

Background: Relaxin activates peroxisome proliferator-activated receptor γ (PPARγ) by an unknown PPARγ ligand-independent mechanism.
Results: Expression of PPARγ coactivator 1α (PGC1α) was increased by relaxin through distinct signaling pathways.
Conclusion: The mechanism for relaxin regulation of PPARγ is through increased PGC1α levels.
Significance: Discovery of new targets of relaxin signaling may provide further understanding of its pleiotropic effects.
Keywords: cAMP Response Element-binding Protein (CREB), p38 MAPK, Peroxisome Proliferator-activated receptor (PPAR), Peroxisome Proliferator-activated Receptor γ Coactivator 1-α (PGC-1a)(PPARGC1A), Protein Kinase A (PKA), Relaxin, Relaxin Family Peptide Receptor 1 (RXFP1)
Abstract
Relaxin activation of its receptor RXFP1 triggers multiple signaling pathways. Previously, we have shown that relaxin activates PPARγ transcriptional activity in a ligand-independent manner, but the mechanism for this effect was unknown. In this study, we examined the signaling pathways of downstream of RXFP1 leading to PPARγ activation. Using cells stably expressing RXFP1, we found that relaxin regulation of PPARγ activity requires accumulation of cAMP and subsequent activation of cAMP-dependent protein kinase (PKA). The activated PKA subsequently phosphorylated cAMP response element-binding protein (CREB) at Ser-133 to activate it directly, as well as indirectly through mitogen activated protein kinase p38 MAPK. Activated CREB was required for relaxin stimulation of PPARγ activity, while there was no evidence for a role of the nitric oxide or ERK MAPK pathways. Relaxin increased the mRNA and protein levels of the coactivator protein PGC1α, and this effect was dependent on PKA, and was completely abrogated by a dominant-negative form of CREB. This mechanism was confirmed in a hepatic stellate cell line stably that endogenously expresses RXFP1. Reduction of PGC1α levels using siRNA diminished the regulation of PPARγ by relaxin. These results suggest that relaxin activates the cAMP/PKA and p38 MAPK pathways to phosphorylate CREB, resulting in increased PGC1α levels. This provides a mechanism for the ligand-independent activation of PPARγ in response to relaxin.
Introduction
Relaxin is a polypeptide hormone that belongs to the insulin-like family of peptides (1, 2). The earliest functions attributed to relaxin involved pregnancy, due to detection of relaxin in the circulation during the first trimester in humans, and its involvement in implantation, and additional functions which vary among species. However, now a number of the relaxin non-reproductive functions have been elucidated, including regulation of vascular tone, vasodilation, fibrosis prevention, and reversal, and action as a neuropeptide (1). The first relaxin receptors identified were the leucine-rich G protein-coupled receptors 7 and 8 (LGR7 and LGR8),3 later renamed relaxin family peptide receptor (RXFP) 1 and 2, respectively (3, 4). Relaxin has higher affinity for RXFP1 as compared with RXFP2 in vitro, but has only been shown to activate RXFP1 in vivo, and therefore RXFP1 is considered the cognate relaxin receptor (5). Upon activation, RXFP1 couples to multiple G-proteins including Gs, Go, and Gi, to modulate cAMP levels through a complex mechanism involving phosphatidylinositide 3′-kinase (PI3K) and protein kinase Cζ (5–8). In addition to increased cAMP production and activation of cAMP-dependent protein kinase (PKA), relaxin activation of RXFP1 has been tied to other signaling pathways, including tyrosine kinase, the mitogen-activated protein kinase ERK1/2, nitric oxide, and glucocorticoid activation (2, 9–11). Thus, relaxin has shown diverse and complex signaling. We recently identified another signaling pathway activated by relaxin, involving peroxisome proliferator-activated receptor gamma (PPARγ) (12).
The peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily. To date, three PPAR isoforms have been identified, including PPARα, PPARβ/δ, and PPARγ (13). The PPARs function as transcription factors with their heterodimerization partners the retinoid X receptors. Upon ligand binding, the DNA binding domains of the PPARs recognize discrete PPAR response elements (PPRE) in target gene promoter regions (14, 15). Collectively, the PPARs act to regulate glucose and lipid metabolism, insulin sensitivity, inflammation, adipogenesis, vasculature function, and tissue remodeling (13). Agonists of PPARγ regulate adipogenesis and insulin sensitivity, and are widely used clinically as antidiabetic medications. However, PPARγ also has anti-inflammatory and antifibrotic effects in several tissues (16–21). Many of these effects are similar to those induced by relaxin. This led us to examine whether PPARγ was involved in the cellular effects of relaxin. We found that relaxin activated PPARγ transcriptional activity and activated PPARγ target genes in fibroblasts expressing RXFP1 (12). More recently, it was found that relaxin stimulation of brain parenchymal arteriole remodeling also involved PPARγ activation (22, 23).
Despite these recent advances, a number of issues remain unclear. The signaling pathways triggered by RXFP1 to activate PPARγ are currently unknown. Furthermore, our studies indicated that in fibroblasts expressing RXFP1, relaxin activated PPARγ through a ligand-independent manner (12), but the mechanism for this activation is unknown. In this study, we sought to define signaling pathways downstream of RXFP1 leading to activation of PPARγ. Moreover, we have explored the mechanism for the ligand-dependent activation of PPARγ by relaxin, and have identified the coactivator protein PPARγ coactivator 1α (PGC1α) as a major player in this activation.
EXPERIMENTAL PROCEDURES
Cells and Transfections
The HEK-RXFP1 cells are HEK-293T cells stably expressing RXFP1 described previously (12). For reporter assays, the HEK-RXFP1 cells were transfected using Fugene6 (Roche) as described previously (12). The plasmids included ACO-PPRE-luc (24) provided by Brian Seed (Harvard University), PGC1α-promoter luciferase reporter (25) and plasmids encoding PKI and inactive mutant (PKImut) (26) from Addgene, dominant-negative CREB (KCREB, Clontech, Mountain View, CA), cAMP response element reporter pGL4.29 (CRE-luc), and Renilla luciferase plasmid (pTK-RL) (all from Promega). For cotransfection of siRNA with reporter plasmids, Dharmafect Duo reagent (Invitrogen) was used according to the manufacturer's instructions. The siRNA used were Silencer Select human PGC1α (s21395), PKA (s11065), p38-MAPK (s3585), or nontargeting negative control (Invitrogen).
