MHV and SARS-CoV has already been well established (31). Combined with our prior work (17), this means that Ubl1, the amino terminus of nsp3, is the first functional domain of the replicase-transcriptase with respect to viral RNA synthesis. Nevertheless, because the N3S mutant had been very difficult to isolate, this raised the possibility that it needed to acquire one or more adaptive mutations in order to be fully viable. The determination of the complete genomic sequence of this mutant revealed that, indeed, it harbored two mutations in the nsp3 coding region. The first of these, E851G, was at the amino-terminal end of PLP2, near the Ubl2 domain. The second mutation, D1791V, fell in the carboxy-terminal Y domain. There were only three additional mutations in the N3S genome outside nsp3. One of these, T220I in nsp15, was at the boundary of a nonessential surface loop on this subunit, and we thus think it is not functionally relevant. Likewise, two mutations in the spike (S) protein, Q99H in the ectodomain and C1280S in the transmembrane domain, could conceivably affect growth but are highly unlikely to signify an interaction between S and nsp3.
The phenotype of the N3S mutant also indicated that it was conditionally impaired. Plaques of N3S were moderately smaller than wild-type plaques at 33°C and 37°C. However, at 39°C, the mutant formed plaques that were tiny compared to those of the wild type (Fig. 4A). To explore the basis for this deficiency, 12 cultures begun from individual plaques of N3S were passaged serially 10 to 13 times at 39°C in L2 cells. Titers of the viral supernatants were then determined at 39°C, and one revertant plaque originating from each culture was purified for analysis. Each chosen revertant formed plaques that were substantially larger than those of the N3S mutant (two examples are shown in Fig. 4A), but none reached the size of wild-type MHV plaques at 39°C. For all 12 independent revertants, we sequenced the 5′ end of the genome, ranging from the end of the leader through the start of the nsp4 gene, as well as the entire N gene and the 3′ UTR. Only one revertant (rev12) had a nucleotide change (U476A) in the 5′ UTR, which occurred in a bulge base of SL A (SLA) (Fig. 2B). Otherwise, no alterations were found in the 5′ or 3′ UTRs of any revertant. This suggested that the changes engineered in the original N3S mutant did not adversely affect the functioning of cis-acting RNA elements at either end of the genome.
FIG 4.

Revertants of the N3S mutant. (A) Plaques of the N3S mutant compared with those of isogenic wild-type virus at 33°C, 37°C, or 39°C, and plaques of two representative revertants of N3S at 39°C. Plaque titrations were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later. (B) Locations of nsp3 mutations in 12 independent revertants of the N3S mutant. The two nsp3 mutations in the original N3S mutant, E851G and D1791V, are shown above the schematic.
In contrast, all revertants exhibited from one to three mutations in the nsp3 coding region, and these were gathered in three main loci (Fig. 4B). Multiple reverting mutations mapped to the amino-terminal Ubl1 and Ac domains. Three of these—A38V, A60T, and D98Y—were positioned within or close to the α2 helix of Ubl1 previously shown to interact with N protein (16). Moreover, A38V was identical to one of the reverting mutations originally seen in the N protein chimeric mutant that led us to discover the Ubl1-N interaction (15). Conversely, though, none of the N3S revertants contained changes in the serine- and arginine-rich region of N protein. An nsp3 mutation found in one revertant, L480I, mapped to the border between the PLP1 and ADRP domains but is not likely to be significant, because most other strains of MHV also have an isoleucine at this position. All other reverting mutations fell in regions already brought to our attention by the two nsp3 mutations in the original N3S mutant (Fig. 4B). One of these, I813T in the Ubl2 domain, was near the E851G mutation of N3S in PLP2. The remainder of the reverting mutations were clustered in the carboxy-terminal Y domain or else in the region adjacent to the first transmembrane domain, which is likely proximal to the Y domain. Notably, most of the revertants contained a further alteration of D1791. This residue, which was changed to valine in the original N3S mutant, was again changed to either alanine or glycine in 8 of the 12 revertants. Collectively, the N3S reverting mutations appear to demarcate the most critical domains of nsp3. We speculate that they point to regions of nsp3 that interact with other proteins or protein domains important for RNA synthesis. However, it is not currently clear why the unique architecture of the N3S mutant genome necessitates its acquisition of these compensatory changes in order to improve nsp3 function.
Initiation of nsp3 by an alternative mechanism.
Owing to our uncertainty about the underlying cause of the mutations in N3S and its revertants, this mutant did not provide an optimal starting point for further dissection of nsp3. We therefore turned to an alternative strategy to circumvent the necessity for the nsp1-nsp2 and nsp2-nsp3 cleavage events, making use of the 2A peptide autoprocessing element from the picornavirus foot-and-mouth disease virus (FMDV). Through an undefined mechanism, the FMDV 2A sequence effectively dictates termination followed by reinitiation of protein synthesis at two adjacent codons in a single ORF (46). Exploiting this device, we constructed a mutant, designated stop-start-NSP3 (SSN3), in which synthesis of the replicase-transcriptase polyprotein began with an amino-terminal segment of nsp1 that was separated from nsp3 by a 2A element (Fig. 5A). The fragment of nsp1 (amino acids 1 to 127) was chosen to preserve the same cis-acting genomic RNA structures as had been retained in the N3S mutant. Autoprocessing of the 2A element would generate a single extra residue, proline, at the amino terminus of nsp3. This was expected to be tolerated, because the first 18 residues of MHV Ubl1 are unstructured and do not contribute to the interaction with N protein (16).
FIG 5.

