

ABSTRACT
NF-κB plays a critical role in the induction and maintenance of innate and adaptive immune transcriptional programs. An associated inhibitor of κB protein (IκB) regulates NF-κB activation and contains a degron motif (DSGΦxS) that undergoes phosphorylation following pathogen recognition or other proinflammatory signals. The E3 ubiquitin ligase SCFβ-TrCP recognizes this phosphodegron through its β-transducin repeat-containing protein (β-TrCP) subunit and induces IκB degradation, allowing NF-κB to translocate to the nucleus and modulate gene expression. Rotavirus (RV), a major cause of pediatric gastroenteritis, can block NF-κB activation through the action of its nonstructural protein NSP1, a putative E3 ubiquitin ligase that mediates the degradation of β-TrCP or other immunomodulatory proteins in a virus strain-specific manner. Here, we show that NSP1 targets β-TrCP by mimicking the IκB phosphodegron. The NSP1 proteins of most human and porcine RV strains conserve a C-terminal phosphodegron-like (PDL) motif, DSGΦS. Deletion of this motif or mutation of its serine residues disrupts NSP1-mediated degradation of β-TrCP and inhibition of NF-κB activation. Additionally, a point mutation within the phosphodegron-binding pocket protects β-TrCP from NSP1-mediated turnover. Fusion of the PDL motif to an NSP1 protein known to target other immunomodulatory proteins generates a chimeric NSP1 protein that can induce β-TrCP degradation and block NF-κB activation. Other viral proteins (Epstein-Barr virus LMP1, HIV-1 Vpu, and vaccinia virus A49) also contain a PDL motif and interact with β-TrCP to inhibit NF-κB activation. Taken together, these data suggest that targeting β-TrCP by molecular mimicry may be a common strategy used by human viruses to evade the host immune response.
IMPORTANCE The transcription factor NF-κB, a central regulator of the host response to infection, is a frequent target of viral antagonism. Pathogen detection activates NF-κB by inducing the phosphorylation of an associated inhibitor protein (IκB), which targets IκB for degradation by the E3 ubiquitin ligase β-TrCP. Rotavirus, a significant cause of childhood gastroenteritis, antagonizes NF-κB through the activity of its NSP1 protein, a putative E3 ubiquitin ligase that mediates β-TrCP turnover. Here, we show that NSP1 functions by mimicking the IκB phosphodegron recognized by β-TrCP. Nearly all human rotavirus strains conserve this motif at the NSP1 C terminus, and its removal disrupts NSP1 antagonist activity. This sequence conserves the biochemical properties of the IκB phosphodegron and can rescue antagonist activity when fused to an NSP1 protein otherwise inactive against β-TrCP. Other viral proteins also mimic IκB to disrupt NF-κB activation, indicating that this is an important immune evasion strategy.
IMPORTANCE
The transcription factor NF-κB, a central regulator of the host response to infection, is a frequent target of viral antagonism. Pathogen detection activates NF-κB by inducing the phosphorylation of an associated inhibitor protein (IκB), which targets IκB for degradation by the E3 ubiquitin ligase β-TrCP. Rotavirus, a significant cause of childhood gastroenteritis, antagonizes NF-κB through the activity of its NSP1 protein, a putative E3 ubiquitin ligase that mediates β-TrCP turnover. Here, we show that NSP1 functions by mimicking the IκB phosphodegron recognized by β-TrCP. Nearly all human rotavirus strains conserve this motif at the NSP1 C terminus, and its removal disrupts NSP1 antagonist activity. This sequence conserves the biochemical properties of the IκB phosphodegron and can rescue antagonist activity when fused to an NSP1 protein otherwise inactive against β-TrCP. Other viral proteins also mimic IκB to disrupt NF-κB activation, indicating that this is an important immune evasion strategy.
INTRODUCTION
The innate immune system is a nonspecific pathogen sensor that utilizes cellular pattern recognition receptors (PRRs)—notably, Toll-like receptor (TLR), retinoic acid-inducible gene 1 (RIG-I)-like receptor (RLR), and nucleotide-binding organization domain (NOD)-like (NLR) receptor—to detect conserved microbial signatures known as pathogen-associated molecular patterns (PAMPs) (1–4). PAMP recognition triggers signaling cascades that activate transcription factors, which in turn upregulate expression of proinflammatory molecules—chemokines, cytokines, and interferons (IFNs)—to activate host defenses and alert neighboring cells (1, 2). Viruses have evolved a wide range of strategies both to conceal their PAMPs and to directly antagonize host innate immune pathways (5). Many of these antagonist functions target nuclear factor kappa B (NF-κB) (6), one of the most broadly influential families of cellular transcription factors (7).
Binding of PRRs to their respective PAMPs and interaction of tumor necrosis factor receptor (TNFR) and interleukin-1 receptor (IL-1R) family members with their cognate cytokines can activate NF-κB (7). NF-κB regulates not only innate and adaptive immunity (8) but also cell differentiation, proliferation, and survival (7). In an uninfected cell, inhibitor of κB (IκB) proteins sequester NF-κB dimers in an inactive state in the cytoplasm (9). Signals from PRRs or other receptors activate the IκB kinase (IKK) complex, which phosphorylates IκBs on a pair of serine residues within a conserved degron motif (DSGΦxS [Φ, hydrophobic residue]) (10). SCFβ-TrCP, an E3 ubiquitin ligase of the Skp-Cullin-F-box (SCF) family, recognizes this phosphodegron through its F-box subunit, β-transducin repeat-containing protein (β-TrCP) (11, 12). In association with the E2 ubiquitin-conjugating enzyme UBCH5, SCFβ-TrCP polyubiquitinates phosphorylated IκB to trigger its degradation by the proteasome (13). This releases dimeric NF-κB to translocate to the nucleus, where it binds κB sites in the promoter and enhancer regions of target genes (14).
Rotavirus (RV), a member of the Reoviridae family of double-stranded RNA (dsRNA) viruses, is a significant cause of viral gastroenteritis in children and accounts for ~450,000 deaths annually (15). The icosahedral RV particle encapsidates an 11-segment genome that encodes six structural and five or six nonstructural proteins (16). RV replicates primarily in the villous tips of the small intestine (16), where it is sensed by the RLRs RIG-I and melanoma differentiation-associated protein 5 (MDA5) (17–19). RIG-I and MDA5 signal through mitochondrial antiviral signaling protein (MAVS) to activate NF-κB and another transcription factor, IFN regulatory factor 3 (IRF3) (20). Nuclear translocation of activated NF-κB and IRF3 increases expression of type I IFN, which induces an antiviral state by activating the JAK-STAT pathway and upregulating IFN-stimulated gene (ISG) expression (21, 22).
RV infection results in elevated levels of type I and II IFNs, which signal in an autocrine and paracrine manner to restrict viral replication and spread (23). However, like many other viruses (22), RV expresses a number of proteins with described IFN antagonist activity (23–25), the best characterized of which is nonstructural protein 1 (NSP1). NSP1 is the most variable RV protein and has evolved several strain-specific strategies to induce the proteasome-mediated degradation of one or more innate immune factors: β-TrCP, IRF3/5/7/9, MAVS, and/or TNF receptor-associated factor 2 (TRAF2) (26–30). Through these activities, NSP1 may act as a virulence determinant, by disrupting innate immune signaling pathways to establish a cellular environment permissible for RV replication and spread (23, 31). NSP1 contains a RING domain near its N terminus (32), which suggests that it acts as an E3 ubiquitin ligase (26, 27, 33), although direct evidence of target polyubiquitination is not yet available. NSP1 is most variable at its C terminus (32, 34, 35), a region known to be critical for association with and degradation of target proteins (36, 37). To date, 22 NSP1 genotypes, distinguished by 79% nucleotide identity, have been recognized by the Rotavirus Classification Working Group (38). However, few of these genetically distinct NSP1 proteins have been evaluated functionally, leaving the evolutionary basis for their dissimilarity largely unknown. Furthermore, the high degree of sequence variability has complicated the identification of motifs within NSP1 that are used to target host innate immune factors.
Through phylogenetic analysis of available NSP1 sequences from RV species A (RVA)—those responsible for the majority of human RV infection—we show that NSP1 proteins from nearly all human and porcine strains conserve a C-terminal DSGΦS sequence that mimics the IκB phosphodegron recognized by SCFβ-TrCP. Importantly, this sequence is present in OSU NSP1, the canonical mediator of β-TrCP degradation (26), and we demonstrate its critical role in the function of OSU NSP1 as an NF-κB antagonist. We also show that this motif mimics the biochemical properties of a cellular phosphodegron and that it can be transferred to a nonfunctional NSP1 to establish NF-κB antagonist activity. A number of unrelated viral proteins—Epstein-Barr virus (EBV) LMP1 (39), human immunodeficiency virus type 1 (HIV-1) Vpu (40), and vaccinia virus (VACV) A49 (41)—also mimic the phosphodegron to associate with β-TrCP and thereby inhibit NF-κB activation. Together, our observations support an increasingly recognized role for viral phosphodegron-like (PDL) motifs in targeting β-TrCP to block NF-κB activation during infection.
