

Abstract
Monogenic diseases are frequent causes of neonatal morbidity and mortality, and disease presentations are often undifferentiated at birth. More than 3500 monogenic diseases have been characterized, but clinical testing is available for only some of them and many feature clinical and genetic heterogeneity. Hence, an immense unmet need exists for improved molecular diagnosis in infants. Because disease progression is extremely rapid, albeit heterogeneous, in newborns, molecular diagnoses must occur quickly to be relevant for clinical decision-making. We describe 50-hour differential diagnosis of genetic disorders by whole-genome sequencing (WGS) that features automated bioinformatic analysis and is intended to be a prototype for use in neonatal intensive care units. Retrospective 50-hour WGS identified known molecular diagnoses in two children. Prospective WGS disclosed potential molecular diagnosis of a severe GJB2-related skin disease in one neonate; BRAT1-related lethal neonatal rigidity and multifocal seizure syndrome in another infant; identified BCL9L as a novel, recessive visceral heterotaxy gene (HTX6) in a pedigree; and ruled out known candidate genes in one infant. Sequencing of parents or affected siblings expedited the identification of disease genes in prospective cases. Thus, rapid WGS can potentially broaden and foreshorten differential diagnosis, resulting in fewer empirical treatments and faster progression to genetic and prognostic counseling.
INTRODUCTION
Genomic medicine is a new, structured approach to disease diagnosis and management that prominently features genome sequence information (1). Whole-genome sequencing (WGS) by next-generation sequencing (NGS) technologies has the potential for simultaneous, comprehensive, differential diagnostic testing of likely monogenic illnesses, which accelerates molecular diagnoses and minimizes the duration of empirical treatment and time to genetic counseling (2–7). Indeed, in some cases, WGS or exome sequencing provides molecular diagnoses that could not have been ascertained by conventional single-gene sequencing approaches because of pleiotropic clinical presentation or the lack of an appropriate molecular test (7–9).
Neonatal intensive care units (NICUs) are especially suitable for early adoption of diagnostic WGS because many of the 3528 monogenic diseases of known cause are present during the first 28 days of life (10). In the United States, more than 20% of infant deaths are caused by congenital malformations, deformations, and chromosomal abnormalities that cause genetic diseases (11–13). Although this proportion has remained unchanged for the past 20 years, the precise prevalence of monogenic diseases in NICUs is poorly understood because ascertainment rates are low. Serial gene sequencing is too slow to be clinically useful for NICU diagnosis. Newborn screens, while rapid, identify only a few genetic disorders for which inexpensive tests and cost-effective treatments exist (14, 15). Further complicating diagnosis is the fact that the full clinical phenotype may not be manifest in newborn infants (neonates), and genetic heterogeneity can be immense. Thus, acutely ill neonates with genetic diseases are often discharged or deceased before a diagnosis is made. As a result, NICU treatment of genetic diseases is usually empirical, may lack efficacy, may be inappropriate, or may cause adverse effects.
NICUs are also suitable for early adoption of genomic medicine because extraordinary interventional efforts are customary and innovation is encouraged. Indeed, NICU treatment is among the most cost-effective of high-cost health care, and the long-term outcomes of most NICU subpopulations are excellent (16–18). In genetic diseases for which treatments exist, rapid diagnosis is critical for timely delivery of interventions that lessen morbidity and mortality (14–17, 19, 20). For neonatal genetic diseases without effective therapeutic interventions, of which there are many (21), timely diagnosis avoids futile intensive care and is critical for research to develop management guidelines that optimize outcomes (22). In addition to influencing treatment, neonatal diagnosis of genetic disorders and genetic counseling can spare parents diagnostic odysseys that instill inappropriate hope or perpetuate needless guilt.
Two recent studies exemplify the diagnostic and therapeutic uses of NGS in the context of childhood genetic diseases. WGS of fraternal twins concordant for 3,4-dihydroxyphenylalanine (dopa)–responsive dystonia revealed known mutations in the sepiapterin reductase (SPR) gene (3). In contrast to other forms of dystonia, treatment with 5-hydroxytryptamine and serotonin reuptake inhibitors is beneficial in patients with SPR defects. Application of this therapy in appropriate cases resulted in clinical improvement. Likewise, extensive testing failed to provide a molecular diagnosis for a child with fulminant pan-colitis (extensive inflammation of the colon) (8), in whom standard treatments for presumed Crohn’s disease—an inflammatory bowel disease—were ineffective. NGS of the patient’s exome, together with confirmatory studies, revealed X-linked inhibitor of apoptosis (XIAP) deficiency. The treating physicians had not entertained this diagnosis because XIAP mutations had not previously been associated with colitis. Hemopoietic progenitor cell transplant was performed, as indicated for XIAP deficiency, with complete resolution of colitis. Last, for ~3700 genetic illnesses for which a molecular basis has not yet been established (10), WGS can suggest candidate genes for functional and inheritance-based confirmatory research (23).
The current cost of research-grade WGS is $7666 (24)—which is similar to the current cost of commercial diagnostic dideoxy sequencing of two or three disease genes. Within the context of the average cost per day and per stay in a NICU in the United States (13), WGS in carefully selected cases is acceptable and even potentially cost-saving (3–7). However, the turnaround time for interpreted WGS results, such as that of dideoxy sequencing, is too slow to be of practical use for NICU diagnoses or clinical guidance (typically ~4 to 6 weeks) (2–4). Here, we report a system that permits WGS and bioinformatic analysis (largely automated) of suspected genetic disorders within 50 hours, a time frame that appears to be promising for emergency use in level 3 NICUs.
RESULTS
Symptom- and sign-assisted genome analysis (SSAGA) is a new clinicopathological correlation tool that maps the clinical features of 591 well-established, recessive genetic diseases with pediatric presentations (table S1) to corresponding phenotypes and genes known to cause the symptoms (2, 10). SSAGA was developed for comprehensive automated performance of the following two tasks: (i) WGS analyses restricted to a superset of gene-associated regions relevant to clinical presentations, in accord with published guidelines for genetic testing in children (25–28), and (ii) prioritization of clinical information to assist in the interpretation of WGS results. SSAGA has a menu of 227 clinical terms arranged in nine symptom categories (fig. S1). Standardized clinical terms (29) have been mapped to 591 genetic diseases on the basis of authoritative databases (10, 30) and expert physician reviews. Each disease gene is represented by an average of 8 terms and at most 11 terms (minimum, 1 term, 15 disease genes; maximum, 11 terms, 3 disease genes).
To validate the feasibility of automated matching of clinical terms to diseases and genes, we entered retrospectively the presenting features of 533 children who have received a molecular diagnosis at our institution [Children’s Mercy Hospital (CMH), Kansas City, MO] within the last 10 years into SSAGA. Sensitivity was 99.3% (529), as determined by correct disease and affected gene nominations. Failures included a patient with glucose-6-phosphate dehydrogenase deficiency who presented with muscle weakness [which is not a feature mentioned in authoritative databases (10, 30)]; a patient with Janus kinase 3 mutations who had the term “respiratory infection” in his medical records rather than “increased susceptibility of infections,” which is the description in authoritative databases; and a patient with cystic fibrosis who had the term “recurrent infections” in his medical records rather than “respiratory infections,” which is the description in authoritative databases. SSAGA nominated an average of 194 genes per patient (maximum, 430; minimum, 5). Thus, SSAGA displayed sufficient sensitivity for the initial selection of known, recessive candidate genes in children with specific clinical presentations.
Rapid WGS
To assess our ability to recapitulate known results, we performed rapid WGS retrospectively on DNA samples from two infants with molecular diagnoses that had previously been identified by clinical testing. Then, to assess the potential diagnostic use of rapid WGS, we prospectively performed WGS in five undiagnosed newborns with clinical presentations that strongly suggested a genetic disorder as well as their siblings.
Automation of the five main components of WGS as well as bioinformatics-based gene-variant characterization and clinical interpretation, all in an integrated workflow, made possible ~50-hour time to differential molecular diagnosis of genetic disorders (Fig. 1). Specifically, sample preparation for WGS was shortened from 16 to 4.5 hours, while a physician simultaneously entered into SSAGA clinical terms that described the neonates’ illnesses (fig. S1). For each sample, rapid WGS [2 × 100 base pair (bp) reads, including on-board cluster generation and paired-end sequencing] was performed in a single run on the Illumina HiSeq 2500 and took ~26 hours. Base calling, genomic sequence alignment, and gene variant calling took ~15 hours. The HiSeq 2500 runs yielded 121 to 139 gigabases (GB) of aligned sequences (34-to 41-fold aligned genome coverage; Table 1). Eighty-eight to 91% of bases had >99.9% likelihood of being correct (quality score >30, using Illumina software equivalent to Phred) (31, 32). We detected 4.00 ± 0.20 million nucleotides that differed from the reference genome sequence (variants) (mean ± SD) in nine samples, one from each of nine infants (Table 1).
Fig. 1.

STAT-Seq. Summary of the steps and timing of STAT-Seq, resulting in an interval of 50 hours between consent and delivery of a preliminary, verbal diagnosis. t, hours.
