

Abstract
Diabetes has been recognized as an important risk factor for a variety of intracellular bacterial infections, but research into the dysregulated immune mechanisms contributing to the impaired host–pathogen interactions is in its infancy. Diabetes is characterized by a chronic state of low-grade inflammation due to activation of pro-inflammatory mediators and increased formation of advanced glycation end products. Increased oxidative stress also exacerbates the chronic inflammatory processes observed in diabetes. The reduced phagocytic and antibacterial activity of neutrophils and macrophages provides an intracellular niche for the pathogen to replicate. Phagocytic and antibacterial dysfunction may be mediated directly through altered glucose metabolism and oxidative stress. Furthermore, impaired activation of natural killer cells contributes to decreased levels of interferon-γ, required for promoting macrophage antibacterial mechanisms. Together with impaired dendritic cell function, this impedes timely activation of adaptive immune responses. Increased intracellular oxidation of antigen-presenting cells in individuals with diabetes alters the cytokine profile generated and the subsequent balance of T-cell immunity. The establishment of acute intracellular bacterial infections in the diabetic host is associated with impaired T-cell-mediated immune responses. Concomitant to the greater intracellular bacterial burden and potential cumulative effect of chronic inflammatory processes, late hyper-inflammatory cytokine responses are often observed in individuals with diabetes, contributing to systemic pathology. The convergence of intracellular bacterial infections and diabetes poses new challenges for immunologists, providing the impetus for multidisciplinary research.
Keywords: cell-mediated immunity, diabetes, inflammation, intracellular bacterial infections, melioidosis, tuberculosis
Introduction
Global socio-economic changes over the last half century have been met with an unprecedented increase in non-communicable diseases such as diabetes.1 According to the most recent statistics from the International Diabetes Federation, the global prevalence of diabetes reached 382 million in 2013 and is predicted to escalate to 592 million by 2035.2 Approximately 85–95% of the global prevalence of diabetes is attributed to type 2 diabetes.2 Although the rising incidence of diabetes is widely recognized in high-income countries, approximately 80% of people with diabetes currently live in low- and middle-income countries, with the largest increases also predicted to occur in these regions.2 This has significant public health and economic implications, given the concurrent high prevalence of infectious diseases and already limited healthcare availability. The convergence of communicable and non-communicable diseases and heightened morbidity and mortality associated with co-morbid disease, raises significant issues regarding infection control and the re-emergence of intracellular bacterial infections.
Diabetes is associated with an increased risk of infectious diseases and their complications, including an average twofold higher risk of mortality compared with non-diabetic individuals.3 Combatting this double burden is challenging given that the mechanisms underlying the increased susceptibility of individuals with diabetes remain ill-defined. Despite renewed research interest over the past decade, findings have been inconsistent, with reports of altered phagocyte function and either augmented, attenuated or unchanged cytokine responses to infection in association with diabetes.4–8 The immunological basis for the synergy between diabetes and intracellular bacterial infections warrants further investigation. Here we review the current clinical and experimental evidence of immunological alterations associated with diabetes and their putative role in the increased susceptibility to intracellular bacterial infections.
Intracellular bacterial infections associated with diabetes
The increased incidence of intracellular bacterial infections is one of many complications associated with diabetes. A clear link between tuberculosis and diabetes has been documented in several cohort studies.9–12 Tuberculosis is the most significant cause of death globally from an intracellular bacterial infection and an estimated one-third of the global population is currently infected with the causative pathogen, Mycobacterium tuberculosis.13 Data from a recent prospective study indicated that individuals with diabetes have a threefold higher risk of developing tuberculosis and at least 10–35% of patients with tuberculosis have co-morbid diabetes (Table1).14
Table 1.
Significant association of tuberculosis and melioidosis with diabetes
| Pathogen | Annual incidence (cases per 100 000) | Relative risk1 |
Diabetes prevalence in infected patients (%) | Population at risk (millions)2 | References | |
|---|---|---|---|---|---|---|
| Infection | Mortality | |||||
| Mycobacterium tuberculosis | 122 | 3 | 5 | 10–35 | 382 | 2,13,19–21,31,33,118,140–142 |
| Burkholderia pseudomallei | 13–20 | 13 | 1·2 | 39–76 | 238 | 15,16,143–145 |
Relative risk of infection and death from infection in individuals with diabetes compared with non-diabetic individuals.
Number of individuals with diabetes living in endemic regions.
The important tropical infection, melioidosis, is also closely linked to diabetes. Melioidosis, caused by the intracellular bacterial pathogen Burkholderia pseudomallei, is a significant cause of morbidity and mortality in northern Australia and Southeast Asia.15,16 In northeast Thailand, melioidosis is the third most common cause of death from an infectious disease.16 Although less prevalent than tuberculosis, melioidosis remains under-reported due to inherent difficulties in diagnosis and limited availability of diagnostic facilities in resource-poor regions of endemnicity.16 For this reason, it is likely that reported cases represent just the ‘tip of the iceberg’. Melioidosis exhibits one of the strongest associations with diabetes, which has been consistently reported as the most significant risk factor (Table1).15 Diabetes is observed in up to 76% of patients with melioidosis in some regions.15,17,18
Diabetes presents new clinical challenges in the control of intracellular bacterial infections. Epidemiological studies have documented an association between diabetes and the severity of clinical presentations and outcomes from both tuberculosis and melioidosis.15,18–21 The majority of immunocompetent hosts infected with M. tuberculosis develop latent infections (Fig.1), characterized by a robust immune response that limits bacterial growth and tissue damage to prevent development of active disease. The transition from latent to active infection is highly dependent on the immune status of the host. Increased mortality has been described in patients with tuberculosis and co-morbid diabetes (Fig.1). There is clinical evidence that patients with tuberculosis and co-morbid diabetes are more likely to have cavitary lung lesions and experience a fourfold increased rate of relapse compared with patients without risk factors (Fig.1).19,22 While there are conflicting reports of a direct correlation between diabetes and increased mortality in patients with melioidosis, diabetes is a strong risk factor for acute bacteraemia and relapse.15,23,24
Figure 1.
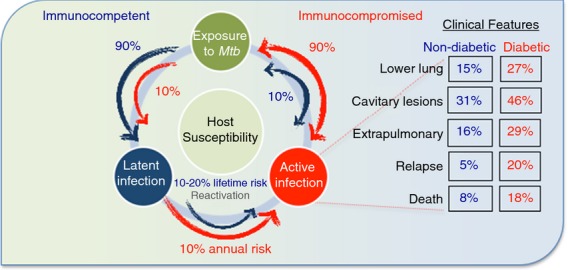
Diabetes is associated with increased progression to active tuberculosis and unfavourable clinical outcomes. Following exposure to Mycobacterium tuberculosis (Mtb), immunocompetent hosts predominantly develop latent infection (90%), with only 10% developing active tuberculosis (blue arrows).160 This is reversed in immunocompromised hosts, such as individuals with diabetes, who predominantly develop active infection (red arrows).160 In immunocompromised hosts, the annual risk of reactivation of latent tuberculosis exceeds 10%, compared with a lifetime risk of only 10–20% in immunocompetent hosts.161 Along with a predisposition for developing active disease, more unfavourable outcomes of tuberculosis, including lower lung involvement, cavitary lesions, extrapulmonary disease, relapse and death, are associated with co-morbid diabetes.20
Protective host immunity to intracellular bacterial infections relies on the appropriate timing and function of a range of immune defences. Invasion of the respiratory epithelium by M. tuberculosis triggers an early inflammatory response necessary for the rapid recruitment of neutrophils, macrophages, natural killer (NK) cells and dendritic cells (DC), involved in the initial containment of infection.25–27 Efficient phagocytosis and antigen presentation are required for the development of cell-mediated adaptive responses elicited by CD4+ Th1 cells and CD8+ cytotoxic T cells.28 Effective interaction between many immune cell populations at sites of infection, where they form dynamic aggregates known as granulomas, prevents active disease by containing bacteria and limiting collateral tissue damage.29 If any of these immune responses are compromised, reactivation of latent infection and development of active disease occurs. Failure to mount a robust immune response to intracellular bacterial infections may contribute to the increased susceptibility of individuals with diabetes and their predisposition to developing active disease.
Greater incidence and re-emergence of intracellular bacterial infections is anticipated as the diabetes epidemic escalates, increasing the population of susceptible individuals. The significance of this is emphasized in regions where the high incidence of diabetes is coupled with an equally high burden of tuberculosis.30 The western Pacific and Southeast Asia regions shoulder 60% of the burden of both diabetes and tuberculosis (Table2). In populations with a high prevalence of diabetes, 15–25% of active tuberculosis cases are attributable to diabetes, comparatively more than are attributed to other risk factors such as HIV (Table3).31 In Mexico, the tuberculosis-attributable fraction due to HIV is just 2%, compared with the 25% attributed to diabetes.32 Meanwhile, a recent study in India has found that up to 50% of patients with tuberculosis either had diabetes (25·3%) or were in a pre-diabetic state (24·5%).33 Despite emphasis being placed on tuberculosis and HIV co-infection, the burden of tuberculosis attributed to diabetes is of equal or greater concern in many regions due to the increasing global prevalence of diabetes.22 While the increasing rate of melioidosis over the past two decades has been attributed in part to improved diagnostic capabilities, it is likely that coinciding increases in the prevalence of diabetes in endemic regions is also a contributing factor.16 Increased travel to and from endemic regions also increases the risk of infection in those residing in other geographical locations and facilitates the global spread of infectious diseases. Combined with the increasing incidence of diabetes, there is an overwhelming need for further research to understand the immunological mechanisms linking diabetes and intracellular bacterial infections.
