

Abstract
Herpes simplex virus (HSV) undergoes a lytic infection in epithelial cells and a latent infection in neuronal cells, and epigenetic mechanisms play a major role in the differential gene expression under the two conditions. Herpes viron DNA is not associated with histones but is rapidly loaded with heterochromatin upon entry into the cell. Viral proteins promote reversal of the epigenetic silencing in epithelial cells while the viral latency-associated transcript promotes additional heterochromatin in neuronal cells. The cellular sensors that initiate the chromatinization of foreign DNA have not been fully defined. IFI16 and cGAS are both essential for innate sensing of HSV DNA, and new evidence shows how they work together to initiate innate signaling. IFI16 also plays a role in the heterochromatinization of HSV DNA, and this review will examine how IFI16 integrates epigenetic regulation and innate sensing of foreign viral DNA to show how these two responses are related.
Keywords: Epigenetics, DNA virus, IFI16, cGAS, Innate immunity
Introduction
When foreign DNA is introduced into mammalian cells, host cell responses recognize it as a potential “danger” and initiate a series of responses that attempt to control its expression and minimize its damage of the cell, including epigenetic silencing of the DNA to reduce its expression, induction of DNA damage responses, induction of innate responses, and recruitment of nuclear domain 10 (ND10) or PML body components that reduce its expression. The mechanism(s) by which foreign DNA is recognized and how it is distinguished from “self” DNA are not known, and the relationships between these responses to foreign DNA have not been defined. Herpes simplex virus (HSV) DNA evokes the same foreign DNA responses in that HSV DNA is not associated with histones in virions but becomes rapidly associated with chromatin upon entry into host cells, DNA damage response proteins and ND10 proteins localize near HSV genomes, and innate responses are induced in response to HSV DNA. However, little has been known until recently about the cellular sensors that recognize HSV DNA and initiate innate responses and chromatinization. Epigenetic mechanisms play a major role in the HSV gene expression during lytic infection of epithelial cells and latent infection of sensory neurons. Recent studies have shown a potential link between the innate responses to HSV and epigenetic regulation. Therefore, in this review I will summarize the epigenetic mechanisms regulating HSV gene expression and discuss the relationships between foreign DNA sensing, epigenetic regulation of that DNA, and innate immune responses. I will also examine how the cellular interferon-inducible protein 16 (IFI16) plays a role in sensing of herpesviral DNA and in integrating the induction of innate responses and epigenetic silencing of the viral DNA.
Epigenetic regulation of herpes simplex virus lytic and latent infection
HSV undergoes a lytic infection at the initial mucosal sites of infection followed by spread to sensory neurons where it establishes a latent infection (Roizman, Knipe, and Whitley, 2013). Lytic infection involves the expression of more than 80 viral genes while latent infection involves the abundant expression of only the latency-associated transcript (LAT) and miRNAs. The basis for the differential expression of HSV genes during lytic infection of epithelial cells versus the expression of only the LAT and miRNAs during latent infection has been an important unanswered question. It has been increasingly recognized that epigenetic mechanisms are central in regulation of eukaryotic gene expression (Gardner, Allis, and Strahl, 2011), and these regulatory mechanisms also apply to HSV gene expression (Knipe and Cliffe, 2008; Knipe et al., 2013).
Herpesviral DNA in the virion is not associated with histones, but instead the negative charges are apparently neutralized by the polyamine spermine. Upon entry into the host cell nucleus of dividing cells, HSV DNA is rapidly associated with histones (Cliffe and Knipe, 2008; Oh and Fraser, 2008), and heterochromatin modifications are rapidly put onto the chromatin by 1–2 hpi (Liang et al., 2009; Raja, Lee, and Knipe, manuscript in preparation). The HSV VP16 virion protein assembles into a transactivator complex with the host cellular proteins host cell factor 1 (HCF-1) and octamer binding factor 1 (Oct-1), and Oct-1 and VP16 bind to sequences in the viral immediate-early promoters, bringing the activator complex to the promoter, which activates transcription of the immediate-early genes (Kristie, article in this issue; Roizman and Zhou, article in this issue). HCF-1 recruits 1) the LSD1 histone demethylase to demethylate H3K9me2 and H3K9me, 2) the JMJD2 demethylases to demethylate H3K9me3, and 3) the H3K4 methyltransferases to put on this euchromatin modification. Thus, VP16 is required for euchromatin modifications as well as reduced histone loading on viral IE promoters (Herrera and Triezenberg, 2004). IE genes are then expressed, including infected cell protein 0 (ICP0). ICP0 promotes histone removal and acetylation of histones on E and L genes (Cliffe and Knipe, 2008), thereby allowing their transcription. ICP0 disrupts the CoRest-HDAC1 complex (Gu et al., 2005) and recruits the CLOCK histone acetyltransferase to the viral genome (Kalamvoki and Roizman, 2010). The mechanisms by which ICP0 affects histone methylation have not been defined yet.