The LX2 cells are a human hepatic stellate cell line (27), and were provided by Dr. S. L. Friedman (Mount Sinai School of Medicine, New York). The LX2 cells were transfected using a Nucleofector 4D device (Lonza) using reagent S.E. and program EW-113. The ACO-PPRE-luc and pTK-RL plasmids described above were found to be unsuitable for the LX2 cells. Therefore, a new PPRE-luciferase reporter (pGL4–3xPPRE-luc) was constructed. Briefly, a DNA fragment containing three tandem copies of the PPAR response element (28) was inserted between the KpnI and HindIII sites of the pGL4.26 vector (Promega) containing a minimal promoter upstream of the firefly luciferase gene. The Renilla luciferase plasmid used was an SV40-driven construct (pGL4.72, Promega).
Cells were treated with purified porcine relaxin (kindly provided by Dr. O. David Sherwood, University of Illinois at Urbana-Champaign) as indicated in the figure legends. Relaxin was used at a concentration of 1 nm, which is in the range sufficient for activation of canonical cAMP-related pathways through RXFP1 (2). Forskolin, the p38 MAPK inhibitor PD-169316, the PKA inhibitor H89, the PI3K inhibitor LY294002, the ERK1/2 inhibitor PD98059, the nitric-oxide synthase inhibitor L-NAME, and pertussis toxin were purchased from EMD Biochemicals. The cyclic nucleotides Sp-6-Bnz-cAMPS and Sp-8-pCPT-2′-O-Me-cAMPS were from Biolog. The nitric oxide donors SNAP (S-nitroso-N-acetyl-d,l-penicillamine) and GSNO (S-nitrosoglutathione) were from Sigma.
Reporter Assays
Cells were cotransfected with firefly and Renilla luciferase reporter plasmids as described above. After 24 h, cells were treated as described for the individual figures, then assays were performed 48 h after transfection. Activation of reporter constructs was monitored using the Dual-Glo Luciferase assay (Promega) and measured in a Spectramax M5 plate reader. The data are expressed as the firefly luciferase activity normalized to the Renilla luciferase in the same sample.
Quantitative Real-time PCR and Conventional RT-PCR
Total cellular RNA was extracted using the Purelink RNA Mini kit (Invitrogen), and treated with RNase-free DNase to remove genomic DNA. The RNA concentration was measured using the Ribogreen assay (Invitrogen). A total of 2 μg of RNA was reverse transcribed to cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA) in a total volume of 20 μl. The cDNA was then subjected to TaqMan real-time PCR. The reaction mixtures were comprised of 2 μl of cDNA (diluted 1:15), 10 μl of Taqman universal PCR master mix, 2.5 μl Taqman primer/probe mix in a final volume of 20 μl per reaction. The human gene expression assays (Applied Biosystems) used were PGC1α (Hs00173304_m1) and TATA binding protein (TBP) (4333769F). Gene expression was normalized to the level of TBP within each sample using the relative ΔΔCT method. Gene expression is shown as relative expression to control. The data shown are representative of three independent experiments. For conventional RT-PCR analysis, total RNA was extracted and cDNA prepared as above from HEK-RXFP1, THP1, HepG2, and LX2 cells. Expression of RXFP1 was determined using intron-spanning primers for human RXFP1 (sense 5′-TCTTGGTATTAATTTGGCCGC-3′, antisense 5′-CATTAACTCAGGTGGCATCTCC-3′, or GAPDH (sense 5′-ACCACAGTCCATGCCATCAC-3′, antisense 5′-TCCACCACCCTGTTGCTGTA-3′) as a loading control.
cAMP and PKA Assays
The production of cAMP production was measured by treating the cells with the indicated concentrations of relaxin in the presence of the phosphodiesterase inhibitors IBMX (0.5 mm) and Ro20–1724 (0.1 mm) for 30 min at room temperature. The level of cAMP was measured using the cAMP-Glo assay (Promega). The level of PKA activation was determined using the Pep-Tag assay (Promega) according to the manufacturer's instructions.
Western Blots
Cells were treated as described for the individual experiments, using mammalian protein extraction reagent (M-PER, Pierce, Thermo Scientific, Rockford, IL) containing a protease & phosphatase inhibitor mixture (Pierce). The lysates were sonicated briefly to rupture the cell and nuclear membranes. The protein concentration was estimated by the BCA assay (Pierce), and lysates were heated in SDS loading buffer at 95 °C for 5 min. The proteins were resolved on 10% polyacrylamide gels and transferred to PVDF membranes. The membranes were blocked with Odyssey blocking buffer (Li-Cor, Lincoln, NE), then incubated with diluted primary antibody overnight at 4 °C. The primary antibodies used and their dilutions were total p38 MAPK (9228, 1:1000), phospho-p38 MAPK (Thr-180/Tyr-182) (4511, 1:1000), CREB (9104, 1:1000), phospho-CREB (Ser-133) (9198, 1:1000), or PKA (4282, 1:1000), all from Cell Signaling Technology, Danvers, MA), PGC1α (101707, 1:500, Cayman Chemical, Ann Arbor, MI), and GAPDH (MAB374, Millipore, Temecula CA). After a 1-hour incubation with fluorescently tagged secondary a-antibodies (Li-Cor, Lincoln, NE), the membranes were scanned on an Odyssey infrared scanner (Li-Cor).
Statistical Analysis
Curve-fitting and statistical analysis was performed using Prism5 software (GraphPad, La Jolla, CA). Differences were analyzed using one-way or two-way analysis of variance as appropriate, with the Bonferonni post-test. Data are expressed as mean ± S.E. of at least three independent determinations as indicated in the figure legends.
RESULTS
Relaxin Activates PKA to Activate PPAR Activity
One major signaling pathway triggered by relaxin is via induction of cAMP production and activation of cAMP-activated protein kinase (PKA). To examine the role of cAMP/PKA in activation of PPARγ, HEK-293 cells stably expressing RXFP1 (HEK-RXFP1 cells) were used. We previously showed that relaxin increased cAMP production and activation of a PPAR response element (PPRE) via activation of PPARγ in these cells, and that these responses were dependent on RXFP1 (12). Relaxin also increased PKA activity in these cells, which was blocked by treatment with the PKA inhibitor H89 (Fig. 1A). Using a PPRE reporter, treatment of HEK-RXFP1 cells with either forskolin, a direct adenylyl cyclase activator, or relaxin, increased transcriptional activity through a PPAR response element (PPRE) reporter, and in both cases this activity was inhibited by the PKA inhibitor H89 (Fig. 1B). These data confirm that PKA is activated in response to relaxin, and that the cAMP/PKA pathway is involved in activation of PPAR transcriptional activity in these cells.