RESULTS
Human and porcine NSP1 proteins conserve a C-terminal phosphodegron-like motif.
NSP1 proteins differentially antagonize the host innate immune response (27). To examine the genetic basis for these activities, 556 RVA NSP1 open reading frames (ORFs) were obtained from GenBank for phylogenetic analysis. These NSP1 sequences formed four distinct clusters that comprised genotypes: (i) A1, A2, and A8; (ii) A3, A11, A12, and A13/A14; (iii) A5, A7, A9, and A6/A10; and (iv) A4/A16 (Fig. 1A). NSP1 genes from human and porcine strains were typically characterized by an A1, A2, or A8 genotype, all of which segregated together according to their phylogenetic history (Fig. 1B). Within this set, the NSP1 genes derived from human strains fell predominantly into the subgroups A1-ST3, A1-Wa, and A2. The NSP1 genes from the remaining subgroups (A1-OSU, A1-R479, and A8) derived from a wider range of host species: predominantly pigs, but occasionally humans, cows, or horses. To date, the RV strains collected from avian hosts carry NSP1 genes, exclusively of the A4/A16 genotypes, that appear only distantly related to those of mammalian strains. The NSP1 genes from the remaining two animal-like genotype clusters—A3, A11, A12, and A13/A14 (UK-like); and A5, A7, A9, and A6/A10 (SA11-4F-like)—derived from a broad range of host species, occasionally including humans. The A12 genotype, which has been observed infrequently and only in humans, appeared to form a cluster phylogenetically distinct from the other UK-like genotypes, but one that bootstrapping could not confidently distinguish. The genetic distance that separated the human/porcine-, animal-, and avian-like NSP1 genes suggested a high level of molecular divergence, which was confirmed by comparing the topology of the phylogenetic tree constructed from the translated sequences (data not shown).
FIG 1 .

Human and porcine RVA strains conserve a PDL motif within the NSP1 C terminus. (A and B) Maximum likelihood phylogenetic trees, assembled from 556 RVA NSP1 sequences collected globally since 1958. Branches are colored according to genotype. (A) Radial topology illustrates distances between the 16 RVA NSP1 genotypes shown. Genotype A1 resolves into four subgroups. The scale bar indicates phylogenetic distance (changes per site). (B) A circular cladogram shows phylogenetic relationships between NSP1 sequences isolated from different species. The inner ring is colored according to host species. The outer ring is colored according to the presence (dark red) or absence (white) of a C-terminal PDL motif (DSGxS). NSP1 sequences used in this study are indicated. (C, top) Arrangement of key NSP1 domains. The consensus sequence of the RING domain is shown. (Bottom) Alignment of NSP1 C termini into three major groups. The PDL motif and N-terminal lysine residue are shaded in grey. TD, targeting domain. (D) Alignment of viral PDL motifs and phosphodegrons from known targets of β-TrCP, shaded as in panel C.
OSU NSP1 (porcine A1) mediates the degradation of β-TrCP (26), and inspection of its sequence identified a C-terminal DSGIS peptide (residues 479 to 483) similar to the β-TrCP recognition motif (DSGΦxS) found in several human (IκBα, IκBβ, β-catenin, NRF2, YAP, and others) and viral (Vpu, LMP1, and A49) proteins (Fig. 1C and D) (42). The site of polyubiquitination on cellular targets of SCFβ-TrCP is a lysine 9 to 14 residues N-terminal to the phosphodegron (12), and a lysine (K465) is also present 14 amino acids N terminal to the DSGIS motif in OSU NSP1. Within the full set of RVA NSP1 genes, a phosphodegron-like motif (DSGΦS) and an N-terminal lysine residue were found in 98% of sequences from human and porcine strains (Fig. 1B and C). The C termini of the NSP1 genes derived from the two animal-like genotype clusters appeared to be conserved among themselves but shared little identity with those of the human/porcine-like strains, suggesting that these sequences have evolved independent strategies of immune antagonism. Studies with the simian RVA strains SA11-4F and SA11-5S (genotype A5)—isogenic except for a 17-residue, C-terminal truncation of SA11-5S NSP1 that arose following serial passage of SA11-4F in cell culture (43)—have demonstrated a critical role for the C terminus in NSP1 function. SA11-5S, unlike SA11-4F, fails to block the induction of IFN-β, and its NSP1 protein cannot mediate the degradation of IRFs (33, 37). The conservation of a C-terminal PDL motif across the majority of human- and porcine-derived RVA strains suggested its importance in NSP1 function, distinct from that described for the other classes of animal RVAs.
OSU NSP1 requires an intact C terminus to mediate its NF-κB antagonist activities.
To explore the role of the PDL motif in OSU NSP1 function, a 13-residue, C-terminal truncation mutant (OSU-ΔC13) was cloned for transient expression. This deletion removed the PDL motif and was analogous by sequence alignment to SA11-5S NSP1 (Fig. 2A). To evaluate the capacity of OSU-ΔC13 NSP1 to block NF-κB activation, HEK293T cells were cotransfected with NSP1 and an NF-κB luciferase reporter. OSU NSP1, but not its ΔC13 mutant, blocked NF-κB activation following stimulation with the cytokine TNF-α (Fig. 2B). OSU-C42A, a point mutant predicted to disrupt the N-terminal RING domain, and SA11-4F NSP1, which mediates the degradation of IRFs but not β-TrCP (27), also failed to block NF-κB activation. NF-κB was nearly twice as active in cells that expressed OSU-C42A versus OSU-ΔC13 NSP1, perhaps indicating that NSP1 mediates the degradation of β-TrCP and another cellular factor(s) to fully block NF-κB activation (Fig. 2B; see Fig. S1A in the supplemental material). Alternatively, disruption of the RING domain may have perturbed the overall structure of NSP1 and affected functions other than its putative E3 ubiquitin ligase activity.
FIG 2 .
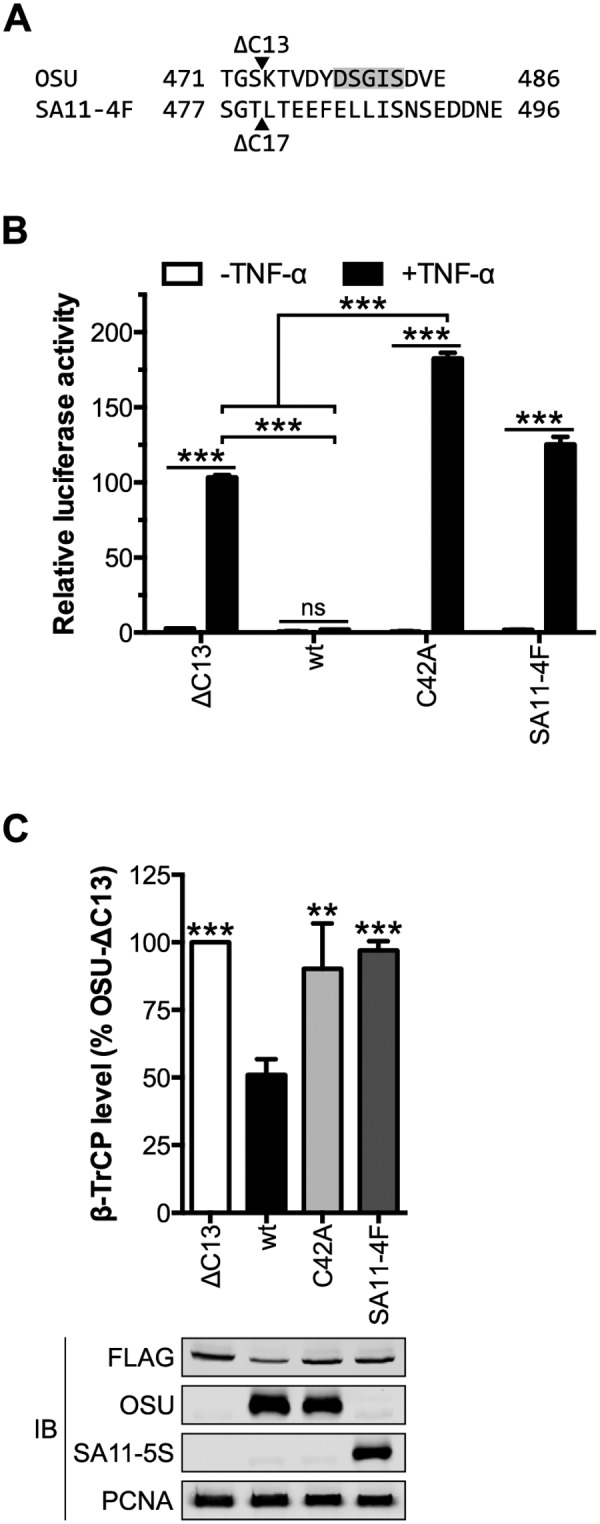
OSU NSP1 requires an intact C terminus to target β-TrCP for degradation. (A) Alignment of the C termini from OSU and SA11-4F NSP1 proteins. Arrowheads indicate the OSU-ΔC13 and SA11-4F-ΔC17 (SA11-5S) truncation mutants. The PDL motif is shaded in grey. (B) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with medium ± 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three experiments performed in triplicate) were analyzed by two-way ANOVA (pairwise as indicated) using Tukey’s multiple comparisons test. (C) HEK293T cells cotransfected with NSP1 and FLAG-β-TrCP were assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). The level of β-TrCP is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD from three independent transfections) were analyzed pairwise with OSU. ns, not significant (P > 0.05); **, P ≤ 0.01; ***, P ≤ 0.001. See also Fig. S1 to S4 in the supplemental material.