In contrast, during latent infection of sensory neurons, the viral lytic genes are associated with heterochromatin (Wang et al., 2005), primarily facultative heterochromatin (Cliffe, Garber, and Knipe, 2009; Kwiatkowski, Thompson, and Bloom, 2009), and only the LAT promoter and enhancer are associated with euchromatin (Kubat et al., 2004a; Kubat et al., 2004b). In neurons, HSV DNA takes several days to become associated with histones (Cliffe, Coen, and Knipe, 2013; Wang et al., 2005), a much longer time than in epithelial cells or fibroblasts, likely because the pool of histones is smaller in the non-dividing neurons. From days 7–14 postinfection, histones accumulate on the viral lytic genes and heterochromatin modifications are put on the histones (Cliffe, Coen, and Knipe, 2013; Wang et al., 2005). Viral lytic gene expression is very inefficient because HCF-1 is in the cytoplasm of sensory neurons (Kristie, Vogel, and Sears, 1999), and VP16 may also not localize into the nucleus of the neurons. A neuron-specific promoter/enhancer drives the expression of LAT (Zwaagstra et al., 1990) and the precursor of a series of miRNAs (Kramer et al., 2011), some of which inhibit ICP4 and ICP0 expression (Umbach et al., 2008). LAT expression reduces lytic gene expression in the neurons (Garber, Schaffer, and Knipe, 1997); furthermore, LAT expression increases H3K9me2, H3K9me3, and H3K27me3 modifications on viral chromatin (Cliffe, Garber, and Knipe, 2009; Wang et al., 2005). One study reported that LAT decreased H3K27me3 modification (Kwiatkowski, Thompson, and Bloom, 2009), but in this study the levels of H3K27me3 reported on cellular genes were different for the wild-type samples versus the LAT promoter mutant samples. In total, the literature supports the concept that LAT reduces lytic gene expression during acute infection (Garber, Schaffer, and Knipe, 1997) and latent infection (Chen et al., 1997) of sensory neurons, promotes heterochromatin on viral lytic genes in sensory neurons (Cliffe, Garber, and Knipe, 2009; Wang et al., 2005), and reduces acute infection death of neurons and increases neuronal survival (Nicoll et al., 2012; Thompson and Sawtell, 2001). Thus, our current working model is that HSV gene products regulate the epigenetic modifications on the HSV genome (Figure 1), with VP16 and ICP0 promoting euchromatin during lytic infection and LAT promoting heterochromatin during latent infection. Many important questions on the mechanisms of epigenetic regulation of HSV gene expression remain as exciting areas for future study.
Figure 1. Model for epigenetic regulation of the lytic versus latent infection decision by HSV.
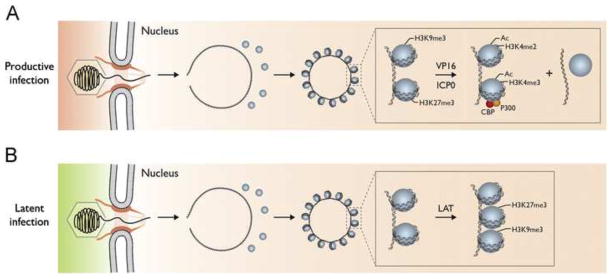
(A). Following infection of epithelial cells, the capsid is transported to the nuclear pore where the viral DNA is released into the nucleus where it rapidly circularizes and becomes associated with histones bearing heterochromatin marks. VP16 from the virion tegument forms a complex with HCF-1 and Oct1 that binds to viral IE promoters and HCF-1 recruits histone modification enzymes and chromatin remodeling complexes that decrease histone association with viral IE genes and increase euchromatin marks on the remaining associated histones. ICP0 is expressed as an IE protein and it promotes similar processes on the rest of the genome. (B). Following infection of neuronal cells, the capsid is also transported to the nuclear pore where the viral DNA is released into the nucleus where it rapidly circularizes and becomes associated with histones. VP16 cannot be transported into the neuronal nucleus, and HCF-1 is not localized in the nucleus so viral IE genes are not transcribed efficiently. Instead, the latency-associated transcript is expressed and it promotes the association of facultative heterochromatin marks on the viral chromatin (Copyright, Lynne Chang, Priya Raja, and David Knipe).
