

Abstract
The first stable complex formed during the assembly of spliceosomes onto pre-mRNA substrates in mammals includes U1 snRNP, which recognizes the 5′ splice site, and the splicing factors SF1 and U2AF, which bind the branch point sequence, polypyrimidine tract, and 3′ splice site. The 5′ and 3′ splice site complexes are thought to be joined together by protein–protein interactions mediated by factors that ensure the fidelity of the initial splice site recognition. In this study, we identified and characterized PRPF40B, a putative mammalian ortholog of the U1 snRNP-associated yeast splicing factor Prp40. PRPF40B is highly enriched in speckles with a behavior similar to splicing factors. We demonstrated that PRPF40B interacts directly with SF1 and associates with U2AF65. Accordingly, PRPF40B colocalizes with these splicing factors in the cell nucleus. Splicing assays with reporter minigenes revealed that PRPF40B modulates alternative splice site selection. In the case of Fas regulation of alternative splicing, weak 5′ and 3′ splice sites and exonic sequences are required for PRPF40B function. Placing our data in a functional context, we also show that PRPF40B depletion increased Fas/CD95 receptor number and cell apoptosis, which suggests the ability of PRPF40B to alter the alternative splicing of key apoptotic genes to regulate cell survival.
Keywords: PRPF40B, alternative splicing, mRNA processing
INTRODUCTION
The expression of protein-coding genes begins when nuclear RNA polymerase II (RNAPII) transcribes a genomic sequence into a precursor messenger RNA (pre-mRNA). The pre-mRNA undergoes splicing, a process that removes intervening sequences (introns), leaving resultant sequences (exons) appropriately aligned and ligated in the messenger RNA (mRNA). In eukaryotes, most genes have multiple introns, and splicing occurs in more than one pattern to form a considerable number of alternatively spliced transcript isoforms. In humans, >90% of genes undergo alternative splicing (Pan et al. 2008; Wang et al. 2008), underscoring the fundamental importance of this regulatory process in expanding protein diversity through evolution. The misregulation of alternative splicing (e.g., by mutations that affect the splicing signals or the splicing machinery itself) is the cause of many human diseases including neurological disorders and cancer (Wang and Cooper 2007; Ward and Cooper 2010). At present, we have a comprehensive understanding of how alternative splicing occurs throughout the genome; however, the molecular mechanisms at work, particularly in pathological situations, are not fully understood.
The splicing reaction is catalyzed by the spliceosome, one of the most complex machines in the cell. The spliceosome is composed of a core of five uridine-rich small nuclear RNAs (snRNAs), termed U1, U2, U4, U5, and U6, and an estimated 200 additional proteins (for review, see Wahl et al. 2009). Each spliceosome is assembled around an intron in an ordered stepwise manner. During the initial steps of splicing, the sequence elements that define the exon–intron boundaries (the 5′ and 3′ splice sites) and associated RNA sequences (the polypyrimidine [Py] tract and branch point sequence [BPS]) are recognized by a network of spliceosome components. In the first step, U1 snRNP binds to the 5′ splice site of the intron, and the essential pre-mRNA splicing factors SF1 and the U2AF65/U2AF35 heterodimer accurately and cooperatively recognize the BPS, Py tract, and 3′ splice site. Isolation of mammalian spliceosomal complexes by gel filtration demonstrated that the formation of this initial complex commits the pre-mRNA for splicing (Reed 1990; Michaud and Reed 1991). The formation of this ATP-independent commitment complex was functionally defined using substrate-competition assays that showed that the 5′ and 3′ splice sites are also required for efficient complex assembly (Jamison et al. 1992; Michaud and Reed 1993). The requirements for the formation of this early spliceosomal complex mirrored the proposed mechanism for functional splice site interactions in yeast (Legrain et al. 1988; Seraphin et al. 1988; Seraphin and Rosbash 1989). Next, the U2 snRNP component of the spliceosome forms an ATP-dependent complex with the BPS, displacing U2AF and SF1. Following these early splice site recognition events, subsequent binding of the rest of the snRNPs and proteins and concomitant RNA–RNA and RNA–protein rearrangements lead to a complex that is ultimately responsible for catalyzing the intron excision and exon–exon ligation reactions (Wahl et al. 2009).
In higher eukaryotes, the correct identification of exons is complex. First, genes have small exons separated by large introns, and second, the splicing signals (the 5′ and 3′ splice sites, BPS, and Py tract) are suboptimally arranged and poorly conserved. We know that additional sequence elements, known as exonic and intronic splicing silencers or enhancers (ISS, ISE, ESS, and ESE), participate in the regulation of alternative splicing. Specific RNA-binding proteins, including heterogeneous nuclear ribonucleoproteins (hnRNPs) and serine/arginine-rich (SR) proteins, recognize these sequences to positively or negatively regulate alternative splicing. These regulators together with an ever-increasing number of additional auxiliary factors facilitating interactions between the 5′ and 3′ splice sites provide the basis for the specificity of this pre-mRNA processing event (Smith and Valcárcel 2000; Singh and Valcárcel 2005; Izquierdo and Valcárcel 2006). The mechanism by which the pairing between the splice sites across an exon occurs is called “exon definition” (Robberson et al. 1990) and is under complex combinatorial control that implicates both the initial recognition of the 5′ and 3′ splice sites and the interplay of other elements such as splicing silencers or enhancer sequences, RNA structure, transcriptional rate, and chromatin modifications, among others (De Conti et al. 2013).
Although we have gained considerable knowledge of the molecular events governing alternative splicing, the initial stabilizing interactions occurring at intron/exon boundaries and the specific factors that modulate these critical early connections by functioning as a scaffold for active site RNAs or proteins are poorly understood.
The yeast splicing factor Prp40 is an essential U1 snRNP-associated protein that contains two WW domains followed by four FF domain repeats and participates as a scaffold in the earlier steps of spliceosome complex formation (Kao and Siliciano 1996). Prp40 binds to Bbp, which is the yeast ortholog of the splicing factor SF1, and Bbp interacts with Mud2p, which is the yeast homolog of human U2AF65. Based on these interactions, Prp40 might help to define a bridging interaction that links both ends of the intron (Abovich and Rosbash 1997). The two putative mammalian orthologs of Prp40 include PRPF40A (HUGO approved gene name: PRP40 pre-mRNA processing factor 40 homolog A), which is also known as HYPA (for Huntingtin-Interacting Protein A) or FBP11 (for Formin-binding protein 11) and PRPF40B (HUGO approved gene name: PRP40 pre-mRNA processing factor 40 homolog B), which is also known as HYPC (for Huntingtin-Interacting Protein C). Based on the Prp40 data, PRPF40A and PRPF40B are thought to participate in early spliceosome assembly, but their functions remain unknown. Notably, PRPF40B has been implicated in the pathogenesis of neurological disorders, including Huntington's disease and Rett syndrome (Faber et al. 1998; Passani et al. 2000; Buschdorf and Strätling 2004). Moreover, mutations in spliceosome components involved in splice site recognition have been recently detected in myelodysplastic syndrome (MDS), a group of disorders that lead to myeloid leukemia (Papaemmanuil et al. 2011; Yoshida et al. 2011). Interestingly, PRPF40B was one of the targets detected with spliceosome mutations. Changes in PRPF40B occurred as missense mutations throughout the open reading frame, suggesting that these mutations might result in a loss of function (Abdel-Wahab and Levine 2011; Yoshida et al. 2011). Herein, we characterized PRPF40B and identified a role for PRPF40B in pre-mRNA splicing. PRPF40B localizes to splicing factor-rich nuclear speckles, binds SF1 and U2AF65, and modulates different splicing events in vivo. In the case of Fas alternative splicing regulation, weak 5′ and 3′ splice sites and exonic sequences are required for PRPF40B action. Placing our data in a functional context, we also show that PRPF40B depletion increased Fas/CD95 receptor number and cell apoptosis, which suggests the ability of PRPF40B to alter the alternative splicing of key apoptotic genes to regulate cell survival. These results support a role for PRPF40B in the early events of alternative splicing that lead to exon definition and might provide novel, important insights regarding the molecular mechanisms involved in PRPF40B-related disorders.
RESULTS
PRPF40B is a protein highly enriched in nuclear speckles
In this study, we initially identified PRPF40B as a member of a subset of factors containing tandem repeats of WW and FF domains. Multiple alignments of these related structural proteins using bioinformatic tools revealed that the primary sequence of PRPF40B contains two WW domains in the amino-terminal half and five FF repeat motifs in the carboxyl-terminal half. Human and murine PRPF40B protein sequences show a high degree of homology with 95% identity and 96.5% similarity. The human PRPF40B protein sequence has 10% homology and 30% identity or 22% and 45% similarity with yeast Prp40, using ungapped or gapped local alignment, respectively. An analysis of the Prp40 sequence revealed the presence of four FF repeat domains, whereas PRPF40B is predicted to encode five of these domains (data not shown). To investigate the subcellular localization of PRPF40B, we performed immunofluorescence experiments with confocal laser microscopy on various cell types using a rabbit IgG-purified polyclonal PRPF40B antibody (see Materials and Methods). We consistently observed a diffuse nucleoplasmic pattern with an increased signal in organized granule-like sites in HEK293T and HeLa cells (Fig. 1A, left panels). The staining pattern of PRPF40B was reminiscent of that of nuclear speckles, which are nuclear compartments enriched in pre-mRNA splicing factors located at the interchromatin region of the nucleoplasm in mammalian cells (Lamond and Spector 1993). To determine whether the nuclear staining pattern of the endogenous PRPF40B protein coincides with that of nuclear speckles, we performed immunofluorescence analyses using an antibody directed against the phosphorylated form of the essential splicing factor SRSF2 (formerly SC35), which is commonly used to define nuclear speckles (Fig. 1A, middle panels). Interestingly, the PRPF40B nuclear dots were found to overlap with speckles in both cell lines (Fig. 1A, right panels). To confirm these data, we performed semiquantitative analysis of the spatial relationship between the relative distribution of PRPF40B and SRSF2 as a way to determine the degree of colocalization (Fig. 1, right). The polyclonal antiserum used here recognizes PRPF40B and several other proteins of slower mobility (we failed to raise peptide-specific antibodies). The mobility of one of these proteins coincides with the predicted mobility of PRPF40A. Given the high homology between PRPF40B and PRPF40A (using EMBOSS global and local alignments, the identity and similarity percentages ranged between 51%–54% and 65–68% for PRPF40A and PRPF40B, respectively) and the fact that both proteins are expressed in the cell lines used in this study (data not shown), the antibodies may be recognizing both proteins in the nuclei. To confirm the spatial localization of PRPF40B, we performed indirect immunofluorescence experiments with green fluorescence protein (GFP)-tagged PRPF40B. A similar pattern of nuclear staining was also observed when enhanced green fluorescence protein (EGFP)-tagged PRPF40B cDNA was transfected into HEK293T and HeLa cells (Fig. 1B). These results confirm the presence of a population of PRPF40B that colocalizes with nuclear speckles and suggest that PRPF40B might be implicated in splicing regulation.
