

Abstract
Werner syndrome is an autosomal recessive genetic instability and cancer predisposition syndrome with features of premature aging. Several lines of evidence have suggested that the Werner syndrome protein WRN plays a role in DNA replication and S-phase progression. In order to define the exact role of WRN in genomic replication we examined cell cycle kinetics during normal cell division and after methyl-methanesulfonate (MMS) DNA damage or hydroxyurea (HU)-mediated replication arrest following acute depletion of WRN from human fibroblasts. Loss of WRN markedly extended the time cells needed to complete the cell cycle after either of these genotoxic treatments. Moreover, replication track analysis of individual, stretched DNA fibers showed that WRN depletion significantly reduced the speed at which replication forks elongated in vivo after MMS or HU treatment. These results establish the importance of WRN during genomic replication and indicate that WRN acts to facilitate fork progression after DNA damage or replication arrest. The data provide a mechanistic basis for a better understanding of WRN-mediated maintenance of genomic stability and for predicting the outcomes of DNA-targeting chemotherapy in several adult cancers that silence WRN expression.
Keywords: Werner syndrome, DNA replication, cell cycle, methyl-methane sulfonate, hydroxyurea
Introduction
Werner syndrome (WS) is an autosomal recessive human genetic instability disorder with features of cancer predisposition and premature aging1. It is caused by mutations in a member of a conserved family of the RecQ helicase genes, WRN (RECQL2,2. Mutations in two other members of this protein family in humans also cause genetic instability and cancer predisposition syndromes. Loss of BLM results in Bloom syndrome3, and mutations in RECQL4 lead to Rothmund–Thomson syndrome with a risk of osteosarcoma4.
RecQ helicases have been implicated in regulating genome stability5, however, the specific mechanisms and pathways in which the WRN protein functions remain to be elucidated. WRN has been shown to interact with proteins involved in DNA repair, replication, and telomere maintenance6, 7. WRN-deficient cells exhibit chromosomal translocations and deletions (ibid.) and these cells are hypersensitive to DNA crosslinkers (cis-platinum, mitomycin-C, or 8-methoxypsoralen + UV light8, 9), and to camptothecin10–12. Our previous work has demonstrated that both spontaneous and damage-induced homologous recombination was reduced in WRN-deficient cells, and that the reduced clonal survival of these cells could be suppressed by expression of the RusA bacterial resolvase, or of a dominant-negative allele of RAD51, SMRAD51, which blocks the generation of recombinant DNA molecules13, 14. These data suggest that WRN may participate in homologous recombination repair, possibly at the stage of resolution of recombination intermediates.
A substantial body of evidence also links WRN to telomere maintenance6, 7, 15, and work by Crabbe et al suggested a defect in the lagging strand DNA replication of telomeric ends in WRN-deficient cells16. Involvement of WRN in replication may extend beyond telomeres. WS patient-derived primary fibroblasts were reported to replicate slowly and have a prolonged S phase17, 18, and a more recent study suggested an elevated rate of spontaneous replication fork inactivation in these cells19. In response to camptothecin or hydroxyurea, WRN can colocalize with sites of DNA synthesis as marked by foci of BrdU incorporation or of the single stranded DNA binding protein, RPA20, 21. WRN can also colocalize with homologous recombination factor RAD51 and RAD51 paralogs, and with the ATR kinase22. WRN is phosphorylated by ATR, the key regulator of the response to disrupted replication fork progression, and/or a related ATM kinase, which responds to DNA breaks23.
The characterization of WRN-deficient cells has been done mostly on patient-derived WRN−/− cells and nonisogenic WRN+/+ controls. Thus it has been hard to assess whether the observed phenotypes are a consequence of WRN absence or a result of adaptation and/or clonal selection of cells to the absence of WRN. In this study we set out to establish whether acute WRN depletion in human cells would result in an increased sensitivity to replication stress induced by DNA damage or replication fork arrest; and if so, to determine whether this would correlate with increased inactivation of replication forks. We found that WRN-depleted human fibroblasts have a marked delay in completing the cell cycle after treatment with methyl methane sulfonate (MMS) or hydroxyurea (HU), e.g. these cells spend more time in late S and/or G2 phases of the cell cycle than controls. Moreover, by measuring the lengths and types of replication tracks in stretched DNA fibers, we found that WRN is required for replication fork elongation on both MMS-damaged DNA and during recovery from HU-mediated fork arrest. These data are the first demonstration, in isogenic human cells, that WRN facilitates global fork progression during recovery from replication stress.
Materials and methods
Cells and culture
Primary human dermal fibroblasts were obtained from Clonetix and used at passages 14–18. SV40-transformed GM639 and GM847 fibroblast cell lines were obtained from the Coriell Institute Cell Repositories (Camden NJ). GM639cc1 is a pNeoA-carrying derivative of GM63924.
Drugs and Dyes
Stock solutions of 5-bromodeoxyuridine (BrdU; 10 mM in water), 5-iododeoxyuridine (IdU, 2mM in water), 5-chlorodeoxyuridine (CldU, 10mM in water), caffeine (100mM in DMSO), nocodazole (0.5mg/ml in DMSO), and hydroxyurea (HU, 1M in PBS) were stored at –20° C until use. MMS was diluted in PBS or water to 1–5% prior to use. Mimosine (10mM in growth media) was stored at 4° C. Propidium iodide (10mg/ml in PBS) was stored at 4° C and 4,6′-diamidino-2-phenylindole (DAPI, 1mg/mL in water) was stored at –20° C. DAPI was obtained from Accurate Chemical and Scientific Corp (Westbury, NY), and the rest of the chemicals from Sigma (St. Louis, MO).
Cell growth, synchronization and drug treatments
All cell strains or lines were grown as adherent monolayers in Dulbecco Modified Minimal Essential Medium (DMEM) supplemented with 2 mM L-glutamine and 10% fetal bovine serum (Hyclone, Ogden, UT) in a humidified 5% CO2, 37°C incubator.
To synchronize cells, subconfluent cultures were treated with 0.5mM mimosine for 12–14 hours25. Cells were then washed in PBS and incubated with fresh media for 8–10 hours. At this time, cells released from the block reached mid to late S phase, and were treated with HU or MMS for replication assays or cell cycle recovery experiments. HU was added at 2mM, and MMS was added at 0.005% (0.55mM) for 1 hr unless stated otherwise. BrdU and other halogenated nucleotides were added to cells to a final concentration of 50μM, caffeine was used at 3mM, and nocodazole at 50ng/ml.
RNAi-mediated depletion of WRN
WRNsi is a hairpin with the stem sequence corresponding to positions 160 to 184 in the WRN ORF (accession # NM_000553; 1 is A in ATG). It was expressed from the pBABEpuro retroviral vector26. WRN2-4 is a hairpin with the stem sequence corresponding to WRN ORF positions 578 to 597. This hairpin was cloned into pLKO.1 lentiviral vector27 between EcoRI and AgeI sites, under the control of the human U6 promoter. Virus was generated by transient transfection of 293T cells. Human fibroblasts were transduced with pBABEpuro-WRNsi, pLKO.1-WRN2-4, or the respective empty vectors, and placed on puromycin selection (1μg/ml for primary fibroblasts and GM847 and 1.5μg/ml for GM639) at 20 hours after infection.
Western blotting
Cells were harvested for Western blot analysis and cell cycle and replication assays at 5–7 days post infection. Western blotting of WRN was done as previously described24, with the rabbit α-WRN (Novus Cat. #NB100472A). For loading controls, α-Chk1 (Santa Cruz Cat. #sc-8408) antibody was used against CHK1 and α-GAPDH (Abcam Cat. # ab9482) was used against GAPDH. Proteins were visualized by ECL (Amersham) and quantified using the Storm Phosphorimager and ImageQuant software (Molecular Dynamics).
Staining for BrdU incorporation and FACS
DNA of ethanol-fixed cells was denatured with 2N HCl and 0.5% Triton X100, and neutralized in 100mM Na borate pH8.5. Cells were next washed in IFA buffer (10mM HEPES pH7.4, 150mM NaCl, 5% normal goat serum, 0.1% Na azide) supplemented with 0.5% Tween 20. Cells were resuspended in IFA containing 10μL of FITC-conjugated mouse αBrdU antibody (Beston-Dickinson) per 106 cells and incubated on ice in the dark for 1 hr. Cells were washed again with IFA/Tween and resuspended for FACS analysis in PBS with 10μg/mL propidium iodide and 100μg/mL RNAse A.
DAPI staining was performed as previously described28.
Data analysis and presentation were done with Summit software (Dako, Carpinteria, CA), and cell cycle phase quantitations were done with Mcycle software (Phoenix Flow Systems, San Diego, CA). To quantify BrdU incorporation, the position of the BrdU-negative population was determined using flow cytometric profiles of relevant negative control samples with no incorporation. Cells with fluorescence above the negative control level were considered positive.