Attempts to cure individuals of latent viruses such as HIV have focused on activating the virus from latency by epigenetic drugs and then treatment with antiviral drugs (Shirakawa et al., 2013). Reactivation of HSV from latent infection in the peripheral nervous system and potentially in the central nervous system has the potential for harm to the individual; therefore, the concept of locking in HSV latency with epigenetic drugs has been raised (Liang et al., 2009). Recent studies have shown that rabbits, guinea pigs, and mice treated with an epigenetic drug that inhibits the LSD1 histone demethylase show reduced reactivation and increased levels of heterochromatin in vivo (Hill et al., 2014). If safe epigenetic drugs can be discovered that block HSV at this very early stage of reactivation, they could have great therapeutic potential.
Sensing of Foreign DNA
Mammalian cells have a number of receptors at various sites within the cell that detect different kinds of foreign nucleic acids and initiate innate immune responses (Iwasaki and Medzhitov, 2013). These include Toll-like receptors (TLRs) in endosomes, RIG-like receptors in the cytosol, and DNA sensors in the cytosol and nucleus. Organisms have also evolved mechanisms to detect foreign DNA and degrade it or restrict its expression. Bacteria have modification-restriction systems to detect foreign DNA and cleave it (Youell and Firman, 2012) as well as CRISPR-CAS systems to cleave and delete sequences from the foreign DNA (Sander and Joung, 2014; Kennedy and Cullen, article in this issue). Mammalian cells assemble chromatin on transfected DNA and silence its expression within a few days (Cereghini and Yaniv, 1984). Similarly, as discussed above, viral DNA genomes, such as those of the herpesviruses, are rapidly associated with histones upon infection of cells, but the viruses encode proteins that combat the epigenetic silencing of their genome (Knipe et al., 2013).
Sensing of HSV DNA, innate responses, and epigenetic regulation of foreign DNA
What are the sensors that detect the foreign DNA and initiate the loading of heterochromatin on that DNA to silence it? The first evidence for DNA sensors came from studies looking at the induction of innate responses by detection of foreign DNA. There are a number of DNA sensors that have been reported to detect viral DNA, and these have been reviewed in a number of excellent reviews (Orzalli and Knipe, 2014; Paludan and Bowie, 2013; Dempsey and Bowie article, this volume), to which the reader is referred for more details about these sensors. The goal of this mini-review is to discuss the relationship between DNA sensing, innate immune responses, and epigenetic regulation.
DNA sensing in general leads to activation of cytokine expression through the STING-IRF-3/NF-κB pathway. Induction of the antiviral type I IFN response is regulated by a cellular signaling cascade that results in the activation of IFN regulatory factor-3 (IRF-3) and nuclear factor–kappa B (NF-κB) transcription factors. A critical component of this signaling cascade is stimulator of IFN genes (STING) (Ishii and Akira, 2006), an endoplasmic reticulum–localized, trans-membrane protein that bridges DNA-sensing mechanisms to downstream signaling events. Upon microbial DNA stimulation, STING re-localizes to distinct intracellular foci (Saitoh et al., 2009) and promotes TANK-binding kinase 1 (TBK1)-dependent phosphorylation of IRF-3 by a direct interaction with both proteins (Tanaka and Chen, 2012). Phosphorylated IRF-3 dimerizes, translocates to the nucleus, and binds to IFN-stimulated regulatory elements (ISREs) in the promoters of IRF-3-responsive genes, including type I IFNs. IFN is subsequently secreted from cells and can act in an autocrine or paracrine fashion to upregulate IFN-stimulated genes (ISGs) via the type I IFN receptor and a JAK-STAT signaling cascade (reviewed in Stark and Darnell, 2012). A subset of ISGs can also be induced independently of IFN, but IFN significantly amplifies this induction (Wathelet, Berr, and Huez, 1992).