Table 2.
Regional prevalence of diabetes and tuberculosis
| WHO regions | Prevalence of diabetes (2013) |
Incidence of tuberculosis (2012) |
||
|---|---|---|---|---|
| Millions | % | Millions | % | |
| WPR | 138 | 36·1 | 2·4 | 20·2 |
| SEA | 72 | 18·8 | 4·8 | 40·3 |
| AMR | 61 | 16·0 | 0·4 | 3·4 |
| EUR | 56 | 14·7 | 0·5 | 4·2 |
| EMR | 35 | 9·2 | 1·1 | 9·2 |
| AFR | 20 | 5·2 | 2·7 | 22·7 |
| Total | 382 | – | 11·9 | – |
Table 3.
Most significant risk factors for tuberculosis
Chronic inflammation in diabetes contributes to immune dysregulation
Diabetes is a multifactorial metabolic disease, characterized by insulin resistance, glucose intolerance and overt hyperglycaemia. This review is focused on type 2 diabetes, which is aetiologically distinct from other types of diabetes and is closely related to the concurrent global epidemic of obesity.34 The aetiology involves a complex interplay between genetic and environmental factors that predispose to insulin resistance and higher circulating levels of blood glucose and free fatty acids (FFA; Fig.2). Alterations in glucose and lipid metabolism in adipocytes and hepatocytes lead to a progressively pro-inflammatory state characterized by expanding populations of classically activated (M1) macrophages (Fig.2).35 Pancreatic beta cell stress, as a result of metabolic and inflammatory changes, leads to increasing insulin deficiency and hyperglycaemia.36,37 Chronic hyperglycaemia accelerates the formation of advanced glycation end products (AGE) produced by non-enzymatic protein glycation.38 Increased levels of AGE and FFA (derived from excessive dietary intake and increased lipolysis secondary to insulin resistance) stimulate production of inflammatory mediators and reactive oxygen species (ROS).38–42 Diabetes-induced ROS formation also occurs from excessive glucose metabolism via oxidative phosphorylation.43
Figure 2.

The aetiopathogenic mechanisms of type 2 diabetes. Excessive dietary consumption of refined carbohydrates and saturated fatty acids, combined with genetic predisposition, leads to dysregulation of glucose and lipid homeostasis. This is associated with metabolic abnormalities, including increasing insulin resistance, lipolysis and hepatic gluconeogenesis, that further contribute to circulating levels of glucose and free fatty acids (FFA) and affect the function of multiple organ systems.156 Inflammation and oxidative stress induced by excessive FFA and formation of advanced glycation end products (AGE) leads to recruitment of pro-inflammatory (M1) macrophages, CD4+ T-helper type 1 (Th1) and type 17 (Th17) cells and CD8+ cytotoxic T cells (CTL), whereas anti-inflammatory (M2) macrophages, CD4+ T-helper type 2 (Th2) and regulatory T (Treg) cells are down-regulated. This systemic chronic inflammation exacerbates insulin resistance, beta cell injury and diabetic complications. CCL2, chemokine CC motif ligand 2; FFA, free fatty acids; IFN-γ, interferon-γ; IL-1β, interleukin-1β; IL-6, interleukin-6; IR, insulin resistance; TNF-α, tumour necrosis factor-α.
In healthy individuals, production of ROS is balanced by an increase in antioxidant activity, primarily mediated by glutathione, the most abundant redox regulator in eukaryotic cells. Glutathione neutralizes ROS by cycling between reduced (GSH) and oxidized (GSSG) states. A decrease in the ratio of GSH : GSSG is indicative of oxidative stress and has been described in patients with poorly controlled diabetes.44,45 This may be directly attributed to the increased production of ROS or indirectly through NADPH consumption. NADPH, which is consumed in the polyol pathway for glucose metabolism under hyperglycaemic conditions, is a co-factor required for regeneration of GSH. Deficiency in the availability of GSH precursors (cysteine and glycine) has also been documented in diabetes, together with decreased activity of γ-glutamylcysteine synthetase, the rate-limiting enzyme responsible for GSH synthesis.45,46 There is strong clinical evidence that elevated activity of γ-glutamyl transferase, involved in the extracellular catabolism of GSH, is also correlated with diabetes.47 Therefore, both consumption and impaired biosynthesis of GSH resulting from altered activity of multiple enzymes may contribute to increased oxidative stress and the exacerbation of chronic inflammatory processes in diabetes.
It is now widely accepted that obesity, particularly excess visceral adipose tissue, is characterized by a chronic state of low-grade inflammation due to the secretion of pro-inflammatory cytokines by stressed adipocytes and adipose tissue macrophages.48–51 Over-expression of tumour necrosis factor-α (TNF-α) in obese adipose tissue was the seminal finding that linked metabolic changes to inflammation and has since been determined as a key feature mediating insulin resistance.52–55 Pro-inflammatory M1 macrophages are recruited to adipose tissue where they secrete high levels of inflammatory mediators, including TNF-α, C-reactive protein (CRP), interleukin-1β (IL-1β), IL-6, IL-8 and IL-12, as reviewed by Donath and Shoelson (Table4).56 Elevated expression of interferon-γ (IFN-γ) in adipose tissue may also play a role in insulin resistance, contributing to the shift from anti-inflammatory (M2) macrophages to the pro-inflammatory M1 subset.57 Increased baseline secretion of TNF-α, IL-6 and IL-8 by neutrophils and monocytes from diabetic individuals has also been described in vitro.58,59 It is proposed that immune activation and systemic spillover of pro-inflammatory cytokines is central to the development of insulin resistance and drives the micro- and macro-vascular changes observed in diabetes.60–62
Table 4.
Effect of tuberculosis and diabetes on innate immune cell function
| Cell type | Function during infection | Effect of tuberculosis | Effect of diabetes | References | |
|---|---|---|---|---|---|
| Neutrophils | Phagocytosis | ↑ Neutrophils | ↑ Neutrophils | 27,41,59,65,67,74 | |
| Bactericidal activity | ↑ TNF-α, IL-8, IL-17, | ↑ TNF-α, IL-6, IL-8, IL-17, | |||
| Acute inflammatory response | CXCL9, ROS, defensins | CCL2, ROS #x2193; NOx, CXCR2, chemotaxis | |||
| Removal of microbes and dead tissue | |||||
| Promote M1 polarization | |||||
| Type 1 (M1) macrophages | Classically activated, | ↑ M1 | ↑ M1 | 86–90,94 | |
| proinflammatory responses | ↑ TNF- α, IL-1β, IL-6, | ↑ TNF-α, IL-1, IL-1β, IL-6, IL-8, | |||
| Bacterial, protozoa and | IL-18, IL-12, IL-23, CCL2, | IL-12, IL-23, CCL2, ROS, MMP-9 | |||
| viral defence | NOx, ROS | ↓ NOx | |||
| Antigen presentation and T cell activation | |||||
| Type 2 (M2) macrophages | Alternatively activated, | ↑↓ M2 | ↓ M2 | 150–154 | |
| anti-inflammatory responses | ↑ TGF-β, MMP-12 | ↑↓ IL-10 | |||
| Antagonise M1 responses | ↑↓ IL-10 | ||||
| Wound healing/fibrosis | |||||
| Natural killer (NK) cells | Defence against intracellular | ↑ NK | ↑↓ NK | 25,100,101,104,105,155 | |
| pathogens | ↑ IFN-γ, TNF-α, IL-22, | ↑ TNF-α, IL-8, IL-22, CCL2 | 105,155 | ||
| Contain intracellular | ICAM-1, Th1 response | ↑/– IFN-γ | |||
| infections prior to adaptive response | |||||
| Release cytotoxic granules | |||||
| Induce apoptosis of infected cells | |||||
| Antibody dependent cellular cytotoxicity | |||||
| Natural killer T (NKT) cells | Shared properties of NK and | ↑ NKT | ↑↓ NKT | 104,106–110 | |
| T cells for regulation of | ↑ IFN-γ, TNF-α, GM-CSF, | ↑↓ IL-4, IL-10 | |||
| immunity | DC maturation, CTL response | ↓ IL-4 | |||
| Respond to lipid antigens | ↑ IFN-γ, TNF-α; ↑↓ IL-10 | ||||
| Cytokines promote either | |||||
| inflammation or tolerance | |||||
| May have cytotoxic functions | |||||
| Dendritic cells (DC) | Antigen presentation | ↑ DC | ↑↓ DC | 88,111–113,156 | |
| Phagocytic when immature | ↑ DC migration | ↑ TNF-α, IL-1β, IL-6, IL-12, | |||
| Antigen uptake and presentation | ↑ Antigen presentation | IL-23, GM-CSF | |||
| T cell activation | ↑ TNF-α, IL-1β, IL-6, IL-12, | ||||
| Initiate adaptive immune response | IL-18, IL-23, IL-27, TGF-β | ||||
| Link between innate and adaptive immunity |
↑, increased; ↓, reduced; ↑↓, increased or reduced (conflicting evidence); –, no change; CCL2, chemokine CC motif ligand 2; CTL, cytotoxic T cells; CXCL9, chemokine C-X-C motif ligand 9; CXCR2, chemokine C-X-C motif receptor 2; GM-CSF, granulocyte macrophage colony-stimulating factor; ICAM-1, intercellular adhesion molecule 1; IL-1β, interleukin-1β; IL-4, interleukin-4; IL-6, interleukin-6; IL-8, interleukin-8; IL-10, interleukin-10; IL-12, interleukin-12; IL-18, interleukin-18; IL-22, interleukin-22; IL-23, interleukin-23; IL-27, interleukin-27; IFN-γ, interferon-γ; MMP-9, matrix metalloproteinase-9; NOx, mono-nitrogen oxides; TGF-β, transforming growth factor-β; TNF-α, tumour necrosis factor-α; ROS, reactive oxygen species.