Table 1.
Sequencing, alignment, and variant statistics of nine samples analyzed by rapid WGS. ACMG category 1 to 4 variants are a subset of gene-associated variants.
| Sample | Run time (hours) | Sequence (GB) | High- quality reads (%) | Mitochondrial genome variants | Nuclear genome variants | Gene- associated variants | ACMG categories 1 to 4 variants | ACMG categories 1 to 4 allele frequency <1% | ACMG categories 1 to 3 allele frequency <1% | Candidate genes | Candidate gene category 1 variants | Candidate gene VUS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UDT002 | 25.5 | 133 | 91 | 33 | 4,014,761 | 1,888,650 | 10,733 | 1,989 | 1,330 | 352 (9) | 2 | 0 |
| UDT173 | 25.5 | 139 | 89 | 40 | 3,977,062 | 1,859,095 | 10,501 | 2,190 | 1,296 | 347 (3) | 0 | 1 |
| CMH064 | 26.6 | 121 | 88 | 41 | 3,985,929 | 1,869,515 | 10,701 | 1,884 | 1,348 | 35 | 0 | 2 |
| CMH076 | 25.7 | 134 | 88 | 34 | 4,498,146 | 2,098,886 | 11,891 | 2,552 | 1,351 | 89 | 0 | 1 |
| CMH172 | 26.5 | 113 | 91 | 39 | 3,759,165 | 1,749,868 | 10,135 | 1,456 | 982 | 174 | 0 | 1 |
| CMH184 | 26.5 | 137 | 90 | 37 | 3,921,135 | 1,840,738 | 10,883 | 1,168 | 833 | 12 | 0 | 0 |
| CMH185 | 40 | 117 | 93 | 37 | 3,922,736 | 1,831,997 | 10,810 | 1,164 | 840 | 14 | 0 | 0 |
| CMH186 | 25.5 | 113 | 93 | 37 | 3,933,062 | 1,827,499 | 10,713 | 1,202 | 868 | 14 | ||
| CMH202 | 40 | 116 | 93 | 39 | 3,947,053 | 1,849,647 | 10,805 | 1,283 | 901 |
Analytical metrics
In three samples, genome variants identified by 50-hour WGS were compared with those identified by deep targeted sequencing of either exons and 20 intron-exon boundary nucleotides of a panel of 525 recessive disease genes [Children’s Mercy Hospital Diagnostic panel 1 (CMH-Dx1)] or the exome (Table 2). CMH-Dx1 comprised 8813 exonic and intronic targets, totaling 2.1 million nucleotides (table S1) (2, 33). The exome and CMH-Dx1 methods, which used Illumina TruSeq enrichment and HiSeq 2000 sequencing, took ~19 days. In contrast, rapid WGS did not use target enrichment, was performed with the HiSeq 2500 instrument, and took ~50 hours. Samples CMH064, UDT002, and UDT173 were sequenced using these three methods, and variants were detected with a single alignment method [the Genomic Short-read Nucleotide Alignment Program (GSNAP)] (34) and variant caller [the Genome Analysis Tool Kit (GATK)] (35). Rapid WGS detected ~96% of the variants identified by a target enrichment method and ~99.5% of the variants identified by both methods had identical genotypes (Table 2), indicating that rapid WGS is highly concordant with established clinical sequencing methods (33). In contrast, analysis of the rapid WGS data set from sample CMH064 with three different alignment and variant detection methods [GSNAP/GATK, the Illumina CASAVA alignment tool, and the Burrows-Wheeler Alignment (BWA) tool] revealed surprising differences between the variants detected. Only ~80% of the variants detected using GATK/GSNAP or BWA were also detected with CASAVA (Table 2 and table S2) (36–41). This suggests that additional studies will be needed to define optimal alignment methods for clinical sequencing. Nevertheless, there was good concordance between the genotypes of variants detected by rapid WGS (using the HiSeq 2500 and CASAVA) and targeted sequencing (using exome enrichment, the HiSeq 2000, and GATK/GSNAP)—99.5% (UDT002), 99.9% (UDT173), and 99.7% (CMH064) (Table 2)—further indicating that rapid WGS is highly concordant with an established genotyping method (33). In subsequent studies, the rapid WGS technique used CASAVA for alignment and variant detection.
Table 2.
Variants and genotypes. Comparisons of variants and genotypes obtained in three samples using three target enrichment methods, two sequencing methods, and two alignment methods. The 50-hour WGS (STAT-Seq) was not enriched and used HiSeq 2500 sequencing. CMH-Dx1 was enriched for 523 genes and HiSeq 2000 sequencing. Average coverage of target nucleotides indicates the average aligned sequence depth over the corresponding target panel. For WGS, the target is the genome; for exome sequencing, the target is the exome; and for CMH-Dx1, the targets are 523 genes.
| Sample | Target enrichment | Sequencing method | Alignment method | Sequence (GB) | Average coverage of target nucleotides | Variants detected by rapid WGS | Genotypes identical to both methods (%) |
|---|---|---|---|---|---|---|---|
| CMH064 | Exome None (WGS) |
HiSeq 2000 HiSeq 2500 |
GATK/GSNAP | 9.8 12.1 |
79 40 |
46,756 (96.0%) | 99.4 |
| UDT173 | CMH-Dx1 None (WGS) |
HiSeq 2000 HiSeq 2500 |
GATK/GSNAP | 4.1 13.9 |
784 46 |
1539 (96.7%) | 99.60 |
| UDT173 | CMH-Dx1 None (WGS) |
HiSeq 2000 HiSeq 2500 |
GATK/GSNAP CASAVA |
4.1 13.9 |
784 46 |
1457 (83.0%) | 99.9 |
| UDT002 | CMH-Dx1 None (WGS) |
HiSeq 2000 HiSeq 2500 |
GATK/GSNAP CASAVA |
4.2 13.3 |
770 44 |
1341 (76.6%) | 99.5 |
Genomic variants were characterized with respect to functional consequence and zygosity with a new software pipeline [Rapid Understanding of Nucleotide variant Effect Software (RUNES), fig. S2] that analyzed each sample in 2.5 hours. Samples contained a mean of 4.00 ± 0.20 million (SD) genomic variants, of which a mean of 1.87 ± 0.09 million (SD) were associated with protein-encoding genes (Table 1). Less than 1% of these variants (mean, 10,848 ± 523 SD) were also of a functional class that could potentially be disease causative (Table 1) (25–27). Of these, ~14% (mean, 1530 ± 518 SD) had an allele frequency that was sufficiently low to be a candidate for being causative in an uncommon disease (<1% allele frequency in 836 individuals sequenced at CMH) (42). Last, of these, ~71% (mean, 1083 ± 240 SD) were also of a functional class that was likely to be disease causative [American College of Medical Genetics (ACMG) categories 1 to 3] (Table 1). This set of variants was evaluated for disease causality in each patient, with priority given to variants within the candidate genes that had been nominated by an individual patient presentation.
Retrospective analyses
Patient UDT002 was a male who presented at 13 months of age with hypotonia, developmental regression. Brain magnetic resonance imaging (MRI) showed diffuse white matter changes suggesting leukodystrophy. Three hundred fifty-two disease genes were nominated by one of the three clinical terms hypotonia, developmental regression, or leukodystrophy; 150 disease genes were nominated by two terms; and 9 disease genes were nominated by all three terms (table S3). Only 16 known pathogenic variants had allele frequencies in dbSNP and the CMH cumulative database that were consistent with uncommon disease mutations. Of these, only two variants mapped to the nine candidate genes; the variants were both compound heterozygous (verified by parental testing) substitution mutations in the gene that encodes the αsubunit of the lysosomal enzyme hexosaminidase A [HEXA Chr 15:72,641,417T>C (gene symbol, chromosome number, chromosome coordinate, reference nucleotide > variant nucleotide), c.986+3A>G (transcript coordinate, reference nucleotide, variant nucleotide), and Chr15:72,640,388C>T, c.1073+1G>A]. The c.986+3A>G alters a 5′ exon–flanking nucleotide and is a known mutation that causes Tay-Sachs disease (TSD), a debilitating lysosomal storage disorder [Online Mendelian Inheritance in Man (OMIM) number 272800]. The variant had not previously been observed in our database of 651 individuals or dbSNP, which is relevant because mutation databases are contaminated with some common polymorphisms, and these can be distinguished from true mutations on the basis of allele frequency (33). The c.1073+1G>A variant is a known TSD mutation that affects an exonic splice donor site (dbSNP rs76173977). The variant has been observed only once before in our database of 414 samples, which is consistent with an allele frequency of a causative mutation in an orphan genetic disease. Thus, the known diagnosis of TSD was confirmed in patient UDT002 by rapid WGS.