FIGURE 1.

The cAMP/PKA pathway activates transcription through a PPRE reporter. A, HEK-RXFP1 cells were pretreated for 30 min in the presence of indicated concentration of the PKA inhibitor H89, then treated 30 min with 1 nm relaxin, and PKA activity was measured by phosphorylation of a PKA substrate. B, HEK-RXFP1 cells transfected with PPRE and Renilla luciferase reporters were pretreated 30 min in the absence and presence of H89 (20 μm), then treated with 10 μm forskolin or 1 nm relaxin for 24 h prior to dual luciferase assay. Data are presented as PPRE luciferase assay relative to control, mean ± S.E. *, p < .05; **, p < .01; ***, p < .001, n = 3. C, white bars: HEK-RXFP1 cells transfected with PPRE and Renilla luciferase reporters were treated for 24 h with 10 μm forskolin, 100 μm Sp-6-Bnz-cAMPS, 10 μm Sp-8CPT-cAMPS, 100 μm SNAP, or 500 μm GSNO, then subject to dual luciferase assay. Black bars: cells were pretreated for 30 min with 20 μm H89, 1 μg/ml PTX, 2 μm LY294002, 20 μm PD169316, 10 μm PD98059, or 100 μm L-NAME, then stimulated with 1 nm relaxin for 24 h. Data are presented as luciferase activity relative to control, mean ± S.E., n = 3. *, p < 0.05 compared with untreated control; †, p < 0.05 compared with relaxin alone. D, HEK-RXFP1 cells were cotransfected with plasmids encoding PKI or an inactive PKI mutant (PKImut) and PPRE and Renilla luciferase reporters, then treated with relaxin as described above, then subject to dual luciferase assay. ***, p < 0.001; n = 3. E, Western blot showing knockdown of PKA or p38 MAPK after treatment with siRNA. F, HEK-RXFP1 cells were cotransfected with PKA siRNA or nontargeting control siRNA and PPRE and Renilla luciferase reporters, then treated with relaxin as described above, then subject to dual luciferase assay. ***, p < 0.001, n = 3.
Because relaxin activates multiple signaling pathways in cells expressing RXFP1, a panel of activators and inhibitors of various pathways was used to screen for potential mechanisms for relaxin-induced PPARγ activation (Fig. 1C). Both forskolin and the PKA-specific activator Sp-6-Bnz-cAMPS caused increased PPRE transcriptional activity, but the exchange protein activated by cAMP (Epac) activator Sp-8-CPT-cAMPS had no effect. The relaxin-stimulated PPRE activation was completely inhibited by H89, and partially inhibited by pertussis toxin and the PI3K inhibitor LY294002. The ERK1/2 inhibitor PD98059 had no influence on the relaxin effect, but the p38 MAPK inhibitor PD169316 partially blocked the effect. The nitric oxide donors (SNAP and GSNO) did not stimulate transcriptional activity, and the nitric-oxide synthase inhibitor L-NAME did not inhibit the ability of relaxin to activate PPARγ, suggesting that the nitric oxide is not a mediator of the relaxin effects.
p38 MAPK Is a Downstream Mediator of PKA-induced PPAR Activity
Because the p38 MAPK inhibitor reduced the relaxin effect (Fig. 1C), this pathway was studied in more detail. The p38 MAPK inhibitor caused a partial decrease in the activation of the PPRE reporter by relaxin (Fig. 2A), but had no significant effect on the baseline levels. To determine whether relaxin causes phosphorylation and activation of p38 MAPK, HEK-RXFP1 were treated with relaxin for various periods of time, and phosphorylation of p38 MAPK was monitored (Fig. 2B). Relaxin caused a rapid and transient increase in p38MAPK phosphorylation that was maximal between 5 and 15 min, with a return to baseline levels with increased time. Furthermore, the relaxin-induced p38 MAPK activation was completely blocked by treatment with H89 (Fig. 2C), suggesting that p38 MAPK is activated downstream of PKA.
FIGURE 2.

p38 MAPK is a downstream mediator of PKA-induced PPAR activity. A, HEK-RXFP1 cells transfected with PPRE and Renilla luciferase reporters were pretreated 30 min with the p38 MAPK inhibitor PD169316 (20 μm) or vehicle, then treated for 24 h with or without 1 nm relaxin then subject to PPRE luciferase assay. The data are expressed as the PPRE luciferase activity relative to untreated cells, mean ± S.E., n = 3. *, p < 0.05; **, p < 0.01. B, HEK-RXFP1 cells were treated with 1 nm relaxin for the indicated times, and lysates were analyzed by Western blot for total and phosphorylated p38 MAPK. C, cells were pretreated 30 min with 20 μm H89 or vehicle, then treated with 1 nm relaxin for 15 min, and lysates were analyzed by Western blot for total and phosphorylated p38 MAPK. D, HEK-RXFP1 cells were cotransfected with p38 MAPK siRNA or nontargeting control siRNA and PPRE and Renilla luciferase reporters, then treated with relaxin as described above, then subject to dual luciferase assay. *, p < .05; ***, p < .001, n = 3. E, cells were cotransfected with PKA siRNA or nontargeting control siRNA, then treated with relaxin for 15 min before Western blot analysis for phosphorylated and total p38 MAPK.
CREB Mediates Relaxin-induced PPAR Activity
A common downstream target of both PKA and p38MAPK activation is cAMP response element-binding protein (CREB). Relaxin treatment of HEK-RXFP1 cells stimulated CREB transcriptional activity of HEK-RXFP1 cells transfected with a CREB luciferase reporter (Fig. 3A). Furthermore, overexpression of the dominant-negative CREB (KCREB) resulted in a complete loss of relaxin-induced PPRE activation (Fig. 3B). Treatment with relaxin resulted in phosphorylation of CREB at Ser-133 (Fig. 3C). The phosphorylation of CREB in response to relaxin was completely blocked by a PKA inhibitor, and partially blocked by the p38 MAPK inhibitor. The partial reduction by the p38 MAPK inhibitor of CREB phosphorylation (36.0 ± 2.1%) compares favorably with the decrease in relaxin-stimulated PPRE activity (47.4 ± 2.7%, Fig. 2A). Together, these data suggest that relaxin causes activation of CREB downstream of PKA and p38 MAPK.
