

Abstract
Purpose
The CYP17A1 inhibitor abiraterone markedly reduces androgen precursors and is thereby effective in castration-resistant prostate cancer (CRPC). However, abiraterone increases progesterone, which can activate certain mutant androgen receptors (ARs) identified previously in flutamide-resistant tumors. Therefore, we sought to determine if CYP17A1 inhibitor treatment selects for progesterone activated mutant ARs.
Experimental Design
AR was examined by targeted sequencing in metastatic tumor biopsies from 18 CRPC patients who were progressing on a CYP17A1 inhibitor (17 on abiraterone, 1 on ketoconazole), alone or in combination with dutasteride, and by whole exome sequencing in residual tumor in one patient treated with neoadjuvant leuprolide plus abiraterone.
Results
The progesterone-activated T878A mutant AR was present at high allele frequency in 3 of the 18 CRPC cases. It was also present in one focus of resistant tumor in the neoadjuvant treated patient, but not in a second clonally related resistant focus which instead had lost one copy of PTEN and both copies of CHD1. The T878A mutation appeared to be less common in the subset of CRPC patients treated with abiraterone plus dutasteride, and transfection studies showed that dutasteride was a more potent direct antagonist of the T878A versus the wildtype AR.
Conclusions
These findings indicate that selection for tumor cells expressing progesterone-activated mutant ARs is a mechanism of resistance to CYP17A1 inhibition.
Introduction
The standard treatment for metastatic prostate cancer (PCa) is surgical or medical castration to abrogate testicular androgen synthesis and androgen receptor (AR) activity. This therapy may also include treatment with a direct AR antagonist such as bicalutamide or flutamide. However, most patients relapse within several years despite castrate androgen levels (castration-resistant prostate cancer, CRPC) (1). One mechanism driving AR activity in CRPC is increased conversion of weak androgens produced by the adrenal glands (DHEA and androstenedione) into potent androgens (testosterone and dihydrotestosterone, DHT) by the tumor cells (2, 3). Moreover, these tumors can also synthesize physiologically significant levels of androgen de novo from cholesterol (4–6). CYP17A1 is the critical enzyme that converts progesterone and related C21 steroids to DHEA and other C19 steroids. CYP17A1 inhibitors can thereby markedly further decrease the levels of testosterone and DHT, and the CYP17A1 inhibitor abiraterone has been approved by the US FDA for treatment of CRPC (7, 8). Unfortunately, patients who respond to abiraterone still generally relapse within 1–2 years, and the molecular mechanisms responsible for these relapses remain to be established.
We showed previously that AR mutations are rare in CRPC patients who relapse after standard medical or surgical castration, while long-term treatment with the AR antagonist flutamide could select for tumor cells expressing mutant AR’s (mutations in codons 875 or 878, codon numbering based on the human reference genome Hg19) that are activated, rather than repressed by hydroxyflutamide (the active metabolite of flutamide) (9–12). Treatment with bicalutamide may similarly select for AR with mutations in codon 742 that can be activated by bicalutamide (13, 14). Interestingly, a mutation in codon 877 that results in AR activation by the AR antagonist enzalutamide also was recently described (15–17). Importantly, AR’s with mutations in codons 875 and 878 can also be strongly stimulated by progesterone, which is only a very weak agonist for the wild-type AR (9, 12). Indeed, we reported previously that the AR transcriptional activity in androgen-starved C4-2 PCa cells, which are a CRPC subline of LNCaP cells that express a T878A mutant AR, was resistant to abiraterone and instead was driven by intratumoral synthesis of progesterone (5).
Significantly, as progesterone is an upstream substrate of CYP17A1, its levels are not decreased by CYP17A1 inhibitors and are generally instead increased in men treated with abiraterone (18). Therefore, we have speculated that CYP17A1 inhibitor therapy may select for tumor cells expressing progesterone activated mutant AR’s (5). To test this hypothesis, in this study we examined the AR in tumors that were resistant to CYP17A1 inhibition in patients treated with abiraterone or ketoconazole (a less specific CYP17A1 inhibitor).
Materials and Methods
Patients and Clinical Samples
All procedures were performed under protocols approved by the Beth Israel Deaconess Medical Center IRB and/or the Dana Farber/Harvard Cancer Center IRB. Tissue samples from radical prostatectomy samples in the neoadjuvant leupron-abiraterone clinical trial were fixed in formalin and processed to paraffin. Needle biopsies of metastatic CRPC lesions in bone or soft tissue (liver or lymph node) were obtained under CT guidance, with up to 6 cores taken at a site. These metastatic CRPC biopsy specimens were immediately fixed in formalin or PaxGene (Qiagen) fixative and processed to paraffin, or frozen in optimal cutting temperature (OCT) medium (Sakura). Metastatic bone specimens frozen in OCT were cut on a −20°C cryostat at 8 μm thickness using a tungsten-carbide blade. Metastatic bone specimens embedded in paraffin were cut on a microtome at 8 μm thickness using a tungsten-carbide blade. The tungsten-carbide blade was cleaned with DNAZap (Ambion) between specimens. Soft tissue specimens embedded in paraffin were cut on a microtome at 6 μm thickness using a stainless steel blade. A new steel blade was used for each specimen. A reference slide was stained with hematoxylin and eosin every 100 μm to confirm the presence of tumor and to evaluate tumor content. All slides were examined by at least two pathologists.