To evaluate the role of the PDL motif in NSP1-mediated turnover of β-TrCP, HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h posttransfection (p.t.) by quantitative anti-FLAG immunoblotting and, as shown in Fig. 2C, it was 50% lower in cells coexpressing OSU versus OSU-ΔC13 NSP1. This effect was relatively stable over a range of DNA concentrations but could be saturated (see Fig. S2 in the supplemental material). OSU-ΔC13 NSP1 elevated β-TrCP expression versus an empty vector control (see Fig. S1B in the supplemental material), despite being expressed at a slightly lower level than that of the wild type (wt) (see Fig. S3 in the supplemental material), and was therefore used as a normalizing control in degradation assays. OSU-ΔC13 NSP1 had no effect on β-TrCP level compared to SA11-4F or OSU-C42A NSP1, suggesting that a 13-residue truncation of OSU NSP1 is sufficient to abolish all β-TrCP degradation activity. Mutation of the lysine residue N-terminal to the PDL motif (K465R) did not affect the steady-state level of OSU NSP1, nor did it disrupt NSP1 function, likely indicating that β-TrCP was not responsible for polyubiquitinating or otherwise regulating NSP1 turnover (see Fig. S4 in the supplemental material). Likewise, degradation of HIV-1 Vpu occurs independently of β-TrCP activity (44), despite the presence of a lysine 14 residues N-terminal to the Vpu PDL motif (Fig. 1D). Together, these data suggest that OSU NSP1 requires an intact C terminus and N-terminal RING domain to block NF-κB activation and to mediate β-TrCP degradation.
Human and porcine NSP1 proteins conserve NF-κB antagonist activity.
Despite the preponderance of available human and human-like RVA NSP1 sequences (Fig. 1B), most functional studies have focused on NSP1 proteins from animal RVAs. To examine if the NF-κB antagonist and β-TrCP degradation activities of OSU NSP1 extended to related NSP1 proteins, particularly those from human RVA strains, a panel of OSU-like NSP1 sequences and their corresponding ΔC13 mutants (representing genotypes A1, A2, and A8) were cloned for transient expression. These sequences absolutely conserved a C-terminal DSGΦS motif (Φ, I/L/V) (Fig. 3A) and were well conserved over their entire length: they shared 81% identity among themselves but only 38% identity with SA11-4F (see Fig. S5 in the supplemental material). HEK293T cells were first transfected with NSP1 (wt or ΔC13) and an NF-κB luciferase reporter. As shown in Fig. 3B, these OSU-like NSP1 proteins invariably blocked NF-κB activation following TNF-α treatment (Fig. 3B). This block required the NSP1 C terminus, as the corresponding ΔC13 mutants, SA11-4F and SA11-5S NSP1, all failed to inhibit NF-κB activation. DS-1 and KU NSP1 proteins (human A2 and A1-Wa, respectively) both contained a nonconservative mutation at Y478 (histidine and aspartic acid, respectively), immediately N-terminal to the PDL motif (Fig. 3A) and in a position contacted by β-TrCP in other phosphodegrons (12); however, reversion to tyrosine had negligible effect on their NF-κB antagonist activities (data not shown).
FIG 3 .

Human and porcine RVA NSP1 proteins conserve NF-κB antagonist activity. (A) Alignment of the C termini from OSU, related RVA, and SA11-4F NSP1 proteins. The last four residues of SA11-4F NSP1 (DDNE) are not shown. Sites of variability in the consensus sequence (excluding SA11-4F) are shaded in gray, dots indicate positions of identity, and asterisks indicate the PDL motif. Hu, human; Po, porcine; Si, simian. (B) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three experiments performed in triplicate) were analyzed by two-way ANOVA (pairwise wt/ΔC NSP1) using Sidak’s multiple comparisons test. (C) HEK293T cells cotransfected with NSP1 and FLAG-β-TrCP were assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). For each NSP1, the level of β-TrCP is expressed as a percentage of β-TrCP in cells cotransfected with the corresponding ΔC mutant. Data (mean ± SD) are from three independent transfections. ***, P ≤ 0.001. See also Fig. S5 and S6 in the supplemental material.
HEK293T cells were then cotransfected with NSP1 (wt or ΔC13) and FLAG-β-TrCP, and the level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting. As shown in Fig. 3C, this group of OSU-like NSP1 proteins did not universally conserve β-TrCP degradation activity, despite all members having a C-terminal PDL motif and potently blocking NF-κB activation. Along with porcine OSU, NSP1 proteins from human D, DS-1, KU, P, and Wa RVA mediated the degradation of β-TrCP. Other human (IAL28, ST3, and WI61) and porcine (Gottfried and YM) RVA NSP1 proteins failed to induce β-TrCP degradation, indicating that this activity was not strictly conserved among NSP1 proteins from either host species. To confirm this finding, transfected cells were treated with the proteasome inhibitor MG132; the level of β-TrCP was rescued in cells that coexpressed OSU NSP1, whereas no effect was observed in cells that coexpressed Gottfried or SA11-4F NSP1 (see Fig. S6 in the supplemental material). Within the panel tested, no residues could be identified that predict the ability of a given NSP1 protein to induce β-TrCP turnover (data not shown). Together, these data suggest that NSP1-mediated degradation of β-TrCP may not be required for NSP1 to inhibit NF-κB activation. This finding is consistent with the action of bovine UK NSP1, which mediates the degradation of IRF3 in a host-cell-specific manner but which can also block IRF3 transcriptional activity without inducing IRF3 degradation (45). Furthermore, EBV LMP1 (39), HIV-1 Vpu (40), and VACV A49 (41) all interact with β-TrCP to block NF-κB activation, without inducing β-TrCP turnover.
The serine residues of the PDL motif are critical for OSU NSP1 function.
Cellular degrons must be phosphorylated on a pair of conserved serine residues to be recognized by β-TrCP (12). All four viral PDL motifs contain this serine pair (Fig. 1D), although only HIV-1 Vpu has been confirmed to undergo phosphorylation, a modification required for its association with β-TrCP (46). The serine residues of the OSU NSP1 PDL motif lie within predicted casein kinase I (CK) I and II sites (Fig. 4A) (47–49). Mutation of the CKII (E486Q), but not CKI (D477N), priming residue almost completely abolished NF-κB antagonism by OSU NSP1 (Fig. 4B). This is consistent with OSU NSP1 being phosphorylated by CKII, which also phosphorylates the Vpu PDL motif (50). Mutation of the serine residues in tandem—to nonphosphorylatable alanine (OSU-AA), phosphomimetic aspartic (OSU-DD) or glutamic (OSU-EE) acid, or phosphorylatable threonine (OSU-TT)—disrupted NF-κB antagonism by OSU NSP1 (Fig. 4C) and indicated a strict requirement for serine (likely, phosphoserine) at these positions.
FIG 4 .