Effect of diabetes on the early immune response to intracellular bacterial infections
Neutrophils
The role of neutrophils in the host immune response to intracellular bacterial infections is still widely debated. As one of the first phagocytic cells to reach sites of infection, neutrophils are adept at destroying invading pathogens through rapid release of ROS and pre-formed proteolytic granules.63 Clinically, neutrophils are the predominant infected cell type in sputum and bronchoalveolar lavage of patients with active tuberculosis.64 There is disparity between the results of in vitro studies regarding the ability of neutrophils to kill M. tuberculosis and B. pseudomallei, probably attributable to both host-specific and organism-specific factors in addition to variability in experimental design.65–67 In experimental animal models, neutrophils are rapidly recruited to sites of infection where they contribute to early defence against M. tuberculosis and B. pseudomallei (Table4).27,68 Neutrophil activation by M. tuberculosis influences the host immune response through regulation of surface receptor expression and secretion of chemokines and cytokines to facilitate early leucocyte migration (Table4).27,40,69 However, while neutrophils may have a beneficial role in the early containment of bacteria, neutrophils harbouring M. tuberculosis may delay the clearance of bacteria during chronic tuberculosis.70–73 This is consistent with the reduced bacterial loads observed following depletion of neutrophils in animal models of chronic tuberculosis.64,72 Therefore, the role of neutrophils may largely depend on the stage of infection and their capacity to respond appropriately depending on the virulence of bacteria.
Increased production of inflammatory cytokines and ROS by unstimulated neutrophils has been described in diabetics (Table4), and attributed to direct activation by AGE.4,41,74 However, neutrophil responses to infection appear to be predominantly suppressed in diabetic hosts.75 Decreased pathogen-stimulated ROS production may be related to impaired glucose metabolism through the pentose-phosphate pathway, which produces NADPH, a requirement for optimal NADPH oxidase activity.75 Furthermore, impaired activity of glutathione reductase, which also regulates neutrophil-based ROS production and phagocytosis, may be a contributing factor to neutrophil dysfunction in diabetic hosts.76 In addition to killing bacteria directly, ROS stimulates the release of neutrophil extracellular traps (NET), another important bactericidal mechanism. Such defects in neutrophil function may favour the ability of intracellular bacteria to ‘hijack’ neutrophils as a means of refuge and dissemination in diabetic hosts.70
Diabetes-induced functional defects in neutrophil responses to B. pseudomallei include impairments in phagocytosis, bacterial killing, neutrophil migration, cytokine production, apoptosis and NET formation.77–79 Diabetes was also associated with attenuated lipopolysaccharide-induced cytokine responses, coinciding with reduced up-regulation of vascular cell adhesion molecule 1 and intercellular adhesion molecule 1, required for leucocyte transmigration into tissue.80 Impaired neutrophil transendothelial migration and production of ROS have been attributed to changes caused by activation of the receptor for AGE.41 The combination of these processes may down-regulate recruitment of phagocytes during the early inflammatory process and impair initial control of bacterial growth. Conversely, increased production of inflammatory cytokines after neutrophil stimulation has also been documented in diabetes.4,66 Differences in experimental design, infective dose and length of co-culture may account for such discrepancies. Human studies have the added confounding influence of variability in the level of hyperglycaemic control and the use of hypoglycaemic agents, which in many cases are not explicitly defined and may have important immunomodulatory effects. Excessive neutrophil involvement is a significant cause of immunopathology in chronic intracellular bacterial infections and this may be exacerbated by the pro-inflammatory milieu involved in driving diabetic complications.
Macrophages
Macrophages play a critical role in providing early host defence against intracellular bacterial infections. Important effector functions of macrophages include the phagocytosis of bacteria and clearance of apoptotic and necrotic neutrophils to contain infection. Recruitment and activation of circulating monocytes to sites of infection, where they differentiate into macrophages, are facilitated by neutrophil-derived cytokines and chemokines, such as TNF-α and CCL2.69 In addition to phagocytic and antibacterial mechanisms, the cytokine profile of macrophages is crucial for driving effective cell-mediated immunity and protection against intracellular bacteria. M1 macrophage polarization in response to intracellular bacterial infections induces up-regulation of co-stimulatory molecules, inducible nitric oxide synthase and inflammatory cytokines, including TNF-α, IL-12 and IL-18. Production of IL-12 and IL-18 is essential for eliciting an IFN-γ response from NK cells and T cells in the establishment of T helper type 1 (Th1) cell-mediated immunity.81 Both IFN-γ and TNF-α activate macrophages and promote killing of intracellular bacteria by stimulating inducible nitric oxide synthase and NADPH oxidase, as recently reviewed by MacMicking.82 Clinical and experimental studies have confirmed the importance of IFN-γ and TNF-α in the control of M. tuberculosis infection.81,83,84 However, excessive cytokine production may contribute to tissue damage, especially if chronically elevated by an unresolved infection.85 Therefore, the inflammatory response requires precise regulation to achieve this balance between protection and injury.
Activated inflammatory macrophages are closely linked to many diabetic complications through the generation of significant levels of pro-inflammatory cytokines and ROS (Table4).86,87 While inflammatory cytokine production by unstimulated macrophages is higher in individuals with diabetes, infection-induced cytokine production tends to be impaired compared with non-diabetic individuals.88,89 This may be associated with reduced macrophage migration to sites of infection as suggested by lower levels of CCL2 in lung lysates in experimental models of diabetes and tuberculosis.88,89 In addition to impaired recruitment, clinical and experimental evidence indicates that monocytes from individuals with diabetes have reduced phagocytic and antibacterial activity against M. tuberculosis and B. pseudomallei in vitro.90–92 Reduced phagocytosis may be associated with defects in complement factors or receptor expression required for bacterial opsonization and internalization.90 As well as providing an intracellular niche that facilitates bacterial persistence, impaired phagocytic and antibacterial activity of macrophages may have downstream effects on the activation of the cell-mediated immune responses necessary for host protection. Reduced secretion of IL-12 and IFN-γ by peripheral blood mononuclear cells from individuals with diabetes has been reported following stimulation with intracellular bacteria.44 This is supported by in vivo evidence of lower levels of IL-12, IFN-γ and TNF-α in experimental animal models of diabetes following acute infection with intracellular bacteria.27,93,94 These diabetes-induced changes in macrophage responses may contribute to poor containment of intracellular bacteria in the critical early stages of infection and subsequent alterations in the type of T-cell response initiated.
It has been suggested that immunological dysregulation associated with diabetes is a direct consequence of impaired glycaemic control.44,95 The epidemiological data linking poor glycaemic control to increased risk of active tuberculosis lends support to this theory.11,96 High glucose concentrations have been shown to inhibit lectin binding, contributing to poor pathogen recognition and impaired bacterial phagocytosis in diabetic hosts.95 Reduced immune recognition of intracellular bacteria and altered cellular interactions potentially facilitate increased bacterial persistence.95 Phagocytic dysfunction may be mediated directly through impaired glucose metabolism or indirectly through increased endoplasmic reticulum stress and accumulation of misfolded proteins.97,98 These mechanisms may also contribute to the decreased expression of cell surface receptors and altered secretion of cytokines and other immunomodulatory proteins, representing an area for further research.
Natural killer cells and natural killer T cells
Natural killer cells play an important role in innate immune responses to pathogens and interest into their contribution to protection against intracellular bacterial infections has gained momentum over the past decade.25 Natural killer cells are regulated by a series of inhibitory and activating receptors.99 Experimental studies of M. tuberculosis infection have demonstrated that NK cells are recruited to sites of infection where they contribute to IFN-γ production and lysis of M. tuberculosis-infected target cells.99–101 The NK cells also modulate T-cell responses to M. tuberculosis, favouring Th1 effector functions and contributing to CD8+ T cell-derived IFN-γ production and cytolytic activity.102 Down-regulation of NK cell activating receptors in patients with tuberculosis coincides with impaired IFN-γ levels and reactivation of disease.103 Although higher numbers of NK cells have been documented in patients with diabetes before infection, decreased expression of activating receptors, NKp46 and NKG2D, has also been observed.104,105 Impaired activation of NK cells may dampen IFN-γ production and the cytolytic activity required for the early containment and killing of intracellular bacteria.105 Given the importance of NK cells in innate immunity, there is a need for research to understand the clinical relevance of diabetes-induced alterations in NK cell function and the direct effect on intracellular bacterial infections.
Natural killer T (NKT) cells are a unique subset of NK cells that also possess T-cell receptors. They respond to glycolipid rather than peptide antigens and have the potential to augment a range of immune responses.106 There is evidence that NKT cells contribute to host protection in M. tuberculosis infection by inhibiting intracellular bacterial growth through cytolytic mechanisms, enhancing maturation and activation of antigen-presenting cells (APC) and modulating the type of immune response generated.106–108 The involvement of NKT cells in adipose tissue inflammation and glucose intolerance has been described in experimental models of diabetes.104,109 Increases in NKT cell numbers are observed in patients with tuberculosis and are higher in the blood and bronchoalveolar lavage of patients with co-morbid diabetes than those without.110 This may be a direct consequence of the increased bacillary burden observed in these patients and has been suggested as a useful marker for active tuberculosis.110 Whether diabetes causes functional defects in NKT cell activity or otherwise biases the immunomodulatory response to intracellular bacterial infections is an area worthy of further research.