Patient UDT173 was a male who presented at 5 months of age with developmental regression, hypotonia, and seizures. Brain MRI showed dysmyelination, hair shaft analysis revealed pili torti (kinky hair), and serum copper and ceruloplasmin were low. On the basis of this clinical presentation, 276 disease genes matched one of these clinical terms and 3 matched three terms (table S4). There were no previously reported disease-causing variants in these 276 genes. However, five of the candidate genes contained either variants of a type that is expected to be disease-causing based on their predicted functional consequence or missense variants of unknown significance (VUS). One of these variants was in a gene that matched all three clinical terms and was a hemizygous substitution mutation in the gene that encodes the α poly-peptide of copper-transporting adenosine triphosphatase (ATP7A Chr X:77,271,307C>T, c.2555C>T, p.P852L), aberrant forms of which are known to cause Menkes disease, a copper-transport disorder. This variant—new to our database and dbSNP—specified a nonconservative substitution in an amino acid that was highly conserved across species and had deleterious SIFT (Sorts Intolerant From Tolerant substitutions), PolyPhen2 (Polymorphism Phenotyping), and BLOSUM (BLOcks SUbstitution Matrix) scores. The known diagnosis of Menkes disease (OMIM number 309400) was recapitulated. As a further assessment of the reliability of variant detection of rapid WGS, samples UDT002 and UDT173 were aligned to the reference genome with three different alignment methods. The causative variants were recovered with each method.
Prospective analyses
Mutations in 35 genes can cause generalized, erosive dermatitis of the type found in CMH064 (table S5). The severe phenotype, negative family history, and absence of consanguinity suggested dominant de novo or recessive inheritance. No known pathogenic mutations were identified in the candidate genes that had low allele frequencies in the CMH cumulative genome and exome sequence database and similar public databases. Average coverage of the genomic regions corresponding to the candidate genes was 38.9-fold, and 98.4% of candidate gene nucleotides had >16× high-quality coverage (sufficient to rule out a heterozygous variant; table S6). Five candidate genes had 100% nucleotides with >16-fold high-quality coverage and, thus, lacked a known pathogenic mutation in an exon or within 20 nucleotides of the intron-exon boundaries. Eighteen of the candidate genes had >99% nucleotides with >16-fold high-quality coverage, and 31 had >95% nucleotides with at least this level of coverage. Furthermore, while 26 of the candidate genes had pseudogenes, paralogs, and/or repeat segments (table S6) that could potentially result in misalignment and variant miscalls, only 0.03% of target nucleotides had poor alignment quality scores.
Among the 35 candidate genes nominated by the phenotype, two rare heterozygous VUS were detected in CMH064; however, dideoxy sequencing of both healthy parents excluded one, in the keratin 14 gene, as a de novo mutation. The exomes of both parents were subsequently sequenced, and variants were examined in the trio at length. Three likely de novo mutations with excellent sequence coverage were identified in disease-causing genes. Of these, one was a candidate gene for CMH064. It was an in-frame deletion of three nucleotides in GJB2, (NM_004004), which encodes the connexin 26 protein. The variant, c.85_87del, p.Phe29del, removes a highly conserved amino acid within the first transmembrane helix (43). Dideoxy sequencing confirmed it to be a de novo mutation. Dominant, de novo GJB2 mutations have been associated with severe neonatal lethal disorders of the skin, such as keratitis-ichthyosis-deafness syndrome (KIDS), that involve the suprabasilar layers of the epidermis (OMIM number 148210) (44). The phenotype of CMH064 was atypical for KIDS, and functional studies are in progress to determine causality definitively.
Diagnoses suggested by the presentation in CMH076 were mitochondrial disorders, organic acidemia, or pyruvate carboxylase deficiency. Together, 75 nuclear genes and the mitochondrial genome cause these diseases (table S7). A negative family history suggested recessive inheritance that resulted from compound heterozygous or hemi-zygous variants or a heterozygous de novo dominant variant. Rapid WGS excluded known pathogenic mutations in the candidate genes. One novel heterozygous VUS was found. However, de novo occurrence of this variant was ruled out by exome sequencing of his healthy parents. No homozygous or compound heterozygous VUS with suitably low allele frequencies were identified in the known disease genes. Potential novel candidates included 929 nuclear genes that encode mitochondrial proteins but have not yet been associated with a genetic disease (45). Only one of these had a homozygous or compound heterozygous VUS with an allele frequency in dbSNP and the CMH database that was sufficiently low to be a candidate for causality in an uncommon inherited disease. Deep exome sequencing of both parents excluded this variant and did not disclose any further potentially causal variants.
A total of 174 genes are known to cause epilepsy of the type found in CMH172 (table S8). A positive family history of neonatal epilepsy and evidence of shared parental ancestry strongly suggested recessive inheritance. No known disease-causing variants or homozygous/compound heterozygous VUS with low allele frequencies were identified in these genes, which largely excluded them as causative in this patient. A genome-wide search of homozygous, likely pathogenic VUS that were novel in the CMH database and dbSNP disclosed a frame-shifting insertion in the BRCA1-associated protein required for ATM activation–1 (BRAT1, Chr 7:2,583,573-2,583,574insATCTTCTC,c.453_454insATCTTCTC, p.Leu152IlefsX70). A literature search yielded a very recent study of BRAT1 mutations in two infants with lethal, multifocal seizures, hypertonia, microcephaly, apnea, and bradycardia (OMIM number 614498) (46). Dideoxy sequencing confirmed the variant to be homozygous in CMH172 and heterozygous in both parents.
Rapid WGS was performed simultaneously on proband CMH184 (male), affected sibling (brother) CMH185, and their healthy parents, CMH186 and CMH202. Twelve genes have been associated with the clinical features of the brothers (heterotaxy and congenital heart disease; table S9). Co-occurrence in two siblings strongly suggested recessive inheritance. No known disease-causing variants or homozygous/compound heterozygous VUS with low allele frequencies were identified in these genes. A genome-wide search of novel, homozygous/compound heterozygous, likely pathogenic VUS that were common to the affected brothers and heterozygous in their parents yielded two nonsynonymous variants in the B cell CLL/lymphoma 9–like gene (BCL9L, Chr 11:118,772,350G>A,c.2102G>A, p.Gly701Asp and Chr 11:118,774,140G>A, c.554C>T, p.Ala185Val). Evidence supporting the candidacy of BCL9L for heterotaxy and congenital heart disease is presented below.
DISCUSSION
Genomic medicine, empowered by WGS, has been heralded as transformational for medical practice (2, 4, 5, 47). Over the last several years, the cost of WGS has fallen markedly, potentially bringing it within the realm of cost-effectiveness for high-intensity medical practice, such as occurs in NICUs (3, 8, 23, 24). Furthermore, experience has been gained with clinical use of WGS that has instructed initial guidelines for its use in molecular diagnosis of genetic disorders (9). However, a major impediment to the implementation of practical genomic medicine has been time to result.
This limitation has always been a problem for diagnosis of genetic diseases: Time to result and cost have greatly constrained the use of serial analysis of single-gene targets by dideoxy sequencing. Hitherto, clinical use of WGS by NGS has also taken at least a month: Sample preparation has taken at least a day; clustering 5 hours; 2 × 100 nucleotide sequencing 11 days; alignment, variant calling, and genotyping 1 day; variant characterization a week; and clinical interpretation at least a week. Although exome sequencing lengthens sample preparation by several days, it decreases computation time somewhat and is less costly. For use in acute care, the turnaround time of molecular diagnosis, including analysis, must match that of medical decision-making, which ranges from 1 to 3 days for most acute medical care. Herein, we described proof of concept for 2-day genome analysis of acutely ill neonates with suspected genetic disorders.
Automating medicine
Rapid WGS was made possible by two innovations. First, a widely used WGS platform has been modified to generate up to 140 GB of sequence in less than 30 hours (HiSeq 2500): Sample preparation took 4.5 hours, and 2 × 100 bp genome sequencing took 25.5 hours (Fig. 1). The total “hands-on” time for technical staff was 5 hours. Modifications included a new flowcell design and faster imaging and chemistry. Previously, NGS has either lacked sufficient sequence quantity, quality, or read lengths for clinical use of WGS or been too slow for use in acute patient care. Rapid WGS generated ~40-fold aligned genome coverage. The sequence quality was very similar to that obtained with its predecessor (HiSeq 2000), as determined by quality scores and alignment rates (48). Genotypes of nucleotide variants were >99.5% concordant with those of very deeply sequenced, partial exomes (33). The accuracy of the latter has been extensively benchmarked and is >99.9% (33).
Second, we automated much of the onerous characterization of genome variation and facilitated interpretation by restricting and prioritizing variants with respect to allele frequency (42), likelihood of a functional consequence (25), and relevance to the prompting illness. Thus, rapid WGS, as described herein, was designed for prompt disease diagnosis rather than carrier testing or newborn screening. SSAGA mapped the clinical features in ill neonates and children to disease genes. Thereby, analysis was limited only to the parts of the genome relevant to an individual patient’s presentation, in accord with guidelines for genetic testing in children (25–28). This greatly decreased the number of variants to be interpreted. In particular, SSAGA caused most incidental (secondary) findings to be masked. In the setting of acute care in the NICU, secondary findings are anticipated to impede facile interpretation, reporting, and communication with physicians and patients greatly (9, 49, 50). SSAGA also assisted in test ordering, permitting a broad selection of genes to be nominated for testing based on entry of the patients’ clinical features with easy-to-use pull-down menus. The version used herein contains ~600 recessive and mitochondrial diseases and has a diagnostic sensitivity of 99.3% for those disorders. SSAGA is likely to be particularly useful in disorders that feature clinical or genetic heterogeneity or early manifestation of partial phenotypes because it maps features to a superset of genetic disorders. SSAGA needs to be expanded to encompass dominant disorders and to the full complement of genetic diseases that meet ACMG guidelines for testing rare disorders (such as having been reported in at least two unrelated families) (26). Although neonatal disease presentations are often incomplete, only one feature is needed to match a disease gene to a presentation. In cases for which SSAGA-delimited genome analysis was negative, such as CMH064 and CMH076, a comprehensive secondary analysis was performed with limitation of variants solely to those with acceptable allele frequencies (42) and likelihood of a functional consequence (25). Nevertheless, secondary analysis was relatively facile, yielding about 1000 variants per sample.