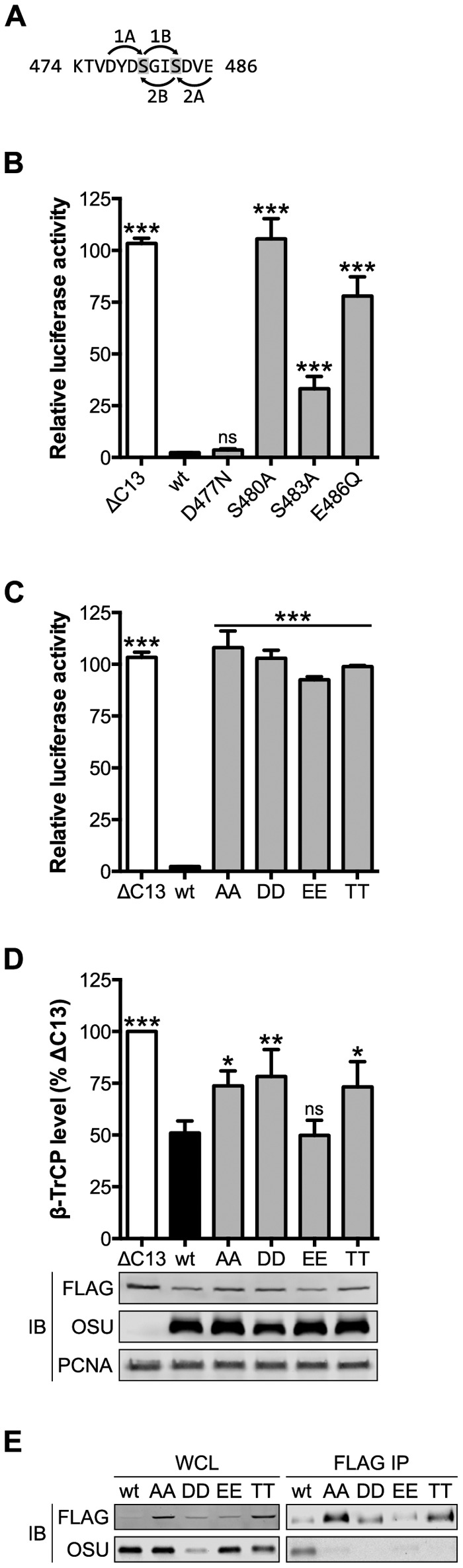
The serine residues of the PDL motif are required for NSP1 function. (A) Model for phosphorylation of the PDL motif of OSU NSP1 by CKI or CKII. Arrows point from the priming residue to the site of serine phosphorylation (shaded). 1A and -B, CKI; 2A and -B, CKII. (B and C) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three experiments performed in triplicate) were analyzed pairwise with OSU. (D) HEK293T cells cotransfected with NSP1 and FLAG-β-TrCP were assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). The level of β-TrCP is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD from three independent transfections) were analyzed pairwise with OSU. (E) HEK293T cells cotransfected with NSP1 and β-TrCP were immunoprecipitated (IP) with anti-FLAG resin. Immunoblots represent 3% clarified whole-cell lysate (WCL) and 10% eluted protein. ns, not significant (P > 0.05); *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
HEK293T cells were then cotransfected with NSP1 and FLAG-β-TrCP, and the level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting. As shown in Fig. 4D, OSU and OSU-EE NSP1 were functionally equivalent in this assay, while OSU-AA, -DD, and -TT NSP1 proteins also mediated a modest degree of β-TrCP degradation. This suggested that transient interaction of the NSP1 RING domain with the E2 subunit of the SCFβ-TrCP complex may have been sufficient to stabilize a likely weak interaction between the mutated PDL motif and the β-TrCP subunit. FLAG immunoprecipitation of β-TrCP:NSP1 complexes from cotransfected HEK293T cells demonstrated that the serine residues were critical for this interaction (Fig. 4E). Together, these data support a critical role for the serine residues within the NSP1 PDL motif and suggest that NSP1 function depends on phosphorylation of these residues.
OSU NSP1 targets the phosphodegron-binding pocket of β-TrCP.
β-TrCP is a member of the F-box protein family that provides specificity to SCF-type E3 ubiquitin ligases (51). These proteins contain an N-terminal F-box domain that interfaces with the other subunits of the SCF complex and a C-terminal WD40 domain that mediates recognition of cellular targets. To identify the domain recognized by OSU NSP1, the F-box (β-TrCP-N287) and WD40 (β-TrCP-288C) domains were separately cloned for transient expression (Fig. 5A). HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP, and the level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting. As shown in Fig. 5B, NSP1 mediated the degradation of β-TrCP-288C, although less efficiently than it did full-length β-TrCP. This suggested preferential recognition of the full-length protein, perhaps in the context of the SCFβ-TrCP complex. The F-box domain was dispensable for degradation, as the level of β-TrCP-N287 was slightly elevated in the presence of OSU versus OSU-ΔC13 NSP1. Despite this, OSU NSP1 coimmunoprecipitated with both fragments of β-TrCP, albeit less efficiently than with full-length β-TrCP (Fig. 5E). This supports a two-pronged interaction between NSP1 and β-TrCP:RING:E2, bridged by β-TrCP-N287, and PDL motif:WD40, mediated by β-TrCP-288C.
FIG 5 .

NSP1 targets the phosphodegron-binding pocket of β-TrCP. (A) Diagram of β-TrCP domain organization and truncations. (B) HEK293T cells cotransfected with OSU NSP1 and FLAG-β-TrCP were assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). The level of β-TrCP is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD) are from three independent transfections. (C) Structure of the β-TrCP WD40 domain (green) and associated β-catenin phosphodegron peptide (yellow) (12). R510 is colored purple. (D) HEK293T cells cotransfected with OSU NSP1 and FLAG-β-TrCP were assayed 24 h p.t. by quantitative immunoblotting (normalized to PCNA). The level of β-TrCP is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD) are from three independent transfections. (E) HEK293T cells cotransfected with NSP1 and β-TrCP were immunoprecipitated (IP) with anti-FLAG resin. Immunoblots represent 3% clarified whole-cell lysate (WCL) and 10% eluted protein.
Phosphodegrons bind the WD40 domain of β-TrCP along its top face, by dipping into the central channel of this β-propeller structure (12). Aspartic acid, which almost invariably marks the phosphodegron start, forms the most extensive network of contacts, in particular with residues R510 and Y524 (β-TrCP isoform 1 numbering) (Fig. 5C). Alanine substitution at either position blocks polyubiquitination of IκBα (12), and it was predicted that such a mutation would also protect β-TrCP from NSP1-mediated degradation. HEK293T cells were cotransfected with OSU NSP1 and an R510A mutant of FLAG-β-TrCP or FLAG-β-TrCP-288C, and the level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting. As shown in Fig. 5D, OSU NSP1 could not mediate the degradation of either β-TrCP-R510A or β-TrCP-288C-R510A, indicating that NSP1 targets the binding pocket used by β-TrCP to interact with cellular phosphodegrons. FLAG-β-TrCP-R510A also failed to coimmunoprecipitate OSU NSP1 (Fig. 5E). Together, these data demonstrate that β-TrCP recognizes cellular phosphodegrons and the NSP1 PDL motif in a similar manner.
The NSP1 PDL motif is a transferrable functional element.
Despite significant sequence variation, all full-length RVA NSP1 proteins are thought to utilize an N-terminal RING domain to interact with a cellular E2 ubiquitin-conjugating enzyme and a C-terminal sequence element to provide binding specificity for a host innate immune target (Fig. 1C) (52). Formation of such a complex is predicted to induce polyubiquitination of the target, followed by its degradation in the proteasome. Sequence variability often masks structural conservation (53), and it was hypothesized that NSP1 conserves its architecture across strains, with one of three C-terminal motifs mediating target specificity (Fig. 1C). To test this possibility, the C terminus of simian SA11-4F NSP1, which specifically targets IRFs (27), was substituted for with that of OSU NSP1. A (D/E)-Φ-(D/E) motif (Φ, aromatic residue) conserved in both proteins was chosen as the site of substitution, in order to keep intact the putative CK sites in OSU NSP1 (Fig. 4A and 6A).
FIG 6 .