FIGURE 3.
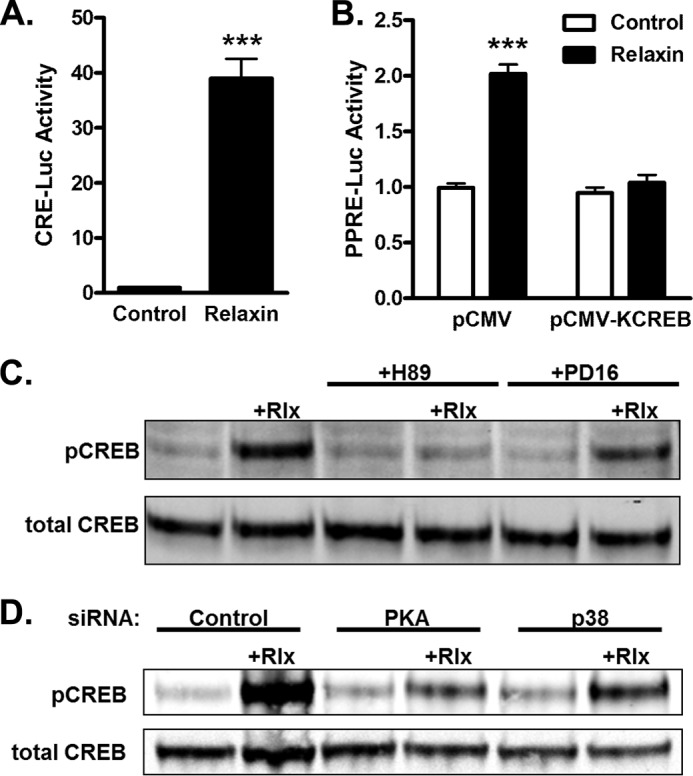
CREB mediates relaxin-induced PPAR activity. A, HEK-RXFP1 cells were transfected with CRE and Renilla luciferase reporters. The cells were treated for 24 h with or without 1 nm relaxin and subjected to dual luciferase assay. B, HEK-RXFP1 cells were transfected with the dominant- negative CREB construct (pCMV-KCREB), or empty vector (pCMV), and the PPRE and Renilla luciferase reporters. The cells were treated for 24 h with or without 1 nm relaxin and subjected to dual luciferase assay. The data are expressed as the luciferase activity relative to untreated cells, mean ± S.E., n = 3. ***, p < 0.001 compared with untreated control. C, HEK-RXFP1 cells were pretreated for 30 min with 20 μm H89, 20 μm PD169316 (PD16), or vehicle, then treated 30 min in the presence or absence of 1 nm relaxin. Lysates were analyzed by Western blotting for total and phosphorylated CREB. C, HEK-RXFP1 cells were transfected with PKA, p38 MAPK, or nontargeting siRNA, then were treated 30 min in the presence or absence of 1 nm relaxin. Lysates were analyzed by Western blotting for total and phosphorylated CREB.
PGC1α Is the Mechanism for Activation of PPARγ Activity by Relaxin
We have shown earlier that relaxin activation of PPARγ in HEK-RXFP1 cells occurs by a ligand-independent mechanism (12). The PPARγ coactivator PGC1α is capable of activating PPARγ in the absence of PPARγ ligand, and furthermore is regulated by both cAMP/PKA and p38 MAPK in a number of cells (29, 30). Therefore, the effect of relaxin on the expression level of PGC1α was tested. Relaxin increased the RNA expression level of PGC1α in HEK-RXFP1 cells (Fig. 4A). The increased RNA expression was evident by 2 h of treatment, and started decreasing by 4 h. This increase in expression level was mirrored in cells expressing a reporter containing the PGC1α promoter upstream of luciferase (Fig. 4B). We also examined the PGC1α protein level, and found that relaxin caused an increase in PGC1α protein by 4.5 h of treatment (Fig. 4C). The increased expression of PGC1α by relaxin was diminished by the PKA inhibitor H89 (Fig. 4D). Furthermore, coexpression of the dominant-negative KCREB construct decreased the relaxin-stimulated increase in PGC1α protein levels (Fig. 4E). The involvement of PGC1α in the ability of relaxin to stimulate PPARγ activation was examined using siRNA-mediated knockdown of PGC1α (Fig. 4, F and G). The PGC1α-siRNA reduced the stimulation of PPARγ activity by relaxin, while control siRNA had no effect, suggesting that PGC1α mediates the effect of relaxin on PPARγ. Overall, these results suggest that relaxin increases PGC1α expression through PKA and CREB activation.
FIGURE 4.

PGC1α is the mechanism for activation of PPARγ activity by relaxin. A, HEK-RXFP1 cells were treated at the indicated times with 1 nm relaxin, and the mRNA for PGC1α was quantified by real time RT-PCR relative to that of TBP. Data are expressed as fold expression of PGC1α compared with untreated cells. B, HEK-RXFP1 cells were transfected with PGC1α-promoter and Renilla luciferase reporters, treated 24 h with 1 nm relaxin, then subject to dual-luciferase assay. C, HEK-RXFP1 cells were treated with 1 nm relaxin for the indicated times, and lysates were analyzed by Western blotting PGC1α and GAPDH as indicated. D, HEK-RXFP1 cells were pretreated 20 μm H89 or vehicle, then incubated for 2 h in the presence or absence of 1 nm relaxin. The mRNA for PGC1α was quantified by real time RT-PCR relative to that of TBP. Data are expressed as fold expression of PGC1α compared with untreated cells. E, HEK-RXFP1 cells were transfected with KCREB or control plasmid, then cells were treated with 1 nm relaxin for 4.5 h. Lysates were analyzed by Western blotting for PGC1α and GAPDH as indicated. F and G, HEK-RXFP1 cells were cotransfected with control or PGC1α siRNA, PPRE and Renilla reporter vectors. After 24 h, the levels of PGC1α were reduced as determined by Western blot (F). Cells were then treated with or without 1 nm relaxin for 24 h and subjected to dual luciferase assay (G). The data are expressed as the luciferase activity relative to untreated cells, mean ± S.E., n = 3. *, p < 0.05; **, p < 0.01.