FIGURE 1.

PRPF40B is enriched in splicing factor-rich nuclear speckles. (A) Endogenous PRPF40B was detected with IgG-purified polyclonal antibodies. HEK293T and HeLa cells were dual-stained with antibodies to detect PRPF40B (green) and SRSF2 (red), and merged images are shown. Line scans with local intensity distributions are represented at the right. The bar in the merged panel indicates the position of the line scans. Bar, 3 mm. (B) Immunofluorescence analysis was performed to determine the location of PRPF40B. HEK293T or HeLa cells were transfected with an EGFP-tagged PRPF40B construct (green) and labeled with antibodies directed against the essential splicing factor SRSF2 (red) to stain nuclear speckles. Individual staining and the superimposition of both images (merge) are shown. Line scans showing local intensity distributions of dual staining are represented to the right of the panels. A bar in the merged panel indicates the position of the line scans. Bar, 3 mm.
To identify the sequence element responsible for the nuclear localization of PRPF40B, the primary amino acid sequence of PRPF40B was analyzed for the presence of a potential nuclear localization signal (NLS) using the WoLF PSORT (an update of PSORT II) (Horton et al. 2007), NucPred (Brameier et al. 2007), and cNLS Mapper (Kosugi et al. 2009) computer programs. This computational analysis identified a consensus NLS, KRRRR, with the highest score at amino acid residues 732–736 in PRPF40B. To test the functionality of this predicted NLS, we generated a truncated derivative of PRPF40B fused to EGFP that lacked this sequence (Fig. 2, EGFP-PRPF40B [1–687]). When analyzed by immunofluorescence, HEK293T cells expressing this mutant construct displayed predominantly cytoplasmic localization (Fig. 2), suggesting that the deleted region contains the signal for PRPF40B nuclear localization. To further characterize PRPF40B, we performed immunofluorescence analysis with a series of EGFP-tagged PRPF40B deletion mutants. The constructs are diagrammed in Figure 2. All mutants retain the putative NLS signal found in the carboxyl region. The PRPF40B mutant with an amino-terminal truncation removing the two WW domains produced a speckled staining pattern within the nucleus that was similar to that observed for the full-length protein (EGFP-PRPF40B [263–871] and EGFP-PRPF40B [1–871], respectively, in Fig. 2). This result strongly implies that the WW domains play no significant role in the nuclear speckle localization of PRPF40B. To determine the importance of the FF domains in targeting PRPF40B to nuclear speckles, we further generated carboxyl-terminal deletions. Removal of the most upstream two FF domain sequences created a fusion protein that significantly accumulated in nuclear speckles (EGFP-PRPF40B [394–871]) (Fig. 2). Deletion of the next FF domain partially disrupted the localization to nuclear speckles (EGFP-PRPF40B [474–871]) (Fig. 2), suggesting that this domain might be required for targeting the protein to nuclear speckles. Further deletions within the carboxyl-terminal region of PRPF40B generated fusion proteins that aggregated in the nucleus (EGFP-PRPF40B [618–871]) (Fig. 2 and data not shown). To further analyze the speckle-targeting capacity of the FF domains, we generated an EGFP chimera with the FF4 domain fused to the carboxyl-terminal region containing the NLS signal. Cells expressing this chimera showed significant nuclear speckle fluorescence (EGFP-PRPF40B [474–553/672–871]) (Fig. 2). The same results were obtained when these EGFP-tagged constructs were transfected in HeLa cells (data not shown). Although PRPF40B is distributed throughout the nucleoplasm with nucleolar exclusion, deletion of the FF domains affected intranuclear distribution with increased accumulation in the nucleoli (Fig. 2). We further examined the distribution of the EGFP-tagged PRPF40B deletion mutants using an antibody against the nucleolar phosphoprotein NOLC1 (Meier and Blobel 1990). We observed that the fusion protein with a deletion of the most upstream two FF domains sequences [EGFP-PRPF40B (394–871)], which did not disrupt the localization to nuclear speckles (Fig. 2), localized to the nucleoli (Fig. 3). Larger deletions [EGFP-PRPF40B (474–871), [EGFP-PRPF40B (618–871)] that affected the speckle distribution of PRPF40B (Fig. 2), resulted in an increased overlapping signal at the nucleoli (Fig. 3). Interestingly, the expression and detection of the EGFP chimera with the FF4 domain showed significant attenuation of nucleolar accumulation and produced a speckled pattern of staining within the nucleus (Fig. 3), which overlaps with the SRSF2 signal (Fig. 2). These results suggest that the putative NLS is essential for PRPF40B nuclear localization and nuclear speckle localization, that the FF domains participate in nucleolar exclusion, and that the FF4 domain is sufficient for localization to speckles. However, other FF domains may also be important for the efficient targeting of PRPF40B to the speckle compartment.
FIGURE 2.

Subcellular localization of PRPF40B. HEK293T cells expressing EGFP-tagged PRPF40B constructs (green) were immunolabeled with SRSF2 antibody (red). Schematic diagrams of the EGFP-PRPF40B fusion proteins are shown at the left of each panel. The numbers in parentheses represent the PRPF40B amino acids contained in the construct. GFP protein, the two WW domains, the putative nuclear localization signal (NLS), and the five FF domains are indicated. Individual and merged images of the cell are shown. Line scans showing the local intensity distribution of the dual staining are represented to the right of the panels. The bars in the merged panels indicate the position of the line scans. Bar, 3 mm.
FIGURE 3.

Role of PRPF40B FF domains in nucleolar exclusion. HEK293T cells expressing EGFP-tagged PRPF40B proteins (green) were immunolabeled with anti-NOLC1 antibodies (red). Individual and merged images of the cells are shown. Line scans showing the local intensity distribution of the dual staining are represented to the right of the panels. Labeling in the figure is the same as in the Figure 2 legend. Bar, 3 mm.
The association of PRPF40B with nuclear speckles is not affected by RNase treatment or transcription inhibition
Nuclear speckles are dynamic structures that are believed to act as storage, assembly, and modification compartments for splicing components, which can supply processing factors to nearby transcription sites. The structural architecture of nuclear speckles depends on interactions that are resistant to RNase treatment (e.g., SRSF2) and interactions that are RNase sensitive (e.g., Sm ribonucleoproteins). To evaluate the nature of the association of PRPF40B with nuclear speckles, we investigated whether RNA interaction was the molecular mechanism involved in concentrating PRPF40B in the nuclear speckle region. For these experiments, HEK293T cells were transiently transfected with EGFP-PRPF40B and treated with RNase A. Under these conditions, the staining pattern of PRPF40B and SRSF2 was unaffected, whereas Sm staining became completely diffuse throughout the nucleoplasm (Fig. 4A). Thus, like SRSF2, PRPF40B is not primarily localized to speckles through its binding to RNA. To determine the effect of transcriptional activity on the distribution of speckle-associated PRPF40B, we treated HEK293T cells with the RNA polymerase II inhibitor α-amanitin. Upon RNA polymerase II inhibition, speckles decrease in number, enlarge, and become rounded because of the accumulation of the splicing machinery (O'Keefe et al. 1994). Indeed, we observed a redistribution of PRPF40B into enlarged foci similar to that observed with SRSF2 upon transcriptional inhibition (Fig. 4B). We conclude that the localization of PRPF40B to nuclear speckles is not dependent on active transcription, and its accumulation in speckles shows a behavior similar to that of splicing factors.
FIGURE 4.

Colocalization of PRPF40B with nuclear speckles is not perturbed following RNase treatment or transcription inhibition. (A) After EGFP-PRPF40B expression, HEK293T cells were treated with RNase A. SRSF2 (left panel) and Sm (right panel) labeling were used as negative and positive controls, respectively. Individual staining and merged images of cells are shown. Bars, 3 mm. (B) HeLa cells were treated with α-amanitin after EGFP-PRPF40B expression. Then, the samples were processed for immunofluorescence analysis. Individual staining of PRPF40B (green), SRSF2 (red), and merged images of untreated (upper panel) or treated (lower panel) cells are shown. Line scans showing the local intensity distribution of PRPF40B in green and SRSF2 in red are shown to the right of the panels. Bars in the merged panels indicate the position of the line scans. Bars, 3 mm.
PRPF40B associates with the splicing factors SF1 and U2AF65
Prp40 directly interacts with Bbp, which is the ortholog of the splicing factor SF1, and associates with Mud2p, which is the ortholog of U2AF65 (Abovich and Rosbash 1997). In addition, SF1 binds cooperatively with U2AF65 to pre-mRNA introns at an early stage of spliceosome assembly (Berglund et al. 1998). Based on these data, we hypothesize that PRPF40B may be associated with SF1 and U2AF65. To address this possibility, we first sought to investigate the spatial distribution of endogenous PRPF40B, SF1, and U2AF65 by confocal microscopy. SF1 and U2AF65 accumulate in speckles and show diffuse nucleoplasmic staining (Fig. 5). When we performed double-labeled immunofluorescence experiments using PRPF40B-specific antibodies and antibodies directed against the splicing factors SF1 (Fig. 5A) and U2AF65 (Fig. 5B) in HEK293T or HeLa cells, we observed a high degree of colocalization of the proteins, thus demonstrating that these factors are in close proximity in the nucleus.