DNA and RNA Extraction
Metastatic specimens containing at least 10% tumor cell content were cut as 8 μm ribbons, and approximately 10 ribbons were isolated per biopsy core. Genomic DNA and total cellular RNA were simultaneously isolated from the same sample using the AllPrep DNA/RNA FFPE Kit (Qiagen) for formalin-fixed samples and the AllPrep DNA/RNA Micro Kit (Qiagen) for fresh-frozen samples. Prior to nucleic acid extraction from neoadjuvant samples, 6 μm sections were stained on glass slides with PIN-4 (Biocare) to identify residual tumor, and samples were cut onto polyethylene naphthalate (PEN) metal frame slides (Molecular Machines & Industries) and lightly stained with Histogene Stain (Life Technologies). Tumor foci were captured from target areas using 20-micron infrared pulses and excised from the adjacent tissue using the ultraviolet laser on an ArcturusXT Nikon Eclipse Ti-E microdissection system, yielding approximately 50–100 ng of genomic DNA per sample. Genomic DNA and total cellular RNA were simultaneously isolated from the same sample using the AllPrep DNA/RNA FFPE Kit (Qiagen). Normal control tissue was microdissected from adjacent areas marked as benign.
Deep Sequencing
10 ng of RNA from metastatic biopsy cores was converted into cDNA using the Sensiscript Reverse Transcription Kit (Qiagen) with an AR-specific primer in the 3′ UTR (5′-AGAGTTATAACAGGCAGAA-3′). The AR ligand binding domain (exons 4-8) and a portion of the DNA binding domain was then amplified with a nested primer in the 3′ UTR and a primer in the DNA binding domain using polymerase chain reaction (PCR) with the HotStart HiFidelity Polymerase Kit (Qiagen): Forward: 5′-GCTGAAGGGAAACAGAAGTACC-3′; Reverse: 5′-GGAAATAGGGTTTCCAATGCTTCA-3′. DNA from PCR amplification reactions was gel-excised, sheared to approximately 200 bp on a Covaris E220 sonicator, and built into multiplexed, barcoded sequencing libraries using the NEBNext Ultra DNA Library Prep Kit for Illumina and NEBNext Multiplex Oligos (New England Biolabs). Libraries were pooled and sequenced on a MiSeq (Illumina) with 100 cycles paired-end.
To validate mutations observed in sequencing cDNA from the metastatic CRPC tumors, PCR was performed from 10 ng of genomic DNA with the following primers flanking the AR exon 8 coding region: Forward: 5′-GTTGGGGAAGAGGCTAGCAG-3′; Reverse: 5′-GGCACTGCAGAGGAGTAGTG-3′. Multiplexed libraries were then prepared from the amplified DNA and sequenced as above. Genomic DNA extracted from tumor foci and control regions in samples from the neoadjuvant trial were similarly amplified with the above primers flanking the exon 8 coding region and sequenced.
To verify that the T878A mutation detected in tumor samples was not the result of contamination with LNCaP derived cells (which carry the same AR mutation), an area of genomic DNA corresponding to a private SNP or mutation (T to A) in the LNCaP cells at ChrX:24078360 within EIF2S3 was also sequenced. PCR primers flanking this SNP were used to amplify 10 ng of genomic DNA from each tumor sample positive for the T878A mutation, and the PCR products were deep sequenced to least 5,000-fold coverage. The following primers were used: Forward: 5′-GCTACTATGCTGAACGGTGC-3′; Reverse: 5′-TTCTCGCAAGCTATCAGCCC-3′.
A neoadjuvant case was also examined by whole exome sequencing using the SureSelectXT Target Enrichment System (Agilent), with the addition of baits covering the entire gene for a group of candidate PCa tumor suppressor genes (PTEN, TP53, RB1, PHLPP1, PHLPP2, KLF6, BRCA1, BRCA2, NKX3.1, CDKN1B, CDKN2A, MSH6, INPP4B, SPOP, CHD1, CHD5, SMAD4, HDAC9, DKK1, and DAB2IP) so that we could more reliably detect single copy losses based on LOH for SNPs. The total sequence captured was ~57 megabases. Multiplex library construction, hybrid capture, and library amplification were performed as described previously (19). The indexed libraries were sequenced on a HiSeq 2500 instrument (Illumina) with 100 paired end (100×100) cycles at the Harvard Medical School Genetics Department Core facility. Sequence data was aligned to the human reference genome (Hg19) using RUM (20), and were processed using SAMtools (21), VarScan (22), and snpEFF (23). All mutations were confirmed by visual inspection using the Integrative Genome Viewer (24). All data was deposited at the Sequence Read Archive (SRA) hosted by the National Center for Biotechnology Information (NCBI) under Accession ID SRP033306.