RUNES performed many laborious steps involved in variant characterization, annotation, and conversion to HGVS (Human Genome Variation Society) nomenclature in ~2 hours. RUNES unified these in an automated report that contained nearly all of the information desirable for variant interpretation, together with a cumulative variant allele frequency and a composite ACMG categorization of variant pathogenicity (fig. S2). ACMG categorization is a particularly useful standard for prioritization of the likelihood of variants being causal (26). In particular, more than 75% of coding variants were of ACMG category 4 (very unlikely to be pathogenic). Removal of such variants allowed rapid interpretation of high-likelihood pathogenic variants in relevant genes. The hands-on time for starting pipeline components and interpretation of known disease genes was, on average, less than 1 hour. Because genomic knowledge is currently limited to 1 to 2% of physicians (physician scientists, medical geneticists, and molecular pathologists), variant characterization, interpretation, and clinical guidance tools are greatly needed, as is training of medical geneticists and genetic counselors in their use.
Return of results
In blinded, retrospective analyses of two patients, rapid WGS correctly recapitulated known diagnoses. In child UDT002, two heterozygous, known mutations were identified in a gene that matched all clinical features. In male UDT173, a hemizygous (X-linked) VUS was identified in the single candidate gene matching all clinical features. The variant, a nonsynonymous nucleotide substitution, was predicted to be damaging. Rapid WGS also provided a definitive diagnosis in one of four infants enrolled prospectively. In CMH172, with refractory epilepsy, rapid WGS disclosed a novel, homozygous frame-shifting insertion in a single candidate gene (BRAT1). BRAT1 mutations were very recently reported in two unrelated Amish infants who suffered lethal, multifocal seizures (46). A molecular diagnosis was reached within 1 hour of WGS data inspection in CMH172, even though extant reference databases [Human Gene Mutation Database (HGMD) and OMIM] had not yet been updated with a BRAT1 disease association. The diagnosis was made clinically reportable by resequencing the patient and her parents. Had this diagnosis been obtained in real time, it may have expedited the decision to reduce or withdraw support. The latter decision was made in the absence of a molecular diagnosis after 5 weeks of ventilatory support, testing, and unsuccessful interventions to control seizures. Given high rates of NICU bed occupancy, accelerated diagnosis by rapid WGS has the potential to reduce the number of neonates who are turned away. The molecular diagnosis was also useful for genetic counseling of the infant’s parents to share the information with other family members at risk for carrying of this mutation. As suggested by recent guidelines (9), this case demonstrates the use of WGS for diagnostic testing when a genetic test for a specific gene of interest is not available.
In four of five affected individuals, prospective, rapid WGS provided a definitive or likely molecular diagnosis in ~50 hours. These cases demonstrated the use of WGS for diagnostic testing when a high degree of genetic heterogeneity exists, as suggested by recent guidelines (9). Confirmatory resequencing, which is necessary for return of results until rapid WGS is compliant with Clinical Laboratory Improvement Amendments (CLIA), took at least an additional 4 days. Until compliance has been established, we suggest preliminary verbal disclosure of molecular diagnoses to the neonatologist of record, followed by formal reporting upon performance of CLIA-conforming resequencing. Staged return of results of broad or complex screening tests, together with considered, expert interpretation and targeted quantification and confirmation, is likely to be acceptable in intensive care. Precedents for rapid return of interim, potentially actionable results include preliminary reporting of histopathology, radiographic, and imaging studies and interim antibiotic selection based on Gram stains pending culture and sensitivity results.
Disease gene sleuthing
Because at least 3700 monogenic disease genes remain to be identified (10), WGS will often rule out known molecular diagnoses and suggest novel candidate disease genes (23, 51). Indeed, in another prospectively enrolled family, WGS resulted in the identification of a novel candidate disease gene, providing a likely molecular diagnosis. The proband was the second affected child of healthy parents. Accurate genetic counseling regarding risk of recurrence had not been possible because the first affected child lacked a molecular diagnosis. We undertook rapid WGS of the quartet simultaneously, allowing us to further limit incidental variants by requiring recessive inheritance. Rapid WGS ruled out 14 genes known to be associated with visceral heterotaxy and congenital heart disease (HTX). Among genes that had not been associated with HTX, rapid WGS of the quartet narrowed the likely pathogenic variants to two in the BCL9L gene. BCL9L had not previously been associated with a human phenotype but is an excellent candidate gene for HTX based on its role in the Wingless (Wnt) signaling pathway, which controls numerous developmental processes, including early embryonic patterning, epithelial-mesenchymal interactions, and stem cell maintenance (51, 52).
Recently, the Wnt pathway was implicated in the left-right asymmetric development of vertebrate embryos, with a role in the regulation of ciliated organ formation and function (53–57). The key effector of Wnt signaling is β-catenin, which functions either to promote cell adhesion by linking cadherin to the actin cytoskeleton via α-catenin or to bind transcriptional coactivators in the nucleus to activate the expression of specific genes (58–60). The protein that controls the switch between these two processes is encoded by BCL9L (also known as BCL9-2) and serves as a docking protein to link β-catenin with other transcription coactivators. BCL9L and α-catenin share competitive overlapping binding sites on β-catenin; phosphorylation of β-catenin determines which pathway is activated. The p.Gly701Asp mutation found in our patients lies within the BCL9L nuclear localization signal, which is essential for β-catenin to perform transcriptional regulatory functions in the nucleus (61).
BCL9L is one of two human homologs of Drosophila legless (lgs), a segment polarity gene required for Wnt signaling during development. lgs-deficient flies die as pharate adults with Wnt-related defects, including absent legs, and antennae and occasional wing defects (62). Fly embryos lacking the maternal lgs contribution display a lethal segment polarity defect. BCL9L-deficient zebrafish exhibit patterning defects of the ventrolateral mesoderm, including severe defects of trunk and tail development (60). Furthermore, inhibition of zebrafish β-catenin results in defective organ laterality (54). Overexpression of constitutively active β-catenin in medaka fish causes cardiac laterality defects (63). β-Catenin–deficient mice have defective development of heart, intestine, liver, pancreas, and stomach, including inverted cell types in the esophagus and posteriorization of the gut (64). Down-regulation of Wnt signaling in mouse and zebrafish causes randomized organ laterality and randomized side-specific gene expression. These likely reflect aberrant Wnt activity on midline formation and function of Kupffer’s vesicle, a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart, and gut (56, 65). The second human homolog of lgs, BCL9, has been implicated in complex congenital heart disease in humans, of the type found in our patients (66–68). BCL9 was originally identified in precursor B cell acute lymphoblastic leukemia with a t(1:14)(q21;q32) translocation (69), linking the Wnt pathway and certain B cell leukemias or lymphomas (62). Finally, it was recently demonstrated that the Wnt/β-catenin signaling pathway regulates the ciliogenic transcription factor foxj1a expression in zebrafish (57). Decreased Wnt signal leads to disruption of left-right patterning, shorter/fewer cilia, loss of ciliary motility, and decreased foxj1a expression. Foxj1a is a member of the forkhead gene family and regulates transcriptional control of production of motile cilia (70). On the basis of this collected evidence, the symbol HTX6 has been reserved for BCL9L-associated autosomal recessive visceral heterotaxy. Additional studies are in progress to show causality definitively. These findings support clinical WGS as being valuable for research in reverse-translation studies (bedside to bench) that reveal new genetically amenable disease models.
Addressing limitations
In one remaining prospective patient, rapid WGS failed to yield a potential or definitive molecular diagnosis. Currently, WGS cannot survey every nucleotide in the genome (71). At 50× aligned coverage of the genome, WGS genotyped at least 95% of the reference genome with greater than 99.95% accuracy, using methods very similar to those used in this study (72). It has been suggested that this level of completeness is applicable for analyzing personal genomes in a clinical setting (72). In particular, GC-rich first exons of genes tend to be underrepresented (33). More complete clinical use of WGS will require higher sequencing depth, multi-platform sequencing and/or alignment methodologies, complementation by exome sequencing, or all three (73). Combined alignments with two methods of sequencing identified ~9% more nucleotide variants than one alone. However, these additions raise the cost of WGS, increase the time to clinical interpretation, and shift the cost-benefit balance.