Relaxin Regulates PGC1α in Cells Endogenously Expressing RXFP1
The signaling pathways activated by relaxin in HEK-RXFP1 cells were tested in cells naturally expressing RXFP1. In the liver, the myofibroblastic hepatic stellate cells are the predominant RXFP1-expressing cells (31, 32). A human hepatic stellate cell line (LX2) that recapitulates many of the properties of activated hepatic stellate cells (27) was used. Relaxin induced an increase in cAMP levels (Fig. 5A) and activated PKA (Fig. 5B), confirming that LX2 cells are responsive to relaxin. Expression of RXFP1 was detected in LX2 cells at a modest level compared with HEK-RXFP1 cells or THP1 monocytes previously shown to express high levels of RXFP1 (Fig. 5C). Relaxin treatment resulted in an increase in transcription through the PPRE reporter, consistent with activation of PPARγ (Fig. 5D). Similar to the results using HEK-RXFP1 cells, relaxin stimulated phosphorylation of p38 MAPK (Fig. 5E). Furthermore, relaxin stimulated phosphorylation of CREB that was reduced by both H89 and the p38 MAPK inhibitor (Fig. 5F). The degree of reduction of relaxin-stimulated CREB phosphorylation with by the p38 MAPK inhibitor (38.4 ± 4.8%) compares favorably to the response seen in HEK-RXFP1 cells. Finally, the expression level of PGC1α was increased in response to relaxin (Fig. 5G). Taken together, these results confirm that the signaling pathways leading to PGC1α expression and PPARγ activation are intact in LX2 cells endogenously expressing RXFP1.
FIGURE 5.

Relaxin regulates PGC1α in LX2 cells endogenously expressing RXFP1. A, LX2 cells were treated with or without 1 nm relaxin in the presence of the phosphodiesterase inhibitors IBMX (0.5 mm) and imidazolidone (0.1 mm) for 30 min, then cAMP levels were determined. B, cells were pretreated with 20 μm H89 for 30 min, then treated 30 min with 1 nm relaxin. PKA activity was measured by phosphorylation of a PKA substrate. C, total RNA was extracted from THP1, HEK-RXFP1, HepG2, and LX2 cells, and subject to RT-PCR for detection of RXFP1 or GAPDH transcripts. D, LX2 cells were transfected with the PPRE and Renilla luciferase reporters, then treated with or without 1 nm relaxin for 24 h and subjected to dual luciferase assay. The data are expressed as the luciferase activity relative to untreated cells. E, cells were treated with 1 nm relaxin for the indicated times, then lysates were analyzed by Western blotting for total and phosphorylated p38 MAPK. F, LX2 cells were pretreated 30 min with 20 μm H89, 20 μm PD169316 (PD16), or vehicle, then treated with or without 1 nm relaxin for 30 min. Lysates were analyzed by Western blotting for total and phosphorylated CREB. G, LX2 cells were treated with 1 nm relaxin for 2 h, and the mRNA for PGC1α was quantified by real time RT-PCR relative to that of TBP. Data are expressed as fold expression of PGC1α compared with untreated cells. Mean ± S.E., n = 3. **, p < .01; ***, p < .001.
DISCUSSION
We have previously shown that relaxin treatment of HEK-RXFP1 cells caused increased transcriptional activity of PPARγ (12). This activation did not require addition of an exogenous PPARγ ligand, nor was it inhibited by a PPARγ ligand-binding inhibitor, suggesting a ligand-independent mechanism of activation. Recently, another study showed relaxin activation of PPARγ in parenchymal brain arterioles, where it regulated hypertrophic outward remodeling (22). In contrast to activation of PPARγ in HEK-RXFP1 cells, the effect in brain arterioles appears to be ligand-dependent, suggesting that the mechanisms of relaxin regulation of PPARγ are tissue-specific. We sought to elucidate the mechanisms for the ligand-independent activation of PPARγ in HEK-RXFP1 cells, and in a cell line naturally expressing RXFP1, the LX2 hepatic stellate cells.
Relaxin binding to RXFP1 activates a number of signal transduction pathways, the most completely studied of which is through cAMP and PKA. Depending on the cell type, RXFP1 couples to multiple G-proteins to activate these pathways. In a number of cells, including the stably transfected HEK-RXFP1 cells, and some cells endogenously expressing RXFP1 such as THP-1 and HeLa cells, RXFP1 alters cAMP levels in a biphasic manner, initially coupling to Gs to activate adenylyl cyclase to increase cAMP levels, followed by coupling to Go to decrease cAMP, followed by a delayed activation of adenylyl cyclase by Gi coupling and activation of phosphoinositide 3′-kinase (PI3K) and protein kinase-Cζ (PKCζ). Other cells retain the coupling to Gs, but do not have the delayed phase of cAMP elevation. We found that the relaxin effect on PPARγ activity was mimicked by direct adenylyl cyclase activation using forskolin, or by the specific PKA activator Sp-6-Bnz-cAMPS, and was inhibited by the PKA inhibitor H89. In contrast, a specific activator of Epac, another mediator of cAMP in some pathways, had no effect, suggesting that PKA alone was involved. Both pertussis toxin and a PI3K inhibitor partially blocked the effect of relaxin, suggesting that the delayed phase of cAMP production mediated by Go coupling by RXFP1 was at least partially responsible for relaxin's effects.
Depending on the cell type, other pathways are triggered by RXFP1, including nitric oxide, ERK1/2 and p38 MAPK (1, 2). Importantly, all of these pathways have been implicated in the regulation of PPARγ (33–35). In a number of cell types, activation results in activation of various nitric-oxide synthases to mediate physiological responses (2, 10, 36). However, in the present study, activation of PPARγ was not stimulated by the addition of nitric oxide donors, nor was the effect of relaxin reduced with a nitric-oxide synthase inhibitor, suggesting that the nitric oxide pathway was not involved in regulation of PPARγ activation. Similarly, ERK1/2 inhibitor had no effect on relaxin-stimulated PPARγ activity, suggesting that this pathway is not involved in this effect. There have been few studies of the role of p38 MAPK in mediating relaxin effects. In human periodontal ligament cells, relaxin regulation of matrix metalloproteinase expression was reduced by a p38 MAPK inhibitor (37), and relaxin potentiated BNP2-mediated p38 MAPK activation in multipotent mesenchymal cells (38). Relaxin induced activation of p38 MAPK in vascular smooth muscle cells, but not HeLa or vascular endothelial cells (39) or renal fibroblasts (40). In the present study, a p38 MAPK inhibitor partially blocked relaxin's effects, and relaxin treatment rapidly and transiently induced phosphorylation of p38 MAPK, which returned to baseline by 30 min. Therefore, it is possible that the earlier studies, which employed a much longer period of treatment (45–60 min), may have missed the peak of p38 MAPK phosphorylation in those cells. The phosphorylation of p38 MAPK in HEK-RXFP1 was downstream of PKA, as it was completely blocked by H89.
