

Abstract
Recently, the interest in natural products for the treatment of cancer is increasing because they are the pre-screened candidates. In the present study, we demonstrate the therapeutic effect of celastrol, a triterpene extracted from the root bark of Chinese medicine on gastric cancer. The proliferation of AGS and YCC-2 cells were most sensitively decreased in six kinds of gastric cancer cell lines after the treatment with celastrol. Celastrol inhibited the cell migration and increased G1 arrest in cell-cycle populations in both cell lines. The treatment with celastrol significantly induced autophagy and apoptosis and increased the expression of autophagy and apoptosis-related proteins. We also found an increase in phosphorylated AMPK following a decrease in all phosphorylated forms of AKT, mTOR and S6K after the treatment with celastrol. Moreover, gastric tumor burdens were reduced in a dose-dependent manner by celastrol administration in a xenografted mice model. Taken together, celastrol distinctly inhibits the gastric cancer cell proliferation and induces autophagy and apoptosis. [BMB Reports 2014; 47(12): 697-702]
Keywords: Apoptosis, Autophagy, Celastrol, Chemo therapy, Gastric cancer
INTRODUCTION
Gastric cancer is the second leading cause of cancer related death and surgical resection is considered the mainstay of curative treatment, though it can only be performed in a small subgroup of patients (1). Most patients diagnosed with advanced gastric cancer or recurrence within five years after surgery will receive chemotherapeutic treatment (2, 3). Chemotherapy clearly improves the gastric cancer survival, but the treatment is limited by the high rates of adverse effects and rapid therapeutic resistance (4). Therefore, there has been an ongoing development of novel therapeutic regimens that can overcome these adverse effects and resistance to cancer therapy.
Recently, numerous studies have indicated that natural remedies have therapeutic effects on inflammatory, metabolic and neoplastic diseases (5). The primary benefit of prescreened candidates of natural compounds is there process of natural selection and that they are already used for some purposes. Of them, celastrol has been of great interest due to its therapeutic potential (6). Celastrol, a quinine methide triterpenoid, is the most abundant bioactive compound derived from the root of Trypterigium wilfordii Hook L, also known as “Thunder of God Vine” (7). It has been reported to be an inhibitor of lipid peroxidation (8) and to have anti-arthritis and anti-Alzheimer effects due to the regulation of cytokine release (9-11). Moreover, celastrol has been found to suppress tumor initiation, progression and metastasis in a wide variety of tumor cells and in vivo cancer models (12). Studies of the therapeutic mechanism have revealed that it inhibits the heat shock protein 90 (Hsp90) and proteasomes (13, 14) and can suppress the NF-κB signaling pathway (15), AKT/mTOR pathway (16, 17), MAPK pathway (18) and VEGFR expression (19) in various cancers.
However, the therapeutic effects of celastrol on gastric cancer have not yet been clearly demonstrated. In this study, we measured the gastric cancer cell proliferation and induction of apoptosis and autophagy after celastrol treatment. We also determined the effects of celastrol on cell-survival-related signaling proteins. Additionally, we used the celastrol treatment as part of a gastric cancer xenograft mouse model and analyzed its tumor suppressive effects in vivo.
RESULTS
Celastrol inhibited cell proliferation and migration and induced cell cycle arrest and apoptosis in gastric cancer cells
determine the cytotoxic effect of celastrol in gastric cancer, we treated six different gastric cancer cell lines with different doses of celastrol (Fig. 1A). After 3 days of treatment, the cell proliferation decreased at concentrations of celastrol up to 1 μM. The IC50 of AGS cells was detected as being as low as 0.25 μM celastrol and the IC50 of YCC-2 was 0.5 μM celastrol. Moreover, the wound-healing properties, representing migration in both AGS and YCC-2 cell lines were found to be significantly impaired at 0.25 μM and 0.5 μM celastrol, respectively (Fig. 1B). The effects of celastrol treatment on the cell cycle were analyzed and the findings are presented in Fig. 1C. After 48 hours of treatment with different doses of celastrol in AGS and YCC-2 cells, a significant increase of cells in the G1 phase was detected at a concentration of 0.25 μM and the SubG1 phase was detected at the concentration of 1 μM after the treatment with celastrol in both cell lines (Fig. 1C). Also an apoptosis induction was detected after celastrol treatment in AGS and YCC-2 cells. Significant apoptosis was induced after the treatment with 0.5 μM celastrol in both cell lines (Fig. 1D).These findings suggest that celastrol is a potent inhibitor of gastric cancer cell proliferation and migration.
