

Abstract
Background
The recently identified RASSF1 locus is located within a 120-kilobase region of chromosome 3p21.3 that frequently undergoes allele loss in lung and breast cancers. We explored the hypothesis that RASSF1 encodes a tumor suppressor gene for lung and breast cancers.
Methods
We assessed expression of two RASSF1 gene products, RASSF1A and RASSF1C, and the methylation status of their respective promoters in 27 non-small-cell lung cancer (NSCLC) cell lines, in 107 resected NSCLCs, in 47 small-cell lung cancer (SCLC) cell lines, in 22 breast cancer cell lines, in 39 resected breast cancers, in 104 nonmalignant lung samples, and in three breast and lung epithelial cultures. We also transfected a lung cancer cell line that lacks RASSF1A expression with vectors containing RASSF1A complementary DNA to determine whether exogenous expression of RASSF1A would affect in vitro growth and in vivo tumorigenicity of this cell line. All statistical tests were two-sided.
Results
RASSF1A messenger RNA was expressed in nonmalignant epithelial cultures but not in 100% of the SCLC, in 65% of the NSCLC, or in 60% of the breast cancer lines. By contrast, RASSF1C was expressed in all nonmalignant cell cultures and in nearly all cancer cell lines. RASSF1A promoter hypermethylation was detected in 100% of SCLC, in 63% of NSCLC, in 64% of breast cancer lines, in 30% of primary NSCLCs, and in 49% of primary breast tumors but in none of the nonmalignant lung tissues. RASSF1A promoter hypermethylation in resected NSCLCs was associated with impaired patient survival (P = .046). Exogenous expression of RASSF1A in a cell line lacking expression decreased in vitro colony formation and in vivo tumorigenicity.
Conclusion
RASSF1A is a potential tumor suppressor gene that undergoes epigenetic inactivation in lung and breast cancers through hypermethylation of its promoter region.
Allelic loss of human chromosome 3p is an early and frequent event in the development of several cancers, including lung and breast cancers (1–5). Identification of a series of nested 3p21.3 homozygous deletions in small-cell lung cancers (SCLCs) directed an intensive effort to positionally clone tumor suppressor genes from a 630-kilobase (kb) region, which was recently narrowed to a 120-kb subregion by identification of a breast cancer homozygous deletion (6–8). Sequencing the entire 630-kb region identified at least 25 genes, several of which may encode tumor suppressor genes for lung cancer (7). Nine genes are located in or on the border of the breast cancer-defined subregion. One of these genes, which spans 7.6 kb of genomic DNA, has a predicted Ras-association domain and homology to the Ras-effector Nore1 (Fig. 1); it has, therefore, been termed “RASSF1” (9,10).
Fig. 1.

Map of the RASSF1 locus, transcripts, and protein domains. A) The exon–intron structure of the RASSF1 locus with the location of the CpG islands in the predicted promoter regions (the locations of which are shown by double-headed arrows) of RASSF1A and RASSF1C. RASSF1A transcription is predicted to come from the most centromeric promoter region located within a CpG island and begins with exon 1A. RASSF1F also commences at this promoter but is missing exon 1C. Transcription of RASSF1C is predicted to begin in the most telomeric promoter region, which is approximately 2 kilobases from that of RASSF1A and begins with exon 1. Blocks represent exons; lines represent introns. B) Schematic of the RASSF1A transcript and predicted protein-sequence domains. The location of the various primers (PKCDF, NF, R182, and R292) used for isoform-specific reverse transcription (RT)–polymerase chain reaction (PCR) analyses are indicated. Tick marks identify the exon boundaries. The potential src homology 3 (SH3)-binding region, putative diacylglycerol (DAG)-binding domain, PEST sequence, Ras-association domain, and ataxia-telangiectasia-mutated (ATM) phosphorylation site are labeled. C) Schematic of the RASSF1C transcript and predicted protein-sequence domains. The locations of the various primers (NOX3, R182, and R292) used for isoform-specific RT–PCR analyses are indicated. D) Schematic of the RASSF1F transcript and predicted protein-sequence domains.
The RASSF1 gene encodes two major transcripts, RASSF1A and RASSF1C, which are produced by alternative promoter selection and alternative messenger RNA (mRNA) splicing. RASSF1A is encoded by RASSF1 exons 1A, 1C, and 2–5. RASSF1C is encoded by RASSF1 exons 1–5 (Fig. 1). The start sites for RASSF1A and RASSF1C are approximately 2 kb apart and have two independent CpG island-containing putative promoter regions. RASSF1A is predicted to encode a 39-kd peptide that contains an N-terminal diacylglycerol (DAG)-binding domain and a Ras-association domain (Fig. 1). RASSF1C is predicted to encode a 32-kd peptide that lacks a DAG-binding domain but contains a Ras-association domain (7,11). Immediately adjacent to the DAG-binding domain of RASSF1A is a sequence PxxP, which is the minimal sequence required for an src homology 3-binding domain. RASSF1A has a central linker that contains a number of prolines, as well as acidic and hydroxyl-bearing residues. These regions, called PEST sequences, are found in proteins that are rapidly turned over by ubiquitination-dependent pathways (12). For RASSF1C, the aminoterminal region unique to this isoform is enriched for these PEST sequences. Within the PEST sequences common to both RASSF1A and RASSF1C is a serine residue that is phosphorylated in vitro by DNA-dependent ataxia-telangiectasia-mutated (ATM) and ataxia-telangiectasia-related kinases (13). The Ras-association domain is more than 50% identical and more than 70% similar to the carboxyl terminal 225 residues of mouse Nore1 (10). The Ras-association domain, consisting of a core of 90 amino acids, is flanked on the amino terminal side by a region homologous with a region found in Nore1 and Caenorhabditis elegans orthologue T24F1.3 protein.
In this article, we characterized RASSF1A and RASSF1C as potential tumor suppressor genes in lung and breast cancers. Because loss of gene expression can be caused by tumor-acquired aberrant methylation, we assessed the methylation status of the RASSF1A promoter region in these tumors (14). In addition, we tested the ability of RASSF1A to suppress the malignant phenotype. Previously, Dammann et al. (15) showed that the RASSF1A promoter is hypermethylated in lung cancer cells and that exogenous expression of RASSF1A suppresses tumorigenesis in nude mice. We have confirmed and extended those findings by analyzing the expression and methylation status of both the RASSF1A and RASSF1C genes in lung and breast cancers.
Methods
Patient Population
Resected lung tumor samples and clinical data were collected from patients after obtaining appropriate institutional review board approval and patients' written informed consent. Primary tumor samples and corresponding noninvolved lung tissues were obtained from 107 patients with non-small-cell lung carcinoma (NSCLC) who had received curative resection surgery at the Prince Charles Hospital, Brisbane, Australia, from June 1990 through March 1993, and for whom clinical and survival data of 5 or more years were available (16,17). Among the 107 patients, there were 76 males and 31 females (range, 28–81 years; mean age, 61 years at diagnosis). Among the patients, 61 had stage I cancers, 21 had stage II, 24 had stage IIIA, and one had stage IIIB (18). Histologically, there were 45 adenocarcinomas, 43 squamous cell carcinomas, 11 adenosquamous carcinomas, four large-cell carcinomas, three atypical carcinoids, and one typical carcinoid. Ninety-eight patients were smokers, with a mean exposure of 31 pack-years, and nine were never smokers or nonsmokers.