A number of foreign DNA sensors have been reported that are located within the cell at various sites within different cells. 1) TLR9, located in endosomes of plasmacytoid dendritic cells, is activated by nonmethylated CpG DNA, and TLR9−/− mice are more susceptible to HSV infection. 2) The DNA-dependent activator of interferon (DAI), located in the cytosol, is required for IFN response to HSV-1 as shown by siRNA depletion (Takaoka et al., 2007) but not in knockout cells (Ishii and Akira, 2006). 3) RNA polymerase III (Ablasser et al., 2009; Chiu, Macmillan, and Chen, 2009) was reported to be required for sensing of HSV-1 DNA (Chiu, Macmillan, and Chen, 2009), but others have not seen a requirement (Melchjorsen et al., 2010; Monroe et al., 2014; Unterholzner et al., 2010). 4) Absent in Melanoma 2 or AIM2 (Fernandes-Alnemri et al., 2009; Hornung et al., 2009) is the prototypic member of the PYHIN family of proteins, named because of their pyrin protein-protein interaction/signaling domain and HIN (hematopoietic IFN inducible nuclear antigen) DNA binding domain. AIM2 is involved in the sensing of vaccinia virus and murine cytomegalovirus DNA but not HSV DNA (Hornung et al., 2009; Rathinam et al., 2010) to activate inflammasomes. 5) Interferon-inducible protein 16 or IFI16 (Unterholzner et al., 2010) is, like AIM2, a member of the PYHIN family of proteins because it has a pyrin domain and two HIN domains. IFI16 was identified originally as a cytoplasmic DNA-binding protein and innate sensor (Unterholzner et al., 2010), but IFI16 can be nuclear or cytoplasmic depending on the cell type (Choubey, Deka, and Ho, 2008). Nuclear localization of IFI16 is now known to be regulated by acetylation (Li et al., 2012). IFI16 is capable of binding DNA directly through sequence-independent contacts with the sugar phosphate backbone (Jin et al., 2012), providing a biochemical basis for DNA sensing. IFI16 is required for both activation of inflammasomes (Johnson, Chikoti, and Chandran, 2013; Kerur et al., 2011) and induction of IFN-β in HSV-infected cells (Li et al., 2012; Orzalli, DeLuca, and Knipe, 2012). 6) Cyclic guanosine monophosphate-adenosine monophosphate synthase or cGAS is required for IRF-3 activation in response to transfected DNA and HSV-1 infection (Sun et al., 2013; Wu et al., 2013). Because IFI16 and cGAS are the most extensively studied and best documented sensors of HSV DNA in infected cells, most of the rest of my discussion will focus on these two DNA sensors.
HSV infection induces innate responses by a number of mechanisms (Roizman, Knipe, and Whitley, 2013). Among these, DNA-sensing plays a role in type I interferon responses to HSV infection. UV-inactivated HSV and HSV recombinants that do not express viral proteins induce interferon-β in human fibroblasts, but expression of viral proteins or just ICP0 prevented induction of IFN-β (Eidson et al., 2002; Mossman et al., 2001; Preston, Harman, and Nicholl, 2001). Consistent with this, infection of human fibroblasts with the HSV-1 d109 recombinant virus that expresses no viral proteins in normal cells leads to IFI16-dependent IFN-β induction, and IFN-β induction is dependent on viral DNA entry into the nucleus (Li et al., 2012; Orzalli, DeLuca, and Knipe, 2012). IFI16 must have a functional nuclear localization signal and be localized into the nucleus to sense HSV-1 DNA and induce IFN-β expression (Li et al., 2012). These results are consistent with a model in which HSV DNA is released from the capsid into the nucleus, and nuclear IFI16 binds to the viral DNA (Figure 2). Interestingly, as an exception to this, in human macrophages HSV capsid proeins are ubiquitinated in the cytoplasm and the capsid is degraded by the proteasome, but IFI16 is cytoplasmic in these cells and required for IFN-β induction (Horan et al., 2013).
Figure 2. Model of nuclear HSV-1 DNA sensing, innate signaling, and epigenetic regulation by IFI16 and inhibition by ICP0.

HSV capsids in the cytoplasm traffic to nuclear pores where viral DNA is released into the nucleus. The viral DNA rapidly circularizes, and nuclear IFI16 binds to the viral DNA and multimerizes, initiating a nuclear-to-cytoplasmic signaling cascade that activates IRF-3, which dimerizes and translocates to the nucleus. Immediate-early expression of ICP0 promotes degradation of IFI16 to inhibit subsequent IRF-3 signaling and IFN-α expression. The multimerized IFI16 on the viral DNA also recruits histone modification complexes that add H3K9me3 modifications to the viral chromatin, resulting in epigenetic silencing of the viral genes. (Copyright, Megan Orzalli and David Knipe).