Effect of diabetes on antigen presentation following infection with intracellular bacteria
Dendritic cells
Dendritic cells represent an important link between innate and adaptive immune responses. Mature DC are potent immune-modulators and APC for priming specific lymphocyte responses.26,111 At the onset of infection with intracellular bacteria, DC accumulate at the site of infection to participate in bacterial uptake and antigen processing. Antigen presentation takes place following migration of mature DC to draining lymph nodes.26,28 Dendritic cells also modulate the lymphocyte profile generated through production of immunoregulatory cytokines, such as IL-12 and IL-18, essential for effective Th1 cell-mediated immune clearance of intracellular bacteria.26,28 Suppression of DC trafficking to lymph nodes has been suggested as a mechanism by which M. tuberculosis evades the early host immune response.26 Defects in DC maturation, migration and interaction with T cells may also contribute to intracellular bacterial persistence within the host.26
Increased expression of activation markers on unstimulated DC from diabetic individuals has been documented.112,113 Despite efficient trafficking of DC to regional lymph nodes, an initial delay in the recruitment of myeloid cells to the pulmonary site of infection was observed in diabetic mice following infection with M. tuberculosis.88 This coincided with reduced levels of CCL2 and CCL5, chemokines involved in the recruitment of macrophages and DC.88 Reduced early recruitment of APC to the primary site of infection may account for delayed induction of protective T-cell-mediated immune responses.114 In an experimental animal model of diabetes, DC phagocytosis of intracellular bacteria was also impaired.91 However, there were no differences in the up-regulation of DC markers involved in antigen presentation and co-stimulation of naive T cells. Impaired phagocytosis and delayed kinetics of antigen presentation at the onset of infection potentially contributes to poor early control and downstream alterations in lymphocyte activation.
Increased oxidative stress in diabetic hosts may also influence the profile of cytokines secreted by APC during intracellular bacterial infections. The reduced intracellular GSH : GSSG ratio in APC from diabetic individuals alters the secreted cytokine profile due to the immunomodulatory properties of GSH.115–117 Consistent with this, peripheral blood mononuclear cells from patients with poorly controlled diabetes had defects in IL-12 production in response to intracellular bacterial infection, which could be reversed by replenishing GSH levels.44 The exact mechanisms by which GSH influences IL-12 production are under investigation but may involve the modulation of intracellular redox status and glutathionylation of signalling intermediates or transcription factors.44 The therapeutic potential of agents that together improve the IL-12/IFN-γ axis and decrease oxidative stress represents an exciting avenue to pursue.
T-cell-mediated immunity is critical to host protection against intracellular bacterial infections. Diabetes-induced alterations in the immunomodulatory nature of DC may influence the type of T-cell response elicited, which is an important determinant in the long-term outcome of intracellular bacterial infections. Effective antigen presentation in secondary lymphoid organs, together with early secretion of IL-12 and IFN-γ, is essential for the priming and differentiation of Th1 cells involved in host protection. Development of active pulmonary or extrapulmonary tuberculosis, a common finding in those with co-morbid diabetes (Fig.1), has been linked to impairments in Th1 cell-mediated immunity.20,118,119 Potential alterations in the development of specific T-cell responses may be a secondary complication of the diabetes-induced innate immune defects already described (Fig.3). In an experimental model of co-morbid tuberculosis and diabetes, reduced levels of chemokines and cytokines were associated with delayed priming of T cells.88,114 This was followed by a higher pulmonary M. tuberculosis burden and an exaggerated inflammatory response during the latter stages of infection after specific adaptive immunity was established (Fig.3).88,114
Figure 3.
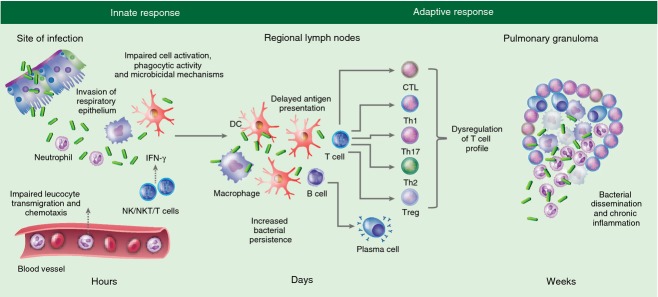
Putative immune mechanisms contributing to the increased susceptibility of diabetic hosts to Mycobacterium tuberculosis. Invasion of the respiratory epithelium by M. tuberculosis triggers an early inflammatory response necessary for the rapid recruitment of neutrophils, macrophages and dendritic cells (DC) to sites of infection. However, defects in bacterial recognition, phagocytic activity and cellular activation lead to impaired production of chemokines and cytokines (CCL2, tumour necrosis factor-α, interleukin-1β, IL-12) in diabetic hosts. Altered activation of natural killer (NK) cells, an important early source of interferon-γ (IFN-γ) to enhance macrophage microbicidal activity, may also facilitate intracellular bacterial persistence. The initiation of adaptive immunity is delayed by impaired antigen-presenting cell (APC) recruitment and function in diabetic hosts and dysregulation of the cytokine profile alters the activation and differentiation of T-cell subsets. B-cell activation and antibody production may also be impaired. The dysregulated inflammatory milieu due to the involvement of different T-cell subsets and impaired killing of intracellular bacteria potentially affects granuloma formation, contributing to increased neutrophil recruitment and central necrosis that facilitates bacterial escape.
Effect of diabetes on the adaptive immune response to intracellular bacterial infections
Lymphocytes
Host protection against intracellular bacterial infections relies on a strong T-cell-mediated response.120 T cells differentiate into a range of subtypes (Th1, Th2, Th17, Treg), which elicit distinct types of immunity fundamentally based on secreted cytokine profiles. An early influx of IFN-γ-producing Th1 cells is a significant determinant of protection against intracellular bacterial infections.121 There is strong evidence to indicate an initial delay in activation of Th1 cell-mediated immunity in diabetic hosts.44,88,114 However, there is also clinical and experimental evidence that the late inflammatory response during chronic tuberculosis is enhanced (Table5), although it may come too late to rescue diabetic hosts from bacterial dissemination.122,123 It is possible that this late hyper-inflammatory response is a direct result of increased antigenic stimulus, as a consequence of impaired innate immune control, or a cumulative build-up adding to the chronic inflammation underlying the immunopathology of diabetes itself.124 Increased circulating levels of Th1- and Th17-associated cytokines have been described in patients with tuberculosis and co-morbid diabetes.125 In vitro stimulation of whole blood with M. tuberculosis antigens resulted in elevated frequencies of CD4+ Th1 cells and Th17 cell subsets.122 However, lower production of IFN-γ by CD4+ T cells from patients with tuberculosis and poorly controlled diabetes has also been documented, consistent with reduced expression of IL-12 by APC.44,126 The inconsistencies between findings may be attributed to differences in study design and cell culture, including the absence of additional leucocyte interactions and removal from hyperglycaemic conditions. Furthermore, the Th1 response in diabetic hosts may have limited efficacy because of impaired cellular interactions and an inability to mount an effective antibacterial response, leading to intracellular bacterial persistence.
Table 5.
Effect of tuberculosis and diabetes on lymphocyte responses
| Cell type | Function during infection | Effect of tuberculosis | Effect of diabetes | References |
|---|---|---|---|---|
| T-helper 1 (Th1) cells | Cell-mediated immune response | ↑ Th1 | ↑ Th1 | 121,124–126,131 |
| Target intracellular pathogens | ↑ IFN-γ, TNF-α, IL-2, NO, LT-α | ↑ IFN-γ, IL-2, TNF-α | ||
| Microbial defence | ↑↓ IL-10 | ↑↓ IL-10 | ||
| Macrophage activation | ↓ NOx | |||
| CTL proliferation | ||||
| T-helper 2 (Th2) cells | Humoral immune response | ↑↓ Th2 | ↓ Th2 | 120,122,124,131 |
| Assist B cells | ↑ TGF-β | ↑↓ IL-10 | ||
| Ig isotype switching | ↑↓ IL4, IL-10 | ↓ IL-4 | ||
| Extracellular pathogen defence | ↓ IL-5 | |||
| Stimulate M2 | ||||
| Eosinophil activation | ||||
| Mast cell activation | ||||
| T-helper 17 (Th17) cells | Defence against fungi and extracellular bacteria | ↑ Th17 | ↑ Th17 | 124,125,127,128 |
| Enhance neutrophil response | ↑ TNF-α, IL-17, IL-22, CXCL9, CXCL10, CXCL11 | ↑ IL-17, IL-22 | ||
| Stimulate resident cells to secrete chemokines | ||||
| Recruit neutrophils and macrophages to sites of inflammation | ||||
| Regulatory T (Treg) cells | Suppress and regulate immune responses | ↑ Treg | ↓ Treg | 119,120,124,125 |
| Decrease immune-mediated damage | ↑ TGF-β | ↑ IFN-γ | ||
| Cytokines inhibit effector T cells and APC | ↑↓ IL-10 | ↑↓ IL-10 | ||
| Prevent pro-inflammatory cytokine secretion | ||||
| Cytotoxic T cells (CTL) | Lysis of infected cells | ↑ CTL | ↑ CTL | 121,157–159 |
| Targets viruses and intracellular bacteria | ↑ IFN-γ, TNF-α, perforin, granulysin | ↑ IFN-γ, TNF-α | ||
| Release of cytolytic granules | ||||
| Induce apoptosis of target cells | ||||
| B cells | Humoral immune response | ↑ B cells | ↑ B cells | 132–134 |
| Antibody production | ↑ IFN-γ, TNF-α, IL-6, IL-12, IgG, IgG1 | ↑ IFN-γ, TNF-α, IL-6, IL-12, IgG2c | ||
| Differentiation into plasma cells | ||||
| Antigen presentation to T cells | ↑↓ IL-4, IL-10 | ↑↓ IL-10 | ||
| Immune modulation | ↓ Ig production |
↑, increased; ↓, reduced; ↑↓, increased or reduced (conflicting evidence); APC, antigen-presenting cells; CXCL10, chemokine C-X-C motif ligand 10; CXCL11, chemokine C-X-C motif ligand 11; Ig, immunoglobulin; IL-2, interleukin-2; IFN-γ, interferon-γ; LT-α, lymphotoxin-α; NOx, mono-nitrogen oxides; TGF-β, transforming growth factor-β; TNF-α, tumour necrosis factor-α.