We also obtained 39 primary breast tumors from patients aged 31–84 years undergoing breast cancer treatment in The University of Texas Southwestern Hospital system. Among the patients, three had stage I cancers, 15 had stage IIA, two had stage IIB, eight had stage IIIA, five had stage IIB, and six had stage IV. Histologically, there were 30 infiltrating ductal carcinomas, four invasive lobular carcinomas, one lobular carcinoma in situ, two ductal carcinomas in situ, and two breast adenocarcinomas at metastatic sites. Clinical information was obtained by retrospective review of clinical records.
Cell Lines and Cell Cultures
Lung and breast tumor cell lines generated by us have been described previously (19–21). Complementary DNAs (cDNAs) and genomic DNAs were obtained from cell lines, most of which have been deposited in the American Type Culture Collection (ATTC) (Manassas, VA), that represented the spectrum of lung cancer histologies. These cell lines include the following: (all Hxxxx lines have the prefix National Cancer Institute [NCI]-) SCLCs (i.e., H69, H82, H128, H146, H182, H187, H196, H209, H249, H289, H290, H345, H378, H524, H526, H592, H735, H738, H740, H748, H774, H841, H847, H862, H889, H1092, H1105, H1184, H1304, H1339, H1450, H1607, H1618, H1672, H1688, H1963, H2028, H2029, H2081, H2108, H2171, H2195, H2227, and HCC970) and NSCLCs (i.e., H23, H28, H125, H157, H226, H358, H720, H727, H838, H920, H1155, H1299, H1437, H1466, H1573, H1648, H1770, H1792, H1819, H1838, H1993, H2009, H2052, H2077, H2087, H2347, H2452, H2882, H2887, HCC44, HCC78, HCC95, HCC193, HCC515, HCC827, and HCC1171). Breast cancer cell lines used in these studies were the following: HTB19, HTB20, HTB22, HTB23, HTB24, HTB25, HTB26, HTB27, HTB121, HTB130, HTB131, HTB132, HTB133 (all HTB lines are available from the ATCC), HCC38, HCC70, HCC202, HCC712, HCC1007, HCC1143, HCC1187, HCC1395, HCC1419, HCC1428, HCC1500, HCC1569, HCC1739, HCC1806, HCC1937, HCC1599, HCC1954, HCC2157, HCC2185, HCC2218, HCC2688, and HCC2713. Normal human bronchial epithelial (NHBE) and small-airway epithelial (SAE) cell cultures were obtained from Clonetics (San Diego, CA) and were grown and harvested as directed by the vendor.
Expression Analysis of RASSF1 Isoforms
The identification of the RASSF1 gene (initially called 123F2) and its major isoforms RASSF1C and RASSF1A was reported as part of the overall characterization of the genes in the larger 630-kb 3p21.3 homozygous-deletion region (7) (Fig. 1, A–C).
Sequence information from exons 1A and 3 was used to design the forward primer PKCDF (5′-GGCGTCGTGCGCAAAGGCC-3′) and the reverse primer R182 (5′-GGGTGGCTTCTTGCTGGAGGG-3′) (Fig. 1, C). This primer pair was used in reverse transcription (RT)-polymerase chain reaction (PCR) screens of lung, heart, and pancreatic tissue-specific cDNA libraries (Clontech Laboratories, Inc., Palo Alto, CA). The conditions used TaqGold (The Perkin-Elmer Corp., Norwalk, CT) with 1× TaqGold buffer adjusted to 2 mM MgCl2. All reactions used a 70 °C–60 °C touchdown, with 5% dimethyl sulfoxide for 35 rounds (denaturation for 30 seconds, annealling for 30 seconds, and extension for 60 seconds) of PCR. The RASSF1A cDNA sequence is identical to that of the RASSF1C cDNA from the second exon to the carboxyl terminus, but the two cDNAs have different 5′ exons (RASSF1A, GenBank Accession #AF102770: exons 1Aand 1C; RASSF1C, GenBank Accession #AF040703: exon 1 [Fig. 1]). We also isolated tissue-specific isoforms from the heart (RASSF1D, GenBank Accession #AF102771) and the pancreas (RASSF1E, GenBank Assession #AF102772) cDNA libraries.
Primers derived from exon–intron junctions (sequences available online at the Journal website) were used for genomic DNA single-strand conformation polymorphism mutation analysis of the coding regions of RASSF1A on a panel of NSCLC, SCLC, and breast cancer cell line DNAs. DNA was prepared from tumors and cell lines by standard methods (22), and aberrantly migrating fragments were sequenced as described previously (16,23).
RNA Analysis
Isoform-specific RT–PCR assays were used for analysis of RASSF1A and RASSF1C expression. Primers for RASSF1C were Nox3 (5′-CTGCAGC-CAAGAGGACTCGG-3′) and R182 and for RASSF1A were either PKCDF or NF (5′-TGCAAGTTCACCTGCCAC-3′) and R182 (Fig. 1, C). Total RNA was isolated from previously described lung and breast cancer cell lines grown in RPMI-1640 medium supplemented with 5% fetal bovine serum (complete medium) (19–21) by Trizol extraction (Life Technologies, Inc. [GIBCO BRL], Rockville, MD). Four micrograms of total RNA was reverse transcribed by use of GIBCO-BRL Superscript First Strand cDNA Kit. All cDNA preparations were tested for the ability to amplify a nontranscribed genomic sequence immediately upstream of the first exon of the RASSF1A transcript. Any cDNAs that produced a product from this sequence were discarded because they were contaminated with genomic DNA.
We also assessed the expression of RASSF1A after exposure to 5-aza-2′-deoxycytidine, a drug that inhibits DNA methylation. We exposed subconfluent cultures of the RASSF1A-nonexpressing NSCLC line NCI-H157 to 0.5 μM 5-aza-2′-deoxycytidine for 48 hours, after which we isolated total RNA and performed RT–PCR for RASSF1A, RASSF1C, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). RT–PCR of GAPDH transcripts was performed with the use of forward primer GAPDH-C (5′-CATGACAACTTTGGTATCGTG-3′) and reverse primer GAPDH-D (5′-GTGTCGCTGTTGAAGTCAGA-3′). RT– PCR products were separated by agarose gel electrophoresis and visualized after staining with ethidium bromide.
Methylation Analysis
The methylation status of the presumed RASSF1A and RASSF1C promoter regions was determined by methylation-specific PCR. Genomic DNAs from lung cancer cell lines not expressing RASSF1A (NCI lines H1299, H1184, H1304, H841, H2108, and H128) or expressing RASSF1A (H1792 and H2009) were modified by sodium bisulfite treatment as described previously (24,25). Bisulfite treatment converts cytosine bases to uracil bases but has no effect on methylcytosine bases. PCR amplification followed by sequencing of the PCR fragments identifies specific CpG dinucleotides in the promoter region that are modified by methylation (24,26,27). PCR primers (sequences available online at the Journal website) were designed to amplify genomic sequences in the presumed promoter regions of RASSF1A (cosmid Luca12; GenBank Accession #AC002481 nucleotides 17 730–18 370) and RASSF1C (GenBank Accession #AC002481 nucleotides 21 022–21 152 and 21 194–21 332). The resulting PCR fragments were sequenced by automated fluorescence-based DNA sequencing to determine the methylation status.