Construction and characterization of the stop-start-nsp3 (SSN3) mutant. (A) Schematics of the genomic 5′ ends of wild-type MHV and the SSN3 and SSN3x mutants. In the wild-type genome, open circles denote cleavage sites for PLP1. In the mutant genomes, the expanded segment details the insertion placed between the nsp1 fragment and the start of the nsp3 coding region, which contains a His7 epitope tag followed by a 58-amino-acid version of the FMDV 2A translational stop-start element (47). The autoprocessing site in the SSN3 sequence is denoted by an arrowhead. Two inactivating mutations in the SSN3x sequence are underlined; the inactivated autoprocessing site is denoted by x. Indicated below each genome are the predicted molecular masses of translation products produced by runoff in vitro transcription and translation of plasmids used to construct full-length genomic cDNAs. (B) Plaques of the SSN3 mutant compared with those of isogenic wild-type virus at 33°C, 37°C, or 39°C. Plaque titrations were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later. (C) Plasmids pA-WT, pA-SSN3, and pASSN3x, used for the 5′-most fragments of the corresponding full-length genomic cDNAs, were truncated with BstZ17I or SpeI for runoff in vitro transcription. The mRNAs were translated in a reticulocyte lysate system, and [35S]methionine-labeled protein products were analyzed by SDS-PAGE, followed by fluorography. Mock, control without added mRNA; [nsp1]-2A and [nsp3], autoprocessed products from pA-SSN3 runoff mRNA; [nsp1]-2A-[nsp3], unprocessed product from pA-SSN3x runoff mRNA; nsp1-[nsp2], product from pA-WT runoff mRNA. RNA transcribed from SpeI-truncated pA-N3SΔ1 (see Fig. 3), translated as a control, encodes an nsp3 fragment identical to that of pA-SSN3/SpeI mRNA, except that it has a methionine instead of a proline at the amino terminus. The [nsp1]-2A, [nsp3], [nsp1]-2A-[nsp3], and nsp1-[nsp2] proteins contain 7, 3, 10, and 10 methionine residues, respectively.
We first implemented this design with the minimal 20-amino-acid FMDV 2A peptide, but no viable virus was obtained. Similarly, inclusion of a homolog of the 2A peptide, from Thosea asigna picornavirus, appeared to be only partially effective, since recovered virus was very impaired. Amino-terminal extensions of the 2A element have been shown to greatly increase stop-start efficiency, consistent with the notion that the stretch of polypeptide occupying the ribosomal exit tunnel causes or contributes to this anomalous translational mechanism (47, 48). Accordingly, the final version of the SSN3 construct incorporated a 20-amino-acid FMDV 2A peptide plus 38 upstream residues from its native polyprotein sequence, preceded by a His7 epitope tag (Fig. 5A). Three independent isolates of the SSN3 mutant were obtained, all having the same phenotype, and one was chosen for further analysis. The sequence of this virus was determined from the 5′ end of the genome through the PLP1 domain and was confirmed to be exactly as expected, with no additional mutations. Plaques formed by the SSN3 mutant were only slightly smaller than wild-type plaques at 37°C and 39°C (Fig. 5). However, at 33°C, we consistently noted heterogeneity in plaque size, with the sporadic appearance of much smaller plaques. The basis for this growth characteristic at 33°C is unknown, but we have observed it for all constructed mutants that contain large deletions in both nsp1 and nsp2, irrespective of the presence of either the 2A element or the His7 epitope tag (data not shown).
Contrary to the direct fashion in which the SSN3 and subsequent derivative mutants were acquired, we were unable to obtain a related mutant, SSN3x (Fig. 5A), that was modified by two changes in the FMDV 2A sequence known to abolish autoprocessing (28). In four separate trials, each with multiple independent samples and robust positive controls, transfected SSN3x full-length RNA produced no signs of infection, indicating that the mutated 2A sequence was lethal. Since the only difference between the SSN3 and SSN3x constructs was the absence of a functional stop-start element in the latter, this strongly suggests that a permanent blockage of the amino terminus of nsp3 results in a nonfunctional replicase-transcriptase.
To seek further evidence for the relative efficacies of the SSN3 and SSN3x stop-start elements, we in vitro-translated synthetic mRNAs that were identical to the 5′ ends of these mutant genomes. Translation of pA-SSN3 mRNA gave rise to two discrete products, the smaller of which was exactly the size expected (21.5 kDa) for the nsp1 fragment fused to the FMDV 2A element (Fig. 5C). The larger pA-SSN3 product, corresponding to the processed nsp3 fragment, migrated more slowly than expected for a 39.5-kDa protein. However, its mobility was identical to that of the product obtained by translation of pA-N3SΔ1 mRNA, which, because of its minimal 5′ UTR, had served to produce a size standard for this nsp3 fragment (see Fig. 3C). (The nsp3 fragments encoded by pA-SSN3 and pA-N3SΔ1 differ only at their amino-terminal residues, proline and methionine, respectively.) In contrast to pA-SSN3, translation of pA-SSN3x mRNA yielded just a single, larger protein with an apparent molecular mass of 65 kDa, bigger than the expected 57.4 kDa by the same differential observed for the other nsp3 products. The total absence of this unprocessed product upon translation of pA-SSN3 mRNA indicated that there was complete efficiency of termination by the incorporated 2A element. Moreover, the relative intensities of the bands for the nsp3 and nsp1-2A fragments (allowing for their different methionine contents) showed that reinitiation was also highly or entirely efficient. Conversely, the lack of production of either the nsp3 or nsp1-2A fragment upon translation of pA-SSN3x mRNA demonstrated that the mutations in the SSN3x construct effectively eliminated autoprocessing by its 2A element. Taken together, these results allowed us to conclude that the SSN3 construct enabled normal initiation of nsp1 with equimolar production of mature nsp3, thereby providing a suitable platform for further dissection of nsp3.
The PLP1 and ADRP domains of nsp3 are entirely dispensable.
Having eliminated the need for the two processing events carried out by PLP1, we were thus able to explore whether this module is required for other roles in the replicase-transcriptase complex. Toward this end, the SSN3 mutant was rebuilt with FLAG and HA epitopes inserted in tandem immediately downstream of the Ac region, yielding a construct designated SSN3HA (Fig. 6A). A PLP1 catalytic knockout, SSN3HAmutP, was then created by mutating an active site residue that has been shown to be essential for MHV PLP1 activity in vitro (18) and in vivo (19). To minimize the probability of spontaneous reversion of this mutation, C289A, we changed all three bases of the wild-type codon (UGU to GCC). Multiple independent isolates of both the SSN3HA and SSN3HAmutP viruses were readily obtained. As no phenotypic differences were noted among them, one isolate of each was chosen for further analysis. Sequence determination from the 5′ end of the genome through most of nsp3 confirmed the presence in each of exactly the engineered composition with no extraneous mutations. Plaques formed by the SSN3HA and SSN3HAmutP mutants were identical to each other and to those of the original SSN3 mutant at 33°C, 37°C, and 39°C (examples at 37°C are shown in Fig. 6B). This showed that, as anticipated, the epitope tags were fully tolerated in what is thought to be an unstructured linker between the Ac region and PLP1 (12). Likewise, we had expected the knockout of PLP1 catalytic activity to have little or no effect in the SSN3 background, based on the prior demonstration of the viability of a PLP knockout mutant and its partial rescue by simultaneous small deletions in the nsp1-nsp2 and nsp2-nsp3 cleavage sites (19).