Following binding to HSV DNA in the nucleus, IFI16 is proposed to undergo a conformational change that allows oligomerization as shown by intranuclear filaments or aggregates (Li, Chen, and Cristea, 2013; Orzalli et al., 2013). The formation of higher-order complexes of these signaling molecules is consistent with the “supramolecular organizing centers” or SMOCs that are assembled with other innate signaling molecules (Kagan, Magupalli, and Wu, 2014). This initiates a signal that activates STING in the cytoplasm, which activates TBK-1 phosphorylation, phosphorylation and dimerization of IRF-3, and IRF-3 localization into the nucleus to bind to the IFN-β gene promoter and activate its transcription (Orzalli, DeLuca, and Knipe, 2012) (Figure 2). The nature of the signal that moves from the nucleus to the cytoplasm to connect IFI16 and STING is not known. In some studies, IFI16 does not move detectably from the nucleus (Li, Chen, and Cristea, 2013; Li et al., 2012; Orzalli, DeLuca, and Knipe, 2012) while in others IFI16 is reported to move to the cytoplasm and co-localize with inflammasome structures (Johnson, Chikoti, and Chandran, 2013; Kerur et al., 2011). Therefore, the mechanism by which IFI16 signals to STING remains a mystery. IFI16 may also play a direct transcriptional role in innate immune induction in that it promotes IFN-α reporter gene expression, increases RNA polymerase II loading on the IFN-α gene promoter, and can bind to the IFN-α promoter sequences (Thompson et al., 2014), although the specificity of the in vitro binding remains to be demonstrated.
Sensing of HSV DNA and epigenetic silencing
Three of the components of the ND10 bodies, PML, hDaxx, and Sp1, which are localized near incoming HSV genomes early during infection, have been shown to restrict viral IE gene expression (Boutell and Everett, 2013). Although the PML-containing ND10 structures or bodies have been described variously as localizing “near” the viral genomes (Maul, Ishov, and Everett, 1996) or “at the site of viral DNA” (Maul, 1998), there no evidence that PML or other ND10 proteins associate directly with viral DNA. Furthermore, the mechanisms of this viral restriction have not been defined. IFI16 acts as a restriction factor for HSV-1 and HCMV replication in human embryonic lung fibroblasts (Gariano et al., 2012). The mouse homolog of IFI16 has not been defined because mice contain ten PYHIN genes while humans encode four, but the closest functional homolog appears to be p204, and depletion of p204 in the mouse cornea increased HSV-1 replication (Conrady et al., 2012).
We found that depletion of IFI16 in HFF cells resulted in increased replication and IE gene expression by an ICP0− mutant virus and that ectopic expression of IFI16 in U2OS cells resulted in decreased IE gene expression (Orzalli et al., 2013). Depletion of IFI16 resulted in decreased H3K9me3 modification and increased H3K4me3 modification on the ICP4 gene promoter, consistent with the increased ICP4 expression. The restriction effect of IFI16 was also observed on transfected plasmids; therefore, the results were consistent with IFI16 recruiting histone modification enzymes that place H3K9 heterochromatin marks on foreign DNA introduced into the cell in an unchromatinized form. These results were recently confirmed by studies in which the IFI16 gene was deleted in U2OS cells, and the IFI16 knockout cells showed enhanced expression of HSV IE genes, reduced heterochromatin, and increased euchromatin and RNA polymerase II loading on IE genes in wildtype virus-infected cells (Johnson et al., 2014).
How does IFI16 distinguish between viral and host DNA in the infected cell nucleus? We initially hypothesized that the IFI16 specificity for viral DNA in the nucleus could involve IFI16 recognizing the underchromatinized viral DNA to initiate innate responses (Orzalli, DeLuca, and Knipe, 2012). Consistent with this, we observed that IFI16 restricted expression of the SV40 large T antigen when expressed from a transfected plasmid but did not restrict T antigen expression in SV40 virus-infected cells (Orzalli et al., 2013). Because SV40 DNA is assembled in a mini-chromosome in the virion, this may make it resistant to IFI16-mediated silencing. A biochemical explanation for this specificity of IFI16 was provided by a recent paper which showed that IFI16 binds cooperatively to double-stranded DNA that is 33 base pairs or longer and does not bind efficiently to DNA the size of the linker DNA between nucleosomes or at a transcription bubble (Morrone et al., 2014). Those authors also speculated that IFI16 would not bind efficiently to cellular DNA that is assembled in nucleosomes.