Microchannel fabrication, DNA fiber stretching and replication track analysis
PDMS microchannels were fabricated using standard photolithography and soft lithography procedures29. SU8-2 was spun on silicon wafers at 500 rpm for 10 sec. and 3000 rpm for additional 30 sec. The heights of microchannels were 3.25 μm as measured with a surface profilometer (KLA-Tencor, model P15, San Jose, CA). DNA stretching was performed as described30, with modifications. Glass cover slips were cleaned in nitric acid:hydrochloric acid 2:1, silanized with a solution of 12.6 μl N-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride (Gelest, SIT8415.0) and 6 μl vinyltrimethoxysilane (Gelest, SIV9220.0) per 50ml of water at 65°C for 17.5 hrs, and stored in ethanol at 4°C. DNAs from cells harvested by trypsinization were isolated in agarose plugs as described in manufacturer recommended protocols for the CHEF-DR II PFGE apparatus (Bio-Rad), with the exception that MMS-treated DNAs and their control counterparts were processed at temperatures at or below 37°C. To release DNA, plugs were briefly heated to 75°C and incubated with β-agarase (NEB). To stretch DNA, oxygen plasma-treated (to decrease hydrophobicity) PDMS matrices with series of microchannels 50–450 μm wide were laid over cover slips, and DNA was loaded into channel space by capillary tension. PDMS matrices were then removed and cover slips were treated with methanol:acetic acid 3:1 for 3 min and air dried. DNA was stained with YOYO-1 (Molecular Probes, 10mM in TE with 20% β-mercaptoethanol) to inspect the quality of stretching. For immunostaining, cover slips were incubated in 2.5N HCL for 40 min, neutralized in 0.1M Na borate pH 8.0 and PBS, and blocked in PBS/5% BSA/0.5% Tween 20. The following antibodies were used, in that order: rat α-CldU/BrdU (Serotec, Cat.# MCA2060), goat α-rat Alexa 594-conjugated (Molecular Probes, Cat.# A11007), mouse α-IdU/BrdU (BD Biosciences, Cat.# 347580), and goat α-mouse Alexa 488-conjugated (Molecular Probes, Cat.# A11001). Antibody dilutions were made in PBS/5% BSA/0.5% Tween 20 with 10% normal goat serum and cover slips were also blocked in this buffer between α-rat secondary antibody and mouse α-IdU/BrdU antibody. Washes between antibodies were done in PBS/1%BSA/0.1% Tween 20. Cover slips were mounted in 10% PBS/90% glycerol/10mM DTT.
Confocal microscopy of stretched DNAs was performed on the Zeiss Axiovert microscope with a 100x objective. Scanning for areas with optimal density of molecules was done with the filter set to the color corresponding to the first label, and then ten to twenty digital images per channel were generated by tracking along their lengths. Lengths of tracks were measured in digital images using the attached Zeiss AxioVision software. Chi square tests were applied to frequency distributions of lengths of tracks. The ratios of first label to second label segment lengths in double-labeled tracks were analyzed with two nonparametric tests: frequency distributions were subjected to chi square tests, and datasets of ratio values were also subjected to Kolmogorov-Smirnov (KS) test. This was done to eliminate the risk that variations between tail end values in the ratio distributions will disproportionately influence confidence.
Results
WRN depletion delays recovery from DNA damage during S-phase
In order to determine the role of WRN in recovery from replication stress we found it necessary to use cell populations maximally enriched for S phase cells. Average tissue culture cell populations are abundant in G1 cells, which are not affected by HU and are differentially affected by camptothecin, cis-platinum, and MMS. Also, many WRN−/− cell line cultures have an increased fraction of G1 cells compared to WRN+/+ lines28, which can further dilute effects produced by replication stress in WRN-deficient cells.
In order to avoid these potential pitfalls, we used assays that focused on S phase cells (see experimental design in Figure 1A). First, we used synchronization to enrich cell populations for S phase fraction. Cell cultures were arrested in late G1 (G1(I) in Figure 1A) by treatment with mimosine. Then mimosine was removed and the cells were incubated for 8–10 hrs. At this time, when the bulk of the population was in S phase, cells were pulse-treated with MMS, and followed through the time course of recovery.
Figure 1.
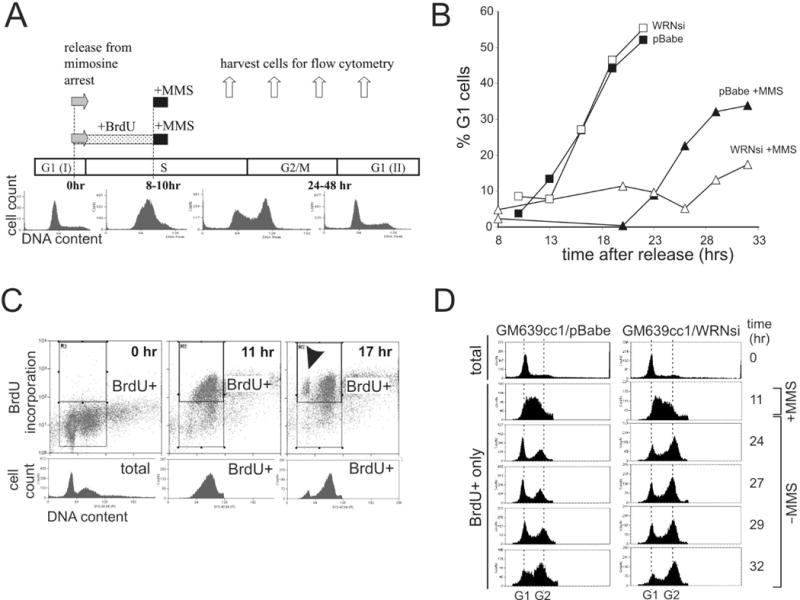
WRN depletion from SV40 transformed fibroblasts causes a cell cycle delay after MMS treatment in S phase. A) Experimental designs. Cells were synchronized in late G1 by a 12–14 hr mimosine arrest. 0 hr was the time of release from mimosine into the cell cycle. Cells were treated with 0.005% MMS for 1 hr beginning at 8–10 hours after release. In some experiments BrdU was added between 0 and 8–10 hrs. Samples were taken throughout the time course to determine cell cycle distributions by FACS as shown in representative cell count vs. DNA content profiles of GM639cc1 cells. B) Cell cycle progression of synchronized GM847 cells with and without MMS treatment. Cell cycle distributions at each time point were determined from cellular DNA content measured by FACS and percent of G1 cells was plotted. C) Examples of FACS profiles of synchronized GM847 cells labeled with BrdU as described in (A). 0 hr, cells arrested with mimosine for 12–14 hrs and then incubated with BrdU for 10 hrs in the presence of mimosine. 11 and 17 hr, cells arrested with mimosine for 12–14 hrs, then released into the cell cycle and incubated for the indicated times. BrdU was present from 0 to 11 hr. Upper rectangles enclose BrdU+ subpopulations. Cell cycle distributions of these populations (BrdU+) were plotted as cell count vs. DNA content profiles beneath the center and right panels. Total (left panel) represents cell cycle distribution of the whole population at 0 hr. BrdU+ cells that reached the G1(II) are marked by an arrowhead. Cell cycle distributions at each time point were determined and percentage of new G1 (G1(II)) cells was plotted. D) Cell count vs. DNA content profiles of total, mimosine arrested (0 hr) and BrdU+ GM639cc1 cells labeled with BrdU for 10 hrs after release from mimosine arrest (BrdU+ only), treated with MMS and then followed for 21 hrs.
To compare isogenic cell lines, we depleted WRN from human fibroblasts. We first used a previously characterized retroviral shRNA, WRNsi26, 28. In addition, we developed a new, lentivirus-expressed shRNA, WRN2-4. Typically, 80–90% of WRN protein was depleted (Supplementary Figure 1A), and this level was maintained throughout the experiment.
In the absence of DNA damage, WRN-depleted and mock-depleted SV40-transformed fibroblasts finished the cell cycle and returned to G1 (G1(II)) with similar kinetics (Figure 1B). Treatment of cells with MMS during S phase induced cell cycle delays in both WRN-depleted and control cells (Figure 1B). For the first 10 hrs after MMS, cells traversed through S and G2 phases (not shown). At later time points, cells began to accumulate in G1(II), with WRN-depleted cells markedly delayed compared to controls (Figure 1B). Addition of a mitosis inhibitor nocodazole to cells recovering from MMS confirmed that not only progression from G2/M to G1 was delayed in WRN-deficient cells, but also the completion of S phase (not shown).
The same experiment was performed using a different shRNA (WRN2-4, Supplementary Figure 1B) and with the addition of BrdU labeling step prior to MMS treatment (see experimental design in Figure 1A). BrdU was present between 0 and 8–10 hrs after release from mimosine, thereby labeling every cell that entered S phase during this interval. This BrdU+ population was identified in FACS profiles by comparison with BrdU− controls (mimosine-arrested cells incubated with BrdU), and the cell cycle progression of this population was followed for about 20 hrs (see an example in Figure 1C). BrdU incorporation allowed distinguishing between BrdU− cells remaining in G1(I), and BrdU+ cells reaching G1(II) (an arrowhead in Figure 1D, right panel), as well as cells in first and second S phases. Once again, WRN-depleted SV40 fibroblasts were slower than controls in completing the cell cycle after MMS treatment in S phase (Supplementary Figure 1B). This effect of WRN depletion on recovery from MMS was even more pronounced in a different SV40 fibroblast line, GM639 (Figure 1D). For example, 13 hrs after MMS treatment, control cells were largely in G1(II), while WRN-depleted cells remained predominantly in the first G2 (time point 24 hrs in Figure 1D). A large proportion of WRN-depleted population remained in the first G2, while the bulk of control cells traversed into the second S phase (time point 32 hrs in Figure 1D).
To rule out a possibility that the difference between WRN-deficient and wild type cells was induced by mimosine, we preformed experiments in an asynchronous population (Figure 2A, B). WRN-depleted and control fibroblasts were pulse-labeled with BrdU for two hours. MMS was added during the second hour of BrdU labeling and then both the drug and the nucleotide analog were removed. Cell cycle distributions prior to (all cells) and during MMS treatment (BrdU+ only cells) in both types of cells were similar (Figure 2A). However, by 19 hrs after MMS, the progression of WRN-depleted cells that were in S phase during MMS treatment lagged behind controls (Figure 2A, B) in the generation of BrdU+ G1 cells. Addition of caffeine during recovery shortened the delay of cell division in both types of cells (Supplementary Figure 1C). Thus MMS-induced delays were at least in part mediated by the ATR/ATM-dependent checkpoint. As before, without MMS treatment, WRN-depleted cells were not slower than controls in their progression from S phase to the next G1 (Supplementary Figure 1C).