Cell culture and AR inhibition studies
C4-2 cells and PC-3 lines stably expressing wild-type or T878A mutant AR were cultured in RPMI-1640 medium with 10% FBS. VCaP and LAPC4 cells were cultured in DMEM medium with 10% FBS. PC-3 and VCaP parental cells were obtained from ATCC and passaged for less than 6 months prior to AR transduction. PC-3 and VCaP cells were authenticated by ATCC by short tandem repeat (STR) profiling. LAPC4 and C4-2 cells were authenticated by STR profiling by DDC Medical (Fairfield, OH). For steroid stimulation and inhibitor studies, cells were grown in medium with 5% charcoal-dextran stripped serum for 3 days and then treated with steroids and antagonists 24 hours. Quantitative real-time RT-PCR amplification was carried out on RNA extracted from cell lines using TRIZOL. 20 ng RNA was used for each reaction and the results were normalized by co-amplification of GAPDH RNA. Reactions were performed on a StepOnePlus PCR system (Applied Biosystems) using Taqman one-step RT-PCR reagents. PCR data are presented as mean ± SD for replicate samples. Primer and probe sequences for PSA, TMPRSS2, and PLZF were previously described (5), and primers for NKX3.1 or FKBP5 were directly purchased as inventoried primer-probe mixes (TaqMan) from Applied Biosystems.
Results
AR mutation in CRPC patients treated with CYP17A1 inhibitors
Bone marrow or soft tissue needle biopsies were obtained from a series of men with CRPC who were progressing during therapy with ketoconazole, abiraterone, or to combination therapy with abiraterone plus dutasteride (dual type 1 and 2 5α-reductase inhibitor). Biopsies in the latter group were obtained as part of a clinical trial (ClinicalTrials.gov identifier NCT01393730), wherein patients with CRPC were treated daily with abiraterone acetate (1000 mg), dutasteride (3.5 mg), and prednisone (5 mg), and biopsies were obtained immediately prior to initiation of therapy and at relapse. Biopsies were sectioned and examined to identify those that contained at least ~10% tumor based on histology, and additional sections were then cut from these positive biopsies for extraction of RNA and genomic DNA. The treatment history for those men with positive biopsies that were analyzed is shown in Table 1. This included one patient treated with ketoconazole (BI-1), three treated with single agent abiraterone (BI-2, 3, and 4), and fourteen treated with combination abiraterone and dutasteride.
Table 1.
Patient Characteristics
| Patient | Age (years) | Gleason Score | Prior ADT (months) | Duration of Abiraterone Therapy (months)* | PSA Response on Abiraterone (initial, nadir, % decline) |
|---|---|---|---|---|---|
| 448-2 | 88 | Unknown | leuprolide 156, bicalutamide | 13 | 50, 2.0, 96% |
| 448-11 | 71 | 4+5 | goserelin 21, bicalutamide 4 | 12 | 812, 37, 95% |
| 448-25 | 69 | 4+3 | leuprolide 15 | 14 | 25, 14, 44% |
| 448-26 | 64 | 4+3 | leuprolide 4 | 6 | 99, 27, 73% |
| 448-36 | 68 | 4+4 | leuprolide 22, ketoconazole 11 | 6 | 75, 31, 59% |
| 448-37 | 86 | 4+3 | leuprolide 60 | 4 | 4.8, 1.8, 63% |
| BI-1** | 64 | 4+3 | leuprolide 4 | 6 (ketoconazole) | 213, 84, 61% |
| BI-2** | 81 | 4+4 | bicalutamide, leuprolide 6 | 40 | 16, 0.2, 99% |
| BI-3 | 61 | 4+5 | leuprolide 48, | 4 | no response |
| BI-4 | 58 | 3+4 | leuprolide 58, nilutamide 4 | 4 | 56, 45, 18% |
| 448-1 | 72 | 4+4 | leuprolide 9, bicalutamide 1, | 14 | 213, 44, 79% |
| 448-4 | 73 | 4+4 | gosereline 16, bicalutamide 4, enzalutamide 6, leuprolide 12 | 12 | 46, 6.0, 87% |
| 448-6** | 61 | unknown | leuprolide 14, bicalutamide 6, enzalutamide 3 | 8 | 40, 13, 68% |
| 448-7 | 57 | 4+3 | finasteride 24, bicalutamdie 6, leuprolide 9 | 5 | 51, 2.9, 94% |
| 448-13 | 80 | 3+3 | leuprolide 32, nilutamdie 6, bicalutamide 19, ketoconazole 1 | 9 | 11, 10, 9% |
| 448-14 | 73 | 5+4 | leuprolide 74, bicalutamide 14, nilutamide 5, ketoconazole 8 | 7 | 22, 21, 5% |
| 448-28 | 84 | unknown | leuprolide 10, bicalutamide 1, nilutamide 3 | 7 | 84, 11, 87% |
| 448-33 | 66 | 4+4 | leuprolide 10, bicalutamide 1 | 8 | 5.4, 2.4, 56% |
| 448-39 | 60 | 4+5 | leuprolide 10, bicalutamide 3 | 9 | 126, 55, 56% |