The data on CpG methylation in RASSF1A-nonexpressing lung cancer cell lines (data available online at the Journal website) were used to design methylation-specific PCR (24) primers for the RASSF1A 5′ promoter region: The primers to detect the methylated form were 5′-GGGTTTTGCGAGAGCGCG-3′ (forward) and 5′-GCTAACAAACGCGAACCG-3′ (reverse), and the primers to detect the unmethylated form were 5′-GGTTTTGTGAGAGTGTGTTTAG-3′ (forward) and 5′-CACTAACAAACACAAACCAAAC-3′ (reverse). Each primer set generated a 169-base-pair (bp) product. Methylation-specific PCR cycling conditions consisted of one incubation of 15 minutes at 95 °C, followed by 40 cycles of a 30-second denaturation at 94 °C, 50 seconds at an annealing temperature (64 °C for methylation-specific and 59 °C for unmethylated-specific primers), a 30-second extension at 72 °C, and a final extension at 72 °C for 10 minutes. PCR products were separated in 2% agarose gels. Lymphocyte DNA, methylated in vitro by CpG (SssI) methylase (New England Biolabs, Inc., Beverly, MA) following the manufacturer's directions, was used as a positive control. A water blank was used as a negative control.
Generation of Transfectants
RASSF1A cDNA was cloned into pcDNA3.1+ (Invitrogen Corp., Carlsbad, CA), resequenced to confirm that the cDNAs were in the correct orientation and reading frame, transcribed, and translated in vitro with commercial kits (Clonetech Laboratories, Inc.). The expression vector containing RASSF1A produced a 42-kd protein, and the vector containing RASSF1C produced a 32-kd protein on sodium dodecyl sulfate–polyacrylamide gels, close to their respective predicted molecular masses of 39 and 32 kd (data not shown). Expression vectors in pcDNA3.1 for mutant and wild-type p53 and their transfection and activity have been described previously (28).
The RASSF1A expression vector was transfected into NSCLC NCI-H1299 cells expressing RASSF1C, but not RASSF1A, by use of Lipofectamine plus (Life Technologies, Inc.) according to the manufacturer's recommendations. For transient transfection studies, approximately 5 × 105 NSCLC NCI-H1299 cells, harvested from 80%–90% confluent cultures in complete medium, were transfected with 1 μg of purified plasmid DNA. Samples were plated in a minimum of triplicate, and cells were collected 48 hours after transfection. Because the pcDNA3.1+ expression vector contains a neomycin resistance gene, clones expressing RASSF1A were selected in complete medium supplemented with G418 (800 μg/mL). Stable clones were maintained in complete medium supplemented with G418 (600 μg/mL). We confirmed that the clones were expressing the transfected RASSF1A gene by isolating total RNA from individual clones and performing RT–PCR as described above. We also transfected NCI-H1299 cells with the vector containing no inserts and isolated stable clones.
The RASSF1A and RASSF1C cDNAs were also cloned in the retroviral vector pBABEpuro and were resequenced to confirm that the genes were in the correct sequence and orientation (29). Virus was prepared in the 293 cell-based Phoenix packaging cell line as described previously (29) from cells infected either with the vector alone or with constructs containing the RASSF1A or RASSF1C cDNA. Culture supernatants were collected by centrifugation at 500g at 37 °C for 10 minutes and used to infect NSCLC NCI-H1299 cells as described previously (29). Because the viral vector contains a puromycin resistance gene, infected cells were selected with 1 μg/mL of puromycin for 7 days. Cells surviving the selection and containing the transgene were pooled, and total cell extracts were made. Western blot analysis was performed as described previously (30) to verify protein expression of the transfected genes. The protein bands were visualized with the Pierce SuperSignal Kit (Pierce Chemical Co., Rockford, IL).
Tumorigenicity Testing
The in vitro growth characteristics of NSCLC NCI-H1299 clones that express RASSF1A were tested for anchorage-dependent and anchorage-independent (soft agar) growth. After 48 hours of growth in nonselective medium, transiently transfected NSCLC NCI-H1299 cells were detached with trypsin and diluted, usually 10- to 25-fold, in complete medium containing 800 μg/mL of G418 and plated into fresh 100-mm dishes. The medium was changed twice weekly. After 14 days, the medium was removed, the plates were washed with phosphate-buffered saline (PBS), and the colonies were stained with 1% methylene blue in 50% (vol/vol) ethanol. For the anchorage-independent, soft agar-growth assays, 1000 RASSF1A-expressing cells were suspended and plated in 0.33% Noble agar (Sigma Chemical Co., St. Louis, MO) in complete medium supplemented with 600 μg/mL G418 and layered over a 0.50% agar base in complete medium. After 21 days, colonies greater than 0.2 mm in diameter were counted.
For retrovirally infected cells, anchorage-independent growth assays were performed as follows: 10 000 viable selected cells from each infection were plated in 0.33% soft agar over a 0.50% agar base in Dulbecco's modified Eagle medium (Life Technologies, Inc.) with 10% heat-inactivated fetal bovine serum. After 21 days, colonies greater than 0.2 mm in diameter were counted.
We also tested the ability of RASSF1A-infected cells to grow in vivo in nude mice. Male BALB/c nude (nu/nu) 3- to 6-week-old mice (Charles River Laboratories, Wilmington, DE) were irradiated on day 0 of the experiment in groups of five animals by a 5-minute exposure to 350 cGy from a cesium source. The next day, each mouse was given an injection subcutaneously on its flank with 0.2 mL of sterile PBS containing 107 viable parental, vector control, or RASSF1A retroviral-infected NSCLC NCI-H1299 tumor cells. Mice were monitored every 2–3 days for tumor size; once tumors reached greater than 1500 mm3, the mice were killed. All animal care was in accord with institutional guidelines.
Antibody Preparation
The entire RASSF1C open reading frame was used to make a glutathione S-transferase (GST) fusion protein, which was expressed in Escherichia coli by use of an established procedure (31), and was used to make rabbit polyclonal antibodies to be described in detail elsewhere. For the western blot analysis, the antiserum was used at a 1 : 1000 dilution in 5% nonfat milk in PBS. Specificity was determined by western blotting of H1299 cells transfected with vector (negative control) and the various RASSF1-expression constructs (e.g., see Fig. 6).
Fig. 6.