There is still a paucity of research on the role of other T-cell subsets in co-morbid diabetes and intracellular bacterial infections. While Th1-mediated immunity plays a crucial role in host protection against intracellular bacteria, the functional significance of Th17 responses is less clear.127 Experimental evidence indicates that Th17 responses may facilitate dissemination of M. tuberculosis, potentially through IL-17 secretion and its role in neutrophil recruitment.128 Bacterial dissemination may be further exacerbated by the functional defects in neutrophil and macrophage bactericidal mechanisms described in diabetic hosts. Without appropriate regulation, exaggerated Th17 responses may also contribute to immune-mediated pathology.129
Regulatory T (Treg) cells are critical for preventing exaggerated inflammatory responses to limit host tissue damage, although this may also limit host immunity and pathogen clearance.130 In experimental models of tuberculosis, Treg cells impair immune protection by delaying recruitment of effector CD4+ and CD8+ T cells to sites of infection.130 The Th2-mediated responses are also correlated with increased susceptibility to intracellular bacterial infections.131 Decreased numbers of Treg and Th2 cells are an underlying feature of diabetes.119,120,122 Interestingly, elevated systemic levels of immunosuppressive Treg and Th2-type cytokines, IL-10, TGF-β and IL-4, and an overall lower Th1 : Th2 ratio have been documented in patients with tuberculosis and diabetes.122,131 This may contribute to susceptibility in diabetic hosts by abrogating the protective mechanisms afforded by Th1-type cytokines and enhancing intracellular bacterial persistence.131 The diabetes-induced dysregulation of T-cell responses to intracellular bacteria is no doubt complex and requires further clarification.
While T-cell activity is critical to the protective immune response to intracellular bacterial infections, the definitive role of B cells is still debated.132 Until recently, B cells were generally considered to be of little benefit during intracellular bacterial infections, given the limited protection afforded by humoral immunity in general. However, it is now appreciated that B cells are present in tuberculosis granulomas in active follicle-like centres, where they influence local inflammatory responses (Table5).133 In particular, B cells have a newly defined role in regulating neutrophil migration to the site of infection. Neutrophilia is a compensatory response to B-cell deficiency during M. tuberculosis infection.133 Hyperglycaemia-induced functional defects in B cells have been described in vitro, specifically resulting in impaired immunoglobulin production, although the clinical relevance of this remains to be shown.134 Whether diabetes also delays the kinetics of B-cell activation and how this may influence the immunoregulatory role of B cells remains to be discerned. In this respect, defects in B-cell function in diabetic hosts may have significant immunomodulatory consequences on the functional response of other leucocytes, although this is yet to be determined.
Granuloma formation
When absolute bacterial clearance cannot be achieved, dynamic cellular interactions at sites of infection lead to the formation of granulomas, a characteristic feature of tuberculosis. Granulomas depend on the organized and complex interaction of many immune cell populations, including macrophages, DC and T and B cells, as recently reviewed by Guirado and Schlesinger.135 The precise balance and kinetics of cytokine and chemokine production and appropriate cellular function are necessary for proper granuloma formation.29,136 For the past decade, granulomas were primarily considered a protective mechanism, providing a barrier against bacterial dissemination and containing inflammatory processes to limit collateral tissue damage.137 However, cavitary granulomatous lesions can increase bacterial dissemination and are associated with destruction of lung parenchyma.138
The influence of diabetes on granuloma formation in tuberculosis is not well understood. Clinical observations of an increased frequency of cavitary lung lesions in patients with tuberculosis and co-morbid diabetes may indicate alterations in granuloma formation.20,32 Larger granulomas were also observed in an experimental model of diabetes and tuberculosis.27 This coincided with reduced production of TNF-α, IL-12 and nitric oxide by alveolar macrophages.27 Up-regulation of IFN-γ and TNF-α is critical for control of M. tuberculosis infection and in facilitating appropriate granuloma formation.81,83,84,136 It is possible that delayed pulmonary migration of macrophages, DC and activated T cells, caused by reduced CCL2 and CCL5, also contributes to altered structural organization of granulomas in individuals with co-morbid diabetes. Cavitary lesions are associated with increased degenerative macrophages and infiltration of neutrophils, which is typical of mice lacking IFN-γ, TNF-α and CCL2.101,139 Activated macrophages play a pivotal role in containment of bacilli within the granuloma, so incomplete macrophage activation or impaired microbicidal mechanisms in diabetes may contribute to M. tuberculosis escape and increased dissemination.
Future perspective
The double burden of diabetes and intracellular bacterial infections represents a significant global challenge. Currently, diagnostic and therapeutic research predominantly uses non-diabetic models and the translatability of this to individuals with diabetes is questionable given the apparent differences in immune responses and disease mechanisms. Although the underlying immunopathology of diabetes is no doubt complex, there is strong clinical and experimental evidence that a delay in inflammatory signals of the innate immune system is followed by altered development of appropriate protective responses against intracellular bacterial infections. While improving glucose control may benefit patients with intracellular infections and co-morbid diabetes, it is likely that the complex immunopathogenesis underlying diabetes will need to be addressed by a more multifactorial therapeutic approach. Understanding the mechanisms underlying co-morbidities like diabetes, which dramatically influence the progression of intracellular bacterial infections, will facilitate improvements in the treatment and management of disease in susceptible populations. Novel, affordable strategies are urgently required, particularly for low- to middle-income countries where the convergence of non-communicable and communicable diseases is unprecedented. Given the ongoing and widespread acceleration of non-communicable diseases, a multidisciplinary approach to research will be vital in addressing current and future challenges of the emerging double burden of co-morbid intracellular bacterial infections.
Disclosures
The authors declare no conflict of interest.
Glossary
- AGE
advanced glycation end products
- APC
antigen-presenting cells
- CCL
chemokine CC motif ligand
- CRP
C-reactive protein
- CTL
cytotoxic T cells
- CXCL
chemokine C-X-C motif ligand
- CXCR
chemokine C-X-C motif receptor
- DC
dendritic cells
- FFA
free fatty acids
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- ICAM-1
intercellular adhesion molecule 1
- IFN
interferon
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IR
insulin resistance
- NADPH
nicotinamide adenine dinucleotide phosphate
- NK
natural killer
- NO
nitric oxide
- NOx
mono-nitrogen oxides
- ROS
reactive oxygen species
- TGF
transforming growth factor
- Th
T-helper
- TNF
tumour necrosis factor
- VCAM-1
vascular cell adhesion molecule 1
References
- Rodriguez-Hernandez H, Simental-Mendia LE, Rodriguez-Ramirez G. Obesity and inflammation: epidemiology, risk factors, and markers of inflammation. Int J Endocrinol. 2013;2013:678159. doi: 10.1155/2013/678159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDF. IDF Diabetes Atlas. 6th edn. Brussels, Belgium: International Diabetes Federation; 2013. URL http://www.idf.org/sites/default/files/EN_6E_Atlas_Full_0.pdf [accessed on 18 March 2014] [PubMed] [Google Scholar]
- Shah BR, Hux JE. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care. 2003;26:510–3. doi: 10.2337/diacare.26.2.510. [DOI] [PubMed] [Google Scholar]
- Hatanaka E, Monteagudo PT, Marrocos MS. Neutrophils and monocytes as potentially important sources of proinflammatory cytokines in diabetes. Clin Exp Immunol. 2006;146:443–7. doi: 10.1111/j.1365-2249.2006.03229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon MR, Vendrell J, Miranda M. Different TNFalpha expression elicited by glucose in monocytes from type 2 diabetes mellitus patients. Atherosclerosis. 2007;194:e18–25. doi: 10.1016/j.atherosclerosis.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Foss-Freitas MC, Foss NT, Donadi EA. In vitro TNF-alpha and IL-6 production by adherent peripheral blood mononuclear cells obtained from type 1 and type 2 diabetic patients evaluated according to the metabolic control. Ann N Y Acad Sci. 2006;1079:177–80. doi: 10.1196/annals.1375.027. [DOI] [PubMed] [Google Scholar]
- Geerlings SE, Brouwer EC, Van Kessel KC. Cytokine secretion is impaired in women with diabetes mellitus. Eur J Clin Invest. 2000;30:995–1001. doi: 10.1046/j.1365-2362.2000.00745.x. [DOI] [PubMed] [Google Scholar]
- Delamaire M, Maugendre D, Moreno M. Impaired leucocyte functions in diabetic patients. Diabet Med. 1997;14:29–34. doi: 10.1002/(SICI)1096-9136(199701)14:1<29::AID-DIA300>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Dobler CC, Flack JR, Marks GB. Risk of tuberculosis among people with diabetes mellitus: an Australian nationwide cohort study. BMJ Open. 2012;2:e000666. doi: 10.1136/bmjopen-2011-000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young F, Wotton CJ, Critchley JA. Increased risk of tuberculosis disease in people with diabetes mellitus: record-linkage study in a UK population. J Epidemiol Community Health. 2012;66:519–23. doi: 10.1136/jech.2010.114595. [DOI] [PubMed] [Google Scholar]