For genetic disease diagnosis, the genomic regions that harbor known or likely disease mutations—the Mendelianome (2, 33)—must be genotyped accurately. In addition to exons and exon-intron boundaries, the Mendelianome includes some regions in the vicinity of genes that have structural variations or rearrangements. NGS of genome regions that contain pseudogenes, paralogs (genes related by genomic duplication), or repetitive motifs can be problematic. CMH064 had fulminant EB. Most EB-associated genes encode large cytoskeletal proteins with regions of constrained amino acid usage, which equate with low nucleotide complexity. In addition, several EB-associated genes have closely related paralogs or pseudogenes. These features impede unambiguous alignment of short reads, which can complicate attribution of variants by NGS. This limitation can prevent definitive exclusion of candidate genes. For example, 4.5% of nucleotides in KRT14, an EB-associated gene, had <16-fold high-quality coverage and, thus, may have failed to disclose a heterozygous variant. In CMH064, however, this possibility was excluded by targeted sequencing of the regions of KRT14 known to contain mutations that cause EB.
Furthermore, WGS is not yet effective for clinical-grade detection of all mutation types. Copy number variations and large deletions require clinical validation of research methods (33). Long, simple sequence–repeat expansions and complex rearrangements are problematic. Nevertheless, with CLIA-type adherence to standard operational processes, the component of the Mendelianome for which WGS is effective is extremely reproducible (33). Thus, the specific diseases, genes, exons, and mutation classes that are qualified for analysis, interpretation, and clinical reporting with WGS can be precisely predicted. This is of critical importance for reporting of differential diagnoses in the genetic disease arena. Thus, although insufficient alone, rapid WGS may still be a cost-effective initial screening tool for differential diagnosis of EB. In our study, all EB-associated genes had >95% nucleotides with high-quality coverage sufficient to exclude heterozygous and homozygous nucleotide variants (>16-fold); 19 of these genes had >99% nucleotides with this coverage. Hence, for rigorous testing of all EB-associated genes and mutation types, additional studies remain necessary, such as immunohistochemistry, targeted sequencing of uncallable nucleotides, and cytogenetic studies. Of 531 disease genes examined, 52 had pseudogenes, paralogs, repetitive motifs, or mutation types that may complicate WGS for comprehensive mutation detection. The comprehensiveness of WGS will be enhanced by longer reads, improved alignment methods, and validated algorithms for detecting large or complex variants (2, 4).
Finally, in singleton (sporadic) cases, such as CMH064, family history is often unrevealing in distinguishing the pattern of inheritance. For example, inheritance of EB can be dominant or recessive. Of two plausible heterozygous VUS detected in candidate genes in CMH064, one was a de novo mutation in connexin 26, which is associated with KIDS, that can be fatal in neonates (43, 44). The phenotype of CMH064 was not typical for KIDS, and functional studies are in progress. For evaluation of dominantly inherited diseases, WGS requires that the parents be concomitantly tested either by rapid WGS, by exome sequencing, or by resequencing of candidate de novo variants.
Rapid WGS failed to yield a definitive molecular diagnosis for CMH076. No known mutations were found in 89 disease-associated nuclear genes or the mitochondrial genome. This was an important negative finding because a molecular diagnosis of several of these genes is “actionable.” That is, specific treatments are indicated (such as pyruvate carboxylase deficiency, thiamine responsive congenital acidosis, biotinidase deficiency, fructose 1,6-bisphosphatase deficiency, and coenzyme Q10 deficiency). Likewise, exclusion of actionable diagnoses can prevent empiric institution of inappropriate treatments. Exclusion of known genetic diseases from a differential diagnosis is also of psychosocial benefit to family members and assists in guiding physicians regarding additional testing. There were no VUS with suitable inheritance patterns, in CMH076 or in either of the healthy parents, in known disease genes or in the remaining 929 nuclear-encoded mitochondrial genes (45).
In contrast to the rapidly declining cost of WGS, the computational cost of genome analysis is largely governed by Moore’s law (74). Sequence alignment, variant calling, and genotyping took 16 hours. Extremely rapid WGS is of practical use in clinical guidance only when married to equally rapid, cost-effective, deployable, and facile interpretation and analysis (2, 4). We are continuing to improve the speed of sequence base calling, alignment, and variant calling. It is likely that this interval can be halved and that HiSeq 2500–based rapid WGS can be performed in fewer than 36 hours by the end of 2012. Clinical validation of rapid WGS, however, will take some time.
MATERIALS AND METHODS
Consent
This study was approved by the Institutional Review Board of CMH. Informed written consent was obtained from adult subjects and parents of living children.
Case selection
CMH is a nonprofit children’s hospital with 314 beds, including 64 level 4 NICU beds. It provides 48% of neonatal intensive care in the Kansas City metropolitan region. In 2011, the NICU had 86% bed occupancy. Retrospective samples, UDT002 and UDT173, were selected from a validation set of 384 samples with known molecular diagnoses for one or more genetic diseases. Seven prospective samples were selected from families with probands that presented in infancy, among 143 individuals without molecular diagnoses who were enrolled between 22 November 2011 and 4 April 2012 for exome or genome sequencing.
Clinicopathological correlation and interpretation
The features of the patients’ diseases were mapped to likely candidate genes. In part, this was performed manually by a board-certified pediatrician and medical geneticist. In part, it was performed automatically by entry of terms describing the patients’ presentations into a new clinicopathological correlation tool, SSAGA (2). It was designed to enable physicians to delimit WGS analyses to genes of causal relevance to individual clinical presentations, in accord with published guidelines for genetic testing in children and with NGS (9, 25, 28). SSAGA has a menu of 227 clinical terms, which are arranged in nine categories (fig. S1). SNOMED CT (Systematized Nomenclature of Medicine—Clinical Terms) (29) map to 591 well-established recessive diseases with known causal genes (table S1). Phenotype-to-disease-to-gene mapping was informed by Gene Reviews (30), OMIM Clinical Synopsis (10), MitoCarta (45), and expert physician reviewers.
Upon entry of the features of an individual patient, SSAGA nominates the corresponding superset of relevant diseases and genes, rank ordered by number of matching terms (fig. S1). It also contains a free-form text box that allows physicians to enter findings for which no SNOMED term exists, clinical term qualifiers, relevant family history, and specific genes of interest. The diagnostic sensitivity of SSAGA improves with use, by manual updating of mappings in cases where nominations failed to include the causal gene. SSAGA is extensible to additional diseases, genes, and clinical terms. Interpretation of results was manual on the basis of ranking of variant reports yielded by RUNES on SSAGA-prioritized candidate genes, supplemented with expert gene nominations (fig. S2). In some pedigrees, the presumed pattern(s) of inheritance allowed additional variant ranking based on obligatory genotypes in affected and unaffected individuals. Aligned sequences containing variants of interest were inspected for veracity in pedigrees with the Integrative Genomics Viewer (32).
Genome and exome sequencing
Isolated genomic DNA was prepared for rapid WGS with a modification of the Illumina TruSeq sample preparation (Illumina). Briefly, 500 ng of DNA was sheared with a Covaris S2 Biodisruptor, end-repaired, A-tailed, and adaptor-ligated. Polymerase chain reaction (PCR) was omitted. Libraries were purified with SPRI beads (Beckman Coulter). Quantitation was carried out by real-time PCR. Libraries were denatured with 0.1 M NaOH and diluted to 2.8 pM in hybridization buffer.
Samples for rapid WGS were each loaded onto two flowcells, followed by sequencing on Illumina HiSeq 2500 instruments that were set to rapid run mode. Cluster generation, followed by 2 × 100 cycle sequencing reads, separated by paired-end turnaround, were performed automatically on the instrument.
Isolated genomic DNA was also prepared for Illumina TruSeq exome or custom gene panel sequencing with standard Illumina TruSeq protocols. Enrichment for the custom gene panel was performed twice by Illumina hybrid selection with 20,477 eighty-nucleotide probes for 8366 genomic regions, representing exons and 20 intron-exon boundary nucleotides. It encompassed 2,158,661 bp, 525 genes, and 591 recessive diseases (2, 33) (table S1). The probes were designed to target 350 nucleotide genomic targets, with an average density of 2.4 probes per target (range, 2 to 56). Custom gene panel–enriched samples were sequenced on HiSeq 2000 instruments with TruSeq v3 reagents to a depth of >3 GB of singleton 100-bp reads in samples UDT173 and UDT002, respectively; 32.9 and 38.3% of base pairs were on target defined with a 0-bp extension, representing 469- and 501-fold enrichment in samples UDT173 and UDT002, respectively. Exome-enriched samples were enriched twice with standard Illumina hybrid selection and were sequenced on HiSeq 2000 instruments with TruSeq v3 reagents to a depth of >8 GB of singleton 100-bp reads per sample.
Genome and exome sequencing were performed as research, not in a manner that complies with routine diagnostic tests as defined by the CLIA guidelines.