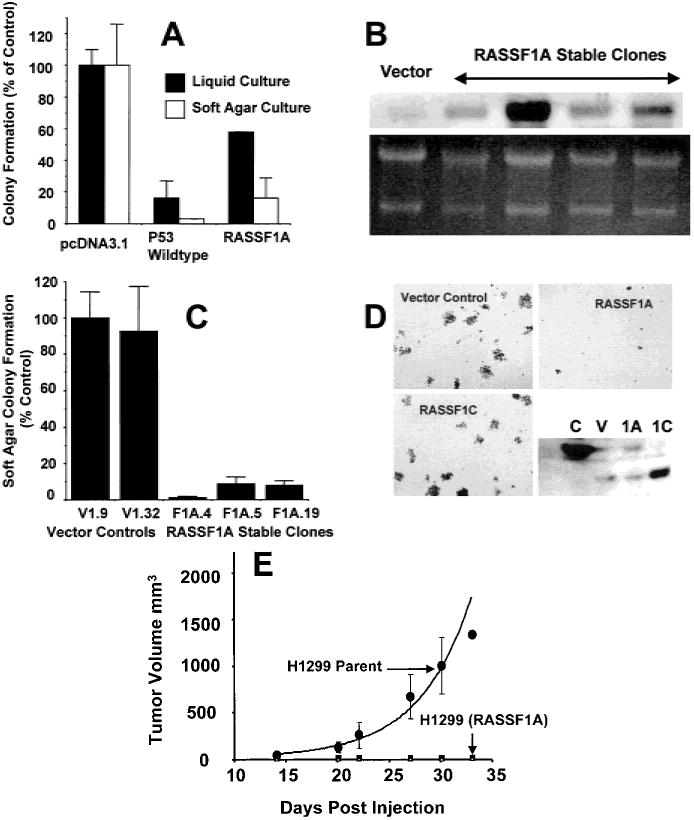
Effect of RASSF1A on the in vitro and in vivo growth of the non-small-cell lung carcinoma (NSCLC) cell line NCI-H1299. A) Anchorage-dependent and anchorage-independent colony formation after transfection of NCI-H1299 cells with the empty vector (pcDNA3.1+) or pcDNA3.1+ expression vectors containing wild-type p53 or RASSF1A. For analysis of anchorage-dependent growth, after 2 days in nonselective growth medium, transfected NCI-H1299 cells were diluted into 100-mm2 dishes with selective medium. Transfected cells were plated in liquid medium (for anchorage-dependent assays) or soft agar (for anchorage-independent assays) containing 800 μg/mL of G418. Colonies were stained with methylene blue in anchorage-dependent experiments after 14 days. Results represent the average of eight to 12 experiments in liquid medium and three soft-agar experiments. Standard deviations are shown or are less than 2%. Solid bars = anchorage-dependent growth (95% confidence interval [CI] = 0 to 36 for wt-p53 (wild-type) and 52 to 60 for RASSF1A); open bars = anchorage-independent growth (95% CI = 0 to 6 for wild-type (wt)-p53 and 0 to 39 for RASSF1A). B) Northern blot analysis of the RASSF1A expression in stable clones of NCI-H1299 cells transfected with the pcDNA3.1+ vector or pcDNA3.1+ containing RASSF1A complementary DNA (cDNA). The vector control (vector) and four separate clones with various RASSF1A messenger RNA levels are shown. Several of these clones were used in the anchorage-independent growth assay shown in D. Ethidium bromide staining of the ribosomal RNA is shown as a loading control. The clones were also verified to express the RASSF1A isoform by reverse transcription–polymerase chain reaction with the use of isoform-specific primers (data not shown). C) Soft-agar (anchorage-independent) colony formation in stable clones of NCI-H1299 cells transfected with the pcDNA3.1+ vector or pcDNA3.1+ containing RASSF1A cDNA. The means and standard deviations are shown. For each of the RASSF1A-expressing clones, the 95% CI = 0 to 4 for F1A.4, 2 to 16 for F1A.5, and 3 to 14 for F1A.19. D) NCI-H1299 cells were infected with the pBABEpuro retrovirus expression vectors containing either the vector control or the RASSF1A or RASSF1C cDNAs. Infected cells (10 000 per plate) were suspended in 0.33% agar, and the suspension was layered over a 0.5% agar base. Colonies greater than 0.2 mm in diameter were counted after 21 days. The lower right panel shows a representative western blot, developed with a rabbit antibody to the RASSF1-glutathione S-transferase fusion protein, to verify the expression of the RASSF1 proteins. C = positive control generated by transient transfection of NCI-H1299 cells with pcDNA3.1+ containing RASSF1A cDNA; V = infection of NCI-H1299 cells with the retroviral vector control (note runover from positive control); 1A = infection of NCI-H1299 cells with the retroviral vector containing RASSF1A; and 1C = infection of NCI-H1299 cells with the retroviral vector containing RASSF1C. E) Effect of RASSF1A on the in vivo growth of NCI-H1299 cells. Approximately 107 viable NCI-H1299 cells expressing RASSF1A were injected into the flanks of each of five previously irradiated BALB/c (nu/nu) nude mice. Tumor size was monitored over time, and size is shown in cubic millimeters. The average volume of tumors grown in more than 20 mice that were given an injection of vector-transfected NCI-H1299 cells is shown (H1299 parent). Mice that were given an injection of RASSF1A-infected NCI-H1299 cells grew no measurable tumors.
Statistical Analysis
Statistical analysis was performed by use of χ2 and Fisher's exact tests for differences between groups. Overall survival curves were calculated by use of the Kaplan–Meier method, and survival curves were compared with the log-rank statistic (32). All analyses, including univariate, multivariate, and Cox analyses, were performed by use of SPSS Windows version 9.0.1 (SPSS Inc., Chicago, IL). All statistical tests were two-sided.
Results
Characterization of the RASSF1 Gene
To determine if the RASSF1A gene was mutated in lung and breast cancers, we performed extensive mutational analysis of the RASSF1A isoform with the use of single-strand conformation polymorphism assays on genomic DNA. We had previously found no RASSF1C mutations in 77 lung cancer cell line samples (7). By use of the RASSF1A sequence as a reference, we found several polymorphisms, including the following: codon 21 (AAG to CAG), Lys to Gln; codon 28 (CGT to CGA), no amino acid change; codon 49 (GGC to GGT), no amino acid change; codon 53 (CGC to TGC), Arg to Cys; codon 129 (GAC to GAG), Asp to Glu; codon 133 (GCT to TCT), Ala to Ser; and codon 325 (TAT to TGT), Tyr to Cys.
Expression of RASSF1A and RASSF1C in Lung and Breast Cancer Cell Lines
RASSF1 is located within a region frequently affected by allele loss during growth of lung, breast, head and neck, kidney, and cervical tumors (1–5). We investigated whether RASSF1A and RASSF1C are expressed in lung and breast cancer cell lines. We used isoform-specific RT–PCR 0 of RASSF1A and RASSF1C in lung and breast tumor cell lines and in normal lung and breast epithelial cultures (Fig. 2). RASSF1A was expressed in normal lung epithelial cultures (NHBE and SAE cultures), in a normal breast epithelial culture (Fig. 2, C), but not in 32 (100%) of 32 SCLC lines, in 17 (65%) of 26 NSCLC cell lines, and in 15 (60%) of 25 (60%) breast cancer cell lines. Representative data are shown in Fig. 2. By contrast, RASSF1C was expressed in nearly all of the lung and breast cancer cell lines tested, with the exceptions of several lung and breast cancer lines with known homozygous deletions that include the RASSF1 locus. In resected lung adenocarcinomas, RASSF1A was expressed in only two of five cancers, while RASSF1C was expressed in all cancers (Fig. 2, C).
Fig. 2.