Patient BI-1 received ketoconazole, BI-2, 3, and 4 received single agent abiraterone, the remaining patients all received abiraterone in combination with dutasteride; relapse was based on development of new lesions on bone scan or increases in measurable disease, which in all cases was associated with an increase in serum PSA
Patients in whom T878A mutation was detected
The AR mRNA was then reverse transcribed using an AR specific primer in the 3′ UTR, PCR amplified, and analyzed by Illumina sequencing. This analysis revealed multiple mutations occurring at low frequency (<5%) that were of unclear functional significance, but the only mutation detected in >5% of reads in any patient sample was a threonine to alanine mutation in exon 8 at codon 878 (T878A) (Table 2). This mutation was found in 67.7% of reads from one patient treated with ketoconazole (patient BI-1), in 60.3% of reads from one patient treated with abiraterone (patient BI-2), and in 6.0% and 18.4% of reads from two liver biopsy cores in a patient treated with abiraterone plus dutasteride (patient 448-6). In this latter patient, similar analysis of a liver biopsy taken prior to starting therapy detected the T878A mutation in 119/135534 reads (.09%) (not shown). Significantly, none of these patients with the T878A mutation had been treated previously with flutamide or nilutamide, which function as agonists for this mutant AR (see Table 1).
Table 2.
AR mutations in cDNA from CRPC patients failing ketoconazole or abiraterone
| Patient | Treatment | Base | Amino Acid | Frequency | Tissue | Read Depth |
|---|---|---|---|---|---|---|
| BI-1 | ketoconazole | A to C | S647R | 2.4% | bone marrow | 18575 |
| A to G | T878A | 67.7% | bone marrow | 75086 | ||
| BI-2 | abiraterone | C to A | C595X | 3.0% | bone marrow | 179286 |
| G to A | V866M | 1.4% | bone marrow | 549428 | ||
| A to G | T878A | 60.3% | bone marrow | 518501 | ||
| 448-6 | abiraterone/dutasteride | A to G | E473G | 1.1% | liver | 5492 |
| C to T | S613F | 2.7% | liver | 146715 | ||
| A to G | T878A | 6.0% | liver* | 31709 | ||
| A to G | T878A | 18.4% | liver* | 341455 |
T878A mutation was detected in cDNA from 2 separate liver needle biopsies
To confirm these findings, we next examined genomic DNA extracted from each tumor biopsy. In cases where multiple tissue cores had been obtained from the same site, DNA from each was extracted and analyzed separately. The genomic DNA was PCR amplified using a combination of exonic and intronic primers encompassing the exon 8 coding region. To rule out the presence of any contaminating DNA from LNCaP or LNCaP derived cells that have the T878A mutation, we identified in LNCaP cells a private SNP (or mutation) in another X chromosome gene, EIF2S3 (ChrX:24078360 T>A), and also amplified each genomic sample with primers flanking this SNP. The genomic sequencing confirmed the presence of the T878A mutation in each biopsy from all three patients, with allelic frequencies from ~14% to ~23% (Supplementary Table S1). As tumor purity in these metastatic samples was <50%, these results indicate that a high frequency of tumor cells contained the T878A AR, consistent with it being a driver mutation. The LNCaP EIF2S3 gene private SNP was not detected in any of the patient tumor derived biopsies, confirming that the mutation was not from contaminating LNCaP cell DNA.
AR mutations in patient treated with neoadjuvant leuprolide and abiraterone
To further assess whether abiraterone treatment may be selecting for tumor cells with progesterone responsive mutant ARs, we examined a radical prostatectomy specimen from a patient enrolled in a neoadjuvant trial who was treated for 24 weeks with leuprolide and abiraterone prior to surgery (ClinicalTrials.gov identifier NCT00924469). Residual tumor was identified in two blocks from the radical prostatectomy specimen (blocks Q and S), and each focus along with an area that did not contain tumor was purified by laser-capture microdissection (Supplementary Figure S1). Genomic DNA from each focus and the nontumor control area were then examined by whole exome sequencing using a custom capture array that included the complete genes for a series of tumor suppressor genes implicated previously in PCa (see Materials and Methods).