FIG 6.

Mutation and deletion of domains of nsp3. (A) Schematics of the genomic 5′ ends of the SSN3HA recombinant and mutants derived therefrom. At the top is a partial diagram of the domains of nsp3, as described in the legend to Fig. 1. In each mutant, HA marks the position of the FLAG and HA epitope tags inserted at the end of the Ac region. The solid rectangle between the nsp1 fragment and the start of the nsp3 coding region represents the FMDV 2A translational stop-start element, as in Fig. 5. In SSN3HAmutP, the point mutation in PLP1 is indicated. In SSN3HAΔP1, SSN3HAΔP2, and SSN3HAΔP3, numbers denote the first and last residues of nsp3 deletions. Residue numbers given are those of mature wild-type nsp3 and do not include the extra amino-terminal proline or the 17-amino-acid epitope tag insertion following Ac in SSN3HA and related constructs. (B) Plaques of the SSN3, SSN3HA, SSN3HAmutP, and SSN3HAΔP3 mutants compared with those of isogenic wild-type virus at 37°C. Plaque titrations were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later.
We next designed deletions of PLP1 in the SSN3HA background (Fig. 6A). Because no structural information is yet available for a betacoronavirus PLP1, other measures were used to infer the limits of this domain. In the first mutant, SSN3HAΔP1, the extent of the deletion (amino acids 234 to 484) was based on the smallest expressed segments of MHV nsp3 that exhibited protease activity in vitro (49, 50), as well as on the boundaries of the upstream Ac (15) and downstream ADRP (23, 51) domains. In the second mutant, SSN3HAΔP2 (deletion of amino acids 274 to 460), the boundaries of the first deletion were moved inward, to the edges of the region of PLP1 that is most conserved among the lineage A betacoronaviruses. In addition, the more conservative downstream boundary preserved residue M475, a mutation of which has very far-reaching deleterious effects in a particular temperature-sensitive mutant, Brts31 (52). In a final construct, the SSN3HAΔP3 mutant (deletion of amino acids 234 to 651), the downstream deletion boundary was extended in the opposite direction, to encompass both PLP1 and ADRP, addressing the possibility that the original deletion juxtaposed the essential Ubl1 domain too close to ADRP. In multiple independent trials, we were unable to isolate SSN3HAΔP1 or SSN3HAΔP2 virus. Remarkably, only the mutant containing the largest deletion was viable. Two independent isolates of the SSN3HAΔP3 virus were recovered. For one of these, we determined the sequence of the entire genome, which revealed exactly the constructed mutations and rearrangements, with no additional mutations. The SSN3HAΔP3 mutant formed plaques only slightly smaller than those of the corresponding SSN3HA parent at 33°C, 37°C, and 39°C (plaques at 37°C are shown in Fig. 6B). This demonstrated that MHV can retain a surprising degree of hardiness despite the loss of half of nsp1, all of nsp2, and one-fifth of nsp3; the sum of the deletions in the SSN3HAΔP3 mutant totals 3,120 nucleotides, nearly one-tenth of the viral genome.
To corroborate the genetic compositions of the SSN3HA virus and its derivatives, we examined the expression of different subunits of the replicase-transcriptase. Infected cell lysates were immunoprecipitated with an antibody to the amino-terminal 347 amino acids of nsp3 (antibody VU164) (31) and then probed by Western blotting with an antibody to the HA epitope tag that had been inserted at the end of the Ac domain (Fig. 6A). (The FLAG epitope tag planted at the same internal position, adjacent to the HA tag, did not work in Western blots.) For cells infected with the SSN3HA and SSN3HAmutP viruses, a protein with an apparent molecular mass of 260 kDa was detected (Fig. 7A). nsp3 has a calculated molecular mass of 222 kDa (or 224 kDa with the epitope tag insertion) and has been described as 210 kDa (30, 31). However, we observed the same band for wild-type, SSN3, SSN3HA, and SSN3HAmutP viruses in direct Western blots with VU164 antiserum (data not shown). We therefore conclude that the apparent 260-kDa band is nsp3, not a precursor, and that its lower mobility is probably attributable to differences between the molecular mass standards used by us and by others. Consistent with this, a single band of 235 kDa was observed for the SSN3HAΔP3 mutant, again larger than the calculated molecular mass of 178 kDa (Fig. 7A). This finding confirms that mature nsp3 of SSN3HAΔP3 was stably produced and that it harbored a large deletion. All of the same protein species were observed when immunoprecipitations were performed with a different anti-nsp3 antiserum (antibody D3) (30), which had been raised against the amino-terminal 205 amino acids of nsp3 (Fig. 7B). Notably, nsp3 of the SSN3HAΔP3 virus was only weakly immunoprecipitated by D3 despite containing the complete region of nsp3 recognized by this antibody. This may indicate that the deleted nsp3 of this mutant is conformationally different from wild-type nsp3.
FIG 7.
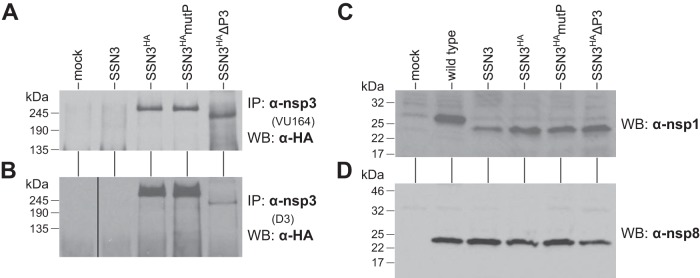
Protein expression by SSN3-related mutants. Infected cell lysates were prepared at the peak of infection with each virus and thus do not represent a quantitative kinetic comparison of protein production. (A, B) Lysates from mock-infected or virus-infected cells were immunoprecipitated with anti-nsp3 polyclonal antibody VU164 (A) or D3 (B). Immunoprecipitated material was then analyzed by Western blots probed with anti-HA monoclonal antibody. (C, D) Lysates from mock-infected or virus-infected cells were analyzed by Western blots probed with anti-nsp1 antibody VU221 (C) or anti-nsp8 antibody VU124 (D).