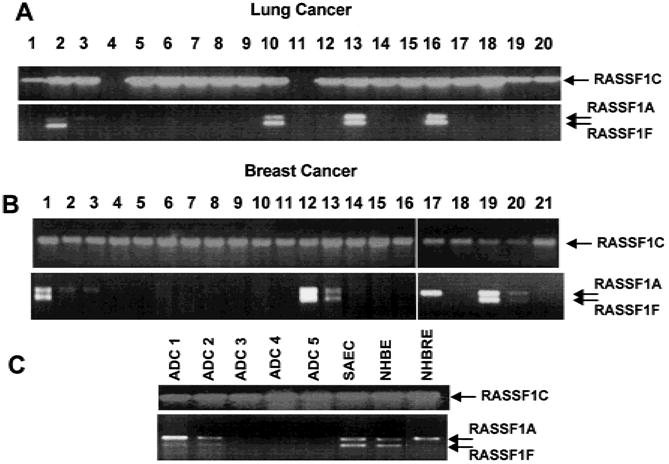
RASSF1A and RASSF1C messenger RNA levels detected by isoform-specific reverse transcription–polymerase chain reaction (RT–PCR) in a sampling of lung cancer cell lines (A), breast cancer lines (B), and resected lung tumors and normal human lung and breast epithelial cultures (C). All RT– PCR products were separated on 2% agarose gels and were identified by staining with ethidium bromide. Arrows indicate location of transcripts. A) Lung cancer lines tested in lanes: 1 = H157; 2 = H358; 3 = H727; 4 = H740; 5 = H748; 6 = H838; 7 = H1184; 8 = H1299; 9 = H1304; 10 = H1437; 11 = H1450; 12 = H1770; 13 = H1792; 14 = H1963; 15 = H1993; 16 = H2009; 17 = H2077; 18 = H2108; 19 = HHCC44; and 20 = HCC78. B) Breast cancer lines tested in lanes: 1 = HCC38; 2 = HCC1187; 3 = HTB19; 4 = HTB20; 5 = HTB22; 6 = HTB23; 7 = HTB24; 8 = HTB25; 9 = HTB26; 10 = HTB27; 11 = HTB121; 12 = HTB129; 13 = HTB130; 14 = HTB131; 15 = HTB132; 16 = HTB133; 17 = HCC1395; 18 = HCC1428; 19 = HCC1569; 20 = HCC1806; and 21 = HCC2157. C) Resected lung adenocarcinoma samples (ADC 1–5) and cultures of normal small-airway epithelial cells (SAECs), normal human bronchial epithelial (NHBE) cultures, and normal human breast epithelial (NHBRE) cultures.
During RT–PCR analysis for RASSF1A, we frequently noted two closely spaced bands in RASSF1A-expressing tumors and in NHBE cultures (Fig. 2). We sequenced these RT–PCR products and found that the larger band corresponded to RASSF1A, while the smaller product represented a different transcript, RASSF1F (GenBank Accession #AF286217). This transcript skips exon 1C to produce an mRNA encoding a predicted truncated peptide of 92 amino acids ending within the DAG-binding domain (Fig. 1, D). The biologic function, if any, of RASSF1F is unknown. In nearly all of the samples, RASSF1F is expressed when RASSF1A is expressed. However, in some breast cancers and normal breast epithelial cultures (see Fig. 2 for examples), RASSF1A is expressed without RASSF1F expression.
Methylation Status of the RASSF1A Promoter Region
Aberrant promoter methylation in tumors has been found to lead to the loss of gene expression of several tumor suppressor genes in human cancers (14). To assess whether the loss of RASSF1A expression in lung cancer was the result of promoter hypermethylation, we determined the CpG methylation status in the 5′ region of RASSF1A (from −800 to +600 bp of the predicted RASSF1A transcript start site) by sequencing sodium bisulfite-modified DNA from eight lung cancer cell lines. All of the six lung cancer cell lines not expressing RASSF1A exhibited methylation of almost all CpG dinucleotide sites in the putative promoter region (data available online at the Journal website). The two lung cancer cell lines that did express RASSF1A either were not methylated at these CpG sites or showed limited methylation. By contrast, no methylation was found in CpG sites in the presumed RASSF1C promoter region of these eight cell lines.
To confirm that promoter hypermethylation contributes to the lack of expression of RASSF1A in the lung cancer cell lines, we assessed the effect of 5-aza-2′-deoxycytidine, a drug that inhibits DNA methylase, on RASSF1A expression. We exposed the RASSF1A-nonexpressing NSCLC line NCI-H157 to 5-aza-2′-deoxycytidine and found re-expression of RASSF1A by this cell line but little or no change in the expression of the housekeeping gene GAPDH or in the expression of RASSF1C (Fig. 3).
Fig. 3.
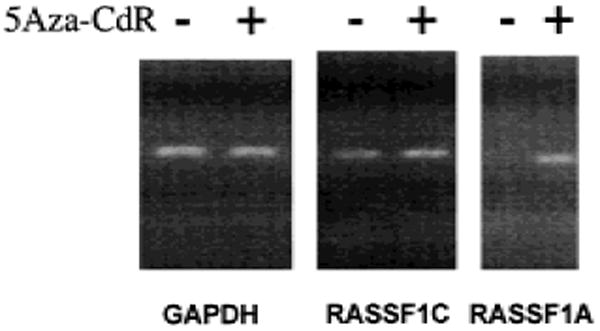
Expression of RASSF1A after treatment of lung cancer cells with 5-aza-2′-deoxycytidine (5Aza-CdR). NCI-H157, a non-small-cell lung carcinoma (NSCLC) cell line that expresses RASSF1C but not RASSF1A, was grown in the presence (+ lanes) and absence (− lanes) of 0.5 μM 5Aza-CdR for 48 hours. Total RNA was isolated, complementary DNA was prepared, and isoform-specific reverse transcription–polymerase chain reaction was performed for RASSF1A, RASSF1C, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control.
Methylation-Specific PCR Analysis of the Promoter Region of RASSF1A in Lung and Breast Cancers
To determine the methylation status of the promoter region of RASSF1A in primary lung and breast cancers, we used methylation-specific PCR analysis. Genomic DNA from a large number of primary resected NSCLCs, paired lung tissues resected from the same patients but not involved with the cancer, primary resected breast cancers, and a large panel of lung and breast cancer cell lines were treated with sodium bisulfite and tested for the presence of methylated and unmethylated CpG dinucleotides in the promoter region of RASSF1A (Fig. 4). All of the primary resected NSCLCs and non-tumor-paired samples contained unmethylated promoter sequences, which were expected because these resected tumors were not microdissected and were contaminated with stromal cells. However, 32 (30%) of 107 primary NSCLCs, 47 (100%) of 47 SCLC lines, and 19 (49%) of 39 primary breast cancers exhibited the methylated RASSF1A allele (Fig. 4; Table 1). By contrast, no methylated alleles were detected in 104 paired resected nonmalignant lung tissues (Fig. 4; Table 1).
Fig. 4.
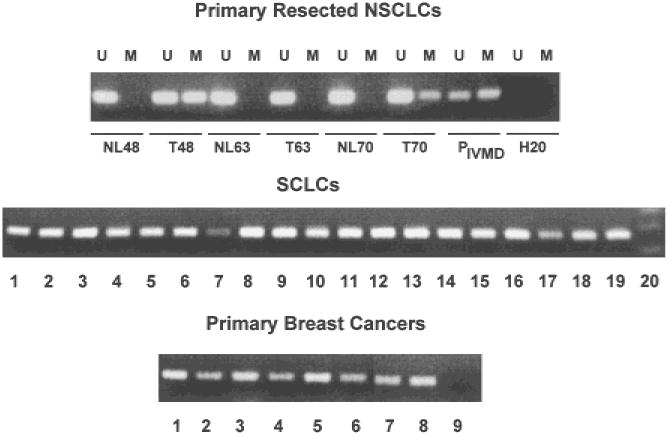
Methylation-specific polymerase chain reaction (PCR) for the detection of methylated RASSF1A 5′ CpG sequences in primary resected non-small-cell lung carcinomas (NSCLCs) and their accompanying normal lung tissue (upper panel), small-cell lung carcinoma (SCLC) cell lines (middle panel), and primary breast cancers (lower panel). Representative samples are shown. For resected NSCLCs, U = results with primers specific for unmethylated sequences; M = results with primers specific for methylated sequences. NL = normal lung tissue; T = tumor; P = results with peripheral blood lymphocyte DNA, which is unmethylated or in vitro methylated (IVMD); and H20 = negative controls with water blanks. For SCLCs, each lane shows the PCR results for the methylated sequences from a different cell line. Lane 20 is negative control. For the breast cancers, each lane shows the PCR results for methylated sequences from a different sample. PCR products were separated on 2% agarose gels, and bands were detected after staining with ethidium bromide.