Viral evasion of IFI16
The herpesviruses have been shown to evade the effects of IFI16 by two mechanisms. First, ICP0 promotes the degradation of IFI16 in HFF cells in a RING domain- and proteasome-dependent manner (Johnson, Chikoti, and Chandran, 2013; Orzalli, DeLuca, and Knipe, 2012). Although one study reported that ICP0 was not necessary or sufficient for IFI16 degradation (Cuchet-Lourenco et al., 2013), most of the experiments in this study used tumor cells where IFI16 is often not functional and not activated by binding to HSV DNA (Li et al., 2012; Orzalli et al., 2013). Further studies in our lab have shown that IFI16 has a short half-life, and HSV infection can lead to both ICP0-dependent and ICP0-independent loss of IFI16, the latter likely involving vhs-dependent degradation of IFI16 mRNA (Orzalli, Broekema, and Knipe, manuscript in preparation). Second, HCMV pUL83 binds to the pyrin domain of IFI16 and prevents its multimerization and initiation of the IRF-3 signaling pathway (Li, Chen, and Cristea, 2013). Therefore, both viruses target IFI16 to remove it or block its signaling, supporting the importance of IFI16 in initiating innate and epigenetic responses to herpesviral infection.
cGAS
In 2013, the Chen laboratory reported the discovery of the cGAS enzyme, whose catalytic activity is activated upon binding to DNA to synthesize cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) (Sun et al., 2013; Wu et al., 2013). cGAMP binds to STING and activates the IRF-3 signaling pathway. This novel sensor and signaling mechanism was required for HSV-1 activation of IRF-3 dimerization (Wu et al., 2013). Cells from cGAS−/− mice show reduced antiviral responses, and the cGAS−/− mice are more susceptible to DNA virus infection (Li et al., 2013). These results showed that cGAS is required for IRF-3 signaling and IFN-β induction in HSV-1 infected cells and mice. Surprisingly, Schoggins et al. (2014) showed that cGAS−/− mice are also more susceptible to RNA virus infection, indicating that cGAS activity may be pan-antiviral (Schoggins et al., 2014). Schoggins et al. (2014) also observed a reduction in the basal levels of IFN-β and several ISGs in cGAS-deficient bone marrow-derived macrophages, implicating cGAS in the basal homeostasis of innate signaling (Schoggins et al., 2014). cGAMP can also spread to neighboring cells via gap junctions to activate STING (Ablasser et al., 2013). Many proposed DNA sensors, including IFI16, are IFN inducible; therefore, cGAS depletion could indirectly affect the ability of additional DNA sensors to induce antiviral cytokine production. No microbial gene products that specifically inhibit the activity of cGAS have been documented in publications, but we have recently identified an HSV gene product that appears to be required for wildtype HSV inhibition of cGAS activity (Broekema and Knipe, manuscript in preparation).
cGAS and IFI16
In combination, these results have raised the question of how the cytosolic cGAS and the nuclear IFI16 DNA sensors are both required for IRF-3 signaling and interferon-β induction in HSV-infected cells. Our own results have shown that both IFI16 and cGAS are required for IFN-β induction by HSV-1 infection in HFFs (Orzalli et al., manuscript in revision). However, cGAS plays a unique role in human HFF cells in that it shows low levels of cGAMP production in HSV-infected cells. Instead, it localizes partially in the nucleus, interacts with IFI16, and stabilizes IFI16 against proteasomal degradation. Therefore, cGAS may have different activities in different cell types and under different conditions.
Role of IFI16 and cGAS in sensing DNA of other viruses
Similar to the situation with HSV, both cGAS and IFI16 have been reported to be required for IRF-3 signaling in HIV-infected cells. Detection of HIV reverse transcriptase products required cGAS for IRF-3 dimerization (Gao et al., 2013). In another study, IFI16 was found to sense HIV DNA and to restrict HIV-1 gene expression and replication (Jakobsen et al., 2013). Induction of pyroptosis in quiescent CD4+ T cells requires IFI16 (Monroe et al., 2014). Therefore, there is evidence that cGAS and IFI16 are both involved in innate responses to and epigenetic silencing of HIV infection, and further studies are needed to define the precise role of each in the various cell types that HIV infects.
Normal roles of IFI16 in human cells
IFI16 has been implicated in innate responses to and restriction of microbial DNA, but what physiological roles does IFI16 play in cells not exposed to microbes? IFI16 has also been implicated in cell senescence and cell growth control. IFI16 levels increase in aging cells (Xin, Pereira-Smith, and Choubey, 2004; Angelova and Knipe, unpublished), and this has been implicated in the aging process. Furthermore, IFI16 is often not expressed or defective in cancer cells (Li et al., 2012; Orzalli et al., 2013; Orzalli, DeLuca, and Knipe, 2012; Xin et al., 2003). When introduced into tumor cells, IFI16 inhibits cell growth (Xin et al., 2003; Tarnita, Orzalli and Knipe, unpublished results), consistent with a role in controlling normal cell growth. It will be interesting to see how the functional mechanisms of these IFI16 activities compare with the IFI16 mechanisms that function during innate sensing and epigenetic silencing of foreign DNA and to see whether they are related or distinct functions of IFI16.