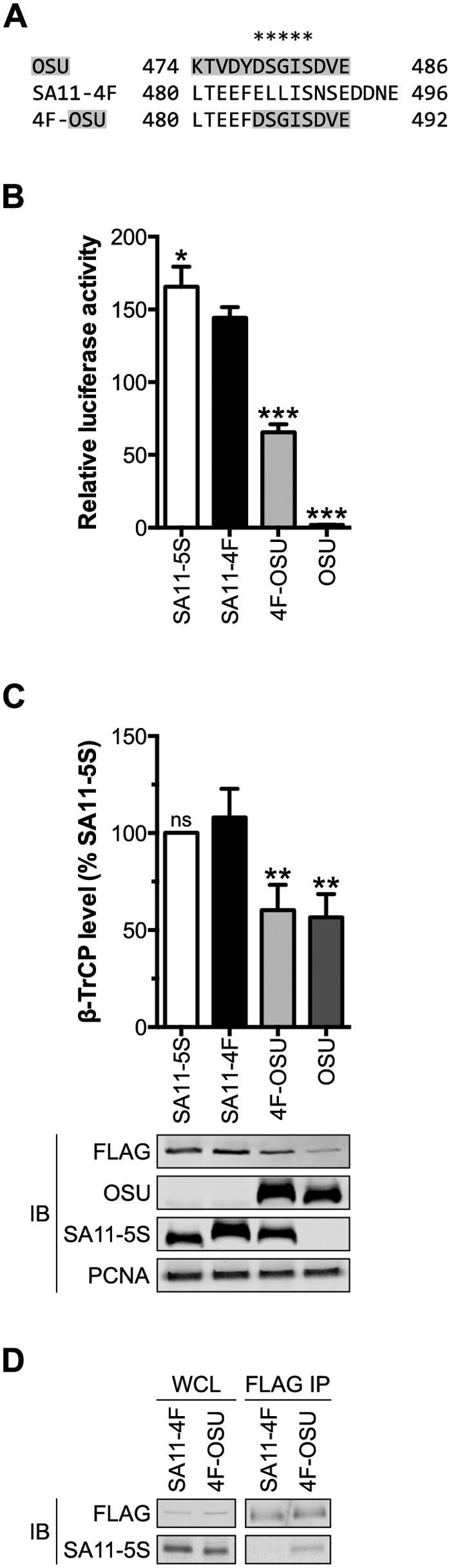
The PDL motif is the minimum sequence required by NSP1 to target β-TrCP. (A) Alignment of the C termini from OSU, SA11-4F, and 4F-OSU NSP1. Residues from OSU NSP1 are shaded in gray, and asterisks indicate the PDL motif. (B) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three experiments performed in triplicate) were analyzed pairwise with SA11-4F. (C) HEK293T cells cotransfected with NSP1 and FLAG-β-TrCP were assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). The level of β-TrCP is expressed as a percentage of β-TrCP in SA11-5S-transfected cells. Data (mean ± SD from three independent transfections) were analyzed pairwise with SA11-4F. (D) HEK293T cells cotransfected with NSP1 and β-TrCP were immunoprecipitated (IP) with anti-FLAG resin. Immunoblots represent 3% clarified whole-cell lysate (WCL) and 10% eluted protein. ns, not significant (P > 0.05); *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
This chimeric NSP1, dubbed 4F-OSU, was cotransfected into HEK293T cells with an NF-κB luciferase reporter and, as shown in Fig. 6B, mediated a 2-fold block in NF-κB activation versus SA11-5S and SA11-4F NSP1. 4F-OSU lacked the potency of OSU NSP1, indicating that regions outside the C-terminal PDL motif likely contribute to NF-κB antagonism by OSU NSP1 and are not fully captured by SA11-4F NSP1. HEK293T cells were then cotransfected with NSP1 and FLAG-β-TrCP, and the level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (Fig. 6C). Unlike SA11-4F NSP1, the 4F-OSU chimera was able to induce β-TrCP turnover comparable to that of OSU NSP1. This finding suggests that the PDL motif, when paired with an intact RING domain, is sufficient to target SCFβ-TrCP and that SA11-4F and OSU NSP1 proteins conserve an architecture and mechanism similar enough to permit this transfer of functionality. This was confirmed by a coimmunoprecipitation assay, in which 4F-OSU, but not SA11-4F, NSP1 associated with FLAG-β-TrCP (Fig. 6D). Together, these data suggest that the C-terminal PDL motif is a transferrable functional element, likely due to structural conservation across NSP1 proteins, but that other sequence elements and/or functions of OSU and OSU-like NSP1 proteins contribute to NF-κB antagonism.
DISCUSSION
β-TrCP was first identified by its association with HIV-1 Vpu in mediating endoplasmic reticulum (ER)-associated degradation of CD4 during viral infection (46). Subsequent studies demonstrated that Vpu (40)—along with EBV LMP1 (39), RV NSP1 (26), and VACV A49 (41)—could block NF-κB activation by interacting with β-TrCP. Vpu, LMP1, and A49 were all shown to act by molecular mimicry, in which they associated with β-TrCP through a motif similar to the IκB phosphodegron. Here, we have shown that NSP1 also utilizes such a sequence, which we generically refer to as a phosphodegron-like (PDL) motif, to target β-TrCP. Within this group, only NSP1 has putative E3 ubiquitin ligase activity and can mediate β-TrCP turnover. Conversely, Vpu uniquely hijacks the E3 ubiquitin ligase activity of SCFβ-TrCP to target other host proteins for degradation, namely, CD4 and tetherin (54). The use of a PDL motif-containing protein by three disparate groups of viruses—dsRNA, dsDNA, and retroviruses—to target β-TrCP emphasizes the critical role of the NF-κB pathway in pathogen surveillance and clearance and the need for viruses to circumvent its activation (Fig. 7).
FIG 7 .

Diverse functions of viral PDL motif-containing proteins. The models depict how RV NSP1, EBV LMP1, HIV-1 Vpu, and VACV A49 disrupt β-TrCP activity. These viral proteins use a PDL motif to (i) induce the turnover of β-TrCP (degradation), (ii) block β-TrCP from recognizing cellular phosphodegrons (interaction), or (iii) induce the turnover of cellular proteins by bridging β-TrCP to these targets (adaptor). Yellow, serine residues of the PDL motif; orange, ubiquitin.
Removal of the PDL motif by C-terminal truncation is sufficient to prevent OSU and related NSP1 proteins from mediating β-TrCP turnover and inhibiting NF-κB activation (Fig. 2 and 3). Similar regions of SA11-4F and SA11-30-19 NSP1 are critical for IRF degradation and to block IFN induction (37), suggesting that the requirement of an intact C terminus for proper NSP1 function is independent of virus strain or host protein target(s). Importantly, sequence analysis has demonstrated that, excluding NSP1 from avian strains, nearly all NSP1 C termini cluster into one of three consensus sequences: the OSU-like [DE]YDSGΦSDΦE, UK-like EYDLELSDΦE, and SA11-4F-like E[FY][DE]LLΦS[DNS]S[DE][DS][DE][DEN][DE], where Φ is a hydrophobic residue and brackets surround possible residues at a given position (Fig. 1) (data not shown). Mutagenesis of these consensus sequences is likely to identify key residues required by NSP1 to target IRFs—frequently targeted by other viral immune antagonists (22)—or to mediate dual specificity for β-TrCP and IRFs, a property of NCDV NSP1 (bovine A3, UK-like) (26, 27).
The NSP1 PDL motif (DSGΦS) closely resembles the canonical phosphodegron (DSGΦxS), which suggested it would interact with β-TrCP in a similar manner. Indeed, the only difference between these motifs is the length of the GΦx spacer, which can vary from 2 to 5 residues (42); a longer spacer is likely accommodated by deeper insertion into the phosphodegron-binding pocket of β-TrCP (55). A hallmark feature of phosphodegrons is a pair of phosphorylated serine residues, with the first being particularly important for recognition by β-TrCP (Fig. 4B) (12). The serine residues of NSP1 are found within putative CKII sites (Fig. 4A and B), and their tandem mutation (to A, D, E, or T) prevents NSP1 from antagonizing NF-κB or interacting with β-TrCP (Fig. 4B and E). Interestingly, these mutants—particularly OSU-EE NSP1—retain some β-TrCP degradation activity (Fig. 4D), indicating that an electronegative sequence might sufficiently stabilize the likely transient interaction required by NSP1 to polyubiqutinate β-TrCP. Indeed, Ser-to-Glu substitution of the VACV A49 PDL motif strengthens A49 interaction with β-TrCP and its NF-κB antagonist activity (41). However, since β-TrCP degradation does not appear necessary for NSP1 to antagonize NF-κB (Fig. 3), a stable interaction (one dependent on NSP1 serine phosphorylation) likely is necessary to block NF-κB activation, by preventing β-TrCP from associating with phosphorylated IκB.
The interaction of NSP1 with β-TrCP appears to be two-pronged, since both the N-terminal F-box and C-terminal WD40 domain of β-TrCP can coimmunoprecipitate NSP1 (Fig. 5E). We predict that interaction of NSP1 with β-TrCP-N287, which assembles into the larger SCF complex (data not shown), is mediated by contacts between the NSP1 RING domain and SCF-associated E2 ubiquitin-conjugating enzyme, whereas the PDL motif makes direct contacts with the WD40 domain of β-TrCP-288C. These data also suggest that NSP1 preferentially recognizes SCFβ-TrCP over free β-TrCP, which may be kinetically favorable with regard to recruitment of an E2 ubiquitin-conjugating enzyme. Addressing these possibilities will likely elucidate not only the E2 enzyme(s) used by NSP1 but also how NSP1 recognizes different populations of β-TrCP in the cell.
Evolutionary pressures have driven human and animal RVA NSP1 proteins, respectively, to principally block the host antiviral response by targeting β-TrCP and IRFs for degradation. The basis for this divergence in strategy remains unclear. Continued sequencing and functional studies of NSP1—focused, respectively, on animal and human RVA strains—will be required to monitor the progress of the RV-host evolutionary arms race and address this key question. Despite significant sequence variability between the two groups, their shared putative E3 ubiquitin ligase activity—mediated by an N-terminal RING domain and a C-terminal targeting motif—enabled us to substitute the C terminus of an animal NSP1 (SA11-4F) with that of OSU NSP1, thereby generating a chimera (4F-OSU) that could mediate the degradation of β-TrCP and block NF-κB activation (Fig. 6). One implication of this finding is that human and animal RVA NSP1 proteins may share a three-dimensional architecture, despite their lack of sequence similarity. Additionally, the differences in activity observed between OSU and 4F-OSU NSP1 likely relate to functions or sequence elements outside the C terminus.
