

Abstract
Virally encoded microRNAs (miRNAs) have recently been discovered in herpesviruses. However, their biological roles are mostly unknown. We developed an algorithm for the prediction of miRNA targets and applied it to human cytomegalovirus miRNAs, resulting in the identification of the major histocompatibility complex class I–related chain B (MICB) gene as a top candidate target of hcmv-miR-UL112. MICB is a stress-induced ligand of the natural killer (NK) cell activating receptor NKG2D and is critical for the NK cell killing of virus-infected cells and tumor cells. We show that hcmv-miR-UL112 specifically down-regulates MICB expression during viral infection, leading to decreased binding of NKG2D and reduced killing by NK cells. Our results reveal a miRNA-based immunoevasion mechanism that appears to be exploited by human cytomegalovirus.
MiRNAs constitute a large family of small noncoding RNAs that regulate gene expression posttranscriptionally, affecting mRNA degradation and translation by base-pairing with the 3′ untranslated regions (3′UTRs) (1). The recent discovery of virally encoded miRNAs, mostly in herpesviruses, intriguingly suggests that miRNAs may function in interspecies regulation involving viral miRNAs and host genes (2–4). Human cytomegalovirus (HCMV) is known to have evolved effective immune evasion strategies, encoding many immunomodulatory proteins that manipulate the immune response (5, 6). It is thus conceivable that miRNAs encoded by HCMV (2) might be exploited during immune evasion. To test this hypothesis, we sought to identify potential human target genes of the HCMV miRNAs by using our newly developed target prediction algorithm, RepTar (7). Briefly, RepTar has its basis in the observation that miRNA binding sites can repeat several times in the target's 3′UTR (1). It therefore searches for repetitive elements in each 3′UTR sequence and evaluates these elements as potential miRNA binding sites. A complementary module of the algorithm, cRepTar, screens for additional single binding sites by using the information obtained by RepTar (7). This algorithm, unlike most other algorithms (8), is independent of evolutionary conservation of the binding sites. Therefore, it is more suitable for predicting targets of the less evolutionary conserved viral miRNAs (2).
We applied RepTar and subsequently cRepTar to all human 3′UTRs, searching for potential binding sites of the 11 HCMV miRNAs listed in miRBase 7.0 (9). MICB, an immunorelated gene, was among the highest ranking predicted targets and the top prediction for hcmv-miR-UL112 (Fig. 1A). MICB is a stress-induced ligand of NKG2D, a natural killer (NK) activating receptor expressed on almost all human NK cells and activated cytotoxic T lymphocytes (CTLs) (10). The importance of MICB in the immune response against HCMV infection is substantiated by the specific down-regulation of MICB surface expression via the UL16 protein of HCMV (11, 12). MICA, another stress-induced ligand of NKG2D, was also ranked among the top predicted targets of hcmv-miR-UL112 (Fig. 1A). The hcmv-miR-UL112 putative binding sites of both genes are almost identical and are located within a highly similar but not evolutionarily conserved (7) 150-nucleotide (nt) region of their 3′UTRs.
Fig. 1.
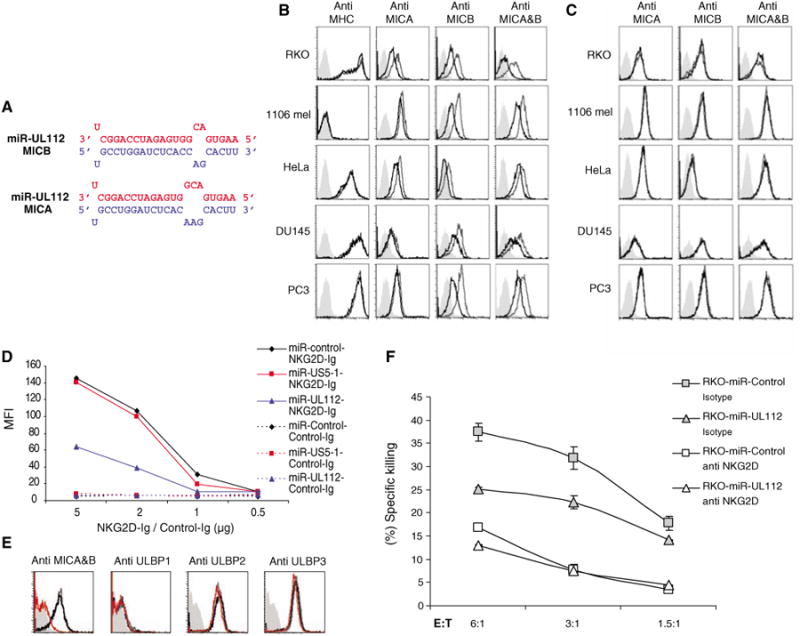
hcmv-miR-UL112 specifically down-regulates MICB expression and reduces NK cytotoxicity. For all panels, one representative experiment is shown out of at least three performed. (A) The predicted duplex of hcmv-miR-UL112 (red) and its target site (blue) in the 3′UTR of MICB (top) and MICA (bottom). (B) Ectopic expression of hcmv-miR-UL112 down-regulates MICB expression. Various human cell lines were transduced with lentiviruses expressing GFP either with hcmv-miR-UL112 (black histogram) or miR-control (open gray histogram). Expression levels of MHC class I, MICA, and MICB were assessed by FACS. (C) Ectopic expression of hcmv-miR-US5-1 does not affect MICB expression. Human cell lines were transduced with lentiviruses expressing GFP and either hcmv-miR-US5-1 (black histogram) or miR-control (open gray histogram). The expression levels of MICA and MICB were assessed by FACS. The histogram plots of (B) and (C) were gated only on the GFP-positive cells. Background levels for (B) and (C) were measured by using only the secondary Cy5-conjugated Ab (gray solid histogram). (D) Reduced binding of NKG2D to cells expressing hcmv-miR-UL112. Binding of NKG2D-Ig to the RKO cells expressing miR-control, hcmv-miR-US5-1, or hcmv-miR-UL112 was assessed by FACS using NKG2D-Ig and the control CD99-Ig (Control-Ig) in various concentrations. (E) The reduced NKG2D-Ig binding is due to reduced MICB expression. The expression level of the various NKG2D ligands was assessed by FACS in RKO cells expressing hcmv-miR-UL112 (open red histogram), miR-control (open gray histogram), or hcmv-miR-US5-1 (open black histogram). The histogram plots are gated only on the GFP-positive cells. The background was measured as in (B) and (C) (solid gray histogram). (F) Reduced killing of RKO cells expressing hcmv-miR-UL112. Bulk NK cells were preincubated either with anti-NKG2D mAb (white) or with isotype-match control mAb (gray). Labeled RKO cells expressing miR-control or hcmv-miR-UL112 