FIGURE 5.

Colocalization of PRPF40B with the splicing factors SF1 (A) and U2AF65 (B). Immunofluorescence analysis of HeLa (lower panels) and HEK293T (upper panels) cells dual-labeled with antibodies to detect PRPF40B and SF1 (A) or U2AF65 (B). The merged images represent a superimposition of PRPF40B (green) and SF1 or U2AF65 (red) labeling. Line scans showing the local intensity distribution of dual staining are shown to the right of the panels. Bars in the merged panels indicate the position of the line scans. Bars, 3 mm.
To test whether PRPF40B could directly interact with SF1, we performed in vitro GST-binding assays. To this end, full-length, amino-terminal (amino acid residues 1–333), and carboxyl-terminal (amino acid residues 446–871) PRPF40B were purified from bacteria as GST fusions (Fig. 6A). As a positive control, we used the amino regions of TCERG1 (Goldstrohm et al. 2001) (TCERG1 Nt in Fig. 6B) and U2AF65 (data not shown) (see also Fig. 6C), which are known to bind SF1, fused to GST. The GST protein and carboxyl region of TCERG1 (TCERG1 Ct in Fig. 6B) were used as negative controls. The proteins were bound to glutathione–agarose beads and incubated with purified T7-tagged SF1 recombinant protein. After extensive washing, the bead-bound proteins were eluted by boiling the samples with SDS-PAGE loading buffer and analyzing by SDS-PAGE and Western blotting to detect SF1. The results of the in vitro binding assay revealed an interaction between PRPF40B and SF1. In the GST-binding assay, the amino portion of PRPF40B displayed strong binding, whereas the carboxyl-terminal region showed weak binding (PRPF40B [1–333] and PRPF40B [446–871], respectively, in Fig. 6B). These results suggest that PRPF40B contains SF1-binding domains mainly in the amino-terminal segment. Further deletion analysis is required to precisely locate the PRPF40B residues involved in the SF1 interaction. To measure the strength of these interactions, we determined the effect of increasing salt concentrations on the binding between PRPF40B and SF1 (Fig. 6C). Recombinant SF1 was incubated with GST, GST-PRPF40B, and GST-U2AF65 (as positive control) and bound to glutathione beads in the presence of 150, 200, 300, and 500 mM NaCl. Beads with bound proteins were extensively washed with a buffer containing the same salt concentration as that in the binding reaction. The bound proteins were eluted by boiling with SDS-PAGE loading buffer and then analyzed by SDS-PAGE and Western blotting. The interaction between PRPF40B and SF1 proved to be stable under high salt concentrations up to 500 mM, although binding was gradually reduced with increasing salt concentrations (Fig. 6C).
FIGURE 6.

PRPF40B associates with the splicing factors SF1 and U2AF65 in vitro. (A) Schematic representations of the GST-PRPF40B fusion proteins are shown. The numbers in parentheses represent the PRPF40B amino acids contained in the constructs. The two WW domains and five FF domains are indicated. (B) Both the amino- and carboxyl-terminal portions of PRPF40B are important for interacting with SF1. Recombinant full-length GST-PRPF40B and its amino- and carboxyl-terminal regions were bound to glutathione–agarose beads and incubated with T7-tagged SF1 partially purified from HEK293T. GST and GST-TCERG1 carboxyl terminus (Ct) were used as negative controls, and the GST-TCERG1 amino-terminal region (Nt) was the positive control. Eluted proteins were separated by SDS-PAGE and analyzed by Western blotting (top) using anti-SF1 to assess the interactions with SF1 or silver staining (bottom) to detect the GST fusion proteins. (C) GST, GST-U2AF65, and GST-PRPF40B were used to bind recombinant SF1. The bound protein was eluted with a linear gradient of 150–500 mM NaCl. After the elution step, the samples were separated by SDS-PAGE and analyzed by Western blotting or silver staining as described above.
To investigate whether these proteins interact in living cells, we transfected HEK293T cells with T7-tagged PRPF40B or an empty vector as a negative control. After immunoprecipitation with anti-T7 antibodies, the samples were analyzed by immunoblotting with specific antibodies directed against endogenous SF1 and U2AF65. The results showed that endogenous SF1 and U2AF65 were detected in the immunoprecipitates, indicating an association between PRPF40B and SF1 and U2AF65 (Fig. 7A). To confirm these results, we transiently transfected a T7-tagged SF1 expression vector and analyzed the immunoprecipitates with antibodies directed against endogenous PRPF40B and U2AF65. We observed the presence of endogenous PRPF40B and U2AF65 associated with SF1, confirming the observation that an association between SF1 and PRPF40B exists (Fig. 7B). In contrast, ERK2, an unrelated mitogen-activated protein kinase, could not be detected in the eluates of the immunoprecipitation experiments (Fig. 7A,B). We conclude that PRPF40B associates with the SF1 and U2AF65 splicing factors. RNase A treatment of the extracts impaired the interaction between PRPF40B and SF1 and U2AF65 (Fig. 7C,D), which may indicate that this interaction is stabilized by concomitant binding to specific mRNAs. To test whether the interaction between PRPF40B and the SF1 and U2AF65 splicing factors was mediated by U1 snRNP, we used an antisense morpholino oligonucleotide (AMO) complementary to U1 snRNA to knock down U1 snRNP in HEK293T cells (Kaida et al. 2010; Berg et al. 2012). We confirmed that the U1 AMO inhibited the activity of U1 snRNP by testing its effect on in vivo Fas alternative splicing (data not shown). We then repeated the immunoprecipitation experiments in the presence of control or U1 AMOs and detected endogenous SF1 and U2AF65 in the immunoprecipitates (Fig. 7E), which suggest that U1 snRNP does not mediate this interaction. Taken together, these data indicate that PRPF40B associates with the splicing factors SF1 and U2AF65, two factors involved in 3′ splice site recognition.
FIGURE 7.

PRPF40B associates with the splicing factors SF1 and U2AF65 in vivo. (A) HEK293T cells were transiently transfected with a plasmid encoding T7-tagged PRPF40B or an empty vector as a negative control. Whole-cell extract (WCE) fractions were prepared and directly analyzed by Western blotting or subjected to immunoprecipitation (IP) with T7-specific antibodies followed by SDS-PAGE and Western blotting analysis using antibodies to detect SF1, U2AF65, and ERK2. (B) The same experimental procedures described in A were performed to overexpress T7-SF1, and immunoprecipitation was performed with PRPF40B-specific antibodies. (C,D) The same experiments described in A and B were performed in the presence of RNaseA, and (E) in the presence of control or U1 AMOs.
PRPF40B influences alternative splice site selection
Based on the results obtained above, we conducted in vivo splicing assays to examine the ability of PRPF40B to influence alternative splicing. Reporter constructs containing alternative exons flanked by constitutive introns were transiently cotransfected with T7-tagged PRPF40B or an empty vector in HEK293T cells. We used minigenes that recapitulate alternative 5′ splice site choice (Bcl-x and Fas) (see Fig. 8 for a schematic representation of the minigenes). Human Bcl-x pre-mRNA undergoes alternative splicing at 5′ splice sites in exon 2 to produce the anti-apoptotic long (Bcl-xL) and proapoptotic short (Bcl-xS) isoforms (Boise et al. 1993). The death receptor Fas/CD95, which encodes a transmembrane death receptor, can be alternatively processed to exclude exon 6 from its mRNA to produce a soluble isoform whose function is to inhibit apoptosis (Cheng et al. 1994; Cascino et al. 1995).
FIGURE 8.

PRPF40B regulates alternative splicing. (A,B) Schematic representation of the human Bcl-x and Fas minigenes and their derived splicing variants. Exons (boxes), introns (horizontal lines), and patterns of alternative splicing events (inclined lines) are represented. HEK293T cells were cotransfected with the Bcl-x (A) or Fas (B) minigenes together with an empty vector (Mock) or a PRPF40B expression plasmid (PRPF40B-OE) for overexpression experiments. For the RNAi experiments, HEK293T cells were cotransfected with the corresponding minigene together with siPRPF40B or siEGFP as a control. The graphs show the densitometric analysis results as the exon skipping average from three independent experiments (means ± SEM). (*) P < 0.05; (***) P < 0.005. A fraction of the cell lysates was analyzed by immunoblotting with the indicated antibodies to detect the PRPF40B and ERK2 proteins. (C,D) PRPF40B regulates the alternative splicing of the endogenous Fas gene. HEK293T cells were transfected with an empty vector (Mock) or a PRPF40B-expressing plasmid (OE). For the RNAi experiments, the HEK293T cells were transfected with siPRPF40B or siEGFP as a control. After total RNA extraction, RT-PCR (C) and RT-qPCR (D) were performed using the primers indicated in the schematic representations of the Fas gene. RT-PCR primers spanned the region between exons 5 and 7, including both the anti- and the proapoptotic isoforms of the Fas gene. RT-qPCR primers amplified the proapoptotic isoform including exon 6. The bar graphs represent the ratio of exon 6 inclusion, which is shown relative to the control that was set at 1. The data are from five independent experiments (means ± SEM). (*) P < 0.05; (**) P < 0.01. A fraction of the cell lysates was analyzed by immunoblotting with the indicated antibodies to detect the PRPF40B and ERK2 proteins.