Western blotting with anti-nsp1 antibody revealed a 26-kDa protein for wild-type-infected cell lysates (Fig. 7C), consistent with the expected molecular mass of 27.4 kDa for nsp1. The same antiserum also detected the 21.5-kDa nsp1-2A fragment of the SSN3 mutant and all of its derivatives, even though this fragment contained only half of the nsp1 molecule. (The His7 epitope tag that had been engineered upstream of the FMDV 2A element [Fig. 5] did not work in Western blots.) This result demonstrated that there was efficient in vivo termination by the 2A element, as we had observed in vitro (Fig. 5C). Additionally, Western blots of infected cell lysates probed with anti-nsp8 antibody detected the same 21.6-kDa species in wild-type- and mutant-infected cell lysates. This showed that, at the peak of infection, nearly equivalent amounts of this downstream replicase subunit were produced, suggesting that reinitiation by the FMDV 2A element was also highly efficient in vivo.
To more completely assess the fitness of the SSN3HA and SSN3HAΔP3 mutants, we evaluated their growth and RNA synthesis capabilities relative to those of the wild type. In infections begun at a multiplicity of 1.0 PFU per cell (limited by the titers that could be achieved for stocks of SSN3HAΔP3), wild-type virus reached peak titers of 7.8 × 107 PFU/ml between 8 and 12 h postinfection (Fig. 8A). In contrast, the SSN3HA and SSN3HAΔP3 mutants reached maximal titers that were 1.6 and 2.0 log10 lower than those of the wild type with somewhat delayed kinetics, peaking between 12 and 16 h postinfection. Data from a second, independent growth experiment were nearly identical to those shown in Fig. 8A. We also noticed that 17Cl1 cell monolayers infected with wild-type MHV exhibited extensive syncytia and cytopathic effect and were almost completely detached by 24 h postinfection. However, 17Cl1 monolayers infected with either of the two mutants underwent a period of partial detachment from 16 to 24 h postinfection but had recovered to roughly 80% confluence by 48 h postinfection. This recovery was not observed with L2 cell monolayers. The differential growth kinetics (Fig. 8A) make clear that the major detriment caused by the engineering of the SSN3-derived mutants was due to the loss of nsp1 and nsp2. The gap between the growth curves for wild-type virus and both the SSN3HA and SSN3HAΔP3 mutants was roughly equal to the sum of the reductions seen previously for an individual nsp1 carboxy-terminal deletion mutant (53) and an individual nsp2 deletion mutant (31). Comparatively, the removal of the PLP1 and ADRP domains in the SSN3HAΔP3 mutant had only a minor effect. Northern blotting of intracellular RNA harvested at the peak of infection revealed a corresponding quantitative drop in RNA synthesis by the SSN3HA and SSN3HAΔP3 mutants relative to that of the wild type (Fig. 8B). However, both mutants produced the same subgenomic RNA species in the same relative proportions as wild-type MHV. This showed that deletion of the PLP1 and ADRP domains of nsp3 did not qualitatively alter the ability of MHV to execute the characteristic pattern of coronavirus RNA replication and transcription.
FIG 8.
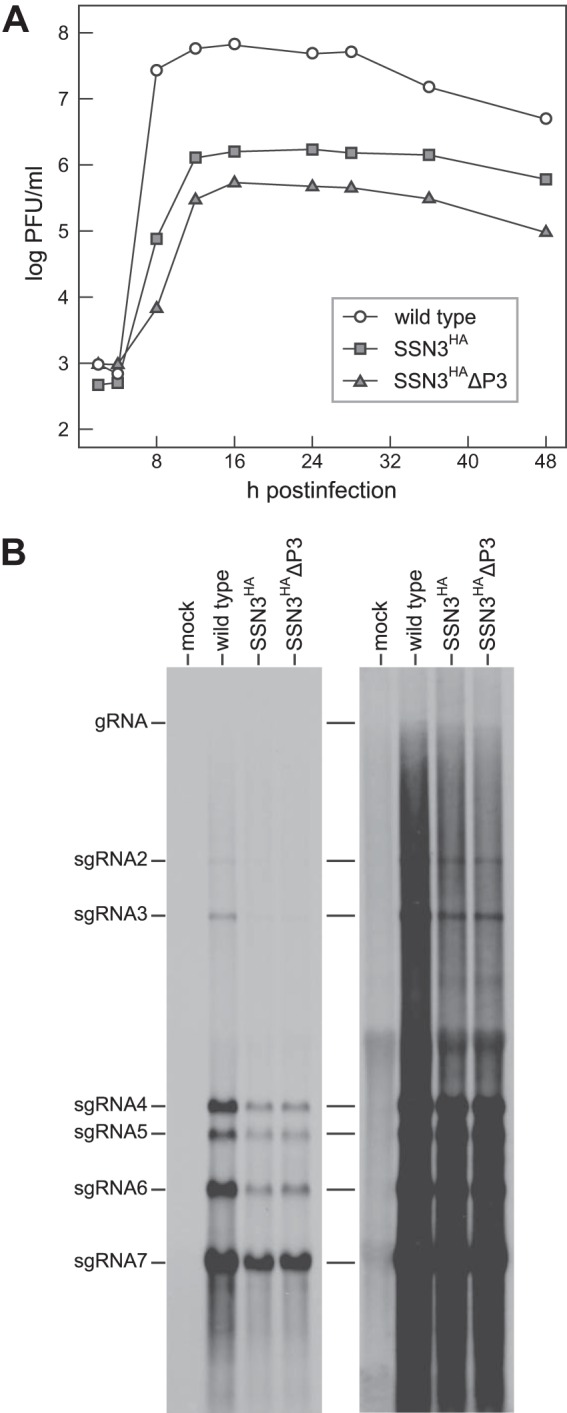
Growth and RNA synthesis by replicase-transcriptase mutants. (A) Growth kinetics of wild-type, SSN3HA, and SSN3HAΔP3 viruses. Confluent monolayers of 17Cl1 cells were infected at a multiplicity of 1.0 PFU per cell. At the indicated times postinfection, aliquots of medium were removed, and infectious titers were determined by plaque assay on L2 cells. (B) Northern blot of total RNA isolated from mock-infected 17Cl1 cells or cells infected at a multiplicity of 1.0 PFU per cell with wild-type virus, the SSN3HA mutant, or the SSN3HAΔP3 mutant. RNA was isolated from infected cells at 8.5, 10, or 12 h postinfection, respectively, for wild-type, SSN3HA, or SSN3HAΔP3 virus. MHV RNA was detected with a probe specific for the 3′ end of the genome. gRNA, genomic RNA; sgRNA, subgenomic RNA. The right panel is an overexposure to allow visualization of the larger RNA species.