Analysis of high confidence mutations revealed that both foci were derived from the same initial tumor, with 71 shared mutations (64 missense, 6 nonsense, and 1 frameshift) (Supplementary Table S2). Table 3 shows a subset of these mutations with allelic frequencies >10% and >20-fold coverage in both blocks. We also detected 245 mutations that were unique to the tumor focus in block Q (Supplementary Table S3), and 1944 mutations that were unique to the tumor in block S (Supplementary Table S4). Subsets of these unique mutations in the block Q focus (with at least 25-fold coverage and 35% allele frequency) and block S focus (at least 35-fold coverage and 45% allele frequency) are shown in Table 3. The latter mutations in the block S focus include a frameshift mutation in a DNA mismatch repair gene (mutL homolog 1, MLH1) that may account for the high mutation rate in this focus, as has been reported previously (25). Significantly, the AR T878A mutation was also found at high allele frequency in the block S tumor focus (15/32 reads, ~47%), but not in block Q (0/17 reads) or adjacent normal prostate (0/37 reads) (Table 3).
Table 3.
Mutations in block Q versus S residual tumor foci in neoadjuvant abiraterone/leuprolide trial
| Tumor Focus | Gene | Type | Change | Block S Focus | Block Q Focus | Normal Depth | COSMIC | ||
|---|---|---|---|---|---|---|---|---|---|
| Frequency | Depth | Frequency | Depth | ||||||
| QS | ABCA7 | Missense | D99N | 19.67% | 61 | 15.56% | 45 | 39 | |
| QS | AMACR | Missense | R171C | 18.75% | 32 | 13.79% | 29 | 29 | |
| QS | ARAF | Missense | M545I | 12.82% | 78 | 27.91% | 129 | 84 | |
| QS | CACNA1F | Frameshift | +1bp codon 588 | 15.00% | 40 | 14.29% | 77 | 51 | |
| QS | CCDC14 | Missense | L804F | 15.22% | 46 | 25.00% | 32 | 62 | |
| QS | CHEK2 | Missense | S110F | 42.11% | 38 | 13.79% | 29 | 83 | |
| QS | DCHS1 | Nonsense | Q769X | 18.87% | 53 | 15.18% | 191 | 51 | |
| QS | DNAH11 | Missense | S508N | 14.29% | 21 | 16.00% | 25 | 70 | |
| QS | GPR125 | Missense | A1181V | 14.29% | 42 | 11.54% | 26 | 37 | |
| QS | PINK1 | Nonsense | Q180X | 23.68% | 38 | 16.51% | 109 | 37 | |
| QS | SLC5A2 | Missense | G488E | 24.24% | 33 | 17.65% | 34 | 28 | |
| QS | TLN1 | Missense | A361V | 11.67% | 60 | 14.29% | 21 | 97 | Yes |
| S | AR | Missense | T878A | 46.88% | 32 | 17 | 37 | Yes | |
| S | MLH1 | Frameshift | −1bp codon 456 | 37.11% | 97 | 133 | 125 | ||
| S | ATM | Missense | R2060H | 46.49% | 114 | 41 | 57 | Yes | |
| S | BAI3 | Missense | R725Q | 46.92% | 130 | 125 | 143 | ||
| S | BOC | Missense | R3H | 45.31% | 64 | 110 | 41 | Yes | |
| S | CNGA2 | Missense | M378I | 46.81% | 47 | 55 | 88 | ||
| S | EHMT1 | Missense | D964N | 49.19% | 124 | 100 | 151 | ||
| S | FN1 | Missense | T1211I | 46.83% | 126 | 160 | 195 | ||
| S | GALNTL1 | Frameshift | −2bp codon 539 | 47.83% | 46 | 59 | 64 | ||
| S | HDAC6 | Missense | R862K | 48.00% | 75 | 159 | 81 | ||
| S | IDH3B | Missense | A282T | 47.13% | 87 | 98 | 168 | ||
| S | MFSD4 | Missense | S156N | 48.65% | 37 | 148 | 70 | Yes | |
| S | MYBPC1 | Missense | G483E | 46.07% | 89 | 56 | 56 | Yes | |
| S | PRICKLE4 | Frameshift | −1bp codon 374 | 45.57% | 79 | 165 | 84 | ||
| S | RMND5B | Missense | T71K | 45.24% | 42 | 134 | 87 | ||
| S | RPS6KL1 | Missense | A270S | 48.84% | 43 | 115 | 119 | ||
| S | TMEM27 | Missense | V81I | 48.65% | 37 | 39 | 39 | ||
| S | UNK | Missense | V761M | 46.75% | 169 | 99 | 272 | ||
| S | WDFY4 | Missense | P3109S | 48.68% | 76 | 86 | 62 | ||
| Q | ADAMTS12 | Missense | P1139S | 37 | 44.12% | 34 | 48 | Yes | |
| Q | AIFM1 | Missense | G138V | 55 | 35.90% | 39 | 140 | Yes | |
| Q | CLC | Missense | P34L | 22 | 43.33% | 30 | 40 | Yes | |
| Q | GREB1L | Missense | T1401I | 55 | 39.13% | 46 | 48 | ||
| Q | LOXL1 | Nonsense | Q44X | 60 | 37.93% | 29 | 61 | ||
| Q | MLLT4 | Missense | V994M | 62 | 37.04% | 27 | 41 | ||
| Q | NLN | Missense | P181L | 35 | 43.90% | 41 | 43 | ||
| Q | RPTN | Missense | S416R | 109 | 44.44% | 27 | 130 | ||
| Q | SASH1 | Nonsense | Q1136X | 175 | 47.12% | 278 | 184 | ||
| Q | SASH1 | Missense | D1140Y | 222 | 41.64% | 305 | 273 | ||
Mutations in the COSMIC database as having been previously identified in cancer are indicated.