Fig. 1. Celastrol inhibited cell proliferation and migration and induced cell cycle arrest and apoptosis in gastric cancer cells. (A) Detection of cell proliferation by MTT assay after the treatment with celastrol using different doses (0, 0.25 0.5, and 1 μM) for 48 hours in six gastric cancer cell lines. (B) AGS cell migration was detected using a wound-healing assay. AGS and YCC-2 cells were scratched and treated with different doses (0, 0.25, 0.5 and 1 μM) for 24 hours and pictures were taken. (C) AGS and YCC-2 cells were treated with celastrol using different doses (0, 0.25, 0.5 and 1 μM) for 48 hours. Cell populations in each phase of the cell cycle were detected by PI staining. Apoptosis induction was detected by annexin V staining.
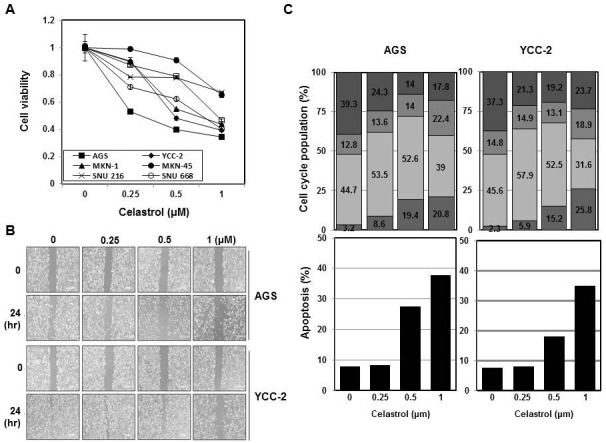
Celastrol increased the expression of cell-cycle and apoptosis related proteins and phosphorylated AMPK
The expression of cell-cycle-related proteins, such as p21 and p27 was markedly increased and the expression of cyclin D1 was decreased by the treatment with celastrol ranging from concentrations of 0.25 μM to 1 μM (Fig. 2A). Moreover, expression levels of apoptosis-related proteins were analyzed in AGS cells (Fig. 2B). Noticeable increases in cytochrome C and Bax were detected after the treatment with 0.5 μM celastrol, whereas apoptosis inhibitory proteins such as survivin, Bcl-2, and Bcl-xl drastically decreased after treatment with the same concentration of celastrol. The activation of caspases was measured by cleavage of the pro-forms and cleaved forms of PARP, caspase-3, caspase-9 and caspase-8 were decreased by the treatment with 0.5 μM celastrol. It is known that celastrol activates AMP-activated protein kinase (AMPK) and inhibits AKT/mammalian target of rapamycin (mTOR) signaling pathways to induce autophagy and apoptosis (20, 21). Therefore, we demonstrated the phosphorylation status of these proteins after the treatment with 0.5 μM celastrol in AGS cells (Fig. 2C). As early as 15 minutes after the initiation of treatment with 0.5 μM celastrol, significant increases in phosphorylated AMPK were detected in both cell lines (the data of YCC-2 were not shown). Substantial downstream targets, such as phosphorylated AKT, phosphorylated mTOR and phosphorylated S6K were also decreased at similar time points. Therefore, celastrol has an effect on the cell-survival-related signaling pathway in gastric cancer cell lines as early as 15 minutes after treatment.
Fig. 2. Celastrol increased expression cell-cycle and apoptosis related proteins and phosphorylated AMPK. AGS and YCC-2 cells were treated with celastrol using different doses (0, 0.25, 0.5 and 1 μM) for 48 hours. (A) p21 and p27 as a CDK inhibitor and cyclin D1 were detected by Western blot analysis. (B) Apoptosis-related proteins from AGS cell lysates were detected by Western blot analysis. (C) AGS cells were treated with 0.5 μM of celastrol at several time points (0, 15, 30 and 120 minutes) and cell lysates were prepared for Western blotting analysis. All experiments were individually performed more than twice and present representative data.
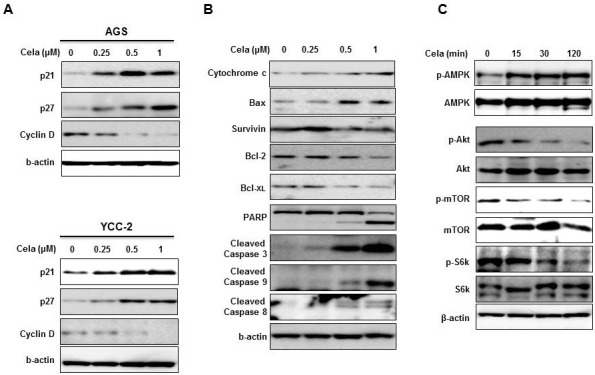
Celastrol induced autophagy and increased expression of the autophagy-related proteins
To determine the autophagy effect in gastric cancer cells (Fig. 3), we prepared stable RFP-tagged LC-3 expressed AGS cells and treated them with different doses of celastrol for 1 day. Compared with the 0.25 μM of celastrol that were needed to induce the G1 arrest, the same concentration of celastrol increased the number of autophagosomes in the cytosol (Fig. 3A).At higher concentrations of 0.5 and 1 μM celastrol, AGS cells were smaller and appeared to be undergoing the apoptotic process. The autophagy-related proteins LC3 I, II, ATG5 and Beclin1 were increased after the treatment with 0.25 μM celastrol, whereas ATG7 was increased at the highest concentration of celastrol (1 μM) in both AGS and YCC-2 cell lines (Fig. 3B). Therefore, it is likely that celastrol induced autophagy to initiate the G1 arrest of gastric cancer cells. Additionally, celastrol induced both autophagy and apoptosis, leading to an inhibition of gastric cancer cell growth.
Fig. 3. Celastrol induced autophagy in gastric cancer cells. (A) AGS cells expressing stable RFP-tagged LC-3 were treated with celastrol at different doses (0, 0.25, 0.5 and 1 μM) for 24 hours. RFP-tagged LC-3 was detected by fluorescent microscopy. DAPI staining was used to detect nuclei. (B) AGS and YCC-2 cells were treated with different doses of celastrol (0, 0.25, 0.5 and 1 μM) for 24 hours. Autophagyinduced proteins were detected by Western blot analysis. All experiments were individually performed more than twice and present representative data.
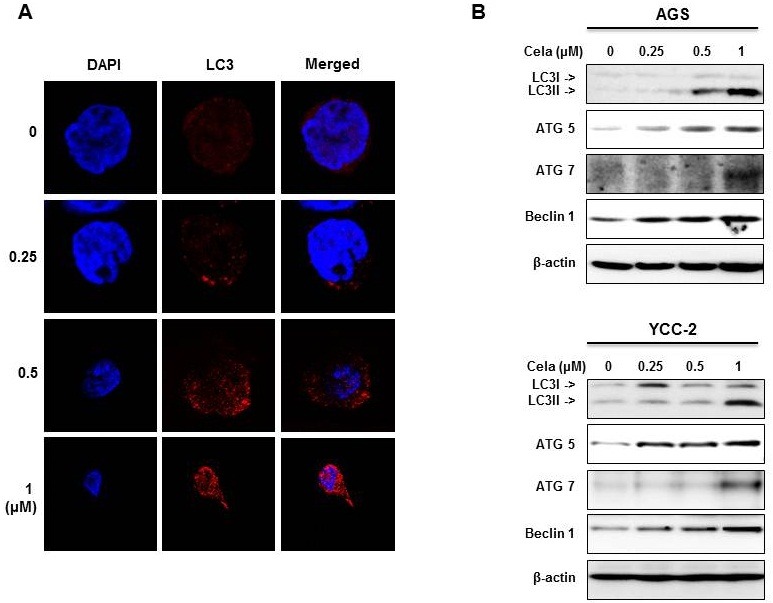
Administration of celastrol reduced gastric tumor burdens in xenografted mice
We confirmed the therapeutic effects of celastrol in vivo in a mouse model (Fig. 4). We prepared AGS cell xenografted mice as described previously (22), and orally administered 1 mg/kg or 2 mg/kg of celastrol until mice were sacrificed. Gastric tumor burdens in xenografted mice were significantly reduced by celastrol administration in a dose-dependent manner (Fig. 4A). After mice were sacrificed, tumors were obtained and measured. Smaller tumor sizes were observed in mice administered with 2 mg/kg celastrol compared to others (Fig. 4B). The immunohistochemical analysis revealed that the celastrol administration increased the phosphorylated AMPK and decreased both phosphorylated AKT and phosphorylated mTOR in gastric tumors (Fig. 4C). Therefore, we confirmed the results of our in vitro studies using an in vivo model and found that celastrol induced the activation of AMPK and deactivation of AKT and mTOR. It is likely that these signaling pathways play a role in the reduction of gastric cancer cell growth.
Fig. 4. Celastrol inhibited the growth of gastric tumors in a mouse xenografted model. AGS cells xenografts were prepared as described in the “Materials and Methods” session. After 10 days of tumor growth to obtain palpable tumors, mice were orally administered 100 μl of control saline, 1 mg/kg/day celastrol or 2 mg/kg/day of celastrol for 12 days and then sacrificed for analyses. (A) Tumor volumes were measured every 2 or 3 days and are graphically presented. (B) Gastric tumors were obtained and pictures were taken. (C) Immunohistochemical analysis of the expression of phosphorylated AMPK, phosphorylated AKT and phosphorylated mTOR. H&E staining was performed.
DISCUSSION
Despite declining incidence rates in the United States, gastric cancer is one of the most common cancers in the world and causes significant morbidity and mortality worldwide (23). Patients diagnosed with advanced gastric cancer have a median survival of only 3 to4 months without chemotherapy (2, 4, 24). The most common chemotherapeutic regimens are a combination of cisplatin and 5-FU and a combination of 5-FU and epirubicin. Response rates to these drugs are between 20 to 40%. However, the duration of responses is quickly lessened and there are very few complete responses. Additionally, it is important to recognize that the toxicity and side effects are not negligible, even in patients with a good functional status. In an effort to improve the cancer treatment, there have been many studies with the goal of exploring new drugs and improving drug side effects (25).
In this study, we studied celastrol as a novel candidate derived from natural compounds for the treatment of gastric cancer. As previously mentioned, celastrol has been vigorously studied to demonstrate its in vitro anti-tumor mechanisms in various cancers. It has also been found to suppress tumors in in vivo models and the therapeutic effects of celastrol were examined on the growth and metastasis of melanoma in syngeneic and xenograft mouse models (26), human prostate tumor xenografts (16), ErbB2-overexpressing human breast cancer cell xenografted mice (27) and human glioma xenografted mice (19). There are two reports on the antitumor effect of celastrol in gastric cancer (21, 28), but direct studies on the effect of celastrol in gastric cancer cells and especially in an in vivo gastric cancer mouse model were necessary. In this study, we identified the inhibition of cell proliferation and the induction of apoptosis and autophagy by the treatment with celastrol. Interestingly, IC50 was achieved using a treatment concentration of 1 μM in most gastric cancer cell lines, suggesting an effective and acceptable concentration of celastrol for the use in clinical trials. Celastrol also induced autophagy at low doses by as low as a 0.25 μM in both cell lines. At present, the role of autophagy in the restriction of cellular proliferation is controversial (29). Nevertheless, it has been reported that autophagy induction sensitizes cells for the induction of apoptosis and cell death and protects against chemotherapy resistance (30). We also determined that celastrol induced autophagy and initiated G1 cell-cycle arrest, which likely enhanced the induction of apoptosis. However, further mechanistic studies are necessary to confirm this conclusion. Another interesting finding is the induction of AMPK phosphorylation by celastrol as early as 15 minutes after the initiation of treatment. It has been reported that celastrol suppresses the cell viability of MCF-7 through AMPK activation which is caused by ROS generation (31). We suggest that celastrol induces AMPK activation and suppresses AKT and mTOR activation, which then leads to autophagy and apoptosis. We are currently under taking mechanistic studies on celastrol-induced AMPK activation in gastric cancer. Additionally, we administered celastrol in a gastric cancer xenografted mouse model and investigated the significant reduction of gastric tumor burdens with an increasing activation of AMPK and decreasing activity of AKT and mTOR. Taken together, we suggest that celastrol-induced AMPK activation plays an important role in the inhibition of gastric cancer growth.
In conclusion, celastrol successfully suppressed the cell proliferation and increased autophagy and apoptosis in gastric cancer cells. Therefore, we have undertaken further studies to elucidate the molecular mechanisms of celastrol in gastric cancer and suggest the further study for a possible application in clinical trials with gastric cancer patients.
MATERIALS AND METHODS
Methods of Western blotting, Preparation of gastric cancer xenografted mice, and Immunochemical staining analysis are available as supplementary materials on BMB Reports online.
Detection of cell proliferation and cell migration
The inhibition of cell proliferation by celastrol was measured using an MTT assay. Celastrol was purchased from Cayman Chemical (Michigan, USA) and dissolved in dimethyl sulfoxide (DMSO). The human stomach adenocarcinoma cell line AGS and the gastric carcinoma cell lines MKN1, MKN45, SNU216, SNU-668, and YCC-2 were purchased from the Korea Cell Line Bank (KCLB, Seoul, Korea). The KCLB authenticates the phenotypes of these cell lines on a regular basis. The analysis of cell proliferation using MTT assay and migration using a woundhealing assay was performed as described previously (32).
Assessment of cell cycle, apoptosis and autophagy
AGS and YCC-2 cells were plated onto culture plates and treated with celastrol. PI staining and annexin V processes were performed as described previously (33). For the detection of autophagy, RFP-tagged LC-3 lenti-virus was kindly obtained from Dr. Ho-Shin Kwak of the National-Cancer-Center of Korea. Then, cells were infected using this virus as described previously (34). Chamber slide cultured cells were fixed and treated with a mounting medium consisting of 4'6-diamidino-2-phenylindole and analyzed by confocal microscopy (LSM 520 META; Carl-Zeiss, Jena, Germany).
Statistical analysis
We employed unpaired t-tests to analyze mean tumor volumes in xenograft mice as described previously (37). Two tumors per mouse were analyzed as different entities. All statistical tests were two-sided and values are expressed as means with ± SD. P-values less than 0.05 were considered statistically significant. Statistical analyses were performed using PASW Statistics software version 18 (SPSS Inc., Chicago, IL, USA).
Acknowledgments
This research was supported by the Cooperative Research Program for Agricultural Science & Technology Development (PJ008462).
References
- 1.Zagouri F., Papadimitriou C. A., Dimopoulos M. A., Pectasides D. Molecularly targeted therapies in unresectable-metastatic gastric cancer: a systematic review. Cancer Treat. Rev. (2011);37:599–610. doi: 10.1016/j.ctrv.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Rivera F., Vega-Villegas M. E., Lopez-Brea M. F. Chemotherapy of advanced gastric cancer. Cancer Treat. Rev. (2007);33:315–324. doi: 10.1016/j.ctrv.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Wagner A. D., Moehler M. Development of targeted therapies in advanced gastric cancer: promising exploratory steps in a new era. Curr. Opin. Oncol. (2009);21:381–385. doi: 10.1097/CCO.0b013e32832c42e0. [DOI] [PubMed] [Google Scholar]
- 4.De Vita F., Giuliani F., Silvestris N., Rossetti S., Pizzolorusso A., Santabarbara G., Galizia G., Colucci G., Ciardiello F., Orditura M. Current status of targeted therapies in advanced gastric cancer. Expert Opin. Ther. Targets. (2012);16:S29–34. doi: 10.1517/14728222.2011.652616. [DOI] [PubMed] [Google Scholar]
- 5.Lourenco A. M., Ferreira L. M., Branco P. S. Molecules of natural origin, semi-synthesis and synthesis with anti-inflammatory and anticancer utilities. Curr. Pharm. Des. (2012);18:3979–4046. doi: 10.2174/138161212802083644. [DOI] [PubMed] [Google Scholar]
- 6.Shanmugam M. K., Nguyen A. H., Kumar A. P., Tan B. K., Sethi G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: potential role in prevention and therapy of cancer. Cancer Lett. (2012);320:158–170. doi: 10.1016/j.canlet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 7.Setty A. R., Sigal L. H. Herbal medications commonly used in the practice of rheumatology: mechanisms of action, efficacy, and side effects. Semin. Arthritis. Rheum. (2005);34:773–784. doi: 10.1016/j.semarthrit.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 8.Sassa H., Takaishi Y., Terada H. The triterpene celastrol as a very potent inhibitor of lipid peroxidation in mitochondria. Biochem. Biophys. Res. Commun. (1990);172:890–897. doi: 10.1016/0006-291X(90)90759-G. [DOI] [PubMed] [Google Scholar]
- 9.He W., Huang F. C., Gavai A., Chan W. K., Amato G., Yu K. T., Zilberstein A. Novel cytokine release inhibitors. Part III: Truncated analogs of tripterine. Bioorg. Med. Chem. Lett. (1998);8:3659–3664. doi: 10.1016/S0960-894X(98)00671-4. [DOI] [PubMed] [Google Scholar]
- 10.Allison A. C., Cacabelos R., Lombardi V. R., Alvarez X. A., Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. (2001);25:1341–1357. doi: 10.1016/S0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 11.Li H., Jia Y. F., Pan Y., Pan D. J., Li D., Zhang L. X. Effect of tripterine on collagen-induced arthritis in rats. Zhongguo Yao Li Xue Bao. (1997);18:270–273. [PubMed] [Google Scholar]
- 12.Kannaiyan R., Shanmugam M. K., Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. (2011);303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 13.Zhang T., Hamza A., Cao X., Wang B., Yu S., Zhan C. G., Sun D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. (2008);7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 14.Wang W. B., Feng L. X., Yue Q. X., Wu W. Y., Guan S. H., Jiang B. H., Yang M., Liu X., Guo D. A. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J. Cell. Physiol. (2012);227:2196–2206. doi: 10.1002/jcp.22956. [DOI] [PubMed] [Google Scholar]
- 15.Sethi G., Ahn K. S., Pandey M. K., Aggarwal B. B. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. (2007);109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 16.Pang X., Yi Z., Zhang J., Lu B., Sung B., Qu W., Aggarwal B. B., Liu M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. (2010);70:1951–1959. doi: 10.1158/0008-5472.CAN-09-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J. H., Won Y. S., Park K. H., Lee M. K., Tachibana H., Yamada K., Seo K. I. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis. (2012);17:1275–1286. doi: 10.1007/s10495-012-0767-5. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H., Liu X. W., Cai T. Y., Cao J., Tu C. X., Lu W., He Q. J., Yang B. Celastrol acts as a potent antimetastatic agent targeting beta1 integrin and inhibiting cell-extracellular matrix adhesion, in part via the p38 mitogen-activated protein kinase pathway. J. Pharmacol. Exp. Ther. (2010);334:489–499. doi: 10.1124/jpet.110.165654. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y., Zhou Y., Fan Y., Zhou D. Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett. (2008);264:101–106. doi: 10.1016/j.canlet.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 20.Hansen J., Palmfeldt J., Vang S., Corydon T. J., Gregersen N., Bross P. Quantitative proteomics reveals cellular targets of celastrol. PloS One. (2011);6:e26634. doi: 10.1371/journal.pone.0026634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kannaiyan R., Manu K. A., Chen L., Li F., Rajendran P., Subramaniam A., Lam P., Kumar A. P., Sethi G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis. (2011);16:1028–1041. doi: 10.1007/s10495-011-0629-6. [DOI] [PubMed] [Google Scholar]
- 22.Ko A., Shin J. Y., Seo J., Lee K. D., Lee E. W., Lee M. S., Lee H. W., Choi I. J., Jeong J. S., Chun K. H., Song J. Acceleration of gastric tumorigenesis through MKRN1-mediated posttranslational regulation of p14ARF. J. Natl. Cancer Inst. (2012);104:1660–1672. doi: 10.1093/jnci/djs424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Y., Ajani J. A. Multidisciplinary management of gastric cancer. Curr. Opin. Gastroenterol. (2010);26:640–646. doi: 10.1097/MOG.0b013e32833efd9b. [DOI] [PubMed] [Google Scholar]
- 24.Cervantes A., Rosello S., Roda D., Rodriguez-Braun E. The treatment of advanced gastric cancer: current strategies and future perspectives. Ann. Oncol. (2008);19(Suppl 5):v103–107. doi: 10.1093/annonc/mdn321. [DOI] [PubMed] [Google Scholar]
- 25.Kim K., Chun K. H., Suh P. G., Kim I. H. Alterations in cell proliferation related gene expressions in gastric cancer. Crit. Rev. Eukaryot. Gene Expr. (2011);21:237–254. doi: 10.1615/CritRevEukarGeneExpr.v21.i3.20. [DOI] [PubMed] [Google Scholar]
- 26.Chen M., Rose A. E., Doudican N., Osman I., Orlow S. J. Celastrol synergistically enhances temozolomide cytotoxicity in melanoma cells. Mol. Cancer Res. (2009);7:1946–1953. doi: 10.1158/1541-7786.MCR-09-0243. [DOI] [PubMed] [Google Scholar]
- 27.Raja S. M., Clubb R. J., Ortega-Cava C., Williams S. H., Bailey T. A., Duan L., Zhao X., Reddi A. L., Nyong A. M., Natarajan A., Band V., Band H. Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol. Ther. (2011);11:263–276. doi: 10.4161/cbt.11.2.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sha M., Ye J., Zhang L. X., Luan Z. Y., Chen Y. B. Celastrol induces apoptosis of gastric cancer cells by miR-146a inhibition of NF-kappaB activity. Cancer Cell Int. (2013);13:50. doi: 10.1186/1475-2867-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain M. V., Paczulla A. M., Klonisch T., Dimgba F. N., Rao S. B., Roberg K., Schweizer F., Lengerke C., Davoodpour P., Palicharla V. R., Maddika S., Los M. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J. Cell. Mol. Med. (2013);17:12–29. doi: 10.1111/jcmm.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun H., Wang Z., Yakisich J. S. Natural products targeting autophagy via the PI3K/Akt/mTOR pathway as anticancer agents. Anticancer Agents Med. Chem. (2012);13:1048–1056. doi: 10.2174/18715206113139990130. [DOI] [PubMed] [Google Scholar]
- 31.Kim J. H., Lee J. O., Lee S. K., Kim N., You G. Y., Moon J. W., Sha J., Kim S. J., Park S. H., Kim H. S. Celastrol suppresses breast cancer MCF-7 cell viability via the AMP-activated protein kinase (AMPK)-induced p53-polo like kinase 2 (PLK-2) pathway. Cellular Signal. (2012);25:805–813. doi: 10.1016/j.cellsig.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Kim S. J., Choi I. J., Cheong T. C., Lee S. J., Lotan R., Park S. H., Chun K. H. Galectin-3 increases gastric cancer cell motility by up-regulating fascin-1 expression. Gastroenterology. (2010);138:1035–1045. doi: 10.1053/j.gastro.2009.09.061. [DOI] [PubMed] [Google Scholar]
- 33.Cheong T. C., Shin J. Y., Chun K. H. Silencing of galectin-3 changes the gene expression and augments the sensitivity of gastric cancer cells to chemotherapeutic agents. Cancer Sci. (2010);101:94–102. doi: 10.1111/j.1349-7006.2009.01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahn Y. H., Yi H., Shin J. Y., Lee K. D., Shin S. P., Lee S. J., Song J., Chun K. H. STAT3 silencing enhances the efficacy of the HSV.tk suicide gene in gastrointestinal cancer therapy. Clin. Exp. Metastasis. (2012);29:359–369. doi: 10.1007/s10585-012-9458-4. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y. G., Kim S. J., Baek J. H., Lee H. W., Jeong S. Y., Chun K. H. Galectin-3 increases the motility of mouse melanoma cells by regulating matrix metalloproteinase-1 expression. Exp. Mol. Med. (2012);44:387–393. doi: 10.3858/emm.2012.44.6.044. [DOI] [PMC free article] [PubMed] [Google Scholar]