A major effector of gene transcription downstream of cAMP/PKA signaling is CREB. Relaxin treatment of human endometrial stromal cells resulted in rapid and transient phosphorylation of CREB (41). In HEK-293 cells stably expressing RXFP1, relaxin induced activation of a reporter plasmid through a CRE response element (42). We found that CREB was phosphorylated within 20–30 min of relaxin treatment, and this effect was completely inhibited by PKA inhibition. In addition, CREB phosphorylation was partially blocked by a p38 MAPK inhibitor, suggesting a bifurcation in signaling at PKA, through p38 MAPK and converging at CREB. This concept was further supported by the use of the dominant negative form of CREB, which completely inhibited PPRE activation in response to relaxin.
Previously, we reported that relaxin activation of PPARγ was ligand-independent, but the mechanism was unknown. The transcriptional activity of PPAR can be regulated by phosphorylation. We found no evidence of altered PPARγ phosphorylation in response to relaxin (data not shown). Another mechanism is by association with coactivator and corepressor proteins. Of particular interest was PGC1α, which was discovered as a coactivator of PPARγ in brown adipose tissue, where it acts to regulate proteins involved in thermogenic programming, such as uncoupling protein-1 (43). PGC1α is capable of ligand-independent binding and activation of PPARγ (44). Furthermore, PKA, p38-MAPK and CREB all participate in activation of PGC1α. In muscle and liver, increased PGC1α expression was triggered by cAMP/PKA or p38 MAPK activation, and in many cases, CREB was a found to be a downstream target of p38 MAPK (25, 45–49). The expression of PGC1α is regulated by CREB through a consensus CRE sequence in the PGC1α promoter (25, 45). Therefore, PGC1α was possible mechanism for ligand-independent activation of PPARγ. We found that PGC1α mRNA and protein were increased in response to relaxin treatment in HEK-RXFP1 cells. This effect was reduced by a PKA inhibitor or a dominant-negative form of CREB. Furthermore, reduction of PGC1α levels with siRNA diminished the ability of relaxin to stimulate PPARγ activity. Taken together, these data strongly suggest that increased PGC1α is the mechanism for the ligand-independent activation of PPARγ by relaxin.
As discussed earlier, cells expressing RXFP1 differ in their signaling properties in response to relaxin, and it is important to compare results in cells with induced expression (such as HEK-RXFP1 cells) with cells endogenously expressing RXFP1 (50, 51). We previously showed that primary rat and mouse hepatic cells expressed RXFP1, and responded to relaxin (31, 52). In this study, we used LX2 cells, a human hepatic stellate cell line that recapitulates many of the properties of primary cells (27). Relaxin treatment caused an elevation of cAMP and activation of PKA, and RXFP1 mRNA was detected in LX2 cells, confirming that RXFP1 is expressed and active in these cells. Relaxin also activated transcription through the PPRE reporter vector. Both p38-MAPK and CREB were phosphorylated in response to relaxin, and CREB phosphorylation was reduced completely by a PKA inhibitor and partially by a p38-MAPK inhibitor. Finally, PGC1α expression was increased by relaxin treatment of LX2 cells. These data suggest that the RXFP1 signaling pathways leading to PGC1α expression are similar in both HEK-RXFP1 and LX2 cells.
In summary, we have provided the first evidence of ligand-independent activation of PPARγ by relaxin. The overall scheme is shown in Fig. 6. Relaxin activates RXFP1 which stimulates cAMP through PI3K-dependent and independent pathways to generation to activate PKA. This leads to phosphorylation of CREB, directly via PKA and indirectly by p38-MAPK. Activation of CREB leads to expression of PGC1α, which associates with PPARγ to increase its transcriptional activity. It is important to note that PGC1α is also phosphorylated by a number of kinases, including PKA and p38-MAPK, to regulate transcriptional activity and protein stability (29, 30). Therefore, although we found that PGC1α gene expression was up-regulated in response to relaxin, we cannot rule out a role for PGC1α phosphorylation in mediating relaxin's effects. Nevertheless, these studies provide the first evidence of PGC1α as a target of relaxin signaling, and provide the mechanism for the ligand-independent activation of PPARγ by relaxin.
FIGURE 6.

Schematic representation of relaxin signaling pathways leading to activation of PPARγ. Relaxin binding to RXFP1 results in a rapid activation of adenylyl cyclase (AC), followed by a delayed phase that involves PI3K. This results in elevated cAMP levels and activation of PKA. The activated PKA phosphorylates CREB, through p38 MAPK-dependent and -independent mechanisms. The phosphorylated CREB then increases PGC1α gene expression. The elevated level of PGC1α then increases the ligand-independent transcriptional activity of PPARγ.
Acknowledgments
We thank Aaron Hsueh (Stanford University), O. David Sherwood (University of Illinois at Urbana-Champaign), Brian Seed (Harvard University), and Scott Friedman (Mt. Sinai School of Medicine) for providing critical reagents and cells.
This work was supported, in whole or in part, by funding through the Dept. of Veterans Affairs Biomedical Laboratory Research & Development Program (BX000849), National Institutes of Health NIAAA (R01 AA015509), the Bly Memorial Research Fund (to R. G. B.), and by a University of Nebraska Medical Center Graduate College Fellowship (to S. S.).
- LGR
- leucine-rich G protein-coupled receptor
- PPARγ
- peroxisome proliferator-activated receptor γ
- PGC1α
- PPARγ coactivator 1α
- RXFP1
- relaxin family peptide receptor 1
- PKA
- cAMP-dependent protein kinase
- MAPK
- mitogen-activated protein kinase
- CREB
- cAMP response element-binding protein
- PI3K
- phosphoinositide 3′-kinase
- PKCζ
- calcium-dependent protein kinase Cζ
- PPRE
- peroxisome proliferator response element
- SNAP
- S-nitroso-N-acetyl-d,l-penicillamine
- GSNO
- S-nitrosoglutathione.