Based on read depth and SNP analyses, we did not detect loss of any of the tumor suppressor genes captured by our bait library (PTEN, TP53, RB1, PHLPP1, PHLPP2, KLF6, BRCA1, BRCA2, NKX3.1, CDKN1B, CDKN2A, MSH6, INPP4B, SPOP, CHD1, CHD5, SMAD4, HDAC9, DKK1, or DAB2IP) in the block S tumor focus. In contrast, in the block Q tumor focus we detected loss of one copy of PTEN and both copies of CHD1 (Supplementary Figure S2). Together these findings reveal that there was substantial heterogeneity in this tumor prior to therapy, with losses of PTEN and CHD1 likely contributing to progression in the block Q focus, and the AR T878A mutation (possibly the result of a hypermutator phenotype) contributing to progression in the block S focus.
T878A mutation enhances the direct AR antagonist activity of dutasteride
More cases must be examined to determine the precise frequency of the T878A mutation, but it was noteworthy that the frequency appeared to be lower in patients on the abiraterone plus dutasteride clinical trial (1/14 cases examined). As progesterone is a substrate for 5α-reductase, we considered that dutasteride might be blocking the generation of a more potent ligand for the T878A AR. Indeed, treatment of C4-2 cells (which express the T878A AR) with dutasteride markedly suppressed the progesterone stimulated expression of multiple AR regulated genes (Fig. 1A). Therefore we next compared the potency of progesterone (Pg) versus its 5α-reduced metabolite, 5α-pregnane-3,20-dione (Pd). However, we found that Pg was more potent at induction of AR regulated genes in C4-2 cells, indicating that dutasteride is not blocking synthesis of a more potent progesterone metabolite (Fig. 1B).
Figure 1. Activation of T878A mutant AR by progesterone and reduced metabolite.
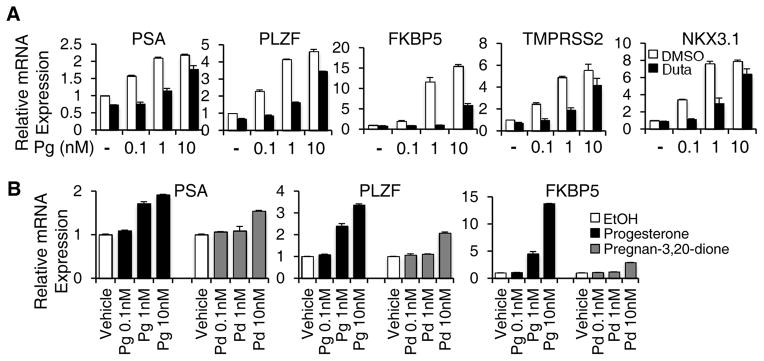
A, C4-2 cells (expressing endogenous T878A AR) cultured in steroid depleted medium containing charcoal-dextran serum were stimulated for 24 hours with progesterone (Pg) at 0.1 – 10 nM, with the addition of dutasteride at 10 μM (black bars) or vehicle control (DMSO, white bars). Expression of the indicated mRNA was then assessed by real time quantitative RT-PCR, with all results being normalized to basal levels. Values shown for all panels are average and S.D. for triplicate PCR reactions and are representative of at least 3 independent experiments. B, C4-2 cells in steroid depleted medium were stimulated with 0.1 – 10 nM progesterone (Pg, black bars) or pregnan-3,20-dione (Pd, grey bars) for 24 hours and mRNA levels were then assessed.
Although dutasteride was developed as a dual its 5α-reductase inhibitor, previous studies indicate that it can also directly inhibit AR activity (26–28). Therefore, we considered that dutasteride might be functioning as a direct AR antagonist independent of its 5α-reductase inhibitory activity. Consistent with these previous studies, dutasteride at 10 μM in C4-2 cells suppressed the expression of multiple AR regulated genes in response to 1 nM DHT (Fig. 2A). Moreover, this inhibition could be substantially overcome at 10 nM DHT, indicating that dutasteride was acting as a competitive AR antagonist. Using lower concentrations of dutasteride, we could also observe inhibitory effects at 1–2 μM (Supplementary Fig. S3). We next compared it to bicalutamide, an AR antagonist that retains its activity against the T878A mutant AR, and found that bicalutamide and dutasteride were comparable in their repression of DHT and progesterone stimulated AR activity in C4-2 cells (Fig. 2B). In contrast, dutasteride was less potent at blocking DHT stimulated AR activity in VCaP cells, which express an amplified wildtype AR (Fig. 2C). Dutasteride was similarly less potent than bicalutamide in LAPC4 cells, which express an unamplified wildtype AR (Supplementary Fig. S4). Finally, we examined an AR negative PCa cell line (PC-3) that was stably transfected with wildtype or T878A mutant AR, and found that dutasteride was again less potent than bicalutamide on the wildype versus the T878A mutant AR (Fig. 2D). Together these findings indicate that treatment with dutasteride may suppress activation of the T878A mutant AR by progesterone, or by other ligands, in patients treated with abiraterone.