Table 1. Frequency of methylation-specific polymerase chain reaction assay for detection of RASSF1A CpG island-methylated alleles in lung and breast cancers.
| DNA sample source* | No. tested | No. of methylation alleles (positive) (%) |
|---|---|---|
| Primary resected NSCLCs | 107 | 32 (30%) |
| Corresponding nonmalignant lung | 104 | 0 (0%) |
| NSCLC lines | 27 | 17 (63%) |
| SCLC lines | 47 | 47 (100%) |
| Primary resected breast cancers | 39 | 19 (49%) |
| Breast cancer lines | 22 | 14 (64%) |
NSCLC = non-small-cell lung carcinoma; SCLC = small-cell lung carcinoma.
We found a high frequency of methylated RASSF1A alleles in the panel of lung and breast cell cancer lines (Table 1). Because the lung and breast cancer cell lines represent essentially clonal populations of cancer cells without contaminating normal cells, we tabulated the frequency of the methylated and unmethylated RASSF1A alleles (Table 2). While the lung and breast cancer lines often derive from clinically more aggressive lesions than the average population of tumors (19–21), our previous studies (20,21) have shown that cancer cell lines continue to retain the genetic alterations found in the uncultured cancer specimens from which they were derived. The presence of only the methylated allele is consistent with either the methylation of both parental alleles or the retention of the methylated allele and the loss of the unmethylated 3p allele. All of the SCLC cell lines showed only the methylated allele or lacked RASSF1A entirely because of a homozygous deletion, consistent with the nearly universal 3p21.3 allele loss in SCLC (1,20,33). Of the NSCLC cell lines, 13 (48%) of 27 (Table 2) had only the methylated RASSF1A allele, and 10 (37%) of 27 had only the unmethylated allele, consistent with a lower rate of 3p21.3 allele loss in this tumor type (1). Likewise, 10 (45%) of 22 samples (Table 2) of breast cancer cell lines had only the methylated allele, and seven (32%) of 22 had only the unmethylated allele, again consistent with the rate of 3p21.3 allele loss found in breast cancer (21). As expected, two tumor lines shown previously to have homozygous deletions involving the 3p21.3 region were negative for both the methylated and the unmethylated allele (Table 2) (7,8).
Table 2. Presence of methylated and unmethylated RASSF1A alleles in 97 lung and breast cancer cell lines*.
| RASSF1A CpG genotype | SCLC | NSCLC | BCCL | Total | |
|---|---|---|---|---|---|
|
| |||||
| Methylated allele | Unmethylated allele | ||||
| + | + | 0 | 4 | 4 | 8 |
| + | − | 47 | 13 | 10 | 70 |
| − | + | 0 | 10 | 7 | 17 |
| − | − | 1 | 0 | 1 | 2† |
| Total | 48 | 27 | 22 | 97 | |
SCLC = small-cell lung cancer; NSCLC = non-small-cell lung cancer; BCCL = breast cancer cell lines.
The two tumor cell lines with methylation-specific polymerase chain reaction genotypes lacking both methylated and unmethylated alleles (SCLC line NCI-H740 and breast cancer line HCC1500) were known to have homozygous deletions including the RASSF1 locus in chromosome region 3p21.3.
For a subset of 61 lung and breast cancer cell lines, we performed both expression and methylation analysis and found a statistically significant association (P<.001, Fisher's exact test) between the presence of methylated RASSF1A alleles and the loss of RASSF1A expression. In 12 samples, RASSF1A was expressed in the absence of a methylated allele; in 44 samples, RASSF1A was not expressed in the presence of a methylated allele; in four samples, RASSF1A was not expressed in the absence of methylated allele (presumably because of some other inactivating mechanism); and in one sample (a breast cancer cell line), RASSF1A was expressed in the presence of both a methylated and an unmethylated allele. These data show the critical association of RASSF1A methylation with loss of RASSF1A expression.
We next assessed whether there was any association between RASSF1A promoter methylation and clinical findings in the patients with primary NSCLC. We found no statistically significant association between RASSF1A methylation and age, sex, tumor-node-metastasis (TNM) pathologic stage (18), or tumor histology in 107 resected NSCLCs (data not shown). In addition, we found no statistically significant association between RASSF1A methylation and age, TNM pathologic stage, tumor histology, estrogen or progesterone receptor status, or HER2/Neu expression in 39 primary resected breast cancers (data not shown).
Survival among lung cancer patients differed by the methylation status of RASSF1A (P = .046) (Fig. 5). Also, by univariate analysis, in this group of 107 patients with NSCLC treated with an attempt at curative surgical resection, tumor (T1, T2, and T3), lymph node stage (N1 and N2), and reported weight loss were statistically significant predictors of adverse survival. Neither smoking history (yes/no or pack-years with 40 pack-year cutoff) nor treatment differences (all patients had surgical resection of lobectomy or pneumonectomy, and only five had prior radiotherapy or chemotherapy) accounted for the adverse survival. Because a multivariate analysis is of limited use with a small sample size, we performed a Cox proportional hazards regression analysis by use of RASSF1A methylation and the main univariate factors (tumor, lymph node stage, and weight loss). RASSF1A methylation was not found to be an independent prognostic factor of survival. However, this result could be due to small numbers because even lymph node stage (a known prognostic factor) was also no longer an independent factor in the analysis. Currently, we are studying a much larger cohort of NSCLC patients to determine whether RASSF1A methylation is an independent prognostic factor of survival.
Fig. 5.

Kaplan-Meier survival curve for 107 patients with resected non-small-cell lung carcinomas based on RASSF1A methylation status (32 methylated and 75 not methylated). For the patients with unmethylated RASSF1A alleles, the number of cases = 75, censored = 39, and events = 36, with a mean overall survival of 52 months (95% confidence interval [CI] = 44 to 59) and a median overall survival of 49 months (95% CI = 44 to 59); for the patients with methylated RASSF1A alleles, the number of cases = 32, censored = nine, and events = 23, with a mean overall survival of 37 months (95% CI = 27 to 46) and a median overall survival of 28 months (95% CI = 9 to 47). The log-rank test statistic for equality of survival distributions for RASSF1A methylation was 3.97, with df 1, P = .0463. The patients at risk for each group were: RASSF1A unmethylated—12 months (n = 63), 36 months (n = 34), and 60 months (n = 16); RASSF1A methylated—12 months (n = 24), 36 months (n = 13), and 60 months (n = 5).
Effect of Exogenous Expression of RASSF1A on Tumor Cell Phenotype
We examined the effect of RASSF1A on the tumor cell phenotype by three methods. We used anchorage-dependent colony formation as a measure of proliferation and anchorage-independent colony formation as a measure of malignant potential. We also directly assessed in vivo tumor formation.