Sequence analysis
CASAVA 1.8.2 (Illumina) performed gapped ELAND alignment of HiSeq 2500 sequences to the reference nuclear and mitochondrial genome sequences [Hg19 and GRCH37 (NC_012920.1), respectively] as well as variant identification. HiSeq 2000 sequences were aligned to the reference nuclear and mitochondrial genome sequences with GSNAP, and variants were identified and genotyped with the GATK (2, 34–36). Sequence analysis used base-call files, FASTQ files that contain sequences and base-call quality scores, the compressed binary version of the Sequence Alignment/Map format (a representation of nucleotide sequence alignments), and Variant Call Format (a format for nucleotide variants). Nucleotide variants were annotated with RUNES (2), our variant characterization pipeline, which incorporated VEP (Variant Effect Predictor) (37), comparisons to NCBI dbSNP, known disease mutations from the HGMD (38), and additional in silico prediction of variant consequences with ENSEMBL and UCSC gene annotations (39, 40) (fig. S2). RUNES assigned each variant an ACMG pathogenicity category (25–27) and an allele frequency on the basis of 722 patients sequenced since October 2011. Rapid WGS in CMH064 and exome sequences of his parents were also analyzed by Clinical Sequence Miner (deCODE Genetics).
Patient 1
CMH064 was a male born at 33 weeks gestation with erosive dermatoses. He was delivered vaginally following induction for preeclampsia. Desquamation and erythroderma from the hairline to occiput were present at birth (Fig. 2A). Denuded, hyperpigmented, and partially scarred lesions were noted above the upper lip, over the mentum, and in place of eyebrows. He had a truncated foreskin. His nails were dystrophic and yellowed. There were no vesicles, pustules, blisters, or mucosal lesions. Family history was positive for psoriasis. His mother had a healthy daughter from a prior union; there was no history of fetal loss. His father was healthy.
Fig. 2.

Skin lesions in patient CMH064. (A) Desquamated lesions with erythroderma on the scalp at birth. (B) Day 30 progression of desquamation. His fingers were edematous and discolored and had retained only three nails.
Cultures and herpesvirus PCR were negative. He developed severe neutropenia by day 3. Skin sloughing worsened. Rigid bronchoscopy and intubation were necessary because of fibrinous oropharyngeal exudate.
Skin biopsy histology revealed acantholysis, loss of cohesion between keratinocytes, and empty lacunae (Fig. 3A). There was focal dermal infiltration with neutrophils and lymphocytes and complete sloughing of the epidermal layer with focal clefting at the suprabasal layer (Fig. 3B). Immunofluorescence staining was negative for IgA (immunoglobulin A), IgM, and IgG except for linear staining for C3. Additional skin immunofluorescence studies revealed slightly reduced plakoglobin and desmoplakin and normal laminin 332; collagen types 4, 7, and 17; and plakophilin-1. Electron microscopy confirmed the absence of dermoepidermal junction (DEJ) separation and showed focally widened spaces between keratinocytes and cell vacuolization from the DEJ to the stratum corneum. Hemidesmosomes were normal. Some keratinocytes had large solitary vacuoles, abnormal condensation of keratin filaments, and perinuclear pallor. Some desmosomes had ragged edges. There were no intracellular inclusions. Negative laboratory studies included karyotype, Ro, La, Smith, ribonucleoprotein, and Scl-70 autoantibodies. Igs were unremarkable apart from an elevated serum IgA.
Fig. 3.
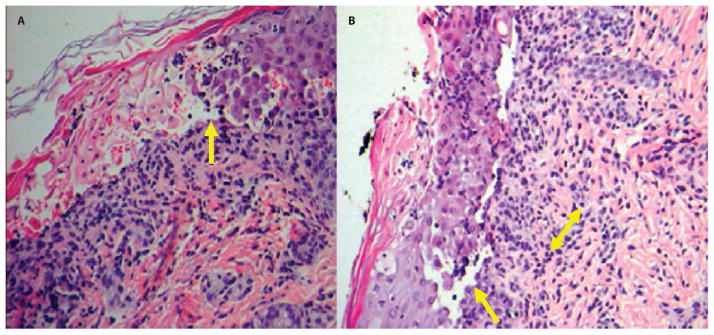
Skin lesion histology for patient CMH064. (A) Dermal acantholysis (loss of intercellular connections resulting in loss of cohesion between ke-ratinocytes) and formation of empty lacunae (cavities; arrow). (B) Focal dermal infiltration of neutrophils and lymphocytes (double-headed arrow). The epidermal layer shows complete sloughing with focal clefting at the suprabasal layer (arrow).
Sloughing of the skin, mucosal surfaces, and cornea continued to worsen, and by day 30, his activity level had markedly decreased (Fig. 2B). His fingers were edematous and discolored and had retained only three nails. On day 39, he developed purulent drainage from facial lesions. Skin cultures were positive for Escherichia coli and Enterococcus faecalis, and blood cultures for E. coli. Antibiotics were administered. He was thrombocytopenic and anemic, necessitating numerous transfusions. On day 47, ultrasound revealed nonocclusive portal vein and left brachiocephalic vein thrombi. By day 54, he developed metabolic acidosis, bloody stools, and persistent tachycardia. Medical interventions were withdrawn and he died on day 54. At autopsy, suprabasal acantholysis was present in the skin and the esophageal mucosa. Dideoxy sequencing of candidate genes KRT5, DSP, JUP, TP63, and KRT14 exons 1, 4, and 6 (the regions harboring most KRT14 mutations) was negative.
Patient 2
CMH076 was a male born at term with lactic acidosis, cardiomyopathy, and corneal clouding. He was born to a primigravid mother whose pregnancy was notable for decreased movements at 35 weeks gestation. His mother and father were healthy. Variable decelerations in heart rate were noted on the day before delivery. Labor was complicated by prolonged rupture of membranes, and delivery was by vacuum extraction for meconium staining. Apgar scores were 2, 3, and 5 at 1, 5, and 10 min, respectively. He had poor respiratory effort and hypotonia and required intubation. Upon transfer to CMH on day 2, he had lactic acidosis (lactate, 12 mmol/dl), coagulopathy, and cloudy corneas. Multiple cultures were negative. Echocardiogram showed chamber enlargement, reduction in biventricular function, noncompaction cardiomyopathy, mild tricuspid insufficiency, and mild aortic insufficiency. Urine testing revealed normal amino acids and elevated 3-methyglutaconic acid, 3-methylglutaric acid, and 2-ethyl-3-hydroxy-propionic acid. Long-chain fatty acids, acyl-carnitines, lysosomal hydrolases, β-galactosidase, β-glucuronidase, sphingomyelinase, glucocerebrosidase, α-L-iduronidase, and α-glucosaminidase were normal. Pressors were required for hypotension, and acidosis increased. He was diagnosed with hypoxic ischemic encephalopathy. On day 3, lactate was 28.2 mmol/dl. On day 5, respiratory distress worsened, accompanied by bloody endotracheal secretions; arterial pH was 7.04 and lactate was 22.0 mmol/dl. Medical interventions were withdrawn at the family’s request, and he expired on day 5. Postmortem testing by array comparative genomic hybridization (aCGH) and sequencing for mitochondrial transfer RNAs and TAZ, associated with Barth syndrome, were normal.
Patient 3
CMH172 was a female with intractable epilepsy. She was delivered at 39 weeks gestation by Cesarean section after an uncomplicated pregnancy. No exposure in utero to drugs, alcohol, or medications was reported. Birth weight was normal, length was 46 cm (<3%), and head circumference was 33 cm (<3%). Amniotic fluid was meconium-stained. Apgar scores were 6, 7, and 8 at 1, 5, and 10 min, respectively. Family history was positive for a female cousin with profound intellectual disability and infrequent seizures, and two cousins by a consanguineous marriage who died at 6 and 8 weeks of age of intractable epilepsy; all were from the same small Mexican town as the proband. Seizures started 1 hour after delivery. Antibiotics were given empirically until cultures and cerebrospinal fluid (CSF) herpesvirus PCR returned negative. Seizures continued despite multiple antiepileptic medications. CSF (including glycine level and CSF/plasma ratio) and brain MRI were normal. Electroencephalogram (EEG) showed focal epileptiform and sharp wave activity. Blood ammonia, electrolytes, pH, and glucose were normal. Oral feeding was poor. She was intubated and required increasing respiratory support for low SaO2 and bradycardia. Ophthalmologic examination and radiologic skeletal survey were normal. An echocardiogram revealed a patent foramen ovale, tricuspid regurgitation, and peripheral pulmonary stenosis. Her karyotype was normal. aCGH was not diagnostic, but multiple tracts of homozygosity suggested shared parental ancestry. A repeat brain MRI at age 3 weeks was normal. Upon transfer to CMH at 5 weeks of age, she was small but symmetric, with bitemporal narrowing, micrognathia, flat nasal bridge, upslanted palpebral fissures, uplifted ear lobes, redundant helices, and fifth finger clinodactyly. She had hypertonia, persistence of cortical thumbs, hyperreflexia, clonus, and facial twitching. B6 challenge improved her EEG transiently, followed by return of multifocal sharp waves. Serum amino acids and urine organic acids were normal. Recurrent seizures continued both clinically and by EEG. After lengthy discussion, the parents requested withdrawal of support.