Figure 2. Dutasteride is more potent antagonist of the T878A mutant versus wildtype AR.

A, C4-2 cells in steroid depleted medium were stimulated for 24 hours with DHT (0.1 – 10 nM) with the addition of dutasteride at 10 μM (black bars) or vehicle control (DMSO, white bars). B, C4-2 cells in steroid depleted medium were stimulated for 24 hours with vehicle (EtOH, white bars), DHT (1 nM, black bars), or progesterone (1 nM, grey bars), in the presence of dutasteride (Duta, 10 μM), bicalutamide (Bic, 10 μM), or vehicle (Veh). C, VCaP cells (amplified wildtype AR) in steroid depleted medium were stimulated for 24 hours with vehicle (EtOH, white bars) or DHT (1 nM, black bars), in the presence of dutasteride (Duta, 10 μM), bicalutamide (Bic, 10 μM), or vehicle (Veh). D, PC-3 cells (AR negative) that were stably transfected with wild-type (WT) or T878A AR were cultured in steroid depleted medium and stimulated for 24 hours with vehicle (EtOH, white bars) or DHT (1 nM, black bars), alone or in the presence of dutasteride (Duta, 10 μM) or bicalutamide (Bic, 10 μM). Expression of the AR regulated endogenous FKBP5 was then assessed. AR protein levels are shown on left.
DISCUSSION
We showed previously that the recurrent tumors in approximately one-third of patients treated long-term with the AR antagonist flutamide contained an AR with mutations in codons 875 or 878 that could be strongly stimulated by hydroxyflutamide, and that these mutant ARs could also be stimulated by progesterone and estradiol (9, 10, 12). In contrast, these mutations were not found in patients treated with surgical or medical castration alone (10, 11), indicating that they were driver mutations occurring in response to strong selective pressure to maintain AR activity. Treatment with abiraterone markedly decreases levels of downstream steroids including testosterone and DHT, but it does not decrease progesterone and related C21 steroids that are the upstream substrates for CYP17A1, which may instead be increased (18). The results in this study demonstrate that progesterone and/or other steroids upstream of CYP17A1, similarly to flutamide, can select for tumor cells with the T878A mutation in vivo, and indicate that this is a mechanism for acquired resistance to abiraterone. Moreover, the selection for this AR mutation indicates that the initial tumor cell clones giving rise to these castration-resistant and abiraterone-resistant tumors were AR positive and dependent on AR transcriptional activity.
It is noteworthy that we did not find other mutations that result in AR activation by progesterone, including H875Y and T878S (12). This may just reflect the modest number of cases examined, but we cannot rule out the possibilities that the progesterone-liganded T878A AR has more favorable functional properties or that another ligand upstream of CYP17A1 can more strongly and selectively drive the T878A mutant AR. Interestingly, while the T878A mutation was found in 3 cases where patients were treated with a single agent CYP17A1 inhibitor (ketoconazole or abiraterone), it was only found in 1/14 of the patients treated with the combination of abiraterone plus dutasteride. Our data indicate that dutasteride may be impairing selection for these mutant cells by functioning as a direct antagonist of the T878A AR. It should be noted that serum concentrations of dutasteride in men treated with the standard 0.5 mg/day dose are approximately 0.1 μM, and would presumably be higher in patients on the abiraterone plus dutasteride trial who received 3.5 mg/day. Therefore, although dutasteride is not a potent antagonist for the T878A AR, and is less potent on the wildtype AR, it may contribute to suppressing the selection of tumor cells with the T878A mutation.
Further studies of patients on abiraterone clinical trials should resolve whether dutasteride or other agents influence the spectrum of mechanisms that drive relapse. In any case, these findings provide a mechanism for abiraterone-resistance and establish that abiraterone-treated tumors are under strong selective pressure to retain AR transcriptional activity. These results have therapeutic implications as the T878A mutant AR can be inhibited by the available AR antagonists bicalutamide and enzalutamide. Indeed, we showed previously that bicalutamide was more effective in patients who developed CRPC after long-term treatment with flutamide, a subset of whom expressed the T878A mutant AR (29). These results also suggest that abiraterone may be more effective if used in conjunction with an AR antagonist, or if used early in conjunction with an LHRH agonist and prior to the selection of multiple castration-resistant clones (although our result in the neoadjuvant study indicates that these resistant clones may emerge early in some tumors). Finally, agents that suppress progesterone synthesis may be effective in a subset of patients who become resistant to abiraterone, both through suppressing mutant ARs and by further reducing downstream substrates for androgen synthesis. Moreover, as progesterone receptor may be increased in CRPC and could possibly contribute to stimulation of some AR regulated genes, lowering progesterone also may be beneficial by decreasing progesterone receptor activity in a subset of patients (30, 31).