The ability of NSP1 to target β-TrCP is genetically economical—a five-residue DSGΦS motif is sufficient to mediate interaction (Fig. 6D)—with potentially far-reaching functional implications. Aside from the IκB family, SCFβ-TrCP mediates the degradation of other proteins involved in NF-κB activation—IFN-α receptor 1 (IFNAR1) (56) and IL-1R-associated kinase 1 (IRAK1) (57)—and of a number of cell cycle and proapoptotic regulatory factors (42). NSP1-mediated degradation of β-TrCP is expected to arrest the turnover of these proteins, which should have manifold implications for RV replication. Understanding if and how RV benefits from these effects will not only inform the relationship between this virus and the cell but may also reveal the pleiotropic effects other PDL motif-containing viral proteins have during infection. β-TrCP deregulation has been implicated in a number of cancers (42), and the unique ability of NSP1 to mediate β-TrCP turnover suggests a potentially therapeutic role for NSP1 in the treatment of such diseases.
In closing, we have shown that rotavirus NSP1 can mimic the phosphodegron recognized by β-TrCP, in order to mediate the degradation of β-TrCP and to block NF-κB activation. This phosphodegron-like motif is conserved among human and porcine RVA NSP1 proteins, and its biochemical properties mimic those of the cellular phosphodegron. Expression of a PDL motif-containing protein to block NF-κB activation is a strategy observed with increasing frequency among human viruses and one that has potentially significant implications. The influence of NF-κB reaches beyond modulation of the immune response and includes regulation of cell proliferation, differentiation, and survival. Teasing apart the multilayered effects of NSP1-mediated β-TrCP degradation and NF-κB inactivation will provide critical insights into virus-host interactions and inform the design of vaccines and antiviral compounds.
MATERIALS AND METHODS
Cells and antibodies.
HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (Quality Biological) supplemented with 10% fetal bovine serum (Gibco). Rabbit polyclonal antisera against simian SA11-5S and porcine OSU NSP1 (27) and mouse monoclonal anti-FLAG M2 antibody (F1804; Sigma) were used at a 1:5,000 dilution. Rabbit polyclonal antibody against PCNA (sc-7907; Santa Cruz Biotechnology) was used at a 1:2,000 dilution. IRDye 800CW-conjugated anti-rabbit IgG (926-32211; LI-COR Biosciences) and IRDye 680-conjugated anti-mouse IgG (926-32220; LI-COR Biosciences) goat polyclonal antibodies were used at a 1:20,000 dilution.
Plasmids and sequences.
The vector pCAGGS (58) (a gift from K. W. Boehme) was modified by standard DNA techniques to introduce a T7 promoter and ligation-independent cloning (LIC) site downstream of the CAG promoter (pLIC6). This vector was further modified to introduce an N-terminal FLAG tag (MADYKDDDDKSG) upstream of the LIC site (pLIC6F). To generate LIC sticky ends, pLIC6 or pLIC6F was digested with NsiI and then treated with T4 DNA polymerase in the presence of dTTP. Full-length or truncated β-TrCP or NSP1 ORFs were PCR amplified from previously described vectors (27) with primers having LIC-compatible ends. Mutations were generated by outward PCR of these original plasmids with suitable primers; ORFs were then PCR amplified with LIC-compatible primers. PCR fragments were purified and treated with T4 DNA polymerase in the presence of dATP to generate LIC sticky ends. Treated insert and pLIC6 or pLIC6F were mixed, incubated at 25°C for at least 20 min, and transformed into Escherichia coli strain TOP10. All constructs were verified by sequencing. Endotoxin-free plasmid DNA was prepared using an EndoFree plasmid kit (Qiagen) and diluted to 250 ng/µl in endotoxin-free Tris-EDTA (TE) buffer before use. The β-TrCP (isoform 1) nucleotide sequence is available in GenBank (accession no. NM_033637). Accession numbers for the NSP1 nucleotide sequences used in functional assays are listed in Table S1 in the supplemental material.
Quantitative immunoblotting.
Transfection complexes (94 µl Opti-MEM [Gibco], 500 ng pLIC6-NSP1, 500 ng pLIC6F-β-TrCP, 2 µl Lipofectamine 2000 [Life Technologies]) were prepared according to the manufacturer’s protocols and transferred to wells of a 12-well plate, unless otherwise specified. Nine hundred microliters of a HEK293T cell suspension (1.3 × 106 cells/ml) was added to each well. Cells were collected 24 h p.t., washed once with phosphate-buffered saline (PBS [pH 7.4]), and lysed in a 100-µl solution of 1× radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate), 1× NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Life Technologies), 25 mM dithiothreitol, 1× phosphatase inhibitor cocktail (1 mM each sodium fluoride, sodium orthovanadate, β-glycerophosphate, and sodium pyrophosphate), and 1× Complete EDTA-free protease inhibitor cocktail (Roche). Lysates were sonicated briefly, electrophoresed on 12% bis-Tris gels, and transferred onto nitrocellulose membranes for immunoblotting. Blots were imaged on an Odyssey Classic and quantified using Image Studio Lite v3.1.4 (LI-COR Biosciences). Bands of interest were normalized to the loading control PCNA. Data are presented as means ± standard deviations (SD) from three independent transfections.
NF-κB reporter assay.
Transfection complexes (9.25 µl Opti-MEM, 92 ng pLIC6-NSP1, 30 ng pGL4.32[luc2P/NF-κB-RE/Hygro] [Promega], 3 ng pGL4.74[hRluc/TK] [Promega], 0.25 µl Lipofectamine 2000) were prepared in triplicate according to the manufacturer’s protocols and transferred to wells of a 96-well plate. Ninety microliters of a HEK293T cell suspension (1.1 × 106 cells/ml) was added to each well. At 24 h p.t., cells were stimulated for 4 h with medium ± 25 ng/ml TNF-α (PeproTech). Following stimulation, cells were lysed in 20 µl 1× passive lysis buffer (Promega), and luciferase activity was measured with the Dual-Glo luciferase assay system (Promega), according to the manufacturer’s protocols. Relative luciferase activity (NF-κB stimulation) was calculated by normalizing firefly to Renilla luciferase activity. The data presented (mean ± SD) are from one of three independent experiments.
Immunoprecipitation.
Transfection complexes (1.45 ml Opti-MEM, 7.25 µg pLIC6-NSP1, 7.25 µg pLIC6F-β-TrCP, 29 µl Lipofectamine 2000) were prepared according to the manufacturer’s protocols and transferred to 10-cm dishes of HEK293T cells. At 40 h p.t., cells were treated for 8 h with 20 µM MG132 (Cayman Chemical). Cells were collected, washed twice with PBS, and resuspended in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1× phosphatase inhibitor cocktail). Cell lysates were clarified by centrifugation and incubated overnight with anti-FLAG M2 magnetic beads (M8823; Sigma). Beads were washed three times with wash buffer (50 mM Tris [pH 7.4], 150 mM NaCl), and bound protein was eluted with 0.1 M glycine (pH 3.0).
Phylogenetic analysis.
All available RVA NSP1 ORFs (833 in total) were downloaded from GenBank on 1 February 2013, and partial sequences were discarded. The GenBank to FASTA tool (http://rocaplab.ocean.washington.edu/tools/genbank_to_fasta/) was used to extract the name, country, host species, date of isolation, and sequence for each strain; missing fields were entered manually. Sequences were omitted whose annotation lacked sufficient detail, which left 556 ORFs with lengths ranging from 1,459 to 1,732 bases. Nucleotide sequences were aligned using the ClustalW plugin of Geneious Pro v5.6.5 (Biomatters). To compare evolutionary history, the alignment was translated into the predicted amino acid sequence (length range, 486 to 557 residues) and aligned using a BLOSUM cost matrix. Linear motifs were annotated using the EMBOSS fuzzpro tool (59). MEGA v5.0.5 (60) was used to select the best available substitution model for each alignment, according to the Bayesian information criterion (BIC) ranking. Phylogenetic trees were constructed using the RAxML BlackBox server (61). One thousand rapid bootstrap replicates were performed, with PO-13 (pigeon A4) NSP1 used as the outgroup. Initial trees were visualized using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/). The NSP1 genotype representing each branch was identified based on clustering and confirmed using the RotaC server (62). The A1 branch was further defined into four clearly resolved subgroups, which were supported by bootstrap values greater than 75. Circular cladograms were generated using the EvolView server (63) and were colored according to host species and conservation of a DSGxS motif.
Statistical analysis.
Prism v6.0f (GraphPad Software) was used to perform all statistical analyses. Unless otherwise stated, data were analyzed with one-way analysis of variance (ANOVA) using Dunnett’s multiple comparisons test.