Conclusion
Host cell mechanisms attempt to silence HSV DNA through epigenetic mechanisms, but HSV and other viruses fight back. In epithelial cells the HSV VP16 virion protein promotes euchromatin and reduced histone loading on viral immediate-early genes to allow their transcription, and the immediate-early ICP0 protein then promotes euchromatin and reduced histone loading on viral early and late genes to along their expression. In neurons, LAT promotes epigenetic silencing of viral lytic genes. As a result, viral gene products drive the epigenetic landscape on the viral genome during both lytic and latent infection. However, little has been known about the host receptor that “senses” the foreign viral DNA and promotes heterochromatin assembly on the viral genome. The nuclear IFI16 protein is required for the major part of the IRF-3 signaling in response to HSV infection in the absence of viral protein expression, identifying it as a potential sensor of HSV DNA. A major remaining question is how innate signaling pathways connect the nuclear IFI16 with the cytoplasmic STING. IFI16 also promotes the attachment of heterochromatic histone H3 K9 trimethylation marks to viral chromatin and the resulting epigenetic silencing of the viral lytic genes. A second major question is the mechanism(s) by which IFI16 recruits the histone modification enzymes to the viral chromatin for this process. It is interesting to note that while IFI16 is hypothesized to bind to unchromatinized viral DNA to initiate innate signaling, it promotes the chromatinization of the viral DNA, so the innate signaling would be down-regulated. Perhaps this provides a mechanism for controlling the levels of innate responses, and an extrapolation of this is that genetic defects in the epigenetic silencing mechanisms could lead to uncontrolled inflammation. In addition to IFI16, cGAS has also been shown to play a role in sensing of HSV DNA, and it has been puzzling how the cytoplasmic cGAS and the nuclear IFI16 could both be required for sensing of HSV DNA. Recent results have shown that in human fibroblasts, cGAS is partially localized to the nucleus, interacts with IFI16, and stabilizes the IFI16 protein. A third major question is why cGAS shows low levels of enzymatic activity in HSV-infected cells and how viral gene products might inhibit its activity. In conclusion, sensing of nuclear HSV DNA plays an important role in the host innate response to viral infection and in epigenetic regulation of viral gene expression, and there are many exciting avenues of research that remain to be explored to fully understand this critical area of interaction of herpesviruses with the host cell.
HSV lytic and latent gene expression is regulated differentially by epigenetic processes.
The sensors of foreign DNA have not been defined fully.
IFI16 and cGAS cooperate to sense viral DNA in HSV-infected cells.
IFI16 plays a role in both innate sensing of HSV DNA and in restricting its expression.
Acknowledgments
I thank Priya Raja and Megan Orzalli for preparation of figures, Megan Orzalli and Nicole Broekema for comments on the manuscript, and Patrick T. Waters for assistance in preparation of the manuscript. Research in the laboratory of the author cited in this review was supported by NIH grants AI063106 and AI098681 from NIAID, and DE023909 from the National Institute of Dental and Cranial Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–72. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–4. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Everett RD. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol. 2013;94:465–81. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- Cereghini S, Yaniv M. Assembly of transfected DNA into chromatin: Structural changes in the origin-promoter-enhancer region upon replication. EMBO J. 1984;3:1243–1253. doi: 10.1002/j.1460-2075.1984.tb01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997;71:5878–84. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell Biochem Biophys. 2009;138:576–91. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choubey D, Deka R, Ho SM. Interferon-inducible IFI16 protein in human cancers and autoimmune diseases. Front Biosci. 2008;13:598–608. doi: 10.2741/2705. [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Coen DM, Knipe DM. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. MBio. 2013:4. doi: 10.1128/mBio.00590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83:8182–90. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Knipe DM. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol. 2008;82:12030–8. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 2012;5:173–83. doi: 10.1038/mi.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, Everett RD. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol. 2013;87:13422–32. doi: 10.1128/JVI.02474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral Infection. J Virol. 2002;76:2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–13. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–6. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber DA, Schaffer PA, Knipe DM. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol. 1997;71:5885–5893. doi: 10.1128/jvi.71.8.5885-5893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariano GR, Dell’Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012;8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A. 2005;102:7571–6. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–96. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Quenelle DC, Cardin RD, Vogel JL, Clement C, Bravo FJ, Foster TP, Bosch-Marce M, Raja P, Lee JS, Bernstein DI, Krause PR, Knipe DM, Kristie TM. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci Transl Med. 2014;6:265ra169. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol. 2013;190:2311–9. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol. 2006;27:525–32. doi: 10.1016/j.it.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Innate Responses to Viral Infections. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 189–213. [Google Scholar]
- Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, Xiao TS, Mikkelsen JG, Mogensen TH, Paludan SR. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A. 2013;110:E4571–80. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, Hornung V, Latz E, Bowie AG, Fitzgerald KA, Xiao TS. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561–71. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B. IFI16 Restricts HSV-1 Replication by Accumulating on the HSV-1 Genome, Repressing HSV-1 Gene Expression, and Directly or Indirectly Modulating Histone Modifications. PLoS Pathog. 2014;10:e1004503. doi: 10.1371/journal.ppat.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol. 2013;87:5005–18. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol. 2014;14:821–6. doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamvoki M, Roizman B. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A. 2010;107:17721–6. doi: 10.1073/pnas.1012991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011;9:363–75. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–21. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. Snapshots: chromatin control of viral infection. Virology. 2013;435:141–56. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology. 2011;417:239–47. doi: 10.1016/j.virol.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Vogel JL, Sears AE. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci U S A. 1999;96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Amelio AL, Giordani NV, Bloom DC. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol. 2004a;78:12508–18. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Tran RK, McAnany P, Bloom DC. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol. 2004b;78:1139–49. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DL, Thompson HW, Bloom DC. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol. 2009;83:8173–81. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen J, Cristea IM. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe. 2013;14:591–9. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A. 2012;109:10558–63. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–4. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–7. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG. Nuclear domain 10, the site of DNA virus transcription and replication. BioEssays. 1998;20:660–667. doi: 10.1002/(SICI)1521-1878(199808)20:8<660::AID-BIES9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Maul GG, Ishov AM, Everett RD. Nuclear Domain 10 as preexisting potential replication start sites of herpes simplex virus type 1. Virology. 1996;217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- Melchjorsen J, Rintahaka J, Soby S, Horan KA, Poltajainen A, Ostergaard L, Paludan SR, Matikainen S. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J Virol. 2010;84:11350–8. doi: 10.1128/JVI.01106-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. 2014;343:428–32. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci U S A. 2014;111:E62–71. doi: 10.1073/pnas.1313577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. Herpes simplex virus triggers and then disarms a host antiviral response. J Virol. 2001;75:750–758. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll MP, Proenca JT, Connor V, Efstathiou S. Influence of herpes simplex virus 1 latency-associated transcripts on the establishment and maintenance of latency in the ROSA26R reporter mouse model. J Virol. 2012;86:8848–58. doi: 10.1128/JVI.00652-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Fraser NW. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J Virol. 2008;82:3530–7. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Conwell SE, Berrios C, Decaprio JA, Knipe DM. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A. 2012;109:E3008–17. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Knipe DM. Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol. 2014;68:477–92. doi: 10.1146/annurev-micro-091313-103409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–80. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CM, Harman AN, Nicholl MJ. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J Virol. 2001;75:8909–8916. doi: 10.1128/JVI.75.19.8909-8916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ. Herpes Simplex Viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 1823–1897. [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–6. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–5. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013;21:277–85. doi: 10.1016/j.tim.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–14. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–5. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA. Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem. 2014;289:23568–81. doi: 10.1074/jbc.M114.554147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol. 2001;75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–3. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A. 2005;102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet MG, Berr PM, Huez GA. Regulation of gene expression by cytokines and virus in human cells lacking the type-I interferon locus. Eur J Biochem. 1992;206:901–10. doi: 10.1111/j.1432-1033.1992.tb16999.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–30. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Curry J, Johnstone RW, Nickoloff BJ, Choubey D. Role of IFI 16, a member of the interferon-inducible p200-protein family, in prostate epithelial cellular senescence. Oncogene. 2003;22:4831–40. doi: 10.1038/sj.onc.1206754. [DOI] [PubMed] [Google Scholar]
- Xin H, Pereira-Smith OM, Choubey D. Role of IFI 16 in cellular senescence of human fibroblasts. Oncogene. 2004;23:6209–17. doi: 10.1038/sj.onc.1207836. [DOI] [PubMed] [Google Scholar]
- Youell J, Firman K. Mechanistic insight into Type I restriction endonucleases. Front Biosci. 2012;17:2122–39. doi: 10.2741/4041. [DOI] [PubMed] [Google Scholar]
- Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsler SL. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol. 1990;64:5019–5028. doi: 10.1128/jvi.64.10.5019-5028.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]