Figure 2.
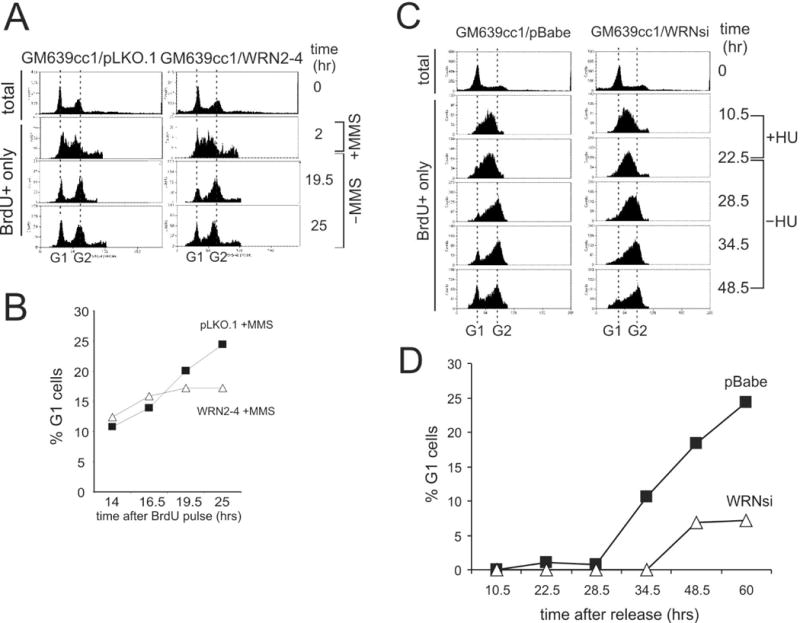
WRN depletion from SV40 transformed fibroblasts (GM639cc1) causes a cell cycle delay after MMS or HU treatment in S phase. A) Unsynchronized GM639cc1 cells that were mock-depleted (pLKO.1) or WRN-depleted (WRN2-4) were labeled with BrdU for 2 hrs. One-half of samples were treated with 0.005% MMS for 1 hr, and cells were followed for additional 25 hrs. FACS profiles of total (0 hr, prior to labeling) and BrdU+ cells were derived as described for Figure 1. B) A quantitation of the experiment shown in (A). C) FACS profiles of total, mimosine-arrested (0 hr) and BrdU+ only cells labeled for the first 10.5 hrs after release from mimosine, incubated with 2mM HU for the next 12.5 hours, and then incubated in the absence of HU for 26 hrs. D) A quantitation of the FACS profiles of BrdU+ cells shown in (C).
Taken together, the data obtained with both synchronized and unsynchronized SV40 fibroblasts indicate that loss of WRN causes a marked extension of the checkpoint-dependent delay of cell division induced by MMS treatment during S phase. This extension arises from prolonged pausing of cells in late S and/or G2 phases of the cell cycle.
WRN depletion impairs recovery from HU-mediated replication arrest
In order to determine if the defect in recovery from replication stress observed in WRN-depleted cells was MMS-specific or more general, we treated WRN-depleted cells with HU. The experimental scheme was as shown in Figure 1A, except that HU was added to BrdU-labeled cells instead of MMS (BrdU was not present during incubation with HU). After 8 hrs of HU arrest, we could detect an approximately 4–5 hr delay in the kinetics of progression of WRN-depleted cells out of S phase and into the next G1, compared to controls (not shown). This WRN-dependent delay became more prominent after 11–13 hrs of HU arrest in S phase (Figure 2C, D). For example, 12 hrs after HU (and 34.5 hrs after the start of the experiment), some control cells entered G1(II) while WRN-depleted cells remained in G2 (Figure 2C, panel 34.5 hr, also see graph in Figure 2D) Thus, WRN loss impairs recovery from replication arrest induced by HU.
To address whether the transformed status of SV40 fibroblast lines affects the WRN-dependent phenotype, we repeated the HU-mediated arrest and recovery experiments in primary human fibroblasts. WRN-depleted primary fibroblast cultures (see Supplementary Figure 1A for depletion data) had significantly fewer cells that entered S phase after mimosine synchronization (Figure 3B, upper panels). This phenotype is unlikely to be caused by mimosine treatment, since untreated WRN-depleted cultures also had fewer S phase cells (not shown). Also, previous work has described an accumulation of G1 cells in WRN-depleted primary fibroblasts28, 31. Interestingly, WRN-depleted primary fibroblasts that entered the cell cycle after mimosine synchronization did so on the same schedule as controls (compare 9 hr panels in Figure 3C). However, the appearance of BrdU+ G1(II) cells that completed the cell cycle was delayed in WRN-depleted fibroblasts, with more cells staying in late S/G2 (Figure 3A, 17 to 22.5 hr panels, also Figure 3D). This phenotype is consistent with the previous observations made with WRN−/− primary fibroblasts19, and suggests that WRN loss caused an extension of S/G2 phases.
Figure 3.
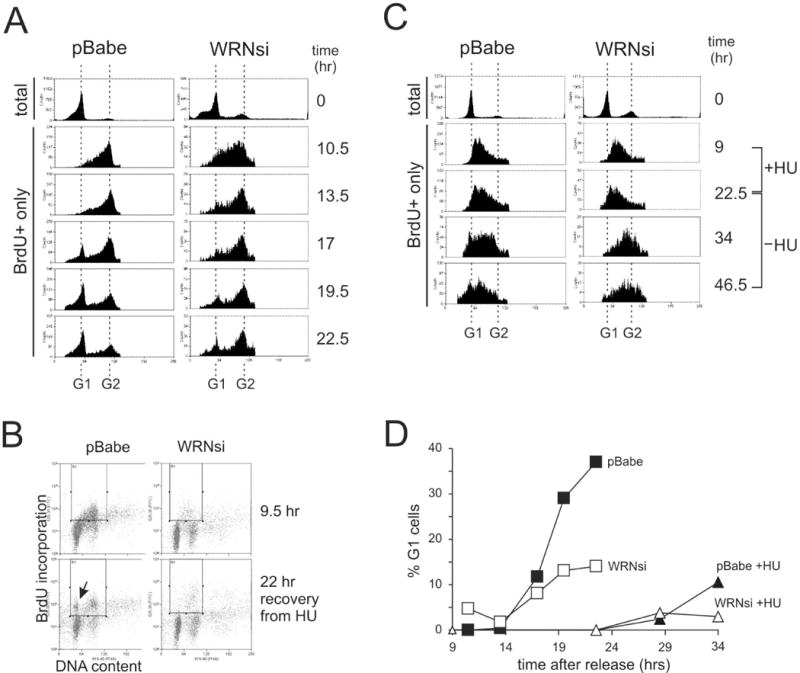
WRN depletion leads to cell cycle delays in primary human fibroblasts. A) A representative Western blot of mock-depleted (pBabe) and WRN-depleted (WRNsi) primary human fibroblasts. CHK1 was used as a loading control. B) FACS profiles of total, mimosine arrested (0 hr) and BrdU+ only cells, labeled during the 10.5 hrs after release from mimosine and then followed for 12 hrs. C) FACS profiles of cells labeled with BrdU for 9.5 hours after release from mimosine (9.5 hr), then incubated for 10 hrs in the presence of 2mM HU and followed for 22 hrs after HU removal. An arrow marks the position of new G1 (G1(II)), BrdU+ cells that have completed the cell cycle after HU arrest in S phase. BrdU+ cells are enclosed in the rectangular gate. D) FACS profiles of total, mimosine arrested (0 hr) and BrdU+ only cells labeled for 9 hrs after release from mimosine, incubated with HU for the next 13.5 hrs and followed for 24 hrs after HU removal. E) FACS profiles of BrdU+ cells shown in (B) and (D) were quantified and fractions of the new G1 cells in populations as a function of time after release from mimosine were plotted.
HU treatment during S phase delayed cell cycle progression of both mock-depleted and WRN-depleted primary fibroblasts (Figure 3B, C, D). However, 11.5 hrs after release from HU block when 10% of control population reached the subsequent G1, WRN-depleted cells remained in late S and/or G2, and no G1(II) cells could be detected (Figure 3C, panel 34hr, also Figure 3D). Thus, WRN depletion from both primary and SV40-transformed human fibroblasts impairs recovery from HU-mediated replication arrest. Taken together, the data obtained thus far suggest a general defect in recovery from replication stress caused by depletion of WRN.
WRN depletion affects fork progression on MMS-damaged DNA
The defect in recovery from replication stress in WRN-depleted cells could have at least two distinct causes. First, WRN may stabilize or restore active replication forks that are slowed or stalled due to DNA damage or nucleotide depletion. In this case, when WRN is depleted, the fraction or activity of forks that are able to elongate after replication stress should be reduced. Alternatively, WRN may be involved in the repair of DNA strand breaks that emerge after irreversible breakdown of replication forks. In this case, WRN depletion should not have impact on the fate of replication forks, but should reduce efficiency of DNA repair in S and G2 phases.