We first cloned RASSF1A cDNA into pcDNA3.1+, an expression vector that contains a selectable marker, and transfected NCI-H1299 cells, which lack endogenous RASSF1A expression. After selection for 14–21 days, we determined colony formation of NCI-H1299 cells in both anchorage-dependent and anchorage-independent assays. Expression of RASSF1A in NCI-H1299 cells resulted in a 40%–60% decrease in anchorage-dependent colony formation and in an approximate 90% decrease in anchorage-independent colony formation compared with cells transfected with the pcDNA3.1 vector alone (Fig. 6, A). Because NCI-H1299 cells have an intragenic p53 homozygous deletion (34), transient expression of wild-type p53 can serve as a positive control for growth inhibition. Indeed, expression of wild-type p53 in NCI-H1299 cells resulted in a 80% and 95% reduction in colony formation in anchorage-dependent and anchorage-independent assays, respectively (Fig. 6, A). Several clones of NCI-H1299 cells transfected with RASSF1A were isolated in selective medium and were found to express RASSF1A by northern blot analysis (Fig. 6, B). Although the clones grew well in vitro, each had reduced anchorage-independent colony formation by approximately 90% compared with the vector-transfected control clones (Fig. 6, C).
To eliminate the possibility that the pcDNA3.1+ vector mediated the growth-suppression effects, we infected NCI-H1299 cells with retroviral-expression vectors containing RASSF1A or RASSF1C and tested the ability of these cells to grow in an anchorage-independent manner. Cells expressing RASSF1A had a marked reduction in the ability to form soft-agar colonies compared with cells infected with the retroviral empty vector or the retroviral vector containing RASSF1C (Fig. 6, D). Cells expressing the retroviral vector formed 3200 colonies per 10 000 cells plated. RASSF1A-expressing cells formed only 19% of the vector control colonies, while RASSF1C formed 108% of the vector control. RASSF1A- and RASSF1C-infected cells grew well in vitro and showed no signs of toxicity or apoptosis (data not shown).
Finally, we tested the ability of the retrovirally infected NCI-H1299 cells to form tumors in nude mice. Cells transfected with the vector (parental cells) formed tumors rapidly (Fig. 6, E). By contrast, cells infected with RASSF1A retroviral vector and expressing the RASSF1A protein had much lower tumorigenicity in vivo (Fig. 6, E).
Discussion
We have found strong evidence that RASSF1A, but not RASSF1C, functions as a tumor suppressor gene that undergoes epigenetic inactivation in cancers by methylation of the CpG islands in the promoter region. Whereas normal lung and breast epithelial cells expressed both RASSF1A and RASSF1C, many lung and breast cancer cell lines did not express RASSF1A, although they did express RASSF1C. These tumor cell lines and uncultured primary lung and breast cancers frequently acquired RASSF1A 5′ CpG island hypermethylation, which was not found in paired lung tissues not involved with cancer from the same patient. Exposure of an NSCLC line to the methylase inhibitor 5-aza-2′-deoxycytidine restored expression of RASSF1A. We found that the loss of RASSF1A expression in a sample of resected NSCLCs was associated with decreased patient survival. Ectopic expression of RASSF1A by transfection by use of several different vectors into a cell line devoid of endogenous RASSF1A suppressed anchorage-independent growth (a measure of metastatic potential) and tumor formation in nude mice. Furthermore, although there was no evidence of in vitro morphologic changes, RASSF1A suppressed proliferation in an anchorage-dependent colony-formation assay. Independently, Dammann et al. (15) have recently reported similar results.
There is mounting evidence that tumor suppressor genes can be inactivated by tumor-acquired methylation of their promoter regions; indeed, this method of tumor suppressor gene inactivation may be more common than amino acid sequence-altering mutations (14). We found only the methylated RASSF1A allele in 45%–100% of the tumor cell lines, depending on the tumor type, which was consistent with either methylation of both parental alleles or loss of the unmethylated allele. Because the 3p21.3 region, where RASSF1 is located, undergoes frequent allele loss in a variety of human tumors, including those of the head and neck, kidney, and cervix (5), it will be important to extend the RASSF1A studies to these types of cancers. Although the methylation studies were prompted by the fact that we did not find any tumor-acquired, amino acid sequence-altering mutations in either RASSF1A or RASSF1C in our earlier study (7), in this study, we did identify several polymorphisms in the RASSF1A-coding region of six NSCLC cell lines, several of which altered the amino acid sequence. Studies are in progress to determine whether any of these polymorphisms have functional consequences.
Analysis of the protein sequence of the RASSF1 isoforms revealed several domains that may aid in identification of specific cellular functions. The presence of a Ras-association domain in both RASSF1 isoforms suggests that these proteins may function as effectors of Ras signaling (or signaling of a Ras-like molecule) in normal cells. If so, the observation that RASSF1A can function as a tumor suppressor gene implies that RASSF1 acts in opposition to Ras-effector pathways that stimulate proliferation. Ras mutations rarely occur in SCLC or in breast cancer and are found in only approximately 30% of NSCLCs (usually in adenocarcinomas) (2). Thus, the observation of RASSF1A methylation with the associated loss of expression in many tumors without Ras mutations suggests that inactivation of RASSF1A expression may be a tumorigenic mechanism that is distinct from the production of Ras mutations that lead to the activation of Ras signaling in tumors. However, it is important to note that, although many proteins have been identified that contain Ras-association domain motifs by database analysis (9), the majority of these proteins have not been validated as bona fide Ras interactors. Therefore, studies are presently under way to assess the role of RASSF1 in Ras-dependent growth control.
Additional clues to the function of RASSF1A may be found in its protein structure. Kim et al. (13) found that both RASSF1A and RASSF1C possess a putative ATM kinase phosphorylation site in their common exon (Fig. 1), based on in vitro phosphorylation studies. Dammann et al. (15) isolated RASSF1 transcripts by use of a yeast two-hybrid assay, with the DNA repair protein xeroderma pigmentosum A as bait. These findings suggest that RASSF1 products may participate in the DNA damage response or in DNA damage-induced regulation of other cellular signaling events. The presence of a putative DAG-binding domain in RASSF1A but not in RASSF1C suggests studies to test the role of tumor promoters that interact with the RASSF1A isoform in a novel light: Tumor promoters act on proteins with DAG-binding domains by facilitating their movement to the cell membrane, thus allowing them to interact with the cell's signaling components. Thus, tumor promoters such as phorbol esters might be expected to move RASSF1A to the membrane, where it may act in its normal function as a growth suppressor until RASSF1A expression is lost.
Our previous work (1,3,4,33) has shown that 3p21.3 allele loss occurs early in lung cancer pathogenesis. Another study (35) has shown that promoter methylation of other tumor suppressor genes (e.g., for p16Ink4A) can be detected in preneoplastic lung tissues or in histologically noninvolved lung tissue. RASSF1A promoter methylation may also represent a potentially important marker for the development of invasive lung and breast cancers. Because many smokers have genetic alterations in their respiratory epithelium as a result of damage by tobacco carcinogens (3,4,33,36), the discovery of a marker such as RASSF1A promoter methylation may be of great use both for early detection and for prognosis in monitoring chemoprevention efforts. Furthermore, RASSF1A may represent another potential target for pharmacologic re-expression as a novel mode for cancer treatment.
Note added in proof
Five (71%) of seven primary resected uncultured SCLCs were positive for RASSF1A methylation by the methylation-specific PCR.
Acknowledgments
Supported by Public Health Service grants CA71618, CA71443, and P50CA70907 and contract N01CO56000 (to M. I. Lerman and F. Latif) from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services; by the Early Detection Research Network for the breast cancer portion; by the G. Harold and Leila Y. Mathers Charitable Foundation; by grants J1658-MED and J1860-MED from the Austrian Science Foundation (to S. Zöchbauer-Müller); by the Association For International Cancer Research, Cancer Research Campaign, Fundacao para a Cientia e a Technologia (to F. Latif); and by grants from the Swedish Cancer Society, Karolinska Institute, Stockholm, and by the Royal Swedish Academy of Science (to E. Zabarovsky).