DISCUSSION
Coronaviruses are unique among RNA viruses in the size and complexity of their RNA-synthetic apparatus. Additionally, coronaviruses are highly unusual among positive-strand RNA viruses in that their genomic RNA is only minimally infectious in tissue culture unless supplemented with a source of N protein. In previous work, we have shown that the latter requirement is due, at least partly and perhaps entirely, to a critical association between N protein and the amino-terminal Ubl1 domain of nsp3 (15, 17). Our working model is that the purpose of the Ubl1-N interaction is to establish a connection between the infecting viral genome and the replicase that is being translated from the genome. This would coordinate the dual roles of genomic RNA, which functions initially as an mRNA and then subsequently as a template for RNA synthesis. Since nsp3 is the first replicase subunit to become anchored in the membrane, its amino terminus would be an advantageous location to dock the distal end of the infecting nucleocapsid, which is not traversed by ribosomes. nsp3 is a huge multidomain protein (222 kDa in MHV) constituting more than a quarter of the mass of the replicase-transcriptase complex (Fig. 1). The structures of many of its constituent modules have been determined, principally for the SARS-CoV homologs (13, 14, 16, 51). However, it is less clear what roles are played by most of these domains, except for the well-characterized processing and deubiquitinating activities of the PLPs (18–20, 30, 49). In the current study, we initiated a dissection of MHV nsp3 aimed at distinguishing which parts of this molecule are directly involved in RNA synthesis, whether in an enzymatic or structural capacity, and which parts are dispensable. Our strategy proceeded from the amino terminus. For Ubl1, we have previously shown that deletion of residues 19 through 111 or mutation of charged residues in the surface loop between the β3 and β4 strands is lethal to the virus (17). These results underlined the importance of the Ubl1-N interaction. In contrast, we previously showed that the Ac region, which is hypervariable even among closely related coronaviruses, can be deleted with no apparent effect on viral phenotype (17).
To continue downstream from Ubl1 and Ac, it was necessary to uncouple nsp3 from the processing of nsp1 and nsp2. The most straightforward approach toward this end was to remove nsp1 and nsp2 and to initiate polyprotein translation with nsp3. We accomplished this in the N3S mutant through a combination of a large deletion and knockout of all remaining start codons, subject to the strict maintenance of potential cis-acting RNA elements embedded in the 5′-most 591 nucleotides of the MHV genome (Fig. 2). The relocation of the replicase start codon to a point downstream of the 5′ cis-acting RNA elements showed that there is no strict requirement that they overlap the replicase ORF, even though this overlap is common to the genomic organization of all alpha- and betacoronaviruses. It is noteworthy that the 5′ UTR created in the N3S mutant was similar in size to those of gamma- and deltacoronaviruses, which have 5′ UTRs ranging from 477 to 606 nt and do not encode a counterpart for nsp1 (54, 55). To our knowledge, the N3S mutant is the first complete knockout of nsp1 (in an alpha- or betacoronavirus) and the first example of a coronavirus in which nsp1 and nsp2 have been simultaneously eliminated. This result confirms and extends prior findings with MHV and SARS-CoV.
MHV nsp1 mutants were previously constructed with carboxy-terminal deletions of 118 (53) or 33 (56) amino acids. These viruses exhibited relatively minor growth defects in cell lines and in primary cells, although the latter virus was shown to be severely attenuated in the mouse host. Extensive recent study of nsp1 has revealed it to be a suppressor of host protein synthesis, both by stimulation of a ribosome-associated endonuclease and by direct inhibition of translation initiation (57, 58). It thus appears that the main role of nsp1 is to institute a favorable cellular environment for the infecting virus. The viability of the N3S mutant rules out any obligatory role for nsp1 in coronavirus RNA synthesis. An earlier study found that certain amino-terminal point mutations in nsp1 were severely debilitating or lethal for MHV (53), but, in retrospect, this may be attributable to disruption of cis-acting RNA structures overlapping the first half of the nsp1 ORF. A very recent analysis of the replication of a DI RNA of bovine coronavirus, which is closely related to MHV, found an absolute requirement for an intact stretch of coding sequence near the amino terminus of nsp1 (35). This nsp1 peptide was proposed to provide a means for DI RNA to be recruited to the preformed replication complexes of the helper virus. Our results argue that, even though this scheme is exploited for DI RNA replication, it does not reflect a mechanism normally used in viral RNA synthesis.
Similarly to nsp1, nsp2 cannot play an essential role in the coronavirus replicase-transcriptase complex. For both MHV and SARS-CoV, it was previously shown that the coding region of nsp2 could be entirely excised, resulting in mutants with roughly 10-fold reductions in peak growth titers (31). Although the nsp2 deletion viruses had a quantitative (2-fold) drop in RNA synthesis relative to wild-type levels, they exhibited no qualitative alteration of RNA species. In contrast to many of the domains of nsp3, where homology can be found across the most divergent genera, nsp2 is very poorly conserved, even among coronaviruses in the same genus. It is therefore unlikely to be specifically involved in the mechanism of RNA synthesis but instead may act in an ancillary capacity during infection. Indirect evidence suggests a role for SARS-CoV nsp2 in the alteration of intracellular signaling (59).
The recovery of the N3S mutant required two adaptive mutations in nsp3, and additional mutations in nsp3 were necessary to allow growth of revertants at 39°C (Fig. 4). These conditions did not provide a strong basis for further dissection of the nsp3 molecule. Consequently, we devised a different strategy, in the SSN3 mutant and its derivatives, to get around the polyprotein processing duties of PLP1. The FMDV 2A element (with an extended upstream peptide) was used to separate a fragment of nsp1 from the amino terminus of nsp3 (Fig. 5A). This device was seen to operate efficiently in vitro and in vivo, both in termination after the partial nsp1 molecule and in reinitiation to produce nsp3 and downstream replicase subunits (Fig. 5C and 7A and B). A minimal (20-amino-acid) version of the 2A element from Thosea asigna picornavirus had been used previously to replace the nsp1-nsp2 cleavage site of the human alphacoronavirus 229E (HCoV-229E) (60). However, it was not clear how effectively it functioned. In the context of the SSN3 mutant, there appeared to be an absolute requirement for processing at the amino terminus of nsp3. The lethality of the nonfunctional 2A element in the SSN3x mutant (Fig. 5A) is a strong indication that, under some circumstances, uncleaved polypeptide upstream of nsp3 can obstruct access to the Ubl1 domain. Our observation is consistent with, although more drastic than, previous demonstrations of the deleterious effects caused by deletion of the nsp2-nsp3 cleavage site (19) or by fusion of an inefficiently cleaved reporter protein to the amino terminus of nsp3 (61).