In order to determine whether WRN affects fork activity, we looked at the behavior of DNA replication in living cells by using immunofluorescent detection of replication tracks in stretched DNA fibers. DNA was stretched on silanized glass with the aid of micro-fabricated capillary channels (see Materials and Methods). The experimental design is outlined in Figure 4A: mimosine synchronized, mid S-phase SV40-transformed fibroblasts were labeled for 40 min with CldU, treated with 0.02% MMS for 20 min, and then labeled for an additional 40 min with IdU (in a separate experiment, we established that virtually no replication can be detected by fiber track analysis if the label is added during incubation with this dose of MMS, not shown). Untreated controls were consecutively labeled with CldU and IdU. DNAs were isolated in agarose plugs and all treatments were conducted at temperatures at or below 37°C to minimize potential breakdown of thermolabile alkylated bases32. Replication was detected by staining with antibodies to IdU or CldU. Since MMS treatment has been shown to reduce the rate of progression of replication forks33, 34, we first focused on measuring the lengths of the IdU and CldU segments in double-labeled tracks (Figure 4A, track type a), as well as the lengths of IdU and CldU single-labeled tracks (Figure 4A, track types b and c). In addition, we determined the ratios of CldU to IdU segment lengths in double-labeled tracks (see35 for an in-depth discussion of the approach).
Figure 4.
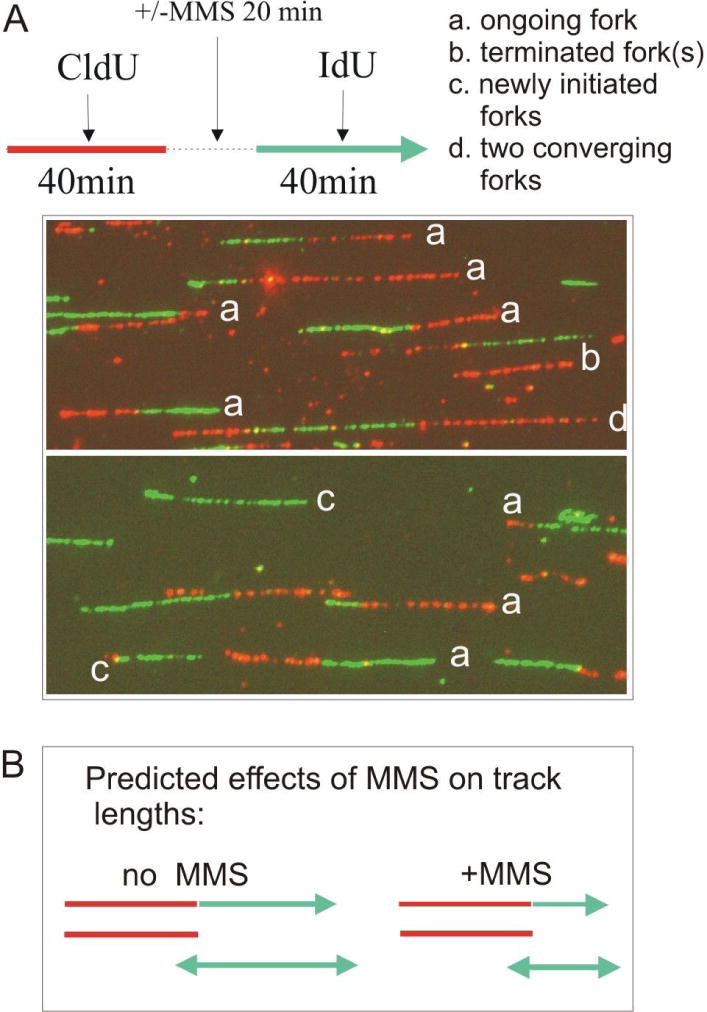
Display of individual replicating DNA molecules visualized by immunofluorescent detection in stretched DNA fibers. A) An experimental design to address effects of MMS on replication fork progression rates and examples of different types of replication tracks as observed in S phase enriched GM639cc1/WRN2-4 fibroblasts, and their simplest interpretations. Replication tracks are generated by sequential pulse-labeling with two different, halogenated nucleotide precursors and visualized by antibody staining. B) Predicted effect of MMS on the fork progression rate. To measure this effect, lengths of red and green segments in type a tracks and lengths of type b and c tracks were determined using AxioVert Software. Triple-labeled tracks such as type d in (A) or more complex tracks were not included in these measurements, because it is not possible to unequivocally assign boundaries between participating forks in such tracks.
In our experimental design (Figure 4A), CldU and IdU track length distributions of untreated cells should be the same, while MMS treatment should shorten the lengths of tracks labeled with IdU (post-damage) but not with CldU (pre-damage, Figure 4B). In addition, CldU track lengths in untreated and MMS-treated DNAs should be similar. All these expectations were met: in untreated WRN-depleted and control fibroblasts, CldU and IdU track lengths were similar, and the ratios of CldU to IdU segment lengths of double-labeled tracks were distributed around 1 (Figure 5A, C). This confirms the assumption that double-labeled tracks are in fact generated by single forks (Figure 4A). Also, single-labeled tracks were overall longer than segments of double-labeled tracks of the same color (Figure 5A). Again, this is consistent with the interpretation that a vast majority of double-labeled tracks is produced by single replication forks, while single-labeled tracks can be produced by two diverging (or converging) forks.
Figure 5.
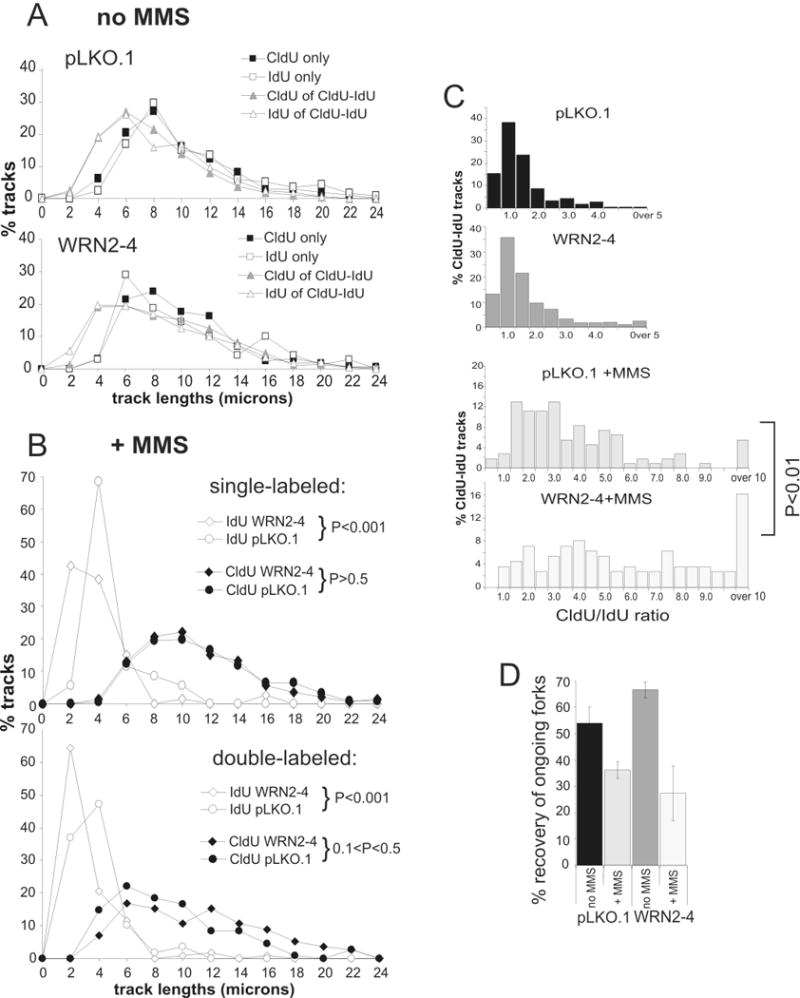
Replication fork progression is impaired in WRN-depleted GM639cc1 fibroblasts after MMS damage. A) Lengths of single-labeled CldU and IdU tracks and of the CldU and IdU segments of double-labeled tracks are equivalent in untreated control (pLKO.1) or WRN-depleted (WRN2-4) GM639cc1 cells. pLKO.1, N= 533, WRN2-4, N=517, where N is the number of tracks analyzed for each experimental condition. Length classes are in 2μm increments (for example, 2 on X axis stands for all lengths between 0 and 2 μm). B) Treatment with 0.02% MMS for 20 min reduces the lengths of IdU tracks (top panel) and the lengths of IdU segments in CldU-IdU double labeled tracks (bottom panel), while CldU track lengths are unaffected. IdU track lengths in WRN-depleted cells (WRN2-4) are affected more severely than in control (pLKO.1) GM639cc1 cells. pLKO.1+MMS, N=354, WRN2-4+MMS, N=629. Confidence was determined in chi square tests. C) Ratio of CldU to IdU track lengths in double-labeled (CldU-IdU) tracks in untreated and MMS-treated cells. X axis values are classes, e.g. 1.0 is all ratios between 0.5 and 1.0. The P value shown was determined in chi square tests. D) Efficiency of recovery of ongoing forks, e.g. the frequency of double-labeled tracks among all tracks containing the first label (for example, CldU-IdU/(CldU-IdU + CldU only)), as a function of WRN status and MMS treatment. Values obtained in two experiments were averaged. The two experiments were done identically except for the order of addition of labels. (CldU, then IdU or: IdU, then CldU). Error bars are standard deviations.