Total RNA was isolated from cells transfected with the minigenes under the conditions of PRPF40B overexpression or gene knockdown, and splicing was analyzed by RT-PCR using specific primers to detect the different transcript isoforms. Different post-transfection times were used between the overexpressed/knockdown samples to visualize the splicing effects under each condition, which affected the alternative splicing in the mock condition (see Fig. 8A,B, siEGFP versus Mock, and Materials and Methods). To note and because of the high homology between PRPF40B and PRPF40, we determined that the siRNAs against PRPF40B did not affect the PRPF40A level using RT-qPCR assays (data not shown). In the case of the Bcl-x minigene, increased levels of PRPF40B produced more efficient utilization of the Bcl-xS 5′ splice site, resulting in a decreased ratio of Bcl-xL to Bcl-xS (Fig. 8A), while the amount of the Bcl-xL isoform was increased upon PRPF40B knockdown (Fig. 8A). For the Fas minigene, PRPF40B enhanced the amount of the short anti-apoptotic Fas isoform (Fig. 8B), while the absence of PRPF40B resulted in a slight increase in the long proapoptotic Fas isoform (Fig. 8B, see below). We obtained the same results using a second siRNA against PRPF40B (data not shown; see Materials and Methods for siRNA sequences). Taken together, these results show that PRPF40B can modulate alternative splicing events.
To further study the function of PRPF40B in regulating Fas alternative splicing, we first analyzed the splicing pattern of the endogenous Fas gene by RT-PCR. Consistent with previous results, we observed that PRPF40B overexpression reduced the level of exon 6 inclusion, giving rise to increased amounts of the short anti-apoptotic isoform. The absence of PRPF40B promoted the inclusion of exon 6, provoking a robust increase in the long proapoptotic Fas isoform (Fig. 8C). We repeated the analysis by qPCR using primers that exclusively amplify the transmembrane receptor isoform and obtained the same results (Fig. 8D).
PRPF40B activity in Fas alternative splicing requires exon 6 sequences and associated 5′ and 3′ splice sites
As a first step to understand the regulation of Fas alternative splicing by PRPF40B, we sought to determine the exonic and intronic regulatory sequences important for PRPF40B function. Although it is assumed that yeast Prp40 bridges the 5′ and 3′ ends of the intron by interacting with Bbp and U1 snRNP without interacting directly with the pre-mRNA, a systematic study of the sequence elements that affect PRPF40B function would provide useful information regarding the mechanism by which PRPF40B modulates alternative splicing. For this goal, we took advantage of minigene constructs in which the exon 6 sequences were divided into three segments of control sequences of approximately the same length (e1, e2, and e3 in Fig. 9A), which were previously used to delineate the responsive elements of the putative tumor suppressor RBM5 within Fas exon 6 (Bonnal et al. 2008). Replacement of the e3 sequence did not compromise PRPF40B function (Fig. 9B, lanes 5,6). Replacement of the e1 and e2 sequences rendered reduced levels of exon skipping by PRPF40B (Fig. 9B, lanes 3,4 and 7–10), which reveals that Fas exon 6 sequences are important for PRPF40B function. Segment e2 corresponds to a uridine-rich exonic silencer (URE6), which is the binding region for the splicing repressor PTB (Izquierdo et al. 2005). Previous data have shown that the URE6 element contains an exonic splicing silencer that mediates repression by the splicing factor PTB and that the silencing effect relies on its pyrimidine-rich content (Izquierdo et al. 2005). To further evaluate the function of PRPF40B through the URE 6 element, we performed RT-PCR experiments with constructs containing three different mutations in URE6 (i.e., a cytidine and adenosine-rich sequence, another uridine-rich sequence that maintains silencer activity, and a scrambled sequence of balanced composition [m0, m1, and m2 in Fig. 9A, respectively]) (Izquierdo et al. 2005; Bonnal et al. 2008). We observed that only the m0 mutation significantly reduced the effects of PRPF40B overexpression (Fig. 9C, lanes 3–4), which supports the previous results with the mutation in the e2 segment and further demonstrate that uridine-rich sequences within the exonic splicing silencer of URE6 are important for PRPF40B function.
FIGURE 9.

Exon 6 sequences and associated 5′ and 3′ splice sites are required for PRPF40B-mediated Fas splicing regulation. (A) Schematic representation of the structure of the Fas gene, including the genomic sequence of exon 6 in the box and the neighboring introns 5 and 6. e1, e2, and e3 represent three segments in exon 6, where e2 corresponds to the uridine-rich exonic silencer (URE6). m0, m1, and m2 represent different RNA sequences replacing the URE6 silencer. The strong Py in intron 5 indicates the substitution of a weak 3′ splice site associated with exon 6 with a strong polypyrimidine tract. The U1C in intron 6 corresponds to a mutant strengthening the 5′ splice site by improving its base-pairing potential with U1 snRNA. (B–D) Analyses of the effects of Fas splice site selection in response to PRPF40B overexpression. HEK293T cells were cotransfected together with the corresponding Fas minigene and a plasmid expressing PRPF40B. RT-PCR was performed to analyze the alternatively spliced forms of the Fas minigenes. The graphs show the densitometric analysis results as the change in exon skipping average from three independent experiments (means ± SEM). (*) P < 0.05; (**) P < 0.01. Cell lysates were analyzed by immunoblotting with the indicated antibodies to detect the PRPF40B and ERK2 proteins. (E) Effect of tethering PTB and PRPF40B to exon 6 through the RNA MS2-binding domains. The sequence of the URE6 element was replaced by two MS2 stem–loop binding sites in tandem. This minigene was cotransfected with plasmids expressing the indicated proteins or empty vector. RT-PCR was performed to assess the alternatively spliced isoforms of Fas. The bar graph shows the densitometric analysis results as the exon skipping average from three independent experiments (means ± SEM). (**) P < 0.01. A fraction of the cell lysates was analyzed by immunoblotting to detect the indicated proteins.
Next, the influence of splice site strength on PRPF40B function was analyzed. Strengthening either the 3′ splice site by inserting a strong polypyrimidine tract (mutant Py) or the 5′ splice site by improving its base-pairing with the U1 snRNA (mutant U1C) splice site associated with exon 6 compromised PRPF40B regulation of Fas alternative splicing (Fig. 9D), suggesting that a strong 5′ or 3′ splice site in exon 6 limits the response to PRPF40B. Taken together, these data indicate that in the case of the regulation of Fas alternative splicing, the presence of exonic sequences and weak 5′ and 3′ splice sites are required for PRPF40B function in alternative exon selection.
To evaluate whether the recruitment of PRPF40B to URE6 is sufficient to mediate the observed effects, we used an MS2 tethering assay. Previous data showed that the substitution of the URE6 sequence with binding sites for the MS2 bacteriophage protein resulted in efficient exon skipping upon MS2-PTB overexpression (Fig. 9E; Izquierdo et al. 2005). PRPF40B overexpression did not induce exon skipping in this minigene construct (Fig. 9E), which further supports the importance of exon 6 sequences in the PRPF40B-mediated regulation of Fas alternative splicing. However, this minigene was insensitive to the expression of an MS2-PRPF40B fusion protein (Fig. 9E). We conclude that PRPF40B recruitment to the URE6 element is insufficient to induce Fas exon 6 skipping.
PRPF40B depletion increases Fas/CD95 receptor number
The absence of PRPF40B promoted the inclusion of the Fas exon 6, causing an increase in the long proapoptotic Fas isoform (Fig. 7B–D), which encodes the membrane-bound form of the Fas receptor. To determine whether the observed change in the alternative splicing of Fas was related to the expression of the transmembrane receptor protein, we examined the proportion of Fas receptors in control and PRPF40B-depleted cells by flow cytometry using a specific anti-Fas/CD95 antibody. The flow cytometric data shown in Figure 10A are displayed as frequency histograms of Fas/CD95 immunofluorescence, detected using an Alexa 647-conjugated secondary antibody. The data are presented on a logarithmic scale and a shift to the right in the histogram indicates an increase in Fas/CD95-associated immunofluorescence. We observed a higher amount of membrane-bound Fas receptor upon PRPF40B-depletion (Fig. 10A), which correlates with previous data regarding the expression of the proapoptotic isoform in these cells. Statistical analyses using the nonparametric Kolmogorov–Smirnov test for comparing two samples revealed a small P value (≤0.001), which indicates that the distribution did not occur due to chance (Fig. 10B).
FIGURE 10.