REFERENCES
- 1. Sherwood O. D. (2004) Relaxin's Physiological Roles and Other Diverse Actions. Endocr. Rev. 25, 205–234 [DOI] [PubMed] [Google Scholar]
- 2. Bathgate R. A., Halls M. L., van der Westhuizen E. T., Callander G. E., Kocan M., Summers R. J. (2013) Relaxin family peptides and their receptors. Physiol. Rev. 93, 405–480 [DOI] [PubMed] [Google Scholar]
- 3. Hsu S. Y., Nakabayashi K., Nishi S., Kumagai J., Kudo M., Sherwood O. D., Hsueh A. J. W. (2002) Activation of orphan receptors by the hormone relaxin. Science 295, 671–674 [DOI] [PubMed] [Google Scholar]
- 4. Bathgate R. A., Ivell R., Sanborn B. M., Sherwood O. D., Summers R. J. (2006) International Union of Pharmacology LVII: Recommendations for the Nomenclature of Receptors for Relaxin Family Peptides. Pharmacol. Rev. 58, 7–31 [DOI] [PubMed] [Google Scholar]
- 5. Halls M. L., Bathgate R. A., Summers R. J. (2006) Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol. Pharmacol. 70, 214–226 [DOI] [PubMed] [Google Scholar]
- 6. Halls M. L., van der Westhuizen E. T., Bathgate R. A. D., Summers R. J. (2007) Relaxin family peptide receptors - former orphans reunite with their parent ligands to activate multiple signalling pathways. Br. J. Pharmacol. 150, 677–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nguyen B. T., Yang L., Sanborn B. M., Dessauer C. W. (2003) Phosphoinositide 3-kinase activity is required for biphasic stimulation of cyclic adenosine 3′,5′-monophosphate by relaxin. Mol. Endocrinol. 17, 1075–1084 [DOI] [PubMed] [Google Scholar]
- 8. Nguyen B. T., Dessauer C. W. (2005) Relaxin stimulates cAMP production in MCF-7 cells upon overexpression of type V adenylyl cyclase. Ann. N.Y. Acad. Sci. 1041, 296–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ivell R., Heng K., Anand-Ivell R. (2007) Diverse signalling mechanisms used by relaxin in natural cells and tissues: the evolution of a “neohormone”. Adv. Exp. Med. Biol. 612, 26–33 [DOI] [PubMed] [Google Scholar]
- 10. Baccari M. C., Bani D. (2008) Relaxin and nitric oxide signalling. Curr. Protein Pept. Sci. 9, 638–645 [DOI] [PubMed] [Google Scholar]
- 11. Chow B. S., Chew E. G., Zhao C., Bathgate R. A., Hewitson T. D., Samuel C. S. (2012) Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One 7, e42714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh S., Bennett R. G. (2010) Relaxin signaling activates peroxisome proliferator-activated receptor γ. Mol. Cell Endocrinol. 315, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michalik L., Auwerx J., Berger J. P., Chatterjee V. K., Glass C. K., Gonzalez F. J., Grimaldi P. A., Kadowaki T., Lazar M. A., O'Rahilly S., Palmer C. N. A., Plutzky J., Reddy J. K., Spiegelman B. M., Staels B., Wahli W. (2006) International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol. Rev. 58, 726–741 [DOI] [PubMed] [Google Scholar]
- 14. Kliewer S. A., Umesono K., Noonan D. J., Heyman R. A., Evans R. M. (1992) Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 358, 771–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dreyer C., Krey G., Keller H., Givel F., Helftenbein G., Wahli W. (1992) Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 68, 879–887 [DOI] [PubMed] [Google Scholar]
- 16. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 17. Sime P. J. (2008) The antifibrogenic potential of PPARγ ligands in pulmonary fibrosis. J. Investig. Med. 56, 534–538 [DOI] [PubMed] [Google Scholar]
- 18. Genovese T., Cuzzocrea S., Di Paola R., Mazzon E., Mastruzzo C., Catalano P., Sortino M., Crimi N., Caputi A. P., Thiemermann C., Vancheri C. (2005) Effect of rosiglitazone and 15-deoxy-{Δ}12,14-prostaglandin J2 on bleomycin-induced lung injury. Eur. Respir. J. 25, 225–234 [DOI] [PubMed] [Google Scholar]
- 19. Iglarz M., Touyz R. M., Viel E. C., Paradis P., Amiri F., Diep Q. N., Schiffrin E. L. (2003) Peroxisome Proliferator-Activated Receptor-{α} and Receptor-{γ} Activators Prevent Cardiac Fibrosis in Mineralocorticoid-Dependent Hypertension. Hypertension 42, 737–743 [DOI] [PubMed] [Google Scholar]
- 20. Wu M., Melichian D. S., Chang E., Warner-Blankenship M., Ghosh A. K., Varga J. (2009) Rosiglitazone Abrogates Bleomycin-Induced Scleroderma and Blocks Profibrotic Responses Through Peroxisome Proliferator-Activated Receptor-{γ}. Am. J. Pathol. 174, 519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsukamoto H., She H., Hazra S., Cheng J., Miyahara T. (2006) Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J. Gastroenterol. Hepatol. 21, S102—S105 [DOI] [PubMed] [Google Scholar]
- 22. Chan S. L., Cipolla M. J. (2011) Relaxin causes selective outward remodeling of brain parenchymal arterioles via activation of peroxisome proliferator-activated receptor-γ. FASEB J. 25, 3229–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan S. L., Sweet J. G., Cipolla M. J. (2013) Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. FASEB J. 27, 3917–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang C., Ting A. T., Seed B. (1998) PPARg agonists inhibit production of monocyte inflammatory cytokines. Nature 391, 82–86 [DOI] [PubMed] [Google Scholar]
- 25. Handschin C., Rhee J., Lin J., Tarr P. T., Spiegelman B. M. (2003) An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc. Natl. Acad. Sci. U.S.A. 100, 7111–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Day R. N., Walder J. A., Maurer R. A. (1989) A protein kinase inhibitor gene reduces both basal and multihormone-stimulated prolactin gene transcription. J. Biol. Chem. 264, 431–436 [PubMed] [Google Scholar]
- 27. Xu L., Hui A. Y., Albanis E., Arthur M. J., O'Byrne S. M., Blaner W. S., Mukherjee P., Friedman S. L., Eng F. J. (2005) Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54, 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forman B. M., Tontonoz P., Chen J., Brun R. P., Spiegelman B. M., Evans R. M. (1995) 15-Deoxy-Δ12,14-Prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 83, 803–812 [DOI] [PubMed] [Google Scholar]