Treatment of control cells with MMS between CldU and IdU pulses resulted in shortening of IdU (post-damage) but not CldU (pre-damage) tracks, as seen both by track length distributions (Figure 5B) and by a shift of a bulk of CldU/IdU ratios of double-labeled tracks to values above 1.5 (Figure 5C, +MMS panels). This indicates a decrease in fork progression rates after MMS, consistent with previous reports33, 34. In WRN-depleted fibroblasts, this effect of MMS on fork rates was exacerbated. All IdU track lengths, whether by newly fired forks (single-labeled, Figure 5B, top panel), or by ongoing forks (double-labeled, Figure 5B bottom panel), were significantly shorter in WRN-depleted cells after MMS than in controls, whereas CldU track lengths were virtually identical. Moreover, WRN-depleted cells had more forks in which post-damage progression was severely depressed. For example, in WRN-depleted cells 16% of ongoing forks had post-damage segments that were 10 or more times shorter than their pre-damage segments, compared to only 6% in control cells (Figure 5C, P<0.01 by chi square test and D=0.325, P<<0.001 by KS test; also see Table 1).
Table 1.
Percentage of ongoing forks, in which second label segments were 5 or more times shorter than the corresponding first label segments.
| Cells | +/−MMS | +/−HU, exp. 1 | +/−HU, exp.2 |
|---|---|---|---|
| GM639cc1/pLKO.1, control | 0.0 | 4.5 | 0.0 |
| GM639cc1/pLKO.1, treated | 21.3 | 7.2 | 6.8 |
| GM639cc1/WRN2-4, control | 2.4 | 10.3 | 3.6 |
| GM639cc1/WRN2-4, treated | 50.0 | 29.0 | 21.4 |
This prompted us to examine whether WRN-depleted cells also had a higher proportion of forks that failed to elongate after MMS damage. Results of experiments performed with the original (CldU, then IdU) and a reversed (IdU, then CldU) order of addition of labels, were averaged to derive a mean value for the recovery of ongoing, e.g. double-labeled, forks. The labeling order was varied in order to eliminate the risk of a potential bias in track type identification caused by antibody staining artifacts. The results consistently showed a more dramatic MMS-dependent reduction in the fraction of ongoing forks in WRN-depleted cells (Figure 5D).
Taken together, our data suggest that MMS damage resulted in more dramatic fork slowing in WRN-depleted cells, as compared with controls. This was evident both in ongoing forks and in forks originated after MMS treatment. Moreover, slowing of ongoing forks in WRN-depleted cells was accompanied by a higher degree of apparent fork inactivation.
WRN depletion impairs fork progression, though not fork reactivation, after HU arrest
Next, we asked whether forks stalled by HU were also inactivated more readily in cells lacking WRN. Synchronized, mid-S phase SV40 fibroblasts were pulse-labeled with the first halogenated nucleotide (CldU or IdU) for 40 minutes, and then HU was added to the media for 4 hrs (in the presence of the first label), prior to transfer to media containing the second label (respectively, IdU or CldU) and no HU (Figure 6). Untreated controls were sequentially labeled with two labels for the same periods of time.
Figure 6.
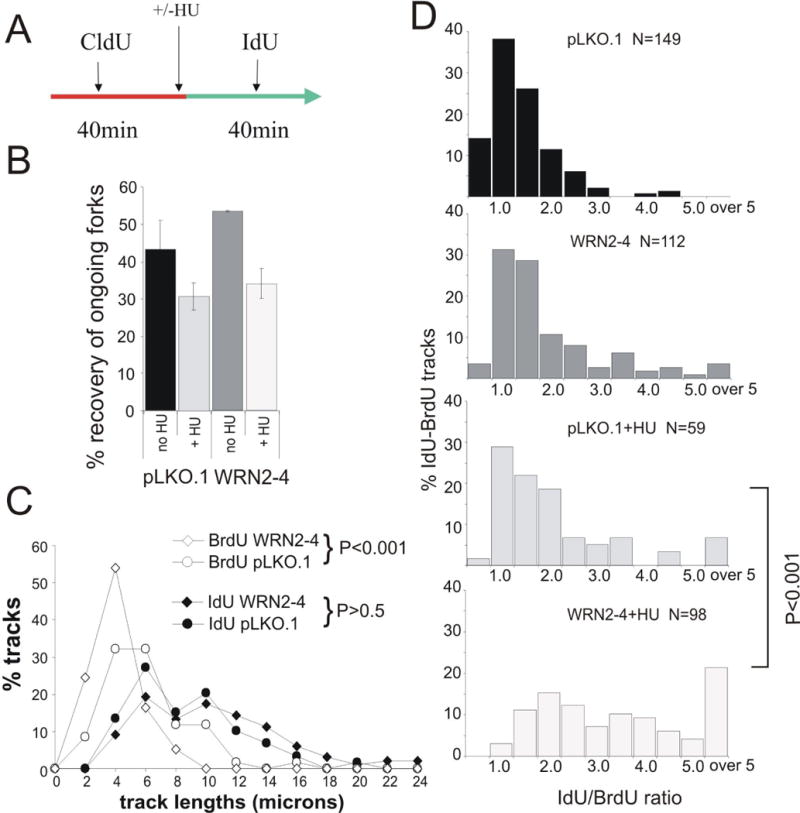
Replication fork progression is impaired in WRN-depleted GM639cc1 fibroblasts after HU-mediated replication arrest. A) An experimental outline for addressing recovery of replication forks after HU-mediated replication fork arrest (2mM HU) for 4 hrs. The shown and the reversed order of labeling were used. B) Independently of WRN status, HU-arrested forks partially lose ability to resume replication once HU is removed. Efficiency of fork recovery after HU-mediated replication arrest was determined as in Figure 5D as the ratio of double-labeled tracks to all tracks containing the first label. Results of two experiments performed with CldU, then IdU or: IdU, then CldU order of labels in GM639cc1 fibroblasts were averaged. 250 to 650 tracks were collected and analyzed for each data point. Error bars are standard deviations. C) WRN-depleted (WRN2-4) GM639cc1 fibroblasts show a HU-dependent shortening of replication tracks. An independent experiment performed as in (A), only with IdU as a first label, BrdU as a second label and HU arrest for 7 hrs. Length distributions in pre-HU (IdU) and post-HU (BrdU) segments of double-labeled tracks in cells released from HU arrest were plotted as in Figure 5B. D) IdU/BrdU ratios determined for the experiment described in (C) were plotted as in Figure 5C. The P values shown in (C) and (D) were determined in chi square tests.
Forks arrested with HU are thought to remain functional, at least for a period of time, unless cells lack protein factors that preserve activity of arrested forks36, 37. Therefore, in order to determine whether WRN plays a role in stabilizing arrested forks, we first determined the percentages of double- and single-labeled tracks in WRN-depleted and control SV40 fibroblasts before and after HU arrest.
Treatment with HU for 4 hrs resulted in a decrease in recovery of ongoing forks, even though a substantial number of forks that were labeled prior to HU addition, was still able to resume DNA synthesis and incorporate the second label (Figure 6B). Remarkably, WRN-depleted cells were not different from controls in their ability to preserve activity of HU-arrested forks. An average 1.6-fold reduction in recovered forks was observed in WRN-depleted cells compared to a 1.4-fold reduction in controls. We next asked if the forks that resumed elongation after HU-mediated arrest progressed slower in WRN-depleted cells than in controls. Track length analysis of the data from one of the experiments analyzed in Figure 6B, showed that this was the case. For example, 29% of the forks that resumed replication after HU in WRN-depleted cells, traveled less than a fifth of the distance they traveled before HU (Table 1). By comparison, in control cells only 7.2% of forks that resumed after HU fell into this category.
We also exposed SV40 fibroblasts to a longer HU arrest (7 hr), and measured the lengths of double-labeled tracks (in this case, IdU-BrdU, Figure 6C, D). The analysis confirmed that forks resuming replication after HU-mediated arrest covered on the average less distance in WRN-depleted cells than in controls. For example, overall length distributions of post-HU segments of double-labeled tracks were significantly shorter in WRN-depleted cells (Figure 6C). Also, the ratios of pre- to post-HU segments in double-labeled tracks were on the average higher in WRN-depleted cells. As seen before (Table 1), a greater proportion of forks in WRN-depleted cells had very short post-HU segments (less than a fifth of the length of their corresponding pre-HU segments; Figure 6D, D=0.436, P<<0.001 for the track segment ratio comparison between WRN-depleted and control cells by KS test; also see Table 1). In the absence of HU treatment, the majority of ongoing forks in both WRN-depleted and control cells progressed at a uniform rate (Figure 6D), though it should be noted that in WRN-depleted cells a slightly higher proportion of forks may have traveled with an irregular rate (D=0.186, P=0.02 for pre HU track comparison between WRN-depleted and control cells by KS test).
In summary, these data indicate that WRN facilitates fork progression after HU-induced replication arrest, which is similar to the case observed for MMS-induced stress. However, WRN does not appear to be required for stabilizing forks during HU arrest, since a similar proportion of forks are able to resume DNA synthesis after HU removal in WRN-depleted and control cells.
Discussion
Several biochemical, cellular and evolutionary lines of evidence have implicated WRN in DNA replication either prior to or after DNA damage (see Introduction and refs. therein). In order to address this issue directly, we examined the replication behavior of intact cycling cells and of single DNA molecules with and without WRN protein. Our findings indicate that acute WRN depletion from human fibroblasts leads to a prolonged delay in cell cycle progression after S-phase MMS damage or HU-mediated replication arrest. WRN-depleted cells spent more time in late S and/or G2 than control cells subjected to the same level of replication stress. This phenotype of WRN depletion was detectable not only in SV40-transformed fibroblast cell lines but also in primary fibroblasts. In fact, WRN-depleted primary fibroblasts were more severely impaired than transformed fibroblasts, and exhibited delays of cell division even in the absence of any exogenous DNA damage or replication arrest, consistent with previous findings of prolonged S phase in primary fibroblasts derived from Werner syndrome (WS) patients19. While it is possible that this phenotype was elicited by a low level of genotoxic stress associated with culturing or handling (for instance, oxygen tension or BrdU incorporation), it is still consistent with the conclusion that WRN is required for a robust and resilient S phase. Overall, these observations suggest that WRN may act either directly at replication forks to ensure fork progression, or on newly replicated DNA to repair S-phase damage.