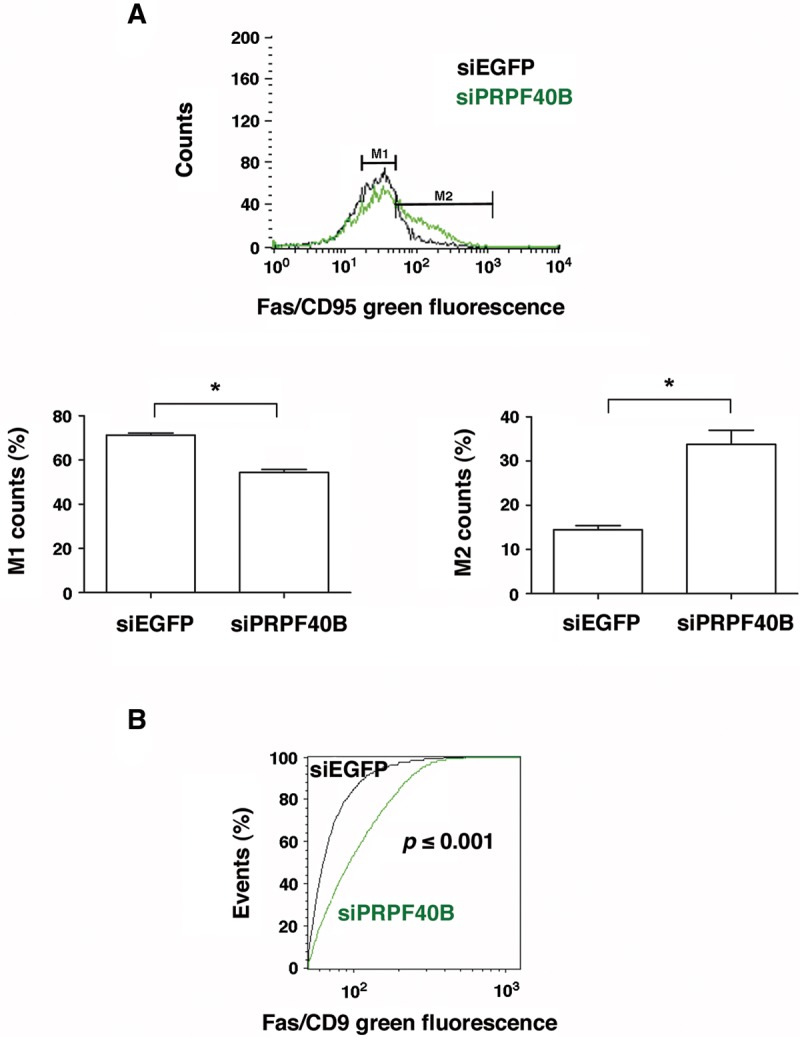
Analysis of the number of Fas receptors on the plasma membrane of control and PRPF40B-depleted cells by flow cytometry. (A) The top panel shows the flow cytometry-generated frequency histogram of Fas/CD95-associated fluorescence in control (siEGFP, black) and siPRPF40B (green) cells. The results shown are representative of three repeat experiments. The bottom panels show the quantification analysis of the Fas/CD95-specific fluorescence in the M1 (left) and M2 (right) populations. The graphs show the data from three independent experiments (means ± SEM). (*) P < 0.05. (B) The graph represents Kolmogorov–Smirnov statistics obtained from comparing the population distributions of the Fas/CD95-associated fluorescence in siEGFP (black) and siPRPF40B (green) cells. The P value was ≤0.001, which indicates that the two groups were sampled from populations with different distributions.
PRPF40B affects cell apoptosis
The previous result indicates that the absence of PRPF40B stimulates cell apoptosis. Several experiments were carried out to determine whether PRPF40B influences apoptosis. We chose to perform the experiments in the same HEK293T cells, although they have high resistance to cell death and therefore the effects obtained are small but significant. First, we measured the percentage of dead cells in control and PRPF40B-depleted cells using propidium iodide (PI). We found that PRPF40B knockdown increased the number of dead cells (Fig. 11A). However, this technique does not distinguish between apoptotic and necrotic cells. To better quantify the apoptotic cells, we performed cell cycle experiments to determine the number of cells in the sub-G1 phase, which corresponds to apoptotic cells. We observed a slight but significant increase in the percentage of cells in the sub-G1 population in PRPF40B-depleted cells compared with control cells (Fig. 11B). These results suggest that PRPF40B depletion increases the proportion of apoptotic cells. To lend additional support to these data, we also performed annexin-V binding assays. Annexin-V specifically binds phosphatidylserine when this molecule is located at the cell surface under apoptotic conditions. We first determined cell viability in control and PRPF40B-depleted cells and observed that the knockdown of PRPF40B resulted in a decrease in the number of viable cells (Fig. 11C). Next, we measured the number of annexin-V-positive cells by flow cytometry in the cells. In agreement with our previous data, we observed significantly augmented annexin-positive cells upon PRPF40B knockdown (Fig. 11C). We also analyzed caspase-3 activity, which is currently used as a marker of apoptosis. To measure active caspase-3 in the cells, we used an assay based on the addition of a luciferase substrate that is cleaved in the presence of active caspase-3, leading to a luminescent signal. The number of luciferase units directly correlates with the amount of active caspase-3 present in the cell. We observed a low (less than twofold) but significant increase in the quantity of active caspase-3 in PRPF40B knockdown cells (Fig. 11D). Caspase-3 is expressed as a 35 kDa inactive precursor from which the 17 kDa (p17) and 11 kDa (p11) subunits are proteolytically generated during apoptosis. The caspase-3 precursor is first cleaved to generate a p11 subunit and a p20 peptide, which is subsequently cleaved to produce the mature p17 subunit. The active caspase-3 enzyme is a heterodimer composed of two p17 peptides and two p11 subunits. Using specific antibodies against caspase-3, we observed significantly more p20 and p17 upon PRPF40B knockdown (Fig. 11E). The amount of the caspase-3 precursor was consequently diminished in PRPF40B-depleted cells (Fig. 11E). These findings confirm that the inhibition of PRPF40B causes an increase in caspase-3 activation. The expression of the anti-apoptotic regulator Bcl-2 was also analyzed. Interestingly, this protein showed reduced expression upon PRPF40B depletion (Fig. 11E). Together, these data show that PRPF40B modulates apoptosis and suggest a functional link between the control of alternative splicing and molecular events leading to cell death by apoptosis.
FIGURE 11.

Effect of PRPF40B depletion on apoptosis. HEK293T cells were transiently transfected with siEGFP (control) or siPRPF40B. (A) Flow cytometry analysis of HEK293T cells stained with PI for 30 min at 37°C. The bar graphs show the data from three independent experiments (means ± SEM). (**) P < 0.01. (B) PRPF40B depletion increases the percentage of the sub-G1 cell population. The cells were fixed prior to RNase treatment and PI staining. The cell cycle phases were determined by flow cytometry. The graph shows the percentage of cells in the sub-G1 phase of the cell cycle. The data are from three independent experiments (means ± SEM). (**) P < 0.01. (C) PRPF40B knockdown increases annexin-V binding. Control and PRPF40B-knockdown HEK293T cells were incubated without or with annexin-V and analyzed by flow cytometry. The bar graphs show the percentage of cells from three independent experiments (means ± SEM). (*) P < 0.05; (**) P < 0.01. (D) Analysis of caspase-3 activation in PRPF40B-depleted cells. Activation of caspase-3 was significantly increased in PRPF40B-knockdown HEK293T cells. Caspase-3 activation was measured by chemiluminescence in control and PRPF40B-depleted cells. The bar graph shows the fold activation from three independent experiments (means ± SEM). (*) P < 0.05. (E) Analysis of PRPF40B, procaspase-3 cleavage (precursor p35 and cleaved fragments p20/p17), and BCL2 levels. Protein expression was analyzed by immunoblotting using specific antibodies in protein extracts obtained from control and PRPF40-depleted cells. ERK2 was used as a loading control. The p20 + p17/procaspase-3 ratio was calculated to determine the values of the caspase isoforms. The quantification of the bands is shown below each panel.
DISCUSSION
In higher eukaryotes, most exons are alternatively spliced to generate distinct transcript isoforms from a single pre-mRNA, thus enhancing the proteome diversity in these organisms. The initial recognition of splice sites is a critical step in the highly regulated splicing process and is responsible for defining the introns that are going to be removed. The initial interactions in the earliest assembly phase of the spliceosome are weak and generally require the assistance of proteins to enhance their stability. Most of the major proteins that form the spliceosomal complex that recognizes the 5′ and 3′ splice sites of an intron have been identified by proteomic approaches (Wahl et al. 2009); however, the full list of alternative splicing regulators that contribute to the assembly of this critical spliceosome is far more complex than previously expected (Makarov et al. 2012). Many of these proteins may contribute to the flexibility and specificity of spliceosome function to explain genome-wide alternative splicing patterns. A number of these regulators are also likely involved in the coupling of alternative splicing to transcription and other RNA processing events (for review, see Moore and Proudfoot 2009; Montes et al. 2012).
Here, our objective was to characterize PRPF40B, a putative homolog of the essential yeast Prp40 protein, which may function to bridge the splice sites of an intron. We sought to provide evidence for a splicing-relevant role of this protein in human cells. Toward this goal, we performed confocal microscopy experiments that indicated that PRPF40B is distributed in dot-like structures throughout the nucleoplasm. PRPF40B was enriched in the splicing factor-rich nuclear speckles (Figs. 1–3), and this localization pattern did not change upon inhibiting transcription (Fig. 4), suggesting that the speckle localization is not the result of an association between PRPF40B and actively transcribed genes in these regions. Upon RNAPII inhibition, speckles decrease in number, enlarge, and become rounded because of the accumulation of splicing machinery (O'Keefe et al. 1994). We found similar dynamic properties of PRPF40B upon transcriptional inhibition, thus demonstrating that this protein shows a behavior similar to that of splicing factors. The localization of PRPF40B was unaffected after RNA degradation (Fig. 4), which indicates that the association of PRPF40B with nuclear speckles is not dependent on RNA binding. Many, if not all, of the pre-mRNA splicing factors localize at least to some extent to speckles. The protein composition of speckles has been assessed by proteomic analyses and many other additional localization studies. This information has revealed that speckles contain other proteins apart from splicing factors, such as transcription factors including a population of the phosphorylated RNAPII, 3′ end-RNA processing factors, proteins involved in translation, and structural proteins (Spector and Lamond 2011). Although not proven by the presented localization data, the detection of PRPF40B in speckles provided the first indication that PRPF40B is involved in RNA metabolism.
Next, we took advantage of previous knowledge of the Prp40 association with splicing factors to examine the binding of PRPF40B with SF1 and U2AF65. We found that PRPF40B associates with these essential splicing factors in vivo and in vitro (Figs. 5–7). These data expand the previous results of Lin and coworkers that showed that the WW domains of PRPF40A are capable of interacting with SF1 and U2AF65 using pull-down experiments (Lin et al. 2004). We also found a tight association between the WW-containing moiety of PRPF40B and SF1, which is not surprising given the high homology between PRPF40A and PRPF40B and between their WW domains in particular. The identity and similarity percentages of the WW1 domains of these proteins are 58% and 78%, respectively, using global and local alignments, and there is 81% identity and 96% similarity for the WW2 domains of PRPF40A and PRPF40B. Treatment of cell extracts with RNase decreased the binding of PRPF40B to SF1 without affecting the interaction of SF1 with U2AF65 (Fig. 7C,D). The association between PRPF40B and these splicing factors remained in AMO-transfected cells (Fig. 7E), which suggest that U1 snRNP does not mediate this interaction. Although PRPF40B may directly interact with SF1 (Fig. 6), the coimmunoprecipitation experiments indicate that the interaction in live cells is dependent on RNA integrity, suggesting that PRPF40B interacts with RNA or that the interaction between the proteins is mediated by another protein bound to the RNA; however, the identity of this putative RNA remains unknown.