- Leung CC, Lam TH, Chan WM. Diabetic control and risk of tuberculosis: a cohort study. Am J Epidemiol. 2008;167:1486–94. doi: 10.1093/aje/kwn075. [DOI] [PubMed] [Google Scholar]
- Perez A, Brown HS, III, Restrepo BI. Association between tuberculosis and diabetes in the Mexican border and non-border regions of Texas. Am J Trop Med Hyg. 2006;74:604–11. [PMC free article] [PubMed] [Google Scholar]
- WHO. Global Tuberculosis Report 2013. Geneva: World Health Organisation; 2013. URL http://apps.who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf?ua=1 [accessed on 18 March 2014] [Google Scholar]
- Restrepo BI, Camerlin AJ, Rahbar MH. Cross-sectional assessment reveals high diabetes prevalence among newly-diagnosed tuberculosis cases. Bull World Health Organ. 2011;89:352–9. doi: 10.2471/BLT.10.085738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie BJ, Ward L, Cheng AC. The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis. 2010;4:e900. doi: 10.1371/journal.pntd.0000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limmathurotsakul D, Wongratanacheewin S, Teerawattanasook N. Increasing incidence of human melioidosis in Northeast Thailand. Am J Trop Med Hyg. 2010;82:1113–7. doi: 10.4269/ajtmh.2010.10-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo TJ, Ang LW, James L. Melioidosis in a tropical city state, Singapore. Emerg Infect Dis. 2009;15:1645–7. doi: 10.3201/eid1510.090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidyalakshmi K, Lipika S, Vishal S. Emerging clinico-epidemiological trends in melioidosis: analysis of 95 cases from western coastal India. Int J Infect Dis. 2012;16:e491–7. doi: 10.1016/j.ijid.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Baker MA, Harries AD, Jeon CY. The impact of diabetes on tuberculosis treatment outcomes: a systematic review. BMC Med. 2011;9:81. doi: 10.1186/1741-7015-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CS, Yang CJ, Chen HC. Impact of type 2 diabetes on manifestations and treatment outcome of pulmonary tuberculosis. Epidemiol Infect. 2009;137:203–10. doi: 10.1017/S0950268808000782. [DOI] [PubMed] [Google Scholar]
- Alisjahbana B, Sahiratmadja E, Nelwan EJ. The effect of type 2 diabetes mellitus on the presentation and treatment response of pulmonary tuberculosis. Clin Infect Dis. 2007;45:428–35. doi: 10.1086/519841. [DOI] [PubMed] [Google Scholar]
- Magee MJ, Foote M, Maggio DM. Diabetes mellitus and risk of all-cause mortality among patients with tuberculosis in the state of Georgia, 2009–2012. Ann Epidemiol. 2014;24:369–75. doi: 10.1016/j.annepidem.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suputtamongkol Y, Chaowagul W, Chetchotisakd P. Risk factors for melioidosis and bacteremic melioidosis. Clin Infect Dis. 1999;29:408–13. doi: 10.1086/520223. [DOI] [PubMed] [Google Scholar]
- Limmathurotsakul D, Chaowagul W, Chierakul W. Risk factors for recurrent melioidosis in northeast Thailand. Clin Infect Dis. 2006;43:979–86. doi: 10.1086/507632. [DOI] [PubMed] [Google Scholar]
- Zufferey C, Germano S, Dutta B. The contribution of non-conventional T cells and NK cells in the mycobacterial-specific IFNgamma response in Bacille Calmette-Guerin (BCG)-immunized infants. PLoS ONE. 2013;8:e77334. doi: 10.1371/journal.pone.0077334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajashree P, Supriya P, Das SD. Differential migration of human monocyte-derived dendritic cells after infection with prevalent clinical strains of Mycobacterium tuberculosis. Immunobiology. 2008;213:567–75. doi: 10.1016/j.imbio.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Sugawara I, Udagawa T, Yamada H. Rat neutrophils prevent the development of tuberculosis. Infect Immun. 2004;72:1804–6. doi: 10.1128/IAI.72.3.1804-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini E, Iona E, Ferroni L. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. 2001;166:7033–41. doi: 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- Khader SA, Rangel-Moreno J, Fountain JJ. In a murine tuberculosis model, the absence of homeostatic chemokines delays granuloma formation and protective immunity. J Immunol. 2009;183:8004–14. doi: 10.4049/jimmunol.0901937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo BI. Convergence of the tuberculosis and diabetes epidemics: renewal of old acquaintances. Clin Infect Dis. 2007;45:436–8. doi: 10.1086/519939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson CR, Forouhi NG, Roglic G. Diabetes and tuberculosis: the impact of the diabetes epidemic on tuberculosis incidence. BMC Public Health. 2007;7:234. doi: 10.1186/1471-2458-7-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584–90. doi: 10.2337/diacare.27.7.1584. [DOI] [PubMed] [Google Scholar]
- Viswanathan V, Kumpatla S, Aravindalochanan V. Prevalence of diabetes and pre-diabetes and associated risk factors among tuberculosis patients in India. PLoS ONE. 2012;7:e41367. doi: 10.1371/journal.pone.0041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AP, Xu G, Brown N. Diabetes and its comorbidities-where East meets West. Nat Rev Endocrinol. 2013;9:537–47. doi: 10.1038/nrendo.2013.102. [DOI] [PubMed] [Google Scholar]
- Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–23. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–70. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- Bensellam M, Duvillie B, Rybachuk G. Glucose-induced O(2) consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS ONE. 2012;7:e29807. doi: 10.1371/journal.pone.0029807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk Z, Ljubic S, Turk N. Detection of autoantibodies against advanced glycation endproducts and AGE-immune complexes in serum of patients with diabetes mellitus. Clin Chim Acta. 2001;303:105–15. doi: 10.1016/s0009-8981(00)00389-2. [DOI] [PubMed] [Google Scholar]
- Vlassara H, Cai W, Crandall J. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci U S A. 2002;99:15596–601. doi: 10.1073/pnas.242407999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrosa J, Saunders BM, Appelberg R. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun. 2000;68:577–83. doi: 10.1128/iai.68.2.577-583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison KS, Parhar RS, Saleh SS. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs) J Leukoc Biol. 2002;71:433–44. [PubMed] [Google Scholar]
- Pal D, Dasgupta S, Kundu R. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–85. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Tan KS, Lee KO, Low KC. Glutathione deficiency in type 2 diabetes impairs cytokine responses and control of intracellular bacteria. J Clin Invest. 2012;122:2289–300. doi: 10.1172/JCI57817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar RV, McKay SV, Patel SG. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care. 2011;34:162–7. doi: 10.2337/dc10-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Ohtsuka Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism. 1989;38:753–8. doi: 10.1016/0026-0495(89)90061-9. [DOI] [PubMed] [Google Scholar]
- Meisinger C, Lowel H, Heier M. Serum gamma-glutamyltransferase and risk of type 2 diabetes mellitus in men and women from the general population. J Intern Med. 2005;258:527–35. doi: 10.1111/j.1365-2796.2005.01572.x. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Deyoung SM, Bodzin JL. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- Furuhashi M, Fucho R, Gorgun CZ. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640–50. doi: 10.1172/JCI34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Vazquez I, Fernandez-Veledo S, Kramer DK. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem. 2008;114:183–94. doi: 10.1080/13813450802181047. [DOI] [PubMed] [Google Scholar]
- Shah PK. Innate immune pathway links obesity to insulin resistance. Circ Res. 2007;100:1531–3. doi: 10.1161/CIRCRESAHA.107.101104. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Moller DE. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000;11:212–7. doi: 10.1016/s1043-2760(00)00272-1. [DOI] [PubMed] [Google Scholar]
- Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia. 1998;41:1241–8. doi: 10.1007/s001250051058. [DOI] [PubMed] [Google Scholar]
- Grimble RF. Inflammatory status and insulin resistance. Curr Opin Clin Nutr Metab Care. 2002;5:551–9. doi: 10.1097/00075197-200209000-00015. [DOI] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Winer S, Chan Y, Paltser G. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–9. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup JC, Mattock MB, Chusney GD. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–92. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- Zozulinska D, Majchrzak A, Sobieska M. Serum interleukin-8 level is increased in diabetic patients. Diabetologia. 1999;42:117–8. doi: 10.1007/s001250051124. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–8. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–90. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Yuan M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord. 2003;27(Suppl. 3):S49–52. doi: 10.1038/sj.ijo.0802501. [DOI] [PubMed] [Google Scholar]
- Romero MM, Basile JI, Lopez B. Outbreaks of Mycobacterium tuberculosis MDR strains differentially induce neutrophil respiratory burst involving lipid rafts, p38 MAPK and Syk. BMC Infect Dis. 2014;14:262. doi: 10.1186/1471-2334-14-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eum SY, Kong JH, Hong MS. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137:122–8. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisich KO, Higgins M, Diamond G. Tumor necrosis factor alpha stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect Immun. 2002;70:4591–9. doi: 10.1128/IAI.70.8.4591-4599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J, Williams N, Rush C. Burkholderia pseudomallei triggers altered inflammatory profiles in a whole-blood model of type 2 diabetes-melioidosis comorbidity. Infect Immun. 2012;80:2089–99. doi: 10.1128/IAI.00212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau AR, Newton SM, Wilkinson KA. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117:1988–94. doi: 10.1172/JCI31097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton A, Haque A, Chu K. A critical role for neutrophils in resistance to experimental infection with Burkholderia pseudomallei. J Infect Dis. 2007;195:99–107. doi: 10.1086/509810. [DOI] [PubMed] [Google Scholar]