Within the framework of the SSN3 mutant, we were then able to address the requirements for the PLP1 and ADRP domains of nsp3. The multiplicity and specificity of nsp3 PLPs vary. Some coronaviruses, like SARS-CoV and MERS-CoV, have a single PLP that efficiently carries out all of the first three cleavage events of replicase polyprotein processing. Others, like MHV, have two PLPs with strictly designated roles; MHV PLP2 cannot be reassigned to perform both PLP1 cleavages (62). Still other coronaviruses, like HCoV-229E, have two PLPs with flexibly overlapping specificities (60). For coronaviruses with two PLPs, PLP1 is thought to have evolved through duplication of PLP2 (26). However, while the PLP1s of alphacoronaviruses remain akin to PLP2s (or single PLPs), the PLP1s of lineage A betacoronaviruses form a separate class with respect to substrate binding site structure (63). In this regard, MHV PLP1 may be considered a feature particular to the lineage A betacoronaviruses, comparable to the nsp15-embedded packaging signal, the hemagglutinin-esterase, and accessory proteins 2a and 5a. As such, PLP1 might not be expected to perform an essential RNA synthesis function conserved across all coronaviruses.
We found that a constructed PLP1 catalytic mutant, SSN3HAmutP, had a phenotype identical to that of its parent viruses, SSN3 and SSN3HA (Fig. 6). This result was anticipated, because Graham and Denison had previously demonstrated the viability of a similar MHV PLP1 catalytic mutant (19). That virus, although severely impaired, was partially rescued when combined with small deletions of both the nsp1-nsp2 and nsp2-nsp3 cleavage sites. Their finding suggested that an inactivated PLP1 could sterically hinder replicase function by nonproductively binding to one or both of its cleavage sites. The SSN3 mutant background could thus be seen as an extreme form of the previous cleavage site deletions, in this case one in which absolutely no vestige of either PLP1 binding site was preserved. The SSN3HAmutP virus showed that PLP1 proteolytic activity was not required for a hypothetical essential function in addition to polyprotein processing. It also provided a positive control for further dissection of nsp3, i.e., an inability to delete PLP1 would have to have been attributed to something other than loss of proteolytic activity.
It remained feasible that the PLP1 domain could harbor enzymatic or structural functions critical for viral RNA synthesis, and these would not be affected by the catalytic knockout mutation. This prompted us to test the possibility of completely deleting PLP1. A previous study of the PLP1 of HCoV-229E proposed that an as-yet undefined nonproteolytic role of PLP1 might be mediated by interactions between its universally conserved zinc finger and other nsp's or RNA (21). Such a prospect, however, is precluded by our isolation of the SSN3HAΔP3 mutant, in which both PLP1 and the adjacent ADRP domain were deleted (Fig. 6). The SSN3HAΔP3 mutant removed 418 amino acids, more than one-fifth of the nsp3 molecule, ruling out any critical secondary role for the PLP1 domain in MHV RNA synthesis. Notably, the deletion in SSN3HAΔP3 (spanning amino acids 234 to 651) encompassed M475, the residue that is mutated in Brts31, a conditionally lethal MHV mutant (52). Paradoxically, at the nonpermissive temperature, Brts31 is defective in processing carried out by the main protease, nsp5, but it is unhindered in processing by PLP1 and PLP2. Our results suggest that the Brts31 mutation is dominant negative, manifesting the acquisition of an aberrant function at the nonpermissive temperature rather than the loss of a normal function.
In contrast to PLP1, the ADRP macrodomain (previously called the X domain [26]) is highly conserved in all coronavirus genera. A similar domain also appears in replicase proteins of alphaviruses, hepatitis E virus, and rubella virus. Moreover, in one alphavirus, Sindbis virus, macrodomain mutations that impair viral RNA synthesis have been identified (64, 65). In (uninfected) cells, ADRP activity comes into play downstream of the pathway of tRNA splicing, and thus a virally encoded ADRP appeared a good candidate to operate in an undefined analogous reaction in coronavirus RNA synthesis (66). Nevertheless, the ADRP enzymatic activity of coronavirus macrodomains has a very low turnover (22, 51, 67). Also, if hydrolysis of ADP-ribose-1″-monophosphate is critical for the virus, it is not clear why host cytoplasmic macrodomain proteins are inadequate for this task. Such considerations raised the possibility that ADP-ribose-1″-monophosphate is not the only substrate for the ADRP (67) or is a surrogate for the relevant substrate, which is possibly mono-ADP-ribosylated protein (68, 69). More to the point, ADRP active-site mutants of HCoV-229E (22) and MHV (23, 68) were found to be unimpaired in growth in tissue culture, although the latter were avirulent with respect to hepatic pathogenesis or induction of encephalitis. The knockout of ADP-ribose-1″-phosphatase clearly shows that this activity is not essential for coronaviruses, but it leaves unaddressed other potential roles of the macrodomain that are not affected by active-site mutations (24). Some structural and biochemical studies have suggested that poly(ADP-ribose) binding and poly(A)-RNA binding are more conserved macrodomain activities that are more likely to be important for viral replication (24, 25). However, the complete deletion of the ADRP domain in the SSN3HAΔP3 mutant unequivocally rules out an essential role for poly(ADP-ribose) binding, poly(A) binding, or any as-yet-unknown activities of the macrodomain in coronavirus RNA synthesis.
In the current study, we have covered nearly half of the MHV nsp3 ectodomain (Fig. 1). In future work, we would like to extend the same methods employed here to dissect the remainder of the nsp3 molecule. In particular, PLP2 and its associated Ubl2 domain (70) have prospects of playing roles in RNA synthesis beyond proteolytic processing. Also, the carboxy-terminal Y domain (22), with its universally conserved clusters of cysteine and histidine residues, presents an intriguing target for potential intermolecular interactions. In addition, it would be of great interest to determine whether infections by SSN3, SSN3HAΔP3, or other mutants created here lead to the formation of the full spectrum of DMVs and other membrane alterations observed during wild-type infection. Such studies may help to elucidate the relationship of these membrane rearrangements to viral RNA synthesis (9).
ACKNOWLEDGMENTS
We thank Cheri Koetzner for sample preparation for viral genome sequencing. We are grateful to Mark Denison (Vanderbilt University, Nashville, TN) and Susan Baker (Loyola University Chicago, Maywood, IL) for generously providing antibodies to the MHV replicase nonstructural proteins. We thank Ralph Baric and Amy Sims (University of North Carolina, Chapel Hill, NC) for making available cloned cDNAs and protocols for the MHV full-length cDNA reverse genetics system. We thank the Applied Genomics Technology Core Facility of the Wadsworth Center for Sanger and next-generation DNA sequencing and, in particular, Matthew Shudt for viral genomic sequencing and Pascal Lapierre of the Wadsworth Center Bioinformatics Core Facility for genomic sequence analysis.