In yeast, the assembly of the earliest splicing complex begins when the 5′ splice site and branch region are initially recognized by U1 snRNP and the splicing factor Bbp followed by recruitment of Mud2 to the polypyrimidine tract. In this context, Prp40 may function to bridge U1 snRNP and Bbp. Based on this hypothesis, mammalian Prp40 proteins have been proposed to bridge the 5′ splice site and the branch region in this early spliceosomal complex (Abovich and Rosbash 1997; Reed 2000; Padgett 2012). Here, we show that the presence of weak 5′ and 3′ splice sites are necessary for efficient PRPF40B function in Fas alternative splicing. In addition, exon 6 sequences near the 3′ splice site, which may serve to stabilize the SF1-U2AF65-mediated recognition of the 3′ splice site, are also required for PRPF40B regulation. The exonic sequences that include or overlap with the PTB-responsive silencer are also important (Fig. 8). However, we could not find a clear sequence motif within these sequences that could be responsible for PRPF40B-mediated regulation of alternative splicing (data not shown). Taken together, these observations are in agreement with a role for PRPF40B during the early protein–protein and/or protein–RNA interactions that lead to exon definition during Fas splicing. Our data show that PRPF40B participates in Fas alternative splicing by promoting exon skipping when the general belief in the field is that prp40-like proteins function to bridge the 5′ and 3′ splice sites of the intron to stabilize the commitment complex and define the exon. However, we do not believe that PRPF40B acts as a splicing repressor (such as PTB) because direct recruitment of PRPF40B through MS2-binding sites did not result in Fas exon 6 skipping (Fig. 8E).
The following findings suggest a role for PRPF40B in pre-mRNA splicing during the early stages of spliceosomal assembly: (i) PRPF40B localizes to splicing factor-rich nuclear speckles; (ii) PRPF40B interacts with SF1 and U2AF65, two splicing factors involved in 3′ splice site recognition, and this interaction appears to be RNA dependent; and (iii) PRP40B regulates the alternative splicing of Bcl-x and Fas. These results are consistent with the documented function of Prp40 in yeast and support the possibility that this factor may be involved in splice site selection in higher eukaryotes; however, this assumption needs to be further tested. Our results do not exclude the possibility that PRPF40B may either alter splicing efficiencies by acting on any general splicing factor or that PRPF40B influences alternative splicing by modulating transcription elongation dynamics (Kornblihtt 2007).
Our results show that PRPF40B modulated the alternative splicing of Fas and Bcl-x, which are key genes in the control of apoptosis. PRPF40B depletion increased the production of the anti-apoptotic Bcl-x isoform and increased the production of the proapoptotic Fas isoform (Fig. 7). These apparently conflicting results have also been described for the splicing modulator RBM10, whose expression enhanced the anti-apoptotic Fas isoform and increased the proapoptotic Bcl-x isoform (Inoue et al. 2014). Nevertheless, our functional experiments have served to evaluate the biological consequences of PRPF40B depletion in promoting apoptosis. In agreement with this result, a higher number of Fas receptors bound to the cell membrane (Fig. 9). We also show that PRPF40B knock down correlates with the induction of apoptosis as determined by assessing dead cells, sub-G1-phase cells, annexin-V binding, cell viability, and caspase-3 cleavage (Fig. 10). In support of our results, PTB depletion, which promotes Fas exon 6 skipping (Izquierdo et al. 2005), resulted in an increase in the percentage of cells in the sub-G1 population and in the annexin-V-positive cells (data not shown). Although more work is clearly needed, our results might indicate the ability of PRPF40B to alter the alternative splicing of Fas to regulate cell survival.
Recently, a striking example of the relationship between splicing factors and human disease has been described. Using whole-exome sequencing for MDS patients, recurrent somatic mutations in spliceosome components have been identified (Papaemmanuil et al. 2011; Yoshida et al. 2011). Notably, the spliceosome proteins that were found to be mutated are involved in the early steps of splice site recognition. One of the frequently mutated splicing proteins was PRPF40B. To date, the mechanism by which these mutated proteins act on splicing regulation, either by a gain of function or dominant negative function, is not known. In addition, the mutated factors might interfere with the balance of the critical early spliceosomal complexes to perturb the normal recognition of splice sites and generate other alternative transcript isoforms that could lead to cell dysfunction and disease. The understanding of the molecular events that function during splice site selection together with the identification of endogenous target genes affected by these mutated proteins are required to fully answer the functional link between splicing and MDS.
MATERIALS AND METHODS
Plasmid constructs
A commercial intermediate vector encoding the human cDNA for PRPF40B (IMAGE clone 664467, Source Bioscience) was used to generate the PRPF40B constructs described below. The mammalian expression vector pEFBOST7-PRPF40B (1–871) and the corresponding deletion expression constructs pEFBOST7-PRPF40B (1–687), pEFBOST7-PRPF40B (263–871), pEFBOST7-PRPF40B (394–871), pEFBOST7-PRPF40B (474–871), and pEFBOST7-PRPF40B (618–871) were obtained by inserting the appropriate PCR fragments using XbaI/ClaI or XbaI/BstBI sites at the ends into the XbaI/BstBI sites of the parental vector pEFBOST7, which encodes the 11-amino acid T7 epitope tag at its amino terminus, using standard cloning procedures (Suñé and Garcia-Blanco 1999). pEFBOS/EGFP/TCERG1 (Sánchez-Álvarez et al. 2010) was used to generate pEFBOS/EGFP/T7PRPF40B (1–871), pEFBOS/EGFP/T7PRPF40B (1–687), and an amino-terminal deletion series by subcloning appropriate EcoRI fragments obtained from pEFBOST7 parental vectors. pEFBOS/EGFP/T7PRPF40B (474–553/672–871) was generated by digesting the expression vector pEFBOS/EGFP/T7PRPF40B (474–871) with XbaI/BamHI and inserting the PCR-amplified fragment PRPF40B (474–553) into the XbaI/BamHI restriction sites. The cDNA encoding human SF1 was amplified from the pGEM-SF1-Bo vector, which was kindly provided by Angela Kramer (University of Geneva, Geneva, Switzerland). PCR fragments containing XbaI and BstBI restriction sites were cloned into corresponding sites in the pEFBOST7 vector. The bacterial expression vectors pGEX2TK-PRPF40B (1–871), pGEX2TK-PRPF40B (1–333), and pGEX2TK-PRPF40B (446–871) were generated by inserting PCR-amplified products digested by BglII/EcoRI into a BamHI/EcoRI-digested pGEX2TK vector (Amersham Pharmacia Biotech). The bacterial expression vectors pGEX2TK-TCERG1 (1–662) and pGEX2TK-TCERG1 (631–1098) have been previously described (Suñé et al. 1997; Carty et al. 2000). The Bcl-x and Fas splicing reporter minigenes were kindly provided by Benoit Chabot (University of Sherbrooke, Québec, Canada) and Juan Valcárcel (Centre for Genomic Regulation, Barcelona, Spain), respectively. The Fas minigene derivatives (e1, e2, e3, m0, m1, m2, Py, and U1C) have been previously described (Izquierdo et al. 2005; Bonnal et al. 2008) and were kindly provided by Juan Valcárcel.
Antibodies
The polyclonal PRPF40B antibody used in this study was generated in rabbits using the GST-PRPF40B (1–871) construct. The GST-PRPF40B fusion protein was expressed as previously reported (Faber et al. 1998). The antiserum was directly used for immunoblotting analysis at a dilution of 1:10,000. For immunofluorescence analysis, IgGs were purified by protein A sepharose columns (GE Healthcare Life Science) under the conditions suggested by the manufacturer, and the purified IgGs were used at 1:1000. An antibody directed against the T7 tag (Bethyl Laboratories) was used at 1:20,000 for immunoblotting. Antibodies directed against ERK2 (C-14, Santa Cruz Biotechnology) were used at 1:2000 for immunoblotting. Antibodies directed against NOLC1 (Abcam) were used at 1:500 for immunofluorescence analysis. Antibodies directed against U2AF65 were used at a dilution of 1:100 for immunofluorescence analysis and were kindly provided by Juan Valcárcel. For immunoblotting, we used a commercially available anti-U2AF65 antibody (sc-48804, Santa Cruz Biotechnology) at a dilution of 1:2000. The mouse monoclonal antibody directed against the splicing factor SF1 (Abnova) was used at 1:100,000 and 1:40,000 for immunoblotting and immunofluorescence analysis, respectively. An antibody directed against the splicing factor SC35 (S4045, Sigma-Aldrich) was used at 1:4000. Antibodies directed against caspase-3 (sc-7148) and Bcl-2 (sc-7382) (Santa Cruz Biotechnology) were used at a dilution of 1:500 for immunoblotting. Antibodies against the Fas/CD-95 receptor (BioLegend) were used at 1:100 for flow cytometry and were kindly provided by Mari Carmen Ruiz (CIBM, Granada). For immunoblotting, primary antibodies were detected using HRP-conjugated secondary antibodies (PerkinElmer Life Science) at 1:5000. Alexa 488-conjugated goat anti-mouse (Molecular Probes), Alexa 647-conjugated goat anti-mouse (Molecular Probes), and Alexa 647-conjugated goat anti-rabbit (Life Technologies) secondary antibodies were used to detect primary antibodies at 1:500 and 1:5000 for immunofluorescence and flow cytometry, respectively.