- 29. Handschin C., Spiegelman B. M. (2006) Peroxisome Proliferator-Activated Receptor {gamma} Coactivator 1 Coactivators, Energy Homeostasis, and Metabolism. Endocr. Rev. 27, 728–735 [DOI] [PubMed] [Google Scholar]
- 30. Fernandez-Marcos P. J., Auwerx J. (2011) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S–890S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bennett R. G., Dalton S. R., Mahan K. J., Gentry-Nielsen M. J., Hamel F. G., Tuma D. J. (2007) Relaxin receptors in hepatic stellate cells and cirrhotic liver. Biochem. Pharmacol. 73, 1033–1040 [DOI] [PubMed] [Google Scholar]
- 32. Fallowfield J. A., Hayden A. L., Snowdon V. K., Aucott R. L., Stutchfield B. M., Mole D. J., Pellicoro A., Gordon-Walker T. T., Henke A., Schrader J., Trivedi P. J., Princivalle M., Forbes S. J., Collins J. E., Iredale J. P. (2014) Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 59, 1492–1504 [DOI] [PubMed] [Google Scholar]
- 33. Lazennec G., Canaple L., Saugy D., Wahli W. (2000) Activation of Peroxisome Proliferator-Activated Receptors (PPARs) by Their Ligands and Protein Kinase A Activators. Mol. Endocrinol. 14, 1962–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ptasinska A., Wang S., Zhang J., Wesley R. A., Danner R. L. (2007) Nitric oxide activation of peroxisome proliferator-activated receptor γ through a p38 MAPK signaling pathway. FASEB J. 21, 950–961 [DOI] [PubMed] [Google Scholar]
- 35. Burns K. A., Vanden Heuvel J. P. (2007) Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 1771, 952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sasser J. M. (2013) The emerging role of relaxin as a novel therapeutic pathway in the treatment of chronic kidney disease. Am. J. Physiol. Regulatory, Integrative Comparative Physiology 305, R559–R565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirate Y., Yamaguchi M., Kasai K. (2012) Effects of relaxin on relapse and periodontal tissue remodeling after experimental tooth movement in rats. Connect Tissue Res. 53, 207–219 [DOI] [PubMed] [Google Scholar]
- 38. Moon J., Oh S., Jeong Y., Kang J., Park J., Son H., Bae S., Park B., Kim M., Koh J., Kim S., Ko H. (2014) Relaxin Augments BMP 2-Induced Osteoblast Differentiation and Bone Formation. J. Bone Mineral Res. 29, 1586–1596 [DOI] [PubMed] [Google Scholar]
- 39. Dschietzig T., Bartsch C., Richter C., Laule M., Baumann G., Stangl K. (2003) Relaxin, a pregnancy hormone, is a functional endothelin-1 antagonist: attenuation of endothelin-1-mediated vasoconstriction by stimulation of endothelin type-B receptor expression via ERK-1/2 and nuclear factor-κB. Circ. Res. 92, 32–40 [DOI] [PubMed] [Google Scholar]
- 40. Heeg M. H., Koziolek M. J., Vasko R., Schaefer L., Sharma K., Müller G. A., Strutz F. (2005) The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 68, 96–109 [DOI] [PubMed] [Google Scholar]
- 41. Zhang Q., Liu S. H., Erikson M., Lewis M., Unemori E. (2002) Relaxin activates the MAP kinase pathway in human endometrial stromal cells. J. Cell Biochem. 85, 536–544 [DOI] [PubMed] [Google Scholar]
- 42. Halls M. L., Bathgate R. A. D., Summers R. J. (2007) Comparison of Signaling Pathways Activated by the Relaxin Family Peptide Receptors, RXFP1 and RXFP2, Using Reporter Genes. J. Pharmacol. Exp. Therap. 320, 281–290 [DOI] [PubMed] [Google Scholar]
- 43. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. (1998) A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 44. Wu Y., Chin W. W., Wang Y., Burris T. P. (2003) Ligand and Coactivator Identity Determines the Requirement of the Charge Clamp for Coactivation of the Peroxisome Proliferator-activated Receptor γ. J. Biol. Chem. 278, 8637–8644 [DOI] [PubMed] [Google Scholar]
- 45. Herzig S., Long F., Jhala U. S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., Spiegelman B., Montminy M. (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183 [DOI] [PubMed] [Google Scholar]
- 46. Collins Q. F., Xiong Y., Lupo E. G., Jr., Liu H., Cao W. (2006) p38 Mitogen-activated Protein Kinase Mediates Free Fatty Acid-induced Gluconeogenesis in Hepatocytes. J. Biol. Chem. 281, 24336–24344 [DOI] [PubMed] [Google Scholar]
- 47. Wright D. C., Geiger P. C., Han D., Jones T. E., Holloszy J. O. (2007) Calcium Induces Increases in Peroxisome Proliferator-activated Receptor γ Coactivator-1α and Mitochondrial Biogenesis by a Pathway Leading to p38 Mitogen-activated Protein Kinase Activation. J. Biol. Chem. 282, 18793–18799 [DOI] [PubMed] [Google Scholar]
- 48. Hong T., Ning J., Yang X., Liu H. Y., Han J., Liu Z., Cao W. (2011) Fine-tuned regulation of the PGC-1alpha gene transcription by different intracellular signaling pathways. Am. J. Physiol. Endocrinol. Metab. 300, E500–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cao W., Collins Q. F., Becker T. C., Robidoux J., Lupo E. G., Jr., Xiong Y., Daniel K. W., Floering L., Collins S. (2005) p38 Mitogen-activated Protein Kinase Plays a Stimulatory Role in Hepatic Gluconeogenesis. J. Biol. Chem. 280, 42731–42737 [DOI] [PubMed] [Google Scholar]
- 50. Halls M. L., Hewitson T. D., Moore X., Du X., Bathgate R. A. D., Summers R. J. (2009) Relaxin Activates Multiple cAMP Signaling Pathway Profiles in Different Target Cells. Ann. N.Y. Acad. Sci. 1160, 108–111 [DOI] [PubMed] [Google Scholar]
- 51. Ivell R., Anand-Ivell R., Bartsch O. (2005) Relaxin signaling from natural receptors. Ann. N.Y. Acad. Sci. 1041, 280–287 [DOI] [PubMed] [Google Scholar]
- 52. Bennett R. G., Kharbanda K. K., Tuma D. J. (2003) Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem. Pharmacol. 66, 867–874 [DOI] [PubMed] [Google Scholar]