Using high-resolution DNA fiber track analyses, we found that WRN can directly affect replication forks. Our results suggest that a higher proportion of forks progressed extremely slowly after MMS damage in WRN-deficient fibroblasts compared to controls. In addition, a greater proportion of forks either did not continue DNA synthesis after MMS damage or may have synthesized tracks too short to be detected (less than 0.5 microns long). These findings are consistent with the conclusion that WRN can facilitate fork progression through MMS-damaged DNA.
When we examined recovery of replication forks from replication arrest mediated by HU, we found that forks stalled by HU can remain stable, but that this stability does not appear to require WRN. What depends on WRN is the progression of replication forks after HU. In WRN-depleted cells the fraction of forks that resumed replication after HU was similar to controls, but these forks generated shorter tracks. This suggests either that the speed of fork progression was diminished, or that more forks terminated shortly after resuming replication. A third conceivable mechanism, that in WRN-depleted cells forks took longer to reactivate, is less likely because it would have manifested in the reduced fraction of forks that resumed replication.
The observation that forks may progress less efficiently after HU may hint that perturbation of dNTP pools during HU arrest has a lasting effect beyond stalling and resumption, which influences the quality of replication. It is possible that post-HU DNA synthesis is associated with the generation of mismatches and/or single-stranded gaps, which may elicit a requirement for factors such as WRN.
We did not detect major differences in fork progression in untreated WRN-depleted versus control SV40 fibroblasts, although a close look at the track length profiles reveals that WRN-depleted cells had more double-labeled tracks in which the ratio of first to second label segment lengths fell in the tail of the ratio distribution (Figures 5C and 6D, also Table 1). This difference may suggest a higher rate of spontaneous pausing or inactivation in a small subset of forks in the absence of WRN. A similar tendency was reported by Rodrigues-Lopez et al. based on single-label replication track analysis of primary fibroblasts from WS patients19. The authors detected differences in mean lengths of replication tracks between WRN+/+ and WRN−/− lines; these and other data were consistent with the conclusion that in WRN−/− cells a fraction of forks was pausing or inactivating prematurely. Arguably, the differences between WRN+ and WRN− cells observed by Rodriguez et al. may be more pronounced than the ones we report in this work. Our data (Figures 2, 3) lead us to think that this can be explained by the transformed status of SV40 fibroblasts (for example, inactivation of p53), which may make them less dependent on WRN. It is also conceivable that some phenotypes of WS cells may be modified by a history of propagation in the absence of WRN and are not recapitulated when WRN is depleted in wild type cells.
What mechanistic models could explain the role of WRN in replication fork progression after treatment with MMS or HU, the two agents with substantially different modes of action? Since WRN absence extends the caffeine-sensitive delay of the cell cycle after replication stress, it is more likely that in WRN-deficient cells the ATR-CHK1 and/or ATM-CHK2 checkpoint signaling is elevated and/or extended, and that WRN is a downstream target of one or both of these checkpoint pathways, rather than an upstream sensor. Consistent with this, the fork progression defects in WRN-depleted cells could be explained by failure to bypass or eliminate the lesions common to MMS and HU.
MMS generates DNA base damage that can be repaired by at least three pathways: base excision repair (BER)38, homologous recombination (HR)39, or lesion bypass36, 40. Lesion bypass may occur by mutagenic translesion synthesis (TLS), or by a non-mutagenic pathway that involves HR-mediated template switching (ibid,41). Loss of the key proteins in each of these pathways confers hypersensitivity to MMS42–46.
BER and HR have also been implicated in the response to HU. Cells defective in the poly(ADP-ribose) polymerase PARP-1 are sensitive to HU and show a cell cycle progression delay after HU arrest47. HU treatment also induces RAD51 focus formation11, 48, and deficiency in the RAD51 paralogs XRCC249 and XRCC350, or mutation of a CHK1 phosphorylation site in RAD5151 render cells hypersensitive to HU. Finally, there is evidence of cross talk between HR and BER at HU-stalled forks47. Few data as yet implicate TLS or specialized polymerases in the recovery from HU arrest52, 53. However, the lesion bypass pathway in general, and TLS in particular, have been suggested to collaborate with HR54–56. Importantly, several findings now indicate that some of these repair proteins can influence fork progression rates. Overexpression of TLS polymerases can slow down the global rates of fork progression57. The key HR protein RAD51 and the RAD51 paralog XRCC3, appear to slow fork progression on DNA containing cis-Pt or BPDE adducts58, 59.
WRN could plausibly participate in each of the above-mentioned pathways. For example, WRN plays a role in RAD51-dependent HR repair13, 14, 24, BER60, and has been shown to interact physically or functionally with RAD5261, BRCA162, TLS polymerases63, FEN-164, PARP-165, WRNIP/Mgs166, and PCNA67.
Thus, one simple model that could explain why WRN-depleted cells exhibit impaired replication fork progression after genotoxic stress is that WRN facilitates the elimination of single-stranded gaps arising behind the fork due to a bypass of an MMS lesion or a replication restart after HU (Figure 7). In order to do that, WRN could participate in and/or cooperate with gap filling by TLS or by HR-mediated template switching, as well as with BER-mediated gap filling or lesion repair36. In each instance, the repair activity immediately behind the fork may affect fork progression rate.
Figure 7.
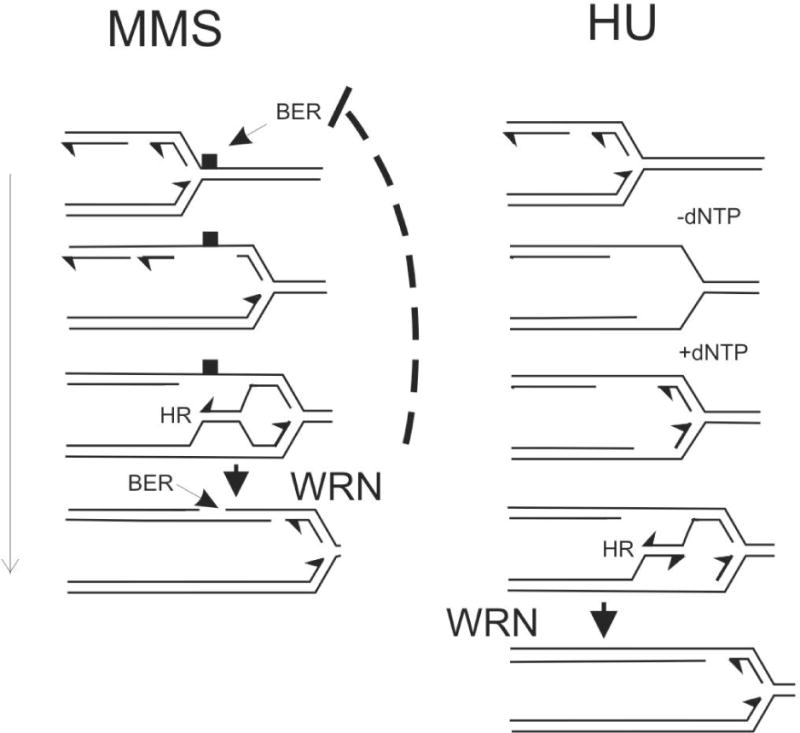
Putative roles of WRN in replication fork progression on MMS-damaged DNA or after HU-mediated replication arrest. Fork progression on MMS-damaged DNA may depend on lesion bypass by template switching, mediated by HR and/or by a TLS polymerase; the lesion is removed by BER. All three reactions may be facilitated by WRN. In one specific version of the model, WRN resolves a D-loop that arose from template switching. WRN may also coordinate BER with fork movement. Fork stalling in the presence of HU may lead to single-stranded gaps as the replicative helicase unwinds a stretch of DNA ahead of the replisome. Once the fork resumes elongation, these gaps may be eliminated by re-initiation of Okazaki fragments and at a stalled though otherwise intact 3′ terminus of a leading strand. However, it is conceivable that these gaps may remain behind as the replisome reinitiates downstream. In this case, gaps may be filled by template switching mediated by HR and/or by TLS polymerases. Shown here is the template-switching scenario, where WRN plays a role similar to the one we show for a fork traversing MMS-damaged DNA.
A specific scenario can be envisioned, where WRN participates in the final stage of template switch-mediated gap filling by resolving a D-loop between daughter DNA strands (Figure 7). This is analogous to the role proposed for the yeast RecQ helicase, Sgs168, and is consistent with the role of WRN in resolution of HR intermediates14. This also agrees with the argument that D-loops may slow fork progression58. WRN could also promote BER specifically behind the fork but not ahead of it, which would eliminate the risk of forks running into BER intermediates – nicked and gapped DNA – and thus would improve fork progression.
Recent work has shown that the human BLM RecQ helicase defective in Bloom syndrome is required for optimal fork velocity during unperturbed S phase69 and for resumption of replication by HU- or aphidicolin-arrested forks70. Our results describe a complementary role for WRN – that of promoting progression of forks that resumed replication – and thus begin to address the mechanistic basis of functional redundancy and cooperation among human RecQ helicases in maintaining genome integrity. It should be possible to further explore which pathways–HR, BER, or lesion bypass–collaborate with WRN, BLM, or other human RecQ helicases at replication forks by measuring fork progression rates in cells that lack individual RecQ helicases, alone or together with one or more of the interacting proteins described above.