This work was supported by National Institutes of Health (National Institute of Allergy and Infectious Diseases) grant R01 AI064603.
REFERENCES
- 1.Masters PS, Perlman S. 2013. Coronaviridae, p 825–858. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY, SARS Study Group. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473-12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawicki SG, Sawicki DL, Siddell SG. 2007. A contemporary view of coronavirus transcription. J Virol 81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulasli M, Verheije MH, de Haan CA, Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol 12:844–861. doi: 10.1111/j.1462-5822.2010.01437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. 2013. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4(4):e00524-13. doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Mulla HM, Turrell L, Smith NM, Payne L, Baliji S, Züst R, Thiel V, Baker SC, Siddell SG, Neuman BW. 2014. Competitive fitness in coronaviruses is not correlated with size or number of double-membrane vesicles under reduced-temperature growth conditions. mBio 5(2):e01107-13. doi: 10.1128/mBio.01107-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oostra M, Hagemeijer MC, van Gent M, Bekker CP, te Lintelo EG, Rottier PJ, de Haan CA. 2008. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol 82:12392–12405. doi: 10.1128/JVI.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanjanahaluethai A, Chen Z, Jukneliene D, Baker SC. 2007. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology 361:391–401. doi: 10.1016/j.virol.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neuman BW, Joseph JS, Saikatendu KS, Serrano P, Chatterjee A, Johnson MA, Liao L, Klaus JP, Yates JR III, Wüthrich K, Stevens RC, Buchmeier MJ, Kuhn P. 2008. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J Virol 82:5279–5294. doi: 10.1128/JVI.02631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serrano P, Johnson MA, Chatterjee A, Neuman BW, Joseph JS, Buchmeier MJ, Kuhn P, Wüthrich K. 2009. NMR structure of the nucleic acid-binding domain of the SARS coronavirus nonstructural protein 3. J Virol 83:12998–13008. doi: 10.1128/JVI.01253-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serrano P, Johnson MA, Almeida MS, Horst R, Herrmann T, Joseph JS, Neuman BW, Subramanian V, Saikatendu KS, Buchmeier MJ, Stevens RC, Kuhn P, Wüthrich K. 2007. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J Virol 81:12049–12060. doi: 10.1128/JVI.00969-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurst KR, Ye R, Goebel SJ, Jayaraman P, Masters PS. 2010. An interaction between the nucleocapsid protein and a component of the replicase-transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J Virol 84:10276–10288. doi: 10.1128/JVI.01287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keane SC, Giedroc DP. 2013. Solution structure of mouse hepatitis virus (MHV) nsp3a and determinants of the interaction with MHV nucleocapsid (N) protein. J Virol 87:3502–3515. doi: 10.1128/JVI.03112-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurst KR, Koetzner CA, Masters PS. 2013. Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. J Virol 87:9159–9172. doi: 10.1128/JVI.01275-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker SC, Yokomori K, Dong S, Carlisle R, Gorbalenya AE, Koonin EV, Lai MM. 1993. Identification of the catalytic sites of a papain-like cysteine proteinase of murine coronavirus. J Virol 67:6056–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham RL, Denison MR. 2006. Replication of murine hepatitis virus is regulated by papain-like proteinase 1 processing of nonstructural proteins 1, 2, and 3. J Virol 80:11610–11620. doi: 10.1128/JVI.01428-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mielech AM, Chen Y, Mesecar AD, Baker SC. 2014. Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res 194:184–190. doi: 10.1016/j.virusres.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herold J, Siddell SG, Gorbalenya AE. 1999. A human RNA viral cysteine proteinase that depends upon a unique Zn2+-binding finger connecting the two domains of a papain-like fold. J Biol Chem 274:14918–14925. doi: 10.1074/jbc.274.21.14918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Putics A, Filipowicz W, Hall J, Gorbalenya AE, Ziebuhr J. 2005. ADP-ribose-1″-monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J Virol 79:12721–12731. doi: 10.1128/JVI.79.20.12721-12731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eriksson KK, Cervantes-Barragán L, Ludewig B, Thiel V. 2008. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1″-phosphatase, a viral function conserved in the alpha-like supergroup. J Virol 82:12325–12334. doi: 10.1128/JVI.02082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neuvonen M, Ahola T. 2009. Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. J Mol Biol 385:212–225. doi: 10.1016/j.jmb.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malet H, Coutard B, Jamal S, Dutartre H, Papageorgiou N, Neuvonen M, Ahola T, Forrester N, Gould EA, Lafitte D, Ferron F, Lescar J, Gorbalenya AE, de Lamballerie X, Canard B. 2009. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J Virol 83:6534–6545. doi: 10.1128/JVI.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ziebuhr J, Thiel V, Gorbalenya AE. 2001. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J Biol Chem 276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yount B, Denison MR, Weiss SR, Baric RS. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J Virol 76:11065–11078. doi: 10.1128/JVI.76.21.11065-11078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahn H, Palmenberg AC. 1996. Mutational analysis of the encephalomyocarditis virus primary cleavage. J Virol 70:6870–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo L, Masters PS. 2010. Evolved variants of the membrane protein can partially replace the envelope protein in murine coronavirus assembly. J Virol 84:12872–12885. doi: 10.1128/JVI.01850-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiller JJ, Kanjanahaluethai A, Baker SC. 1998. Processing of the coronavirus MHV-JHM polymerase polyprotein: identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a. Virology 242:288–302. doi: 10.1006/viro.1997.9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham RL, Sims AC, Brockway SM, Baric RS, Denison MR. 2005. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J Virol 79:13399–13411. doi: 10.1128/JVI.79.21.13399-13411.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockway SM, Lu XT, Peters TR, Dermody TS, Denison MR. 2004. Intracellular localization and protein interactions of the gene 1 protein p28 during mouse hepatitis virus replication. J Virol 78:11551–11562. doi: 10.1128/JVI.78.21.11551-11562.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bost AG, Carnahan RH, Lu XT, Denison MR. 2000. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J Virol 74:3379–3387. doi: 10.1128/JVI.74.7.3379-3387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madhugiri R, Fricke M, Marz M, Ziebuhr J. 2014. RNA structure analysis of alphacoronavirus terminal genome regions. Virus Res 194:76–89. doi: 10.1016/j.virusres.