Patient 4
CMH184 was a male with visceral heterotaxy and congenital heart disease (dextro-transposition of the great arteries, total anomalous pulmonary venous return with pulmonary veins connecting to the right atrium, a large ventricular septal defect, pulmonary valve and main pulmonary artery atresia, mildly hypoplastic branch pulmonary arteries, patent ductus arteriosus with ductal-dependant left-to-right flow, and large atrial septal defect with obligate right-to-left flow). There was situs inversus of the spleen, liver, and stomach, with the aorta on the right of the spine and inferior vena cava on the left. Family history was positive for a 6-year-old brother (CMH185) with the same findings (dextrocardia, ventricular inversion, double outlet right ventricle, pulmonary stenosis, small pulmonary arteries, interrupted inferior vena cava with azygous continuation, and situs inversus of the liver and spleen). His parents (mother, CMH186, and father, CMH202) and two other siblings (one male and one female) were healthy. Testing of ZIC3, associated with X-linked recessive heterotaxy 1, was normal. Patient 4 remains in the NICU and is undergoing cardiac surgery.
Supplementary Material
Acknowledgments
We thank R. Cohen, V. Corbin, G. Richards, and S. Bale for helpful comments, as well as A. Keithly and M. Clifton for their technical help. We thank P. Sulem, H. Gudbjartsson, S. A. Gudjonsson, and K. Stefansson from deCODE Genetics for assistance with sequence analysis. A deo lumen, ab amicis auxilium.
Funding: This work was supported by the Marion Merrell Dow Foundation, Children’s Mercy Hospital, and Illumina Inc.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/4/154/154ra135/DC1
Materials and Methods
Fig. S1. Candidate gene selection by SSAGA for automated variant characterization and interpretation guidance.
Fig. S2. Automated variant characterization by RUNES.
Table S1. Five hundred ninety-one recessive diseases and genes for which clinical terms and a targeted enrichment panel (CMH-Dx1) have been developed.
Table S2. Comparison of the rapid WGS data set from sample CMH064 with three different alignment and variant detection methods (GSNAP/GATK, the Illumina CASAVA alignment tool, and BWA tool).
Table S3. Disease genes nominated by SSAGA on the basis of the clinical features of patient UDT002.
Table S4. Disease genes nominated by SSAGA on the basis of the clinical features of patient UDT173.
Table S5. Disease genes nominated by SSAGA on the basis of the clinical features of patient CMH064.
Table S6. Candidate genes for EB with pseudogenes, paralogs, or segments with reduced sequence complexity.
Table S7. Disease genes nominated by SSAGA on the basis of the clinical features of patient CMH076.
Table S8. Disease genes nominated by SSAGA on the basis of the clinical features of patient CMH172.
Table S9. Disease genes nominated by SSAGA on the basis of the clinical features of patient CMH184.
Author contributions: C.J.S., N.A.M., S.E.S., and D.L.D. undertook analysis of data and confirmatory studies and helped write the manuscript; A.N. developed the tool for identification of variants that affect splicing; N.A.A. compiled patient information and assisted in data analysis; N.A. and N.P.S. performed histopathology; M.L.P., L.A.K., and E.G.F. undertook panel, exome, and dideoxy sequencing and data analysis; J.F., S.H., P.S., Z.K., J.C.W., J.B., R.J.G., E.H.M., and K.P.H. developed the HiSeq 2500 and undertook the genomic sequencing on that instrument; M.A. had the idea to provide next-generation sequences for ill neonates; J.E.P. was the neonatologist of record for the patients; S.F.K. oversaw the work and wrote the manuscript.
Competing interests: J.F., S.H., P.S., Z.K., J.C.W., J.B., R.J.G., E.H.M., and K.P.H. are employees of Illumina Inc., which manufactures the HiSeq 2500 instrument. The other authors declare that they have no competing interests.
Data and materials: The genomic sequence data for this study have been deposited in the database dbGAP. Please contact authors for accession numbers.
REFERENCES AND NOTES
- 1.Green ED, Guyer MS. National Human Genome Research Institute, Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470:204–213. doi: 10.1038/nature09764. [DOI] [PubMed] [Google Scholar]
- 2.Kingsmore SF, Dinwiddie DL, Miller NA, Soden SE, Saunders CJ. Adopting orphans: Comprehensive genetic testing of Mendelian diseases of childhood by next-generation sequencing. Expert Rev Mol Diagn. 2011;11:855–868. doi: 10.1586/erm.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, Reid JG, Fink JK, Morgan MB, Gingras MC, Muzny DM, Hoang LD, Yousaf S, Lupski JR, Gibbs RA. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kingsmore SF, Saunders CJ. Deep sequencing of patient genomes for disease diagnosis: When will it become routine? Sci Transl Med. 2011;3:87ps23. doi: 10.1126/scitranslmed.3002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DCY, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, Halperin JJ, Yang C, Gehman C, Guo D, Irikat RK, Tom W, Fantin NJ, Muzny DM, Gibbs RA. Whole-genome sequencing in a patient with Charcot–Marie–Tooth Neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR, Veith RL, Basehore MJ, Broeckel U, Tomita-Mitchell A, Arca MJ, Casper JT, Margolis DA, Bick DP, Hessner MJ, Routes JM, Verbsky JW, Jacob HJ, Dimmock DP. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 9.American College of Medical Genetics and Genomics (ACMG) Policy Statement. Points to Consider in the Clinical Application of Genomic Sequencing. 2012 May 15; doi: 10.1038/gim.2012.74. available at http://www.acmg.net/StaticContent/PPG/Clinical_Application_of_Genomic_Sequencing.pdf. [DOI] [PubMed]
- 10.Online Mendelian Inheritance in Man. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; Baltimore, MD: [accessed 13 July 2012]. available at www.omim.org/statistics. [Google Scholar]
- 11.Hauck FR, Tanabe KO, Moon RY. Racial and ethnic disparities in infant mortality. Semin Perinatol. 2011;35:209–220. doi: 10.1053/j.semperi.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Lynberg MC, Khoury MJ. Contribution of birth defects to infant mortality among racial/ethnic minority groups, United States, 1983. MMWR CDC Surveill Summ. 1990;39:1–12. [PubMed] [Google Scholar]
- 13.Kochanek KD, Kirmeyer SE, Martin JA, Strobino DM, Guyer B. Annual summary of vital statistics: 2009. Pediatrics. 2012;129:338–348. doi: 10.1542/peds.2011-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander D, Hanson JW. NICHD research initiative in newborn screening. Ment Retard Dev Disabil Res Rev. 2006;12:301–304. doi: 10.1002/mrdd.20131. [DOI] [PubMed] [Google Scholar]
- 15.American College of Medical Genetics’ Newborn Screening Expert Group. Newborn screening: Toward a uniform screening panel and system. Executive summary. Genet Med. 2006;8:1S–11S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Couce ML, Baña A, Bóveda MD, Pérez-Muñuzuri A, Fernández-Lorenzo JR, Fraga JM. Inborn errors of metabolism in a neonatology unit: Impact and long-term results. Pediatr Int. 2011;53:13–17. doi: 10.1111/j.1442-200X.2010.03177.x. [DOI] [PubMed] [Google Scholar]
- 17.Downing GJ, Zuckerman AE, Coon C, Lloyd-Puryear MA. Enhancing the quality and efficiency of newborn screening programs through the use of health information technology. Semin Perinatol. 2010;34:156–162. doi: 10.1053/j.semperi.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Lantos JD, Meadow WL. Costs and end-of-life care in the NICU: Lessons for the MICU? J Law Med Ethics. 2011;39:194–200. doi: 10.1111/j.1748-720X.2011.00588.x. [DOI] [PubMed] [Google Scholar]
- 19.Therrell B, Lorey F, Eaton R, Frazier D, Hoffman G, Boyle C, Green D, Devine O, Hannon H. Impact of Expanded Newborn Screening—United States, 2006. MMWR Morb Mortal Wkly Rep. 2008;57:1012–1015. [PubMed] [Google Scholar]
- 20.Health Resources and Services Administration. Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. 2011 Annual Report to Congress; Rockville, MD. 2011. http://www.hrsa.gov. [Google Scholar]
- 21.Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:CD004426. doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh RH, Rohr F, Splett PL. Bridging evidence and consensus methodology for inherited metabolic disorders: Creating nutrition guidelines. J Eval Clin Pract. 2011 doi: 10.1111/j.1365-2753.2011.01807.x. [DOI] [PubMed] [Google Scholar]
- 23.Sobreira NL, Cirulli ET, Avramopoulos D, Wohler E, Oswald GL, Stevens EL, Ge D, Shianna KV, Smith JP, Maia JM, Gumbs CE, Pevsner J, Thomas G, Valle D, Hoover-Fong JE, Goldstein DB. Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet. 2010;6:e1000991. doi: 10.1371/journal.pgen.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wetterstrand KA. [accessed 13 July 2012];DNA Sequencing Costs: Data from the NHGRI Large-Scale Genome Sequencing Program. available at www.genome.gov/sequencingcosts.