The results reported here have implications for understanding the pathogenesis of Werner syndrome as well as the driving force behind silencing of WRN in a subset of human cancers71. It is accepted that WRN-deficient cells are genetically unstable and that this instability drives the development of clinical symptoms of the syndrome over a lifetime of an individual, or promotes tumor progression. Our data contribute the view that genomic instability of WRN−/− cells may result from a defect in the response of replication forks to DNA damage or arrest, whether due to endogenous insults such as reactive oxygen species, or to environmental genotoxins or chemotherapeutic agents.
Supplementary Material
Supplementary Figure 1. A) Representative Western blots of mock-depleted (pBabe, pLKO.1) or shWRN-depleted (WRNsi, WRN2-4) primary human fibroblasts and GM639cc1 WRN+ SV40 fibroblasts. CHK1 or GAPDH were used as loading controls. Note that different batches of the GAPDH antibody were used for primary and SV40-transformed fibroblast Western blots. B) WRN depletion by the new lentiviral shRNA WRN2-4 results in delayed recovery from MMS treatment in S phase, similar to the data obtained with a retroviral shRNA, WRNsi. Mock-depleted (pLKO.1) or WRN-depleted (WRN2-4) GM847 SV40 fibroblasts were labeled with BrdU for the first 8 hrs after release from mimosine arrest and treated with 0.005% MMS for 1 hr. MMS and BrdU were removed and cells were followed for 25 hrs. BrdU-positive subpopulation was identified as described in Figure 1C, and percent of BrdU-positive G1 cells was plotted as a function of time after release from mimosine. C) Caffeine reduces delay of cell cycle after MMS pulse in S phase. Unsynchronized mock-depleted (pLKO.1) or WRN-depleted (WRN2-4) GM639cc1 SV40 fibroblasts were labeled with BrdU for 2 hrs. MMS was added to some samples at 0.005% during the second hour of BrdU labeling. BrdU and MMS were removed and cells followed for up to 25 hrs. Shown are cell cycle distributions of BrdU+ subpopulations of cells that were recovering in the presence or absence of caffeine (3mM) for 16.5 hrs after a pulse of MMS. For comparison, bottom panels show cell cycle distributions of BrdU+ subpopulations of cells at 14 hrs after a pulse of BrdU and no MMS.
Acknowledgments
We thank Dr. Peter Rabinovitch for support and for the use of the FACS facility, Jeanne Fredrickson, Thong Pham and Mike Shen for assistance with FACS. We are grateful to Drs. David Schwartz, Dalia Dhingra, and Paolo Norio for advice on microchannels and glass derivatization, to Dr. Brian Kennedy for the use of the confocal microscope, to Drs. Carla Grandori, Wenyi Feng and Bonita Brewer for support, discussions, and critique, and to Alden Hackman for help with graphics. This work was supported by the grants P01 CA77872 from NCI to R. J.M. Jr, P20 CA103728-04, Subaward 000618167 from NCI to J.M.S., and R21/R33 EB003307 from NIBIB to A.F.
References
- 1.Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Yu CE, Oshima J, Wijsman EM, Nakura J, Miki T, Piussan C, Matthews S, Fu YH, Mulligan J, Martin GM, Schellenberg GD. Mutations in the consensus helicase domains of the Werner syndrome gene. Werner’s Syndrome Collaborative Group. Am J Hum Genet. 1997;60:330–41. [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis NA, Groden J, Ye T-Z, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–66. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 4.Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22:82–4. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- 5.Hickson ID. RecQ helicases: caretakers of the genome. Nature Reviews Cancer. 2003;3:169–78. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 6.Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;577:237–51. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Lee JW, Harrigan J, Opresko PL, Bohr VA. Pathways and functions of the Werner syndrome protein. Mech Ageing Dev. 2005;126:79–86. doi: 10.1016/j.mad.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Poot M, Yom JS, Whang SH, Kato JT, Gollahon KA, Rabinovitch PS. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB Journal. 2001;15:1224–6. doi: 10.1096/fj.00-0611fje. [DOI] [PubMed] [Google Scholar]
- 9.Poot M, Gollahon KA, Emond MJ, Silber JR, Rabinovitch PS. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB Journal. 2002;16:757–8. doi: 10.1096/fj.01-0906fje. [DOI] [PubMed] [Google Scholar]
- 10.Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Human Genetics. 1999;104:10–4. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- 11.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s Syndrome Protein Is Required for Correct Recovery after Replication Arrest and DNA Damage Induced in S-Phase of Cell Cycle. Mol Biol Cell. 2001;12:2412–21. doi: 10.1091/mbc.12.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2000;456:45–57. doi: 10.1016/s0027-5107(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 13.Prince PR, Emond MJ, Monnat RJ., Jr Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001;15:933–8. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous Recombination Resolution Defect in Werner Syndrome. Mol Cell Biol. 2002;22:6971–8. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Multani AS, Chang S. WRN at telomeres: implications for aging and cancer. J Cell Sci. 2007;120:713–21. doi: 10.1242/jcs.03397. [DOI] [PubMed] [Google Scholar]
- 16.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective Telomere Lagging Strand Synthesis in Cells Lacking WRN Helicase Activity. Science. 2004;306:1951–3. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 17.Fujiwara Y, Higashikawa T, Tatsumi M. A retarded rate of DNA replication and normal level of DNA repair in Werner’s syndrome fibroblasts in culture. J Cell Physiol. 1977;92:365–74. doi: 10.1002/jcp.1040920305. [DOI] [PubMed] [Google Scholar]
- 18.Poot M, Hoehn H, Runger TM, Martin GM. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Experimental Cell Research. 1992;202:267–73. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Lopez A, Jackson D, Iborra F, Cox L. Asymmetry of DNA replication fork progression in Werner’s syndrome. Aging Cell. 2002;1:30–9. doi: 10.1046/j.1474-9728.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 20.Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Reports. 2000;1:80–4. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes to Cells. 2001;6:421–30. doi: 10.1046/j.1365-2443.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 22.Otterlei M, Bruheim P, Ahn B, Bussen W, Karmakar P, Baynton K, Bohr VA. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J Cell Sci. 2006;119:5137–46. doi: 10.1242/jcs.03291. [DOI] [PubMed] [Google Scholar]
- 23.Pichierri P, Rosselli F, Franchitto A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- 24.Swanson C, Saintigny Y, Emond MJ, Monnat J, Raymond J. The Werner syndrome protein has separable recombination and survival functions. DNA Repair. 2004;3:475–82. doi: 10.1016/j.dnarep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Krude T. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Experimental Cell Research. 1999;247:148–59. doi: 10.1006/excr.1998.4342. [DOI] [PubMed] [Google Scholar]
- 26.Grandori C, Wu K-J, Fernandez P, Ngouenet C, Grim J, Clurman BE, Moser MJ, Oshima J, Russell DW, Swisshelm K, Frank S, Amati B, Dalla-Favera R, Monnat RJ., Jr Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003;17:1569–74. doi: 10.1101/gad.1100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK. A Lentiviral RNAi Library for Human and Mouse Genes Applied to an Arrayed Viral High-Content Screen. Cell. 2006;124:1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 28.Dhillon KK, Sidorova J, Saintigny Y, Poot M, Gollahon K, Rabinovitch PS, Monnat RJ. Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell. 2007;6:53–61. doi: 10.1111/j.1474-9726.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen C, Hirdes D, Folch A. Gray-scale photolithography using microfluidic photomasks. Proceedings of the National Academy of Sciences. 2003;100:1499–504. doi: 10.1073/pnas.0435755100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimalanta ET, Lim A, Runnheim R, Lamers C, Churas C, Forrest DK, dePablo JJ, Graham MD, Coppersmith SN, Goldstein S, Schwartz DC. A Microfluidic System for Large DNA Molecule Arrays. Anal Chem. 2004;76:5293–301. doi: 10.1021/ac0496401. [DOI] [PubMed] [Google Scholar]
- 31.Szekely AM, Bleichert F, Numann A, Van Komen S, Manasanch E, Ben Nasr A, Canaan A, Weissman SM. Werner Protein Protects Nonproliferating Cells from Oxidative DNA Damage. Mol Cell Biol. 2005;25:10492–506. doi: 10.1128/MCB.25.23.10492-10506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman ASH, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucl Acids Res. 2005;33:3799–811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tercero JA, Diffley JFX. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–7. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 34.Merrick CJ, Jackson D, Diffley JFX. Visualization of Altered Replication Dynamics after DNA Damage in Human Cells. J Biol Chem. 2004;279:20067–75. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 35.Conti C, Seiler JA, Pommier Y. The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle. 2007;6:2760–7. doi: 10.4161/cc.6.22.4932. Epub 007 Aug 22. [DOI] [PubMed] [Google Scholar]