SUPPLEMENTAL MATERIAL
OSU NSP1 function compared to vector control. (A) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with medium ± 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three independent experiments performed in triplicate) were analyzed by two-way ANOVA (pairwise as indicated) using Tukey’s multiple comparisons test. EV, empty vector (pLIC6). (B) HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA) and is expressed as a percentage of β-TrCP in EV-transfected cells. Data (mean ± SD from three independent transfections) were analyzed by one-way ANOVA (pairwise as indicated) using Tukey’s multiple comparisons test. Download
Effect of plasmid ratio on OSU NSP1-mediated degradation of β-TrCP. (A and B) HEK293T cells were transfected as described in Materials and Methods, with the following transfection complex: 91 µl Opti-MEM, 1,500 ng DNA, 3 µl Lipofectamine 2000. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). Data (mean ± SD from three independent transfections) were analyzed by two-way ANOVA (pairwise as indicated) using Sidak’s multiple comparisons test. (A) The DNA mixture contains 500 ng OSU NSP1, 0 to 1,000 ng FLAG-β-TrCP (in increments of 250 ng), and empty vector (pLIC6) to 1,500 ng. The level of β-TrCP is expressed as a percentage of β-TrCP in cells transfected with 500 ng FLAG-β-TrCP. (B) The DNA mixture contains 0 to 1,000 ng OSU NSP1 (in increments of 250 ng), 500 ng FLAG-β-TrCP, and empty vector (pLIC6) to 1,500 ng. The level of β-TrCP is expressed as a percentage of β-TrCP in cells transfected with 500 ng OSU-ΔC13 NSP1. Download
Expression and function of FLAG-tagged OSU NSP1. (A) HEK293T cells were cotransfected with FLAG-NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three independent experiments performed in triplicate) were analyzed with an unpaired t test (pairwise with OSU). (B) HEK293T cells were cotransfected with FLAG-NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB)(normalized to PCNA) and is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD from three independent transfections) were analyzed with an unpaired t test (pairwise with OSU). Download
The K465R mutation does not affect OSU NSP1 turnover or function. (A) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three independent experiments performed in triplicate) were analyzed pairwise with OSU. (B) HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA) and is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD from three independent transfections) were analyzed pairwise with OSU. Download
Alignment of human and porcine RVA NSP1. Amino acid sequences from the NSP1 genes examined in Fig. 3 were aligned using the T-Coffee server (P. Di Tommaso, S. Moretti, I. Xenarios, M. Orobitg, A. Montanyola, et al., Nucleic Acids Res 39:W13–W17, 2011, doi:10.1093/nar/gkr245; C. Notredame, D. G. Higgins, and J. Heringa, J. Mol. Biol. 302:205-217, 2000, doi:10.1006/jmbi.2000.4042). The image was generated using the ESPript 3.0 server (P. Gouet, X. Robert, and E. Courcelle, Nucleic Acids Res 31:3320–3323, 2003, doi:10.1093/nar/gkg556). Yellow, 100% sequence identity; blue, ≥70% sequence similarity; red, <70% sequence similarity or dissimilar residue; arrowhead, Zn2+-coordinating residue within the N-terminal RING domain; circle, K465; star, DSGΦS motif residue; dot, sequence gap; Hu, human; Po, porcine; Si, simian. Download
Role of the proteasome in NSP1 activity. HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. At 8 h p.t., cells were treated for 16 h with dimethyl sulfoxide (DMSO) or 10 µM MG132. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA) and is expressed as a percentage of β-TrCP in ΔC-transfected cells. Data (mean ± SD from three independent transfections) were analyzed by two-way ANOVA (pairwise as indicated) using Sidak’s multiple comparisons test. Download
GenBank accession numbers for the NSP1 genes used in this study.
ACKNOWLEDGMENTS
This study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
We thank Elif Eren, Xiang Liu, and Kristen Ogden for helpful discussions and critical review of the manuscript.
Footnotes
Citation Morelli M, Dennis AF, Patton JT. 2015. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-κB activation by using molecular mimicry to target β-TrCP. mBio 6(1):e02490-14. doi:10.1128/mBio.02490-14.
REFERENCES
- 1.O’Neill LA, Bowie AG. 2010. Sensing and signaling in antiviral innate immunity. Curr Biol 20:R328–R333. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Wilkins C, Gale M Jr.. 2010. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol 22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoneyama M, Fujita T. 2010. Recognition of viral nucleic acids in innate immunity. Rev Med Virol 20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 5.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Negrate G. 2012. Viral interference with innate immunity by preventing NF-κB activity. Cell Microbiol 14:168–181. doi: 10.1111/j.1462-5822.2011.01720.x. [DOI] [PubMed] [Google Scholar]
- 7.Hayden MS, Ghosh S. 2012. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. 2011. NF-κB in immunobiology. Cell Res 21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinz M, Arslan SÇ, Scheidereit C. 2012. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol Rev 246:59–76. doi: 10.1111/j.1600-065X.2012.01102.x. [DOI] [PubMed] [Google Scholar]
- 10.Kanarek N, Ben-Neriah Y. 2012. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol Rev 246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 11.Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y. 1998. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature 396:590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 12.Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. 2003. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell 11:1445–1456. [DOI] [PubMed] [Google Scholar]
- 13.Spencer E, Jiang J, Chen ZJ. 1999. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev 13:284–294. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann A, Natoli G, Ghosh G. 2006. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 15.Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD, WHO-Coordinated Global Rotavirus Surveillance Network . 2012. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect Dis 12:136–141. [DOI] [PubMed] [Google Scholar]
- 16.Desselberger U. 2014. Rotaviruses. Virus Res 190:75–96. doi: 10.1016/j.virusres.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 17.Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. 2011. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol 186:1618–1626. doi: 10.4049/jimmunol.1002862. [DOI] [PubMed] [Google Scholar]
- 18.Sen A, Pruijssers AJ, Dermody TS, García-Sastre A, Greenberg HB. 2011. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J Virol 85:3717–3732. doi: 10.1128/JVI.02634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uzri D, Greenberg HB. 2013. Characterization of rotavirus RNAs that activate innate immune signaling through the RIG-I-like receptors. PLoS One 8:e69825. doi: 10.1371/journal.pone.0069825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirata Y, Broquet AH, Menchén L, Kagnoff MF. 2007. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J Immunol 179:5425–5432. doi: 10.4049/jimmunol.179.8.5425. [DOI] [PubMed] [Google Scholar]
- 21.Samuel CE. 2001. Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor KE, Mossman KL. 2013. Recent advances in understanding viral evasion of type I interferon. Immunology 138:190–197. doi: 10.1111/imm.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold MM, Sen A, Greenberg HB, Patton JT. 2013. The battle between rotavirus and its host for control of the interferon signaling pathway. PLoS Pathog 9:e1003064. doi: 10.1371/journal.ppat.1003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langland JO, Pettiford S, Jiang B, Jacobs BL. 1994. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon-induced protein kinase PKR. J Virol 68:3821–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci U S A 110:13114–13119. doi: 10.1073/pnas.1306917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graff JW, Ettayebi K, Hardy ME. 2009. Rotavirus NSP1 inhibits NFκB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5:e1000280. doi: 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold MM, Patton JT. 2011. Diversity of interferon antagonist activities mediated by NSP1 proteins of different rotavirus strains. J Virol 85:1970–1979. doi: 10.1128/JVI.01801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnold MM, Barro M, Patton JT. 2013. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol 87:9813–9821. doi: 10.1128/JVI.01146-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bagchi P, Bhowmick R, Nandi S, Kant Nayak M, Chawla-Sarkar M. 2013. Rotavirus NSP1 inhibits interferon induced non-canonical NFκB activation by interacting with TNF receptor associated factor 2. Virology 444:41–44. doi: 10.1016/j.virol.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Nandi S, Chanda S, Bagchi P, Nayak MK, Bhowmick R, Chawla-Sarkar M. 2014. MAVS protein is attenuated by rotavirus nonstructural protein 1. PLoS One 9:e92126. doi: 10.1371/journal.pone.0092126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angel J, Franco MA, Greenberg HB. 2012. Rotavirus immune responses and correlates of protection. Curr Opin Virol 2:419–425. doi: 10.1016/j.coviro.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hua J, Mansell EA, Patton JT. 1993. Comparative analysis of the rotavirus NS53 gene: conservation of basic and cysteine-rich regions in the protein and possible stem-loop structures in the RNA. Virology 196:372–378. doi: 10.1006/viro.1993.1492. [DOI] [PubMed] [Google Scholar]