Footnotes
Notes: The content of the publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Contributor Information
David G. Burbee, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas
Eva Forgacs, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Sabine Zöchbauer-Müller, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Latha Shivakumar, Department of Cell Biology, The University of Texas Southwestern Medical Center at Dallas.
Kwun Fong, Department of Thoracic Medicine, The Prince Charles Hospital, Queensland, Australia.
Boning Gao, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Dwight Randle, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Masashi Kondo, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Arvind Virmani, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Scott Bader, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Yoshitaka Sekido, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Farida Latif, Department of Reproductive and Child Health, University of Birmingham, U.K.
Sara Milchgrub, Department of Pathology, The University of Texas Southwestern Medical Center at Dallas.
Shinichi Toyooka, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas.
Adi F. Gazdar, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas
Michael I. Lerman, Laboratory of Immunobiology, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD
Eugene Zabarovsky, Karolinska Institute, Stockholm, Sweden.
Michael White, Department of Cell Biology, The University of Texas Southwestern Medical Center at Dallas.
John D. Minna, Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center at Dallas
References
- 1.Wistuba I, Behrens C, Virmani A, Mele G, Milchgrub S, Girard L, et al. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000;60:1949–60. [PubMed] [Google Scholar]
- 2.Sekido Y, Fong KM, Minna JD. Progress in understanding the molecular pathogenesis of human lung cancer. Biochim Biophys Acta. 1998;1378:F21–59. doi: 10.1016/s0304-419x(98)00010-9. [DOI] [PubMed] [Google Scholar]
- 3.Wistuba II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, et al. Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst. 1997;89:1366–73. doi: 10.1093/jnci/89.18.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wistuba II, Behrens C, Milchgrub S, Bryant D, Hung J, Minna JD, et al. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene. 1999;18:643–50. doi: 10.1038/sj.onc.1202349. [DOI] [PubMed] [Google Scholar]
- 5.Kok K, Naylor SL, Buys CH. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv Cancer Res. 1997;71:27–92. doi: 10.1016/s0065-230x(08)60096-2. [DOI] [PubMed] [Google Scholar]
- 6.Wei MH, Latif F, Bader S, Kashuba V, Chen JY, Duh FM, et al. Construction of a 600-kilobase cosmid clone contig and generation of a transcriptional map surrounding the lung cancer tumor suppressor gene (TSG) locus on human chromosome 3p21.3: progress toward the isolation of a lung cancer TSG. Cancer Res. 1996;56:1487–92. [PubMed] [Google Scholar]
- 7.Lerman MI, Minna JD. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res. 2000;60:6116–33. [PubMed] [Google Scholar]
- 8.Sekido Y, Ahmadian M, Wistuba II, Latif F, Bader S, Wei MH, et al. Cloning of a breast cancer homozygous deletion junction narrows the region of search for a 3p21.3 tumor suppressor gene. Oncogene. 1998;16:3151–7. doi: 10.1038/sj.onc.1201858. [DOI] [PubMed] [Google Scholar]
- 9.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vavvas D, Li X, Avruch J, Zhang XF. Identification of Nore1 as a potential Ras effector. J Biol Chem. 1998;273:5439–42. doi: 10.1074/jbc.273.10.5439. [DOI] [PubMed] [Google Scholar]
- 11.Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997;6:477–80. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–8. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 13.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–43. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 14.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–96. [PubMed] [Google Scholar]
- 15.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25:315–9. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 16.Fong K, Biesterveld EJ, Virmani A, Wistuba I, Sekido Y, Bader SA, et al. FHIT and FRA3B 3p14.2 allele loss are common in lung cancer and preneoplastic bronchial lesions and are associated with cancer-related FHIT cDNA splicing aberrations. Cancer Res. 1997;57:2256–67. [PubMed] [Google Scholar]
- 17.Geradts J, Fong KM, Zimmerman PV, Maynard R, Minna JD. Correlation of abnormal RB, p16ink4a, and p53 expression with 3p loss of heterozygosity, other genetic abnormalities, and clinical features in 103 primary non-small cell lung cancers. Clin Cancer Res. 1999;5:791–800. [PubMed] [Google Scholar]
- 18.Mountain CF. Revisions in the International System for Staging Lung Cancer. Chest. 1997;111:1710–7. doi: 10.1378/chest.111.6.1710. [DOI] [PubMed] [Google Scholar]
- 19.Phelps RM, Johnson BE, Ihde DC, Gazdar AF, Carbone DP, McClintock PR, et al. NCI-Navy Medical Oncology Branch cell line data base. J Cell Biochem Suppl. 1996;24:32–91. doi: 10.1002/jcb.240630505. [DOI] [PubMed] [Google Scholar]
- 20.Wistuba II, Bryant D, Behrens C, Milchgrub S, Virmani AK, Ashfaq R, et al. Comparison of features of human lung cancer cell lines and their corresponding tumors. Clin Cancer Res. 1999;5:991–1000. [PubMed] [Google Scholar]
- 21.Gazdar AF, Kurvari V, Virmani A, Gollahon L, Sakaguchi M, Westerfield M, et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int J Cancer. 1998;78:766–74. doi: 10.1002/(sici)1097-0215(19981209)78:6<766::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E, Maniatis T, editors. Molelcular cloning: a laboratory manual. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 23.Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekita T. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA. 1989;86:2766–70. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61:249–55. [PubMed] [Google Scholar]
- 26.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–7. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka H, Shimada Y, Harada H, Shinoda M, Hatooka S, Imamura M, et al. Methylation of the 5′ CpG island of the FHIT gene is closely associated with transcriptional inactivation in esophageal squamous cell carcinomas. Cancer Res. 1998;58:3429–34. [PubMed] [Google Scholar]
- 28.Chen JY, Funk WD, Wright WE, Shay JW, Minna JD. Heterogeneity of transcriptional activity of mutant p53 proteins and p53 DNA target sequences. Oncogene. 1993;8:2159–66. [PubMed] [Google Scholar]
- 29.Claudio PP, Howard CM, Pacilio C, Cinti C, Romano G, Minimo C, et al. Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res. 2000;60:372–82. [PubMed] [Google Scholar]
- 30.Gao B, Sekido Y, Maximov A, Saad M, Forgacs E, Latif F, et al. Functional properties of a new voltage-dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2) J Biol Chem. 2000;275:12237–42. doi: 10.1074/jbc.275.16.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 33.Wistuba II, Barry J, Behrens C, Maitra A, Shivapurkar N, Milchgrub S, et al. Molecular changes in the bronchial epithelium of patients with small cell lung cancer. Clin Cancer Res. 2000;6:2604–10. [PMC free article] [PubMed] [Google Scholar]
- 34.Unger T, Nau MM, Segal S, Minna JD. p53: a transdominant regulator of transcription whose function is ablated by mutations occurring in human cancer. EMBO J. 1992;11:1383–90. doi: 10.1002/j.1460-2075.1992.tb05183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. 1998;95:11891–6. doi: 10.1073/pnas.95.20.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park IW, Wistuba II, Maitra A, Milchgrub S, Virmani AK, Minna JD, et al. Multiple clonal abnormalities in the bronchial epithelium of patients with lung cancer. J Natl Cancer Inst. 1999;91:1863–8. doi: 10.1093/jnci/91.21.1863. [DOI] [PubMed] [Google Scholar]