Cell culture and transfections
HEK293T and HeLa cells were grown and maintained as previously described (Sánchez-Álvarez et al. 2006). For immunoblotting, reverse transcriptase-PCR (RT-PCR) analysis, and apoptosis experiments, transfections were performed in 35-mm plates (Falcon, Fisher Scientific). Each plate was seeded with ∼1 × 106 cells 20 h prior to transfection. Cells were grown to ∼60%–70% confluence and transfected with appropriate amounts of the indicated constructs using calcium phosphate. Approximately 40 h after transfection, the cells were harvested and processed. For RNA interference (RNAi) knockdown experiments, ∼0.5 × 106 HEK293T cells were seeded in 35 mm plates. The cells were transfected using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol with 60 nM (final concentration) of either of the following small interfering RNA (siRNA) duplexes: siEGFP: 5′-CUACAACAGCCACAACGE-3′, siPRPF40B: 5′-GCAGUUCUGGACAGCAUCA-3′, and siPRPF40B(2): 5′-AAUCAGAGACCACCAGCUAUCUU-3′. The cells were harvested and processed 48 or 72 h after transfection for RT-PCR or apoptosis experiments, respectively. For double transfections, the cells were transfected with the reporter minigene 48 h after siRNA transfection using the LipoD 293T reagent (SignaGen Laboratories) according to the manufacturer's protocols. Cells were harvested at 24 h after transfection for the Fas experiments and at 48 h for the Bcl-x experiments. After total RNA extraction, the splice variants were amplified by RT-PCR. For immunoprecipitation analysis, transfections were performed in 100 mm plates (Falcon). Each plate was seeded with ∼1.5 × 106 cells 20 h prior to transfection. The cells were grown to ∼80%–90% confluence and transfected with 6 mg of DNA using calcium phosphate. The cells were harvested and processed ∼40 h later. For U1 snRNA blocking, we transfected the AMOs at a final concentration of 3 μM at ∼24 h after DNA transfection using the EndoPorter reagent (Gene Tools), according to the manufacturer's protocol. The sequence of U1 AMO and control AMO (Gene Tools) are as follows: 5′-GGTATCTCCCCTGCCAGGTAAGTAT-3′ and 5′-CCTCTTACCTCAGTTACAATTTATA-3′, respectively. For immunofluorescence, cells were grown on coverslips to 50%–60% confluence, transfected 20 h later using calcium phosphate, and processed ∼24 h after transfection.
Immunofluorescence analysis and treatments
Immunofluorescence studies were performed as previously described (Sánchez-Hernández et al. 2012). To analyze the endogenous SF1 and U2AF65 proteins, a specific protocol was performed: The cells were washed twice in phosphate-buffered saline (PBS) (pH 7.4) and permeabilized with 0.5% Triton X-100 in CSK buffer (100 mM NaCl, 300 mM sucrose, 10 mM PIPES, 3 mM MgCl2, and 1 mM EGTA [pH 6.8]) for 1 min on ice. Subsequently, the cells were fixed with 3.5% paraformaldehyde in PBS for 5 min at room temperature and rinsed with PBS. The cells were incubated with blocking buffer (1% fetal bovine serum in PBS) for 10 min at room temperature and subsequently with primary antibodies in blocking buffer for 1 h at room temperature. After three 15-min washes in PBS, the cells were incubated with the appropriate secondary antibody in blocking buffer for 1 h at room temperature. After three washes with PBS, the coverslips were mounted in ProLong Gold Antifade Reagent (Molecular Probes) onto glass slides. Treatment with 25 mg/mL α-amanitin was performed for 6 h at 37°C prior to immunofluorescence processing. RNase was used at a concentration of 0.1 mg/mL for 2 h at 37°C after the fixation and permeabilization steps. Images were acquired using a Leica SP5 spectral laser confocal microscope and processed using the LAS AF software v2.3.6. The images were digitally processed for presentation using the Adobe Photoshop CS3 extended v10.0 software.
Preparation of cell extracts and immunoprecipitation
In vivo interaction experiments were performed with whole-cell extract (WCE). Cells were washed in cold PBS, pelleted, and lysed in 500 mL of T7 buffer (20 mM HEPES [pH 7.9], 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor mixture [Complete Roche]) for 30 min at 4°C. The cell extracts were centrifuged at maximum speed for 5 min. For RNase A treatment, the cell lysates were incubated with 100 mg/mL of RNase A for 1 h at 4°C. Ten percent fractions of WCE were boiled in SDS-PAGE loading buffer and saved for immunoblotting analysis. The remaining cell lysates were diluted to a 1-mL final volume with T7 buffer. Diluted extracts were added to 50 µL of anti-T7 tag monoclonal antibody covalently coupled to cross-linked agarose beads (Novagen) and incubated with end-over-end rotation for 4–6 h at 4°C. After five washes with T7 buffer, the proteins bound to the antibody resin were eluted by boiling the samples with SDS-PAGE loading buffer, separated on 10% SDS-PAGE gels, and analyzed by Western blotting.
Protein purification and in vitro interaction assay
GST fusion proteins were expressed in BL21 (D3) cells and purified on glutathione–agarose beads (Sigma) following standard procedures. The elution of bound proteins was performed by affinity competition using saturating levels of reduced L-glutathione (Sigma) in a final volume of 1 mL. Proteins were dialyzed against PBS containing 1 mM PMSF and concentrated with an Ultra 0.5 Centrifugal Filter (Millipore). The SF1 protein was expressed in HEK293T cells and purified as previously described (Cazalla et al. 2005). In vitro binding of SF1 to GST fusion proteins was performed as previously described (Xiao and Manley 1997; Rain et al. 1998) with minor modifications. Briefly, 1 mg of GST or GST fusion proteins was bound to 50 mL of glutathione–agarose beads in 300 mL of NETN buffer (20 mM Tris–HCl at pH 8.0, 150 mM NaCl, 0.5% NP-40, and 0.5 mM EDTA) for 30 min with end-over-end rotation at 4°C. The beads were washed twice with 500 mL of NETN and incubated with 1 mg of SF1 in 200 mL of NETN for 30 min with end-over-end rotation at 4°C. After six washes with 200 µL NETN, the bound proteins were eluted by boiling in SDS-PAGE loading buffer for 5 min. Two independent fractions of each sample were separated by 10% SDS-PAGE and analyzed by silver staining and Western blotting.
Western blot analysis
A fraction of the transfected cells was lysed in cold T7 buffer. The proteins were separated by 10% or 12.5% SDS-PAGE, transferred to a nitrocellulose membrane (Amersham Biosciences), and then incubated with specific antibodies. After washing, the membrane was incubated with peroxidase-conjugated secondary antibodies, and the bound antibodies were detected by enhanced chemiluminescence (PerkinElmer Life Science). Densitometric analysis was performed using the QuantityOne software v4.6.5 (Bio-Rad). The intensity of the bands was calculated relative to the internal control (ERK-2) for each lane and each sample.
RNA extraction and RT-PCR analysis
Total RNA was extracted from transfected cells with peqGOLD TriFast (peQlab) according to the manufacturer's protocol. For plasmid-derived RNA, 800 ng of RNA was reverse transcribed by the Moloney murine leukemia virus RT (Invitrogen) for 1 h at 37°C using the primers csnPT2 (5′-AAGCTTGCATCGAATCAGTAG-3′) and RT-SVeda (5′-GGGAAGCTAGAGTAAGTAG-3′) for the Fas and Bcl-x reporter minigenes, respectively. One quarter of the resulting cDNA was amplified by PCR using the oligonucleotides csnPT1 (5′-GTCGACACTTGCTCAAC-3′) and csnPT2 for Fas and X34 (5′-AGGGAGGCAGGCGACGGCGACGAGTTT-3′) and XAgeIR (5′-GTGGATCCCCCGGGCTGCAGGAATTCGAT-3′) for Bcl-x. The intensity of the bands was quantified using the Quantity One software. For endogenous Fas RNA, 1 µg of RNA was reverse transcribed using the qScript cDNA Supermix (Quanta Biosciences) following the manufacturer's protocol. Fas endogenous transcripts were analyzed by conventional PCR, using the oligonucleotides FasE5Fw (5′-GTGAACATGGAATCATCAAGG-3′) and FE7 (5′-TCCTTTCTGTGCTTTCTGCAT-3′). The intensity of the bands was quantified using the Quantity One software. Quantification of Fas endogenous transcripts by real-time PCR was performed by using the iQ SYBR Green Supermix (Bio-Rad) and the iCycler thermal cycler station (Bio-Rad) with the oligonucleotides FE6 (5′-TAACTTGGGGTGGCTTTGTC-3′) and FE7. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control gene and was amplified with the oligonucleotides GAPDH-F (5′-ATGGGGAAGGTGAAGGTCG-3′) and GAPDH-R (5′-GGGTCATTGATGGCAACAATATC-3′).
The statistical analysis was performed using the Prism 5.0 software (GraphPad). A two-tailed Student's t-test was used to compare the means between samples and their respective controls. The P values are represented in the figures by asterisks (* P < 0.05; ** P < 0.01; *** P < 0.005). The absence of an asterisk indicates that the change was not statistically significant.
Cell death and cell cycle assays
HEK293T cells were trypsinized, collected in polystyrene tubes and washed twice with PBS. For the cell death assays, one half of the cells were incubated in PBS containing 40 μg/mL propidium iodide (Sigma) at 37°C for at least 20 min. For the cell cycle analysis, the remainder of the cells was fixed with 70% ethanol at 4°C for at least 15 min. After washing with PBS, the cells were incubated for 20 min at 37°C in a solution of PBS containing 100 μg/mL RNase A (Roche) and 40 μg/mL propidium iodide. The samples were analyzed by flow cytometry. The data were processed using the FlowJo v7.6.5 software.
Annexin-V binding assay
HEK293T cells were trypsinized, collected in polystyrene tubes and washed with PBS. The cells were incubated in 100 mL of a solution containing 5 mL of annexin-V conjugated to fluorescein isothiocyanate (FITC) (Immunostep S.L.) for 15 min at room temperature. The fluorescence was measured by flow cytometry. The data were analyzed using the FlowJo v7.6.5 software. The statistical analysis was performed using the Prism 5.0 software.