- 33.Barro M, Patton JT. 2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol 81:4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunn SJ, Cross TL, Greenberg HB. 1994. Comparison of the rotavirus nonstructural protein NSP1 (NS53) from different species by sequence analysis and Northern blot hybridization. Virology 203:178–183. doi: 10.1006/viro.1994.1471. [DOI] [PubMed] [Google Scholar]
- 35.Xu L, Tian Y, Tarlow O, Harbour D, McCrae MA. 1994. Molecular biology of rotaviruses. IX. Conservation and divergence in genome segment 5. J Gen Virol 75:3413–3421. [DOI] [PubMed] [Google Scholar]
- 36.Graff JW, Mitzel DN, Weisend CM, Flenniken ML, Hardy ME. 2002. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J Virol 76:9545–9550. doi: 10.1128/JVI.76.18.9545-9550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc Natl Acad Sci U S A 102:4114–4119. doi: 10.1073/pnas.0408376102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachsenröder J, Braun A, Machnowska P, Ng TF, Deng X, Guenther S, Bernstein S, Ulrich RG, Delwart E, Johne R. 2014. Metagenomic identification of novel enteric viruses in urban wild rats and genome characterization of a group A rotavirus. J Gen Virol 95:2734–2747. doi: 10.1099/vir.0.070029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang W, Pavlish OA, Spiegelman VS, Parkhitko AA, Fuchs SY. 2003. Interaction of Epstein-Barr virus latent membrane protein 1 with SCFHOS/beta-TrCP E3 ubiquitin ligase regulates extent of NF-kappaB activation. J Biol Chem 278:48942–48949. doi: 10.1074/jbc.M307962200. [DOI] [PubMed] [Google Scholar]
- 40.Bour S, Perrin C, Akari H, Strebel K. 2001. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem 276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 41.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, Ren H, Strnadova P, Bowie AG, Smith GL. 2013. Poxvirus targeting of E3 ligase beta-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog 9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frescas D, Pagano M. 2008. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patton JT, Taraporewala Z, Chen D, Chizhikov V, Jones M, Elhelu A, Collins M, Kearney K, Wagner M, Hoshino Y, Gouvea V. 2001. Effect of intragenic rearrangement and changes in the 3′ consensus sequence on NSP1 expression and rotavirus replication. J Virol 75:2076–2086. doi: 10.1128/JVI.75.5.2076-2086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Estrabaud E, Le Rouzic E, Lopez-Vergès S, Morel M, Belaïdouni N, Benarous R, Transy C, Berlioz-Torrent C, Margottin-Goguet F. 2007. Regulated degradation of the HIV-1 Vpu protein through a betaTrCP-independent pathway limits the release of viral particles. PLoS Pathog. 3:e104. doi: 10.1371/journal.ppat.0030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. 2009. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J Virol 83:10322–10335. doi: 10.1128/JVI.01186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1:565–574. doi: 10.1016/S1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 47.Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. 1990. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem 265:14264–14269. [PubMed] [Google Scholar]
- 48.Meggio F, Pinna LA. 2003. One-thousand-and-one substrates of protein kinase CK2? FASEB J 17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 49.Pinna LA, Ruzzene M. 1996. How do protein kinases recognize their substrates? Biochim Biophys Acta 1314:191–225. doi: 10.1016/S0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 50.Schubert U, Henklein P, Boldyreff B, Wingender E, Strebel K, Porstmann T. 1994. The human immunodeficiency virus type 1 encoded Vpu protein is phosphorylated by casein kinase-2 (CK-2) at positions Ser52 and Ser56 within a predicted alpha-helix-turn-alpha-helix-motif. J Mol Biol 236:16–25. doi: 10.1006/jmbi.1994.1114. [DOI] [PubMed] [Google Scholar]
- 51.Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 52.Graff JW, Ewen J, Ettayebi K, Hardy ME. 2007. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J Gen Virol 88:613–620. doi: 10.1099/vir.0.82255-0. [DOI] [PubMed] [Google Scholar]
- 53.Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. 2002. SCOP: a structural classification of proteins database. Nucleic Acids Res 28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. 2009. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog 5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanarek N, London N, Schueler-Furman O, Ben-Neriah Y. 2010. Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb Perspect Biol 2:a000166. doi: 10.1101/cshperspect.a000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar KG, Krolewski JJ, Fuchs SY. 2004. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem 279:46614–46620. doi: 10.1074/jbc.M407082200. [DOI] [PubMed] [Google Scholar]
- 57.Cui W, Xiao N, Xiao H, Zhou H, Yu M, Gu J, Li X. 2012. Beta-TrCP-mediated IRAK1 degradation releases TAK1-TRAF6 from the membrane to the cytosol for TAK1-dependent NF-κB activation. Mol Cell Biol 32:3990–4000. doi: 10.1128/MCB.00722-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 59.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 60.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 62.Maes P, Matthijnssens J, Rahman M, Van Ranst M. 2009. RotaC: a web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol 9:238. doi: 10.1186/1471-2180-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H, Gao S, Lercher MJ, Hu S, Chen WH. 2012. EvolView, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res 40:W569–W572. doi: 10.1093/nar/gks576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
OSU NSP1 function compared to vector control. (A) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with medium ± 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three independent experiments performed in triplicate) were analyzed by two-way ANOVA (pairwise as indicated) using Tukey’s multiple comparisons test. EV, empty vector (pLIC6). (B) HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA) and is expressed as a percentage of β-TrCP in EV-transfected cells. Data (mean ± SD from three independent transfections) were analyzed by one-way ANOVA (pairwise as indicated) using Tukey’s multiple comparisons test. Download
Effect of plasmid ratio on OSU NSP1-mediated degradation of β-TrCP. (A and B) HEK293T cells were transfected as described in Materials and Methods, with the following transfection complex: 91 µl Opti-MEM, 1,500 ng DNA, 3 µl Lipofectamine 2000. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA). Data (mean ± SD from three independent transfections) were analyzed by two-way ANOVA (pairwise as indicated) using Sidak’s multiple comparisons test. (A) The DNA mixture contains 500 ng OSU NSP1, 0 to 1,000 ng FLAG-β-TrCP (in increments of 250 ng), and empty vector (pLIC6) to 1,500 ng. The level of β-TrCP is expressed as a percentage of β-TrCP in cells transfected with 500 ng FLAG-β-TrCP. (B) The DNA mixture contains 0 to 1,000 ng OSU NSP1 (in increments of 250 ng), 500 ng FLAG-β-TrCP, and empty vector (pLIC6) to 1,500 ng. The level of β-TrCP is expressed as a percentage of β-TrCP in cells transfected with 500 ng OSU-ΔC13 NSP1. Download
Expression and function of FLAG-tagged OSU NSP1. (A) HEK293T cells were cotransfected with FLAG-NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three independent experiments performed in triplicate) were analyzed with an unpaired t test (pairwise with OSU). (B) HEK293T cells were cotransfected with FLAG-NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB)(normalized to PCNA) and is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD from three independent transfections) were analyzed with an unpaired t test (pairwise with OSU). Download
The K465R mutation does not affect OSU NSP1 turnover or function. (A) HEK293T cells were cotransfected with NSP1 and NF-κB firefly and HSV-tk Renilla luciferase reporters. At 24 h p.t., cells were stimulated for 4 h with 25 ng/ml TNF-α. Relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity. Data (mean ± SD from one of three independent experiments performed in triplicate) were analyzed pairwise with OSU. (B) HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA) and is expressed as a percentage of β-TrCP in OSU-ΔC13-transfected cells. Data (mean ± SD from three independent transfections) were analyzed pairwise with OSU. Download
Alignment of human and porcine RVA NSP1. Amino acid sequences from the NSP1 genes examined in Fig. 3 were aligned using the T-Coffee server (P. Di Tommaso, S. Moretti, I. Xenarios, M. Orobitg, A. Montanyola, et al., Nucleic Acids Res 39:W13–W17, 2011, doi:10.1093/nar/gkr245; C. Notredame, D. G. Higgins, and J. Heringa, J. Mol. Biol. 302:205-217, 2000, doi:10.1006/jmbi.2000.4042). The image was generated using the ESPript 3.0 server (P. Gouet, X. Robert, and E. Courcelle, Nucleic Acids Res 31:3320–3323, 2003, doi:10.1093/nar/gkg556). Yellow, 100% sequence identity; blue, ≥70% sequence similarity; red, <70% sequence similarity or dissimilar residue; arrowhead, Zn2+-coordinating residue within the N-terminal RING domain; circle, K465; star, DSGΦS motif residue; dot, sequence gap; Hu, human; Po, porcine; Si, simian. Download
Role of the proteasome in NSP1 activity. HEK293T cells were cotransfected with NSP1 and FLAG-β-TrCP. At 8 h p.t., cells were treated for 16 h with dimethyl sulfoxide (DMSO) or 10 µM MG132. The level of β-TrCP was assayed 24 h p.t. by quantitative immunoblotting (IB) (normalized to PCNA) and is expressed as a percentage of β-TrCP in ΔC-transfected cells. Data (mean ± SD from three independent transfections) were analyzed by two-way ANOVA (pairwise as indicated) using Sidak’s multiple comparisons test. Download
GenBank accession numbers for the NSP1 genes used in this study.