Supplementary Material
Statement of Translational Relevance.
The CYP17A1 inhibitor abiraterone markedly reduces androgen precursors and is thereby effective in castration-resistant prostate cancer (CRPC). However, abiraterone increases progesterone, which can activate certain mutant androgen receptors (ARs) found previously in flutamide-resistant tumors. We identified the progesterone-activated T878A mutant AR in metastatic tumor biopsies from 3/18 CRPC patients who were relapsing on a CYP17A1 inhibitor, and in residual tumor from one patient treated with neoadjuvant leuprolide plus abiraterone. These findings indicate that selection for tumors cells with a progesterone-activated mutant AR is a mechanism for resistance to CYP17A1 inhibitors. Targeting this mechanism with AR antagonists or agents that suppress progesterone synthesis may be effective in a subset of abiraterone-resistant tumors.
Acknowledgments
Grant Support
This work was supported by grants from the National Institutes of Health (P01 CA163227, K99 CA166507 Dana-Farber/Harvard Cancer Center SPORE P50 CA090381, Pacific Northwest Prostate Cancer SPORE P50 CA097186, and T32 CA081156), the Department of Defense Prostate Cancer Research Program (W81XWH-13-1-0267, W81XWH-11-1-0295, W81XWH-10-1-0590, W81XWH-08-1-0414, and W81XWH-07-1-0443), a DF/HCC Mazzone Award, and by generous support through Challenge Awards from the Prostate Cancer Foundation. The abiraterone clinical trials were supported by Ortho Biotech Oncology Research & Development (unit of Cougar Biotechnology, now Janssen Research & Development).
We thank Drs. Jerome P. Richie, Martin G. Sanda, Bruce L. Dalkin, John W. Davis, and Christopher J. Logothetis for urological support and patient accrual.
Footnotes
Conflict of Interest Statement: The authors do not have any conflicts of interest to report.
References
- 1.Yuan X, Cai C, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2013;33:2815–25. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 3.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 5.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, et al. Intratumoral De Novo Steroid Synthesis Activates Androgen Receptor in Castration-Resistant Prostate Cancer and Is Upregulated by Treatment with CYP17A1 Inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 10.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–5. [PubMed] [Google Scholar]
- 11.Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673–8. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 12.Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997;3:1383–8. [PubMed] [Google Scholar]
- 13.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–53. [PubMed] [Google Scholar]
- 14.Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005;65:9611–6. doi: 10.1158/0008-5472.CAN-05-0817. [DOI] [PubMed] [Google Scholar]
- 15.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–43. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 16.Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 18.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 19.Sowalsky AG, Ye H, Bubley GJ, Balk SP. Clonal progression of prostate cancers from Gleason grade 3 to grade 4. Cancer Res. 2013;73:1050–5. doi: 10.1158/0008-5472.CAN-12-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–28. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087–92. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazier CB, Thomas LN, Douglas RC, Vessey JP, Rittmaster RS. Dutasteride, the dual 5alpha-reductase inhibitor, inhibits androgen action and promotes cell death in the LNCaP prostate cancer cell line. Prostate. 2004;58:130–44. doi: 10.1002/pros.10340. [DOI] [PubMed] [Google Scholar]
- 27.Chhipa RR, Halim D, Cheng J, Zhang HY, Mohler JL, Ip C, et al. The direct inhibitory effect of dutasteride or finasteride on androgen receptor activity is cell line specific. Prostate. 2013;73:1483–94. doi: 10.1002/pros.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Godoy A, Azzouni F, Wilton JH, Ip C, Mohler JL. Prostate cancer cells differ in testosterone accumulation, dihydrotestosterone conversion, and androgen receptor signaling response to steroid 5alpha-reductase inhibitors. Prostate. 2013;73:1470–82. doi: 10.1002/pros.22694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joyce R, Fenton MA, Rode P, Constantine M, Gaynes L, Kolvenbag G, et al. High dose bicalutamide for androgen independent prostate cancer: effect of prior hormonal therapy. J Urol. 1998;159:149–53. doi: 10.1016/s0022-5347(01)64039-4. [DOI] [PubMed] [Google Scholar]
- 30.Bonkhoff H, Fixemer T, Hunsicker I, Remberger K. Progesterone receptor expression in human prostate cancer: correlation with tumor progression. Prostate. 2001;48:285–91. doi: 10.1002/pros.1108. [DOI] [PubMed] [Google Scholar]
- 31.Latil A, Bieche I, Vidaud D, Lidereau R, Berthon P, Cussenot O, et al. Evaluation of androgen, estrogen (ER alpha and ER beta), and progesterone receptor expression in human prostate cancer by real-time quantitative reverse transcription-polymerase chain reaction assays. Cancer Res. 2001;61:1919–26. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.