- 36.Tourriere H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair Replication Fork Repair Processes. 2007;6:900–13. doi: 10.1016/j.dnarep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Paulsen RD, Cimprich KA. The ATR pathway: Fine-tuning the fork. DNA Repair Replication Fork Repair Processes. 2007;6:953–66. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Wilson DM, III, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair Repair of small base lesions in DNA–from molecular biology to phenotype. 2007;6:544–59. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 39.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis DNA Replication, the Cell Cycle and Genome Stability. 2003;532:103–15. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 40.Andreassen PR, Ho GPH, D’Andrea AD. DNA damage responses and their many interactions with the replication fork. Carcinogenesis. 2006;27:883–92. doi: 10.1093/carcin/bgi319. [DOI] [PubMed] [Google Scholar]
- 41.Eppink B, Wyman C, Kanaar R. Multiple interlinked mechanisms to circumvent DNA replication roadblocks. Experimental Cell Research DNA Repair and Cell Cycle Control. 2006;312:2660–5. doi: 10.1016/j.yexcr.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 42.Matsuzaki Y, Adachi N, Koyama H. Vertebrate cells lacking FEN-1 endonuclease are viable but hypersensitive to methylating agents and H2O2. Nucl Acids Res. 2002;30:3273–7. doi: 10.1093/nar/gkf440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shibata Y, Nakamura T. Defective Flap Endonuclease 1 Activity in Mammalian Cells Is Associated with Impaired DNA Repair and Prolonged S Phase Delay. J Biol Chem. 2002;277:746–54. doi: 10.1074/jbc.M109461200. [DOI] [PubMed] [Google Scholar]
- 44.Johnson RE, Yu S-L, Prakash S, Prakash L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol Cell Biol. 2007:MCB.01079-07. doi: 10.1128/MCB.01079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M, Takeda S. Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J. 2003;22:3188–97. doi: 10.1093/emboj/cdg308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nikiforov A, Svetlova M, Solovjeva L, Sasina L, Siino J, Nazarov I, Bradbury M, Tomilin N. DNA damage-induced accumulation of Rad18 protein at stalled replication forks in mammalian cells involves upstream protein phosphorylation. Biochemical and Biophysical Research Communications. 2004;323:831–7. doi: 10.1016/j.bbrc.2004.08.165. [DOI] [PubMed] [Google Scholar]
- 47.Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 2004;23:3872–82. doi: 10.1038/sj.onc.1207491. [DOI] [PubMed] [Google Scholar]
- 48.Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–70. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu N, Lim C-S. Differential roles of XRCC2 in homologous recombinational repair of stalled replication forks. Journal of Cellular Biochemistry. 2005;95:942–54. doi: 10.1002/jcb.20457. [DOI] [PubMed] [Google Scholar]
- 50.Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Molecular & Cellular Biology. 2002;22:5869–78. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nature Cell Biology. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 52.Kannouche PL, Wing J, Lehmann AR. Interaction of Human DNA Polymerase [eta] with Monoubiquitinated PCNA: A Possible Mechanism for the Polymerase Switch in Response to DNA Damage. Molecular Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 53.Yuasa MS, Masutani C, Hirano A, Cohn MA, Yamaizumi M, Nakatani Y, Hanaoka F. A human DNA polymerase eta complex containing Rad18, Rad6 and Rev1; proteomic analysis and targeting of the complex to the chromatin-bound fraction of cells undergoing replication fork arrest. Genes to Cells. 2006;11:731–44. doi: 10.1111/j.1365-2443.2006.00974.x. [DOI] [PubMed] [Google Scholar]
- 54.Saberi A, Hochegger H, Szuts D, Lan L, Yasui A, Sale JE, Taniguchi Y, Murakawa Y, Zeng W, Yokomori K, Helleday T, Teraoka H, Arakawa H, Buerstedde J-M, Takeda S. RAD18 and Poly(ADP-Ribose) Polymerase Independently Suppress the Access of Nonhomologous End Joining to Double-Strand Breaks and Facilitate Homologous Recombination-Mediated Repair. Mol Cell Biol. 2007;27:2562–71. doi: 10.1128/MCB.01243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szuts D, Simpson LJ, Kabani S, Yamazoe M, Sale JE. Role for RAD18 in Homologous Recombination in DT40 Cells. Mol Cell Biol. 2006;26:8032–41. doi: 10.1128/MCB.01291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N, Takeda S. Dual Roles for DNA Polymerase [eta] in Homologous DNA Recombination and Translesion DNA Synthesis. Molecular Cell. 2005;20:793–9. doi: 10.1016/j.molcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 57.Pillaire MJ, Betous R, Conti C, Czaplicki J, Pasero P, Bensimon A, Cazaux C, Hoffmann JS. Upregulation of error-prone DNA polymerases beta and kappa slows down fork progression without activating the replication checkpoint. Cell Cycle. 2007;6:471–7. doi: 10.4161/cc.6.4.3857. Epub 2007 Feb 19. [DOI] [PubMed] [Google Scholar]
- 58.Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Molecular Cell. 2003;11:1109–17. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 59.Johansson F, Lagerqvist A, Erixon K, Jenssen D. A method to monitor replication fork progression in mammalian cells: nucleotide excision repair enhances and homologous recombination delays elongation along damaged DNA. Nucl Acids Res. 2004;32:e157. doi: 10.1093/nar/gnh154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrigan JA, Wilson DM, III, Prasad R, Opresko PL, Beck G, May A, Wilson SH, Bohr VA. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase {beta} Nucl Acids Res. 2006;34:745–54. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baynton K, Otterlei M, Bjoras M, von Kobbe C, Bohr VA, Seeberg E. WRN Interacts Physically and Functionally with the Recombination Mediator Protein RAD52. J Biol Chem. 2003;278:36476–86. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- 62.Cheng WH, Kusumoto R, Opresko PL, Sui X, Huang S, Nicolette ML, Paull TT, Campisi J, Seidman M, Bohr VA. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34:2751–60. doi: 10.1093/nar/gkl362. Print 006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kamath-Loeb AS, Lan L, Nakajima S, Yasui A, Loeb LA. Werner syndrome protein interacts functionally with translesion DNA polymerases. PNAS. 2007;104:10394–9. doi: 10.1073/pnas.0702513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, Kao H-I, Bambara RA, Brosh RM., Jr WRN helicase and FEN-1 form a complex upon replication arrest and together process branch-migrating DNA structures associated with the replication fork. Mol Biol Cell. 2003:E03-8-0567. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng W-H, Bohr VA. Central Role for the Werner Syndrome Protein/Poly(ADP-Ribose) Polymerase 1 Complex in the Poly(ADP-Ribosyl)ation Pathway after DNA Damage. Mol Cell Biol. 2003;23:8601–13. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawabe Y-i, Seki M, Seki T, Wang W-S, Imamura O, Furuichi Y, Saitoh H, Enomoto T. Covalent Modification of the Werner’s Syndrome Gene Product with the Ubiquitin-related Protein, SUMO-1. J Biol Chem. 2000;275:20963–6. doi: 10.1074/jbc.C000273200. [DOI] [PubMed] [Google Scholar]
- 67.Rodriguez-Lopez AM, Jackson DA, Nehlin JO, Iborra F, Warren AV, Cox LS. Characterisation of the interaction between WRN, the helicase/exonuclease defective in progeroid Werner’s syndrome, and an essential replication factor, PCNA. Mechanisms of Ageing and Development. 2003;124:167–74. doi: 10.1016/s0047-6374(02)00131-8. [DOI] [PubMed] [Google Scholar]
- 68.Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair Replication Fork Repair Processes. 2007;6:994–1003. doi: 10.1016/j.dnarep.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 69.Rao VA, Conti C, Guirouilh-Barbat J, Nakamura A, Miao Z-H, Davies SL, Sacca B, Hickson ID, Bensimon A, Pommier Y. Endogenous {gamma}-H2AX-ATM-Chk2 Checkpoint Activation in Bloom’s Syndrome Helicase Deficient Cells Is Related to DNA Replication Arrested Forks. Mol Cancer Res. 2007;5:713–24. doi: 10.1158/1541-7786.MCR-07-0028. [DOI] [PubMed] [Google Scholar]
- 70.Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol. 2007;14:677–9. doi: 10.1038/nsmb1267. Epub 2007 Jun 24. [DOI] [PubMed] [Google Scholar]
- 71.Agrelo R, Cheng W-H, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, Guerrero D, Castells A, von Kobbe C, Bohr VA, Esteller M. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proceedings of the National Academy of Sciences. 2006;103:8822–7. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. A) Representative Western blots of mock-depleted (pBabe, pLKO.1) or shWRN-depleted (WRNsi, WRN2-4) primary human fibroblasts and GM639cc1 WRN+ SV40 fibroblasts. CHK1 or GAPDH were used as loading controls. Note that different batches of the GAPDH antibody were used for primary and SV40-transformed fibroblast Western blots. B) WRN depletion by the new lentiviral shRNA WRN2-4 results in delayed recovery from MMS treatment in S phase, similar to the data obtained with a retroviral shRNA, WRNsi. Mock-depleted (pLKO.1) or WRN-depleted (WRN2-4) GM847 SV40 fibroblasts were labeled with BrdU for the first 8 hrs after release from mimosine arrest and treated with 0.005% MMS for 1 hr. MMS and BrdU were removed and cells were followed for 25 hrs. BrdU-positive subpopulation was identified as described in Figure 1C, and percent of BrdU-positive G1 cells was plotted as a function of time after release from mimosine. C) Caffeine reduces delay of cell cycle after MMS pulse in S phase. Unsynchronized mock-depleted (pLKO.1) or WRN-depleted (WRN2-4) GM639cc1 SV40 fibroblasts were labeled with BrdU for 2 hrs. MMS was added to some samples at 0.005% during the second hour of BrdU labeling. BrdU and MMS were removed and cells followed for up to 25 hrs. Shown are cell cycle distributions of BrdU+ subpopulations of cells that were recovering in the presence or absence of caffeine (3mM) for 16.5 hrs after a pulse of MMS. For comparison, bottom panels show cell cycle distributions of BrdU+ subpopulations of cells at 14 hrs after a pulse of BrdU and no MMS.