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su YP, Fan YH, Brian DA. 2014. Dependence of coronavirus RNA replication on an NH2-terminal partial nonstructural protein 1 in cis. J Virol 88:8868–8882. doi: 10.1128/JVI.00738-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang H, Feng M, Schroeder ME, Giedroc DP, Leibowitz JL. 2006. Putative cis-acting stem-loops in the 5′ untranslated region of the severe acute respiratory syndrome coronavirus can substitute for their mouse hepatitis virus counterparts. J Virol 80:10600–10614. doi: 10.1128/JVI.00455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown CG, Nixon KS, Senanayake SD, Brian DA. 2007. An RNA stem-loop within the bovine coronavirus nsp1 coding region is a cis-acting element in defective interfering RNA replication. J Virol 81:7716–7724. doi: 10.1128/JVI.00549-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Kang H, Liu P, Makkinje N, Williamson ST, Leibowitz JL, Giedroc DP. 2008. Structural lability in stem-loop 1 drives a 5′ UTR-3′ UTR interaction in coronavirus replication. J Mol Biol 377:790–803. doi: 10.1016/j.jmb.2008.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen SC, Olsthoorn RC. 2010. Group-specific structural features of the 5′-proximal sequences of coronavirus genomic RNAs. Virology 401:29–41. doi: 10.1016/j.virol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guan BJ, Su YP, Wu HY, Brian DA. 2012. Genetic evidence of a long-range RNA-RNA interaction between the genomic 5′ untranslated region and the nonstructural protein 1 coding region in murine and bovine coronaviruses. J Virol 86:4631–4643. doi: 10.1128/JVI.06265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y-N, Jeong YS, Makino S. 1993. Analysis of cis-acting sequences essential for coronavirus defective interfering RNA replication. Virology 197:53–63. doi: 10.1006/viro.1993.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luytjes W, Gerritsma H, Spaan WJM. 1996. Replication of synthetic interfering RNAs derived from coronavirus mouse hepatitis virus-A59. Virology 216:174–183. doi: 10.1006/viro.1996.0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozak M. 1987. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu HY, Guan BJ, Su YP, Fan YH, Brian DA. 2014. Reselection of a genomic upstream open reading frame in mouse hepatitis coronavirus 5′-untranslated-region mutants. J Virol 88:846–858. doi: 10.1128/JVI.02831-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. 2001. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip.’ J Gen Virol 82:1013–1025. http://vir.sgmjournals.org/content/82/5/1013.long. [DOI] [PubMed] [Google Scholar]
- 47.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. 2001. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol 82:1027–1041. http://vir.sgmjournals.org/content/82/5/1027.long. [DOI] [PubMed] [Google Scholar]
- 48.Minskaia E, Nicholson J, Ryan MD. 2013. Optimisation of the foot-and-mouth disease virus 2A co-expression system for biomedical applications. BMC Biotechnol 13:67. doi: 10.1186/1472-6750-13-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonilla PJ, Hughes SA, Piñón JD, Weiss SR. 1995. Characterization of the leader papain-like proteinase of MHV-A59: identification of a new in vitro cleavage site. Virology 209:489–497. doi: 10.1006/viro.1995.1281. [DOI] [PubMed] [Google Scholar]
- 50.Teng H, Piñón JD, Weiss SR. 1999. Expression of murine coronavirus recombinant papain-like proteinase: efficient cleavage is dependent on the lengths of both the substrate and the proteinase polypeptides. J Virol 73:2658–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P. 2005. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure 13:1665–1675. doi: 10.1016/j.str.2005.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stokes HL, Baliji S, Hui CG, Sawicki SG, Baker SC, Siddell SG. 2010. A new cistron in the murine hepatitis virus replicase gene. J Virol 84:10148–10158. doi: 10.1128/JVI.00901-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brockway SM, Denison MR. 2005. Mutagenesis of the murine hepatitis virus nsp1-coding region identifies residues important for protein processing, viral RNA synthesis, and viral replication. Virology 340:209–223. doi: 10.1016/j.virol.2005.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. 2006. Nidovirales: evolving the largest RNA virus genome. Virus Res 117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol 86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Züst R, Cervantes-Barragán L, Kuri T, Blakqori G, Weber F, Ludewig B, Thiel V. 2007. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog 3:e109. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S. 2009. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat Struct Mol Biol 16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lokugamage KG, Narayanan K, Huang C, Makino S. 2012. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol 86:13598–13608. doi: 10.1128/JVI.01958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cornillez-Ty CT, Liao L, Yates JR III, Kuhn P, Buchmeier MJ. 2009. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol 83:10314–10318. doi: 10.1128/JVI.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziebuhr J, Schelle B, Karl N, Minskaia E, Bayer S, Siddell SG, Gorbalenya AE, Thiel V. 2007. Human coronavirus 229E papain-like proteases have overlapping specificities but distinct functions in viral replication. J Virol 81:3922–3832. doi: 10.1128/JVI.02091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Freeman MC, Graham RL, Lu X, Peek CT, Denison MR. 2014. Coronavirus replicase-reporter fusions provide quantitative analysis of replication and replication complex formation. J Virol 88:5319–5327. doi: 10.1128/JVI.00021-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gadlage MJ, Denison MR. 2010. Exchange of the coronavirus replicase polyprotein cleavage sites alters protease specificity and processing. J Virol 84:6894–6898. doi: 10.1128/JVI.00752-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sulea T, Lindner HA, Purisima EO, Ménard R. 2006. Binding site-based classification of coronaviral papain-like proteases. Proteins 62:760–775. doi: 10.1002/prot.20802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dé I, Fata-Hartley C, Sawicki SG, Sawicki DL. 2003. Functional analysis of nsP3 phosphoprotein mutants of Sindbis virus. J Virol 77:13106–13116. doi: 10.1128/JVI.77.24.13106-13116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park E, Griffin DE. 2009. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 388:305–314. doi: 10.1016/j.virol.2009.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol 331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. 2006. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J Virol 80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fehr AR, Athmer J, Channappanavar R, Phillips JM, Meyerholz DK, Perlman S. 2015. The nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J Virol 89:1523–1536. doi: 10.1128/JVI.02596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Daugherty MD, Young JM, Kerns JA, Malik HS. 2014. Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet 10:e1004403. doi: 10.1371/journal.pgen.1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. 2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci U S A 103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]