- 25.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee, ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 26.Maddalena A, Bale S, Das S, Grody W, Richards S. ACMG Laboratory Quality Assurance Committee, Technical standards and guidelines: Molecular genetic testing for ultra-rare disorders. Genet Med. 2005;7:571–583. doi: 10.1097/01.gim.0000182738.95726.ca. [DOI] [PubMed] [Google Scholar]
- 27.Zoccoli MA, Chan M, Erker JC, Ferreira-Gonzalez A, Lubin IM. NCCLS document MM9-A. NCCLS; PA, USA: 2004. Nucleic Acid Sequencing Methods in Diagnostic Laboratory Medicine; Approved Guideline. [Google Scholar]
- 28.American Society of Human Genetics Board of Directors; American College of Medical Genetics Board of Directors. Points to consider: Ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 1995;57:1233–1241. [PMC free article] [PubMed] [Google Scholar]
- 29.http://www.nlm.nih.gov/research/umls/Snomed/snomed_main.html.
- 30.GeneReviews at GeneTests: Medical Genetics Information Resource (database online) University of Washington; Seattle: 1997–2011. available at http://www.genetests.org. [Google Scholar]
- 31.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 32.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DePristo M, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. The Human Gene Mutation Database: Providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4:69–72. doi: 10.1186/1479-7364-4-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Gordon L, Hendrix M, Hourlier T, Johnson N, Kähäri AK, Keefe D, Keenan S, Kinsella R, Komorowska M, Koscielny G, Kulesha E, Larsson P, Longden I, McLaren W, Muffato M, Overduin B, Pignatelli M, Pritchard B, Riat HS, Ritchie GR, Ruffier M, Schuster M, Sobral D, Tang YA, Taylor K, Trevanion S, Vandrovcova J, White S, Wilson M, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I, Durbin R, Fernández-Suarez XM, Harrow J, Herrero J, Hubbard TJ, Parker A, Proctor G, Spudich G, Vogel J, Yates A, Zadissa A, Searle SM. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, Pohl A, Malladi VS, Li CH, Learned K, Kirkup V, Hsu F, Harte RA, Guruvadoo L, Goldman M, Giardine BM, Fujita PA, Diekhans M, Cline MS, Clawson H, Barber GP, Haussler D, James Kent W. The UCSC Genome Browser database: Extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cotton RG, Scriver CR. Proof of “disease causing” mutation. Hum Mutat. 1998;12:1–3. doi: 10.1002/(SICI)1098-1004(1998)12:1<1::AID-HUMU1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 43.Richard G. Connexin disorders of the skin. Clin Dermatol. 2005;23:23–32. doi: 10.1016/j.clindermatol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Sbidian E, Feldmann D, Bengoa J, Fraitag S, Abadie V, de Prost Y, Bodemer C, Hadj-Rabia S. Germline mosaicism in keratitis-ichthyosis-deafness syndrome: Pre-natal diagnosis in a familial lethal form. Clin Genet. 2010;77:587–592. doi: 10.1111/j.1399-0004.2009.01339.x. [DOI] [PubMed] [Google Scholar]
- 45.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, Cassidy RP, Fiorentini CJ, Heiken KF, Lawrence JJ, Mahoney MH, Miller CJ, Nair DT, Politi KA, Worcester KN, Setton RA, Dipiazza R, Sherman EA, Eastman JT, Francklyn C, Robey-Bond S, Rider NL, Gabriel S, Morton DH, Strauss KA. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Topol E. The Creative Destruction of Medicine. Basic Books, Perseus Books Group; New York, NY: 2012. [Google Scholar]
- 48.Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, Zhang L, Farmer AD, Bell CJ, Kim RW, May GD, Woodward JE, Caillier SJ, McElroy JP, Gomez R, Pando MJ, Clendenen LE, Ganusova EE, Schilkey FD, Ramaraj T, Khan OA, Huntley JJ, Luo S, Kwok PY, Wu TD, Schroth GP, Oksenberg JR, Hauser SL, Kingsmore SF. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohane IS, Masys DR, Altman RB. The incidentalome: A threat to genomic medicine. JAMA. 2006;296:212–215. doi: 10.1001/jama.296.2.212. [DOI] [PubMed] [Google Scholar]
- 50.Cassa CA, Savage SK, Taylor PL, Green RC, McGuire AL, Mandl KD. Disclosing pathogenic genetic variants to research participants: Quantifying an emerging ethical responsibility. Genome Res. 2012;22:421–428. doi: 10.1101/gr.127845.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asharani PV, Keupp K, Semler O, Wang W, Li Y, Thiele H, Yigit G, Pohl E, Becker J, Frommolt P, Sonntag C, Altmüller J, Zimmermann K, Greenspan DS, Akarsu NA, Netzer C, Schönau E, Wirth R, Hammerschmidt M, Nürnberg P, Wollnik B, Carney TJ. Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. Am J Hum Genet. 2012;90:661–674. doi: 10.1016/j.ajhg.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Es JH, Barker N, Clevers H. You Wnt some, you lose some: Oncogenes in the Wnt signaling pathway. Curr Opin Genet Dev. 2003;13:28–33. doi: 10.1016/s0959-437x(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 53.Cadigan KM, Nusse R. Wnt signaling: A common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 54.Zhang M, Zhang J, Lin SC, Meng A. β-Catenin 1 and β-catenin 2 play similar and distinct roles in left-right asymmetric development of zebrafish embryos. Development. 2012;139:2009–2019. doi: 10.1242/dev.074435. [DOI] [PubMed] [Google Scholar]
- 55.Schneider I, Schneider PN, Derry SW, Lin S, Barton LJ, Westfall T, Slusarski DC. Zebrafish Nkd1 promotes Dvl degradation and is required for left-right patterning. Dev Biol. 2010;348:22–33. doi: 10.1016/j.ydbio.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin X, Xu X. Distinct functions of Wnt/β-catenin signaling in KV development and cardiac asymmetry. Development. 2009;136:207–217. doi: 10.1242/dev.029561. [DOI] [PubMed] [Google Scholar]
- 57.Caron A, Xu X, Lin X. Wnt/β-catenin signaling directly regulates Foxj1 expression and ciliogenesis in zebrafish Kupffer’s vesicle. Development. 2012;139:514–524. doi: 10.1242/dev.071746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kemler R. From cadherins to catenins: Cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993;9:317–321. doi: 10.1016/0168-9525(93)90250-l. [DOI] [PubMed] [Google Scholar]
- 59.Korinek K, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler RW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin–Tcf complex in APC−/ − colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 60.Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 61.Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004;18:2225–2230. doi: 10.1101/gad.317604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Züllig S, Basler K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell. 2002;109:47–60. doi: 10.1016/s0092-8674(02)00679-7. [DOI] [PubMed] [Google Scholar]
- 63.Bajoghli B, Aghaallaei N, Soroldoni D, Czerny T. The roles of Groucho/Tle in left-right asymmetry and Kupffer’s vesicle organogenesis. Dev Biol. 2007;303:347–361. doi: 10.1016/j.ydbio.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 64.Grigoryan T, Wend P, Klaus A, Birchmeier W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss- and gain-of-function mutations of β-catenin in mice. Genes Dev. 2008;22:2308–2341. doi: 10.1101/gad.1686208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakaya MA, Biris K, Tsukiyama T, Jaime S, Rawls JA, Yamaguchi TP. Wnt3a links left-right determination with segmentation and anteroposterior axis elongation. Development. 2005;132:5425–5436. doi: 10.1242/dev.02149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tomita-Mitchell A, Mahnke DK, Struble CA, Tuffnell ME, Stamm KD, Hidestrand M, Harris SE, Goetsch MA, Simpson PM, Bick DP, Broeckel U, Pelech AN, Tweddell JS, Mitchell ME. Human gene copy number spectra analysis in congenital heart malformations. Physiol Genomics. 2012;44:518–541. doi: 10.1152/physiolgenomics.00013.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla-Dieck L, de M, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Christiansen J, Dyck JD, Elyas BG, Lilley M, Bamforth JS, Hicks M, Sprysak KA, Tomaszewski R, Haase SM, Vicen-Wyhony LM, Somerville MJ. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res. 2004;94:1429–1435. doi: 10.1161/01.RES.0000130528.72330.5c. [DOI] [PubMed] [Google Scholar]
- 69.Willis TG, Zalcberg IR, Coignet LJ, Wlodarska I, Stul M, Jadayel DM, Bastard C, Treleaven JG, Catovsky D, Silva ML, Dyer MJ. Molecular cloning of translocation t(1;14)(q21;q32) defines a novel gene (BCL9) at chromosome 1q21. Blood. 1998;91:1873–1881. [PubMed] [Google Scholar]
- 70.Yu X, Ng CP, Habacher H, Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat Genet. 2008;40:1445–1453. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]
- 71.1000 Genome Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011;21:1498–1505. doi: 10.1101/gr.123638.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sampson J, Jacobs K, Yeager M, Chanock S, Chatterjee N. Efficient study design for next generation sequencing. Genet Epidemiol. 2011;35:269–277. doi: 10.1002/gepi.20575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mardis ER. The $1,000 genome, the $100,000 analysis? Genome Med. 2010;2:84. doi: 10.1186/gm205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang MQ. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 76.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee, ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.