- Sawant KV, McMurray DN. Guinea pig neutrophils infected with Mycobacterium tuberculosis produce cytokines which activate alveolar macrophages in noncontact cultures. Infect Immun. 2007;75:1870–7. doi: 10.1128/IAI.00858-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eruslanov EB, Lyadova IV, Kondratieva TK. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73:1744–53. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Hoffmann R, Lang R. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun. 2006;74:4295–309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Majlessi L, Deriaud E. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity. 2009;31:761–71. doi: 10.1016/j.immuni.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Marzo E, Vilaplana C, Tapia G. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis (Edinb) 2014;94:55–64. doi: 10.1016/j.tube.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Talukdar S, da Oh Y, Bandyopadhyay G. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–12. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba-Loureiro TC, Hirabara SM, Mendonca JR. Diabetes causes marked changes in function and metabolism of rat neutrophils. J Endocrinol. 2006;188:295–303. doi: 10.1677/joe.1.06438. [DOI] [PubMed] [Google Scholar]
- Yan J, Meng X, Wancket LM. Glutathione reductase facilitates host defense by sustaining phagocytic oxidative burst and promoting the development of neutrophil extracellular traps. J Immunol. 2012;188:2316–27. doi: 10.4049/jimmunol.1102683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanchamroen S, Kewcharoenwong C, Susaengrat W. Human polymorphonuclear neutrophil responses to Burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun. 2009;77:456–63. doi: 10.1128/IAI.00503-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riyapa D, Buddhisa S, Korbsrisate S. Neutrophil extracellular traps exhibit antibacterial activity against burkholderia pseudomallei and are influenced by bacterial and host factors. Infect Immun. 2012;80:3921–9. doi: 10.1128/IAI.00806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kewcharoenwong C, Rinchai D, Utispan K. Glibenclamide reduces pro-inflammatory cytokine production by neutrophils of diabetes patients in response to bacterial infection. Sci Rep. 2013;3:3363. doi: 10.1038/srep03363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen AS, Pedersen-Skovsgaard T, Berg RM. Type 2 diabetes mellitus is associated with impaired cytokine response and adhesion molecule expression in human endotoxemia. Intensive Care Med. 2010;36:1548–55. doi: 10.1007/s00134-010-1845-1. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Chan J, Triebold KJ. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–54. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12:367–82. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Chan J. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–72. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- Solovic I, Sester M, Gomez-Reino JJ. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: a TBNET consensus statement. Eur Respir J. 2010;36:1185–206. doi: 10.1183/09031936.00028510. [DOI] [PubMed] [Google Scholar]
- Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–34. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orie NN, Zidek W, Tepel M. Increased intracellular generation of reactive oxygen species in mononuclear leukocytes from patients with diabetes mellitus type 2. Exp Clin Endocrinol Diabetes. 2000;108:175–80. doi: 10.1055/s-2000-7740. [DOI] [PubMed] [Google Scholar]
- Devaraj S, Venugopal SK, Singh U. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta} Diabetes. 2005;54:85–91. doi: 10.2337/diabetes.54.1.85. [DOI] [PubMed] [Google Scholar]
- Vallerskog T, Martens GW, Kornfeld H. Diabetic mice display a delayed adaptive immune response to Mycobacterium tuberculosis. J Immunol. 2010;184:6275–82. doi: 10.4049/jimmunol.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stew SS, Martinez PJ, Schlesinger LS. Differential expression of monocyte surface markers among TB patients with diabetes co-morbidity. Tuberculosis (Edinb) 2013;93(Suppl):S78–82. doi: 10.1016/S1472-9792(13)70015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez DI, Twahirwa M, Schlesinger LS. Reduced Mycobacterium tuberculosis association with monocytes from diabetes patients that have poor glucose control. Tuberculosis (Edinb) 2013;93:192–7. doi: 10.1016/j.tube.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NL, Morris JL, Rush C. Impact of streptozotocin-induced diabetes on functional responses of dendritic cells and macrophages towards Burkholderia pseudomallei. FEMS Immunol Med Microbiol. 2011;61:218–27. doi: 10.1111/j.1574-695X.2010.00767.x. [DOI] [PubMed] [Google Scholar]
- Hodgson KA, Morris JL, Feterl ML. Altered macrophage function is associated with severe Burkholderia pseudomallei infection in a murine model of type 2 diabetes. Microbes Infect. 2011;13:1177–84. doi: 10.1016/j.micinf.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Chin CY, Monack DM, Nathan S. Delayed activation of host innate immune pathways in streptozotocin-induced diabetic hosts leads to more severe disease during infection with Burkholderia pseudomallei. Immunology. 2012;135:312–32. doi: 10.1111/j.1365-2567.2011.03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, Kawakami K, Uezu K. Lower expression of Th1-related cytokines and inducible nitric oxide synthase in mice with streptozotocin-induced diabetes mellitus infected with Mycobacterium tuberculosis. Clin Exp Immunol. 2005;139:57–64. doi: 10.1111/j.1365-2249.2005.02677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas R, Wallis R, Soilleux EJ. High glucose disrupts oligosaccharide recognition function via competitive inhibition: a potential mechanism for immune dysregulation in diabetes mellitus. Immunobiology. 2011;216:126–31. doi: 10.1016/j.imbio.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablos-Mendez A, Blustein J, Knirsch CA. The role of diabetes mellitus in the higher prevalence of tuberculosis among Hispanics. Am J Public Health. 1997;87:574–9. doi: 10.2105/ajph.87.4.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha B, Samuel SM, Triggle CR. Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Exp Diabetes Res. 2012;2012:481840. doi: 10.1155/2012/481840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankayalapati R, Garg A, Porgador A. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol. 2005;175:4611–7. doi: 10.4049/jimmunol.175.7.4611. [DOI] [PubMed] [Google Scholar]
- Portevin D, Via LE, Eum S. Natural killer cells are recruited during pulmonary tuberculosis and their ex vivo responses to mycobacteria vary between healthy human donors in association with KIR haplotype. Cell Microbiol. 2012;14:1734–44. doi: 10.1111/j.1462-5822.2012.01834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng CG, Kaviratne M, Rothfuchs AG. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol. 2006;177:7086–93. doi: 10.4049/jimmunol.177.10.7086. [DOI] [PubMed] [Google Scholar]
- Vankayalapati R, Klucar P, Wizel B. NK cells regulate CD8+ T cell effector function in response to an intracellular pathogen. J Immunol. 2004;172:130–7. doi: 10.4049/jimmunol.172.1.130. [DOI] [PubMed] [Google Scholar]
- Bozzano F, Costa P, Passalacqua G. Functionally relevant decreases in activatory receptor expression on NK cells are associated with pulmonary tuberculosis in vivo and persist after successful treatment. Int Immunol. 2009;21:779–91. doi: 10.1093/intimm/dxp046. [DOI] [PubMed] [Google Scholar]
- Guo H, Xu B, Gao L. High frequency of activated natural killer and natural killer T-cells in patients with new onset of type 2 diabetes mellitus. Exp Biol Med. 2012;237:556–62. doi: 10.1258/ebm.2012.011272. [DOI] [PubMed] [Google Scholar]
- Berrou J, Fougeray S, Venot M. Natural killer cell function, an important target for infection and tumor protection, is impaired in type 2 diabetes. PLoS ONE. 2013;8:e62418. doi: 10.1371/journal.pone.0062418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataswamy MM, Baena A, Goldberg MF. Incorporation of NKT cell-activating glycolipids enhances immunogenicity and vaccine efficacy of Mycobacterium bovis bacillus Calmette-Guerin. J Immunol. 2009;183:1644–56. doi: 10.4049/jimmunol.0900858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian A, Alt J, Perera V. Activation of NKT cells protects mice from tuberculosis. Infect Immun. 2002;70:6302–9. doi: 10.1128/IAI.70.11.6302-6309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sada-Ovalle I, Chiba A, Gonzales A. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 2008;4:12. doi: 10.1371/journal.ppat.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Andoh Y, Clingan CS. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS ONE. 2012;7:22. doi: 10.1371/journal.pone.0030568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Xiao HP, Cui HY. Significant increase in natural-killer T cells in patients with tuberculosis complicated by type 2 diabetes mellitus. J Int Med Res. 2011;39:105–11. doi: 10.1177/147323001103900113. [DOI] [PubMed] [Google Scholar]
- Mihret A, Mamo G, Tafesse M. Dendritic cells activate and mature after infection with Mycobacterium tuberculosis. BMC Res Notes. 2011;4:247. doi: 10.1186/1756-0500-4-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musilli C, Paccosi S, Pala L. Characterization of circulating and monocyte-derived dendritic cells in obese and diabetic patients. Mol Immunol. 2011;49:234–8. doi: 10.1016/j.molimm.2011.08.019. [DOI] [PubMed] [Google Scholar]