Active caspase-3 measurement
Approximately 1 × 104 HEK293T cells were collected to measure caspase-3 activation. Quantifications were performed using the Caspase-Glo 3/7 assay (Promega) according to the manufacturer's protocol. The luminescent signal (relative light units, RLUs), which was directly proportional to caspase activation, was measured using an Infinite F200 TECAN Luminometer.
Analysis of the expression of membrane-bound Fas/CD95 receptor
HEK293T cells were trypsinized, collected in polystyrene tubes and washed with PBS. The cells were incubated with the primary antibody anti-Fas/CD95 in PBS for 30 min at 4°C in the dark. After washing twice with PBS, the cells were incubated with the corresponding secondary antibody for 30 min at 4°C in the dark. The cells were washed with PBS, and the fluorescence was measured on a FACSCalibur flow cytometer (BD Biosciences Clontech). The data were analyzed using the Cell Quest Pro v1.b.1f4b software.
ACKNOWLEDGMENTS
We are grateful to members of the laboratories for their helpful suggestions, critical discussions, and comments. This work was supported by grants from the Spanish Ministry of Science and Innovation (BFU2011-24577 to C.S. and BFU2013-44660R to C.H.M.), and the Andalusian Government (Excellence Projects CVI-4626 and BIO-2515 to C.S., and CTS-6587 to C.H.M.). Support from the European Region Development Fund, ERDF (FEDER) is also acknowledged. S.B. was supported by a fellowship from the Andalusian Government through the Excellence Project CVI-4626.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.047258.114.
REFERENCES
- Abdel-Wahab O, Levine R 2011. The spliceosome as an indicted conspirator in myeloid malignancies. Cancer Cell 20: 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abovich N, Rosbash M 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89: 403–412. [DOI] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. 2012. U1 snRNP determines mRNA length and regulates isoform expression. Cell 150: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Abovich N, Rosbash M 1998. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev 12: 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB 1993. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74: 597–608. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Martínez C, Förch P, Bachi A, Wilm M, Valcárcel J 2008. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell 32: 81–95. [DOI] [PubMed] [Google Scholar]
- Brameier M, Krings A, MacCallum RM 2007. NucPred—predicting nuclear localization of proteins. Bioinformatics 23: 1159–1160. [DOI] [PubMed] [Google Scholar]
- Buschdorf JP, Strätling WH 2004. A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J Mol Med (Berl) 82: 135–143. [DOI] [PubMed] [Google Scholar]
- Carty SM, Goldstrohm AC, Suñé C, Garcia-Blanco MA, Greenleaf AL 2000. Protein-interaction modules that organize nuclear function: FF domains of CA150 bind the phosphoCTD of RNA polymerase II. Proc Natl Acad Sci 97: 9015–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascino I, Fiucci G, Papoff G, Ruberti G 1995. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol 154: 2706–2713. [PubMed] [Google Scholar]
- Cazalla D, Sanford JR, Cáceres JF 2005. A rapid and efficient protocol to purify biologically active recombinant proteins from mammalian cells. Protein Expr Purif 42: 54–58. [DOI] [PubMed] [Google Scholar]
- Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD 1994. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 263: 1759–1762. [DOI] [PubMed] [Google Scholar]
- De Conti L, Baralle M, Buratti E 2013. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA 4: 49–60. [DOI] [PubMed] [Google Scholar]
- Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME 1998. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet 7: 1463–1474. [DOI] [PubMed] [Google Scholar]
- Goldstrohm AC, Albrecht TR, Suñé C, Bedford MT, Garcia-Blanco MA 2001. The transcription elongation factor CA150 interacts with RNA polymerase II and the pre-mRNA splicing factor SF1. Mol Cell Biol 21: 7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K 2007. WoLF PSORT: protein localization predictor. Nucleic Acids Res 35: W585–W587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Yamamoto N, Kimura M, Nishio K, Yamane H, Nakajima K 2014. RBM10 regulates alternative splicing. FEBS Lett 588: 942–947. [DOI] [PubMed] [Google Scholar]
- Izquierdo JM, Valcárcel J 2006. A simple principle to explain the evolution of pre-mRNA splicing. Genes Dev 20: 1679–1684. [DOI] [PubMed] [Google Scholar]
- Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigó R, Bilbao D, Valcárcel J 2005. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell 19: 475–484. [DOI] [PubMed] [Google Scholar]
- Jamison SF, Crow A, Garcia-Blanco MA 1992. The spliceosome assembly pathway in mammalian extracts. Mol Cell Biol 12: 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G 2010. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468: 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HY, Siliciano PG 1996. Identification of Prp40, a novel essential yeast splicing factor associated with the U1 small nuclear ribonucleoprotein particle. Mol Cell Biol 16: 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR 2007. Coupling transcription and alternative splicing. Adv Exp Med Biol 623: 175–189. [DOI] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H 2009. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci 106: 10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond AI, Spector DL 1993. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4: 605–612. [DOI] [PubMed] [Google Scholar]
- Legrain P, Seraphin B, Rosbash M 1988. Early commitment of yeast pre-mRNA to the spliceosome pathway. Mol Cell Biol 8: 3755–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KT, Lu RM, Tarn WY 2004. The WW domain-containing proteins interact with the early spliceosome and participate in pre-mRNA splicing in vivo. Mol Cell Biol 24: 9176–9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov EM, Owen N, Bottrill A, Makarova OV 2012. Functional mammalian spliceosomal complex E contains SMN complex proteins in addition to U1 and U2 snRNPs. Nucleic Acids Res 40: 2639–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier UT, Blobel G 1990. A nuclear localization signal binding protein in the nucleolus. J Cell Biol 111: 2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud S, Reed R 1991. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev 5: 2534–2546. [DOI] [PubMed] [Google Scholar]
- Michaud S, Reed R 1993. A functional association between the 5′ and 3′ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes Dev 7: 1008–1020. [DOI] [PubMed] [Google Scholar]
- Montes M, Becerra S, Sánchez-Álvarez M, Suñé C 2012. Functional coupling of transcription and pre-mRNA processing. Gene 501: 104–117. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136: 688–700. [DOI] [PubMed] [Google Scholar]
- O'Keefe RT, Mayeda A, Sadowski CL, Krainer AR, Spector DL 1994. Disruption of pre-mRNA splicing in vivo results in reorganization of splicing factors. J Cell Biol 124: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett RA 2012. New connections between splicing and human disease. Trends Genet 28: 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. 2011. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passani LA, Bedford MT, Faber PW, McGinnis KM, Sharp AH, Gusella JF, Vonsattel JP, MacDonald ME 2000. Huntingtin's WW domain partners in Huntington's disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington's disease pathogenesis. Hum Mol Genet 9: 2175–2182. [DOI] [PubMed] [Google Scholar]
- Rain JC, Rafi Z, Rhani Z, Legrain P, Krämer A 1998. Conservation of functional domains involved in RNA binding and protein–protein interactions in human and Saccharomyces cerevisiae pre-mRNA splicing factor SF1. RNA 4: 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed R 1990. Protein composition of mammalian spliceosomes assembled in vitro. Proc Natl Acad Sci 87: 8031–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed R 2000. Mechanisms of fidelity in pre-mRNA splicing. Curr Opin Cell Biol 12: 340–345. [DOI] [PubMed] [Google Scholar]
- Robberson BL, Cote GJ, Berget SM 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol Cell Biol 10: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Álvarez M, Goldstrohm AC, Garcia-Blanco MA, Suñé C 2006. Human transcription elongation factor CA150 localizes to splicing factor-rich nuclear speckles and assembles transcription and splicing components into complexes through its amino and carboxyl regions. Mol Cell Biol 26: 4998–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Álvarez M, Montes M, Sánchez-Hernández N, Hernández-Munain C, Suñé C 2010. Differential effects of sumoylation on transcription and alternative splicing by transcription elongation regulator 1 (TCERG1). J Biol Chem 285: 15220–15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Hernández N, Ruiz L, Sánchez-Álvarez M, Montes M, Macias MJ, Hernández-Munain C, Suñé C 2012. The FF4 and FF5 domains of transcription elongation regulator 1 (TCERG1) target proteins to the periphery of speckles. J Biol Chem 287: 17789–17800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraphin B, Rosbash M 1989. Identification of functional U1 snRNA-pre-mRNA complexes committed to spliceosome assembly and splicing. Cell 59: 349–358. [DOI] [PubMed] [Google Scholar]
- Seraphin B, Kretzner L, Rosbash M 1988. A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J 7: 2533–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcárcel J 2005. Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol 12: 645–653. [DOI] [PubMed] [Google Scholar]
- Smith CW, Valcárcel J 2000. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci 25: 381–388. [DOI] [PubMed] [Google Scholar]
- Spector DL, Lamond AI 2011. Nuclear speckles. Cold Spring Harb Perspect Biol 3: a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suñé C, Garcia-Blanco MA 1999. Transcriptional cofactor CA150 regulates RNA polymerase II elongation in a TATA-box-dependent manner. Mol Cell Biol 19: 4719–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suñé C, Hayashi T, Liu Y, Lane WS, Young RA, Garcia-Blanco MA 1997. CA150, a nuclear protein associated with the RNA polymerase II holoenzyme, is involved in Tat-activated human immunodeficiency virus type 1 transcription. Mol Cell Biol 17: 6029–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wang GS, Cooper TA 2007. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8: 749–761. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AJ, Cooper TA 2010. The pathobiology of splicing. J Pathol 220: 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SH, Manley JL 1997. Phosphorylation of the ASF/SF2 RS domain affects both protein–protein and protein–RNA interactions and is necessary for splicing. Genes Dev 11: 334–344. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. [DOI] [PubMed] [Google Scholar]