- Surendar J, Mohan V, Pavankumar N. Increased levels of serum granulocyte-macrophage colony-stimulating factor is associated with activated peripheral dendritic cells in type 2 diabetes subjects (CURES-99) Diabetes Technol Ther. 2012;14:344–9. doi: 10.1089/dia.2011.0182. [DOI] [PubMed] [Google Scholar]
- Martens GW, Arikan MC, Lee J. Tuberculosis susceptibility of diabetic mice. Am J Respir Cell Mol Biol. 2007;37:518–24. doi: 10.1165/rcmb.2006-0478OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam K, Ghousunnissa S, Nair S. Glutathione-redox balance regulates c-rel-driven IL-12 production in macrophages: possible implications in antituberculosis immunotherapy. J Immunol. 2010;184:2918–29. doi: 10.4049/jimmunol.0900439. [DOI] [PubMed] [Google Scholar]
- Dobashi K, Aihara M, Araki T. Regulation of LPS induced IL-12 production by IFN-gamma and IL-4 through intracellular glutathione status in human alveolar macrophages. Clin Exp Immunol. 2001;124:290–6. doi: 10.1046/j.1365-2249.2001.01535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Shimamura T, Tagami T. The skewing to Th1 induced by lentinan is directed through the distinctive cytokine production by macrophages with elevated intracellular glutathione content. Int Immunopharmacol. 2002;2:673–89. doi: 10.1016/s1567-5769(01)00212-0. [DOI] [PubMed] [Google Scholar]
- Jeon CY, Murray MB. Diabetes mellitus increases the risk of active tuberculosis: a systematic review of 13 observational studies. PLoS Med. 2008;5:e152. doi: 10.1371/journal.pmed.0050152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussiotis VA, Tsai EY, Yunis EJ. IL-10-producing T cells suppress immune responses in anergic tuberculosis patients. J Clin Invest. 2000;105:1317–25. doi: 10.1172/JCI9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XY, Xiao L, Chen HB. T regulatory cells and Th1/Th2 cytokines in peripheral blood from tuberculosis patients. Eur J Clin Microbiol Infect Dis. 2010;29:643–50. doi: 10.1007/s10096-010-0908-0. [DOI] [PubMed] [Google Scholar]
- Chackerian AA, Perera TV, Behar SM. Gamma interferon-producing CD4+ T lymphocytes in the lung correlate with resistance to infection with Mycobacterium tuberculosis. Infect Immun. 2001;69:2666–74. doi: 10.1128/IAI.69.4.2666-2674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar NP, Sridhar R, Banurekha VV. Type 2 diabetes mellitus coincident with pulmonary tuberculosis is associated with heightened systemic type 1, type 17, and other proinflammatory cytokines. Ann Am Thorac Soc. 2013;10:441–9. doi: 10.1513/AnnalsATS.201305-112OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo BI, Fisher-Hoch SP, Pino PA. Tuberculosis in poorly controlled type 2 diabetes: altered cytokine expression in peripheral white blood cells. Clin Infect Dis. 2008;47:634–41. doi: 10.1086/590565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Xiao C, Wang P. The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Hum Immunol. 2014;75:289–96. doi: 10.1016/j.humimm.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Kumar NP, Sridhar R, Banurekha VV. Expansion of pathogen-specific T-helper 1 and T-helper 17 cells in pulmonary tuberculosis with coincident type 2 diabetes mellitus. J Infect Dis. 2013;208:739–48. doi: 10.1093/infdis/jit241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukaguchi K, Okamura H, Matsuzawa K. [Longitudinal assessment of IFN-gamma production in patients with pulmonary tuberculosis complicated with diabetes mellitus] Kekkaku. 2002;77:409–13. [PubMed] [Google Scholar]
- Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- Redford PS, Boonstra A, Read S. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol. 2010;40:2200–10. doi: 10.1002/eji.201040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FC, Bianco MV, Meikle V. Increased IL-17 expression is associated with pathology in a bovine model of tuberculosis. Tuberculosis (Edinb) 2011;91:57–63. doi: 10.1016/j.tube.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Shafiani S, Tucker-Heard G, Kariyone A. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med. 2010;207:1409–20. doi: 10.1084/jem.20091885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Attiyah RJ, Mustafa AS. Mycobacterial antigen-induced T helper type 1 (Th1) and Th2 reactivity of peripheral blood mononuclear cells from diabetic and non-diabetic tuberculosis patients and Mycobacterium bovis bacilli Calmette-Guerin (BCG)-vaccinated healthy subjects. Clin Exp Immunol. 2009;158:64–73. doi: 10.1111/j.1365-2249.2009.04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zheng X, Zhang J. CD19(+)CD1d(+)CD5(+) B cell frequencies are increased in patients with tuberculosis and suppress Th17 responses. Cell Immunol. 2012;274:89–97. doi: 10.1016/j.cellimm.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol. 2007;178:7222–34. doi: 10.4049/jimmunol.178.11.7222. [DOI] [PubMed] [Google Scholar]
- Sakowicz-Burkiewicz M, Kocbuch K, Grden M. High glucose concentration impairs ATP outflow and immunoglobulin production by human peripheral B lymphocytes: involvement of P2X7 receptor. Immunobiology. 2013;218:591–601. doi: 10.1016/j.imbio.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Guirado E, Schlesinger LS. Modeling the Mycobacterium tuberculosis granuloma – the critical battlefield in host immunity and disease. Front Immunol. 2013;4:98. doi: 10.3389/fimmu.2013.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algood HM, Lin PL, Yankura D. TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J Immunol. 2004;172:6846–57. doi: 10.4049/jimmunol.172.11.6846. [DOI] [PubMed] [Google Scholar]
- Bean AG, Roach DR, Briscoe H. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol. 1999;162:3504–11. [PubMed] [Google Scholar]
- Dheda K, Booth H, Huggett JF. Lung remodeling in pulmonary tuberculosis. J Infect Dis. 2005;192:1201–9. doi: 10.1086/444545. [DOI] [PubMed] [Google Scholar]
- Maus UA, Waelsch K, Kuziel WA. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol. 2003;170:3273–8. doi: 10.4049/jimmunol.170.6.3273. [DOI] [PubMed] [Google Scholar]
- Dooley KE, Chaisson RE. Tuberculosis and diabetes mellitus: convergence of two epidemics. Lancet Infect Dis. 2009;9:737–46. doi: 10.1016/S1473-3099(09)70282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder JF, Chaulk CP, Dalvi M. A high tuberculosis case-fatality rate in a setting of effective tuberculosis control: implications for acceptable treatment success rates. Int J Tuberc Lung Dis. 2002;6:1114–7. [PubMed] [Google Scholar]
- Oursler KK, Moore RD, Bishai WR. Survival of patients with pulmonary tuberculosis: clinical and molecular epidemiologic factors. Clin Infect Dis. 2002;34:752–9. doi: 10.1086/338784. [DOI] [PubMed] [Google Scholar]
- Shih HI, Chuang YC, Cheung BM. Sporadic and outbreak cases of melioidosis in southern Taiwan: clinical features and antimicrobial susceptibility. Infection. 2009;37:9–15. doi: 10.1007/s15010-008-7324-8. [DOI] [PubMed] [Google Scholar]
- Hassan MR, Pani SP, Peng NP. Incidence, risk factors and clinical epidemiology of melioidosis: a complex socio-ecological emerging infectious disease in the Alor Setar region of Kedah, Malaysia. BMC Infect Dis. 2010;10:302. doi: 10.1186/1471-2334-10-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- How SH, Ng KH, Jamalludin AR. Melioidosis in Pahang, Malaysia. Med J Malaysia. 2005;60:606–13. [PubMed] [Google Scholar]
- UNAIDS. 2013. Global report: UNAIDS report on the global AIDS epidemic 2013 . Joint United Nations Programme on HIV/AIDS. URL http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf [accessed on 18 March 2014]
- Marais BJ, Lonnroth K, Lawn SD. Tuberculosis comorbidity with communicable and non-communicable diseases: integrating health services and control efforts. Lancet Infect Dis. 2013;13:436–48. doi: 10.1016/S1473-3099(13)70015-X. [DOI] [PubMed] [Google Scholar]
- Getahun H, Gunneberg C, Granich R. HIV infection-associated tuberculosis: the epidemiology and the response. Clin Infect Dis. 2010;50(Suppl. 3):S201–7. doi: 10.1086/651492. [DOI] [PubMed] [Google Scholar]
- FAO, IFAD, WFP. The State of Food Insecurity in the World. The Multiple Dimensions of Food Security. Rome: FAO; 2013. [Google Scholar]
- Kahnert A, Seiler P, Stein M. Alternative activation deprives macrophages of a coordinated defense program to Mycobacterium tuberculosis. Eur J Immunol. 2006;36:631–47. doi: 10.1002/eji.200535496. [DOI] [PubMed] [Google Scholar]
- Ji Y, Sun S, Xu A. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem. 2012;287:13561–71. doi: 10.1074/jbc.M112.350066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Molofsky AB, Liang HE. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida AS, Lago PM, Boechat N. Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J Immunol. 2009;183:718–31. doi: 10.4049/jimmunol.0801212. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch LA, O'Connell JM, Kwasnik AK. Are natural killer cells protecting the metabolically healthy obese patient? Obesity. 2009;17:601–5. doi: 10.1038/oby.2008.565. [DOI] [PubMed] [Google Scholar]
- Bertola A, Ciucci T, Rousseau D. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61:2238–47. doi: 10.2337/db11-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong YH, Jeon BY, Gu SH. Differentiation of antigen-specific T cells with limited functional capacity during Mycobacterium tuberculosis infection. Infect Immun. 2014;82:132–9. doi: 10.1128/IAI.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AO, Mazzaccaro RJ, Russell RG. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc Natl Acad Sci U S A. 2000;97:4204–8. doi: 10.1073/pnas.97.8.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzigeorgiou A, Seijkens T, Zarzycka B. Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc Natl Acad Sci U S A. 2014;111:2686–91. doi: 10.1073/pnas.1400419111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufariello JM, Chan J, Flynn JL. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect Dis. 2003;3:578–90. doi: 10.1016/s1473-3099(03)00741-2. [DOI] [PubMed] [Google Scholar]
- Corbett EL, Watt CJ, Walker N. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med. 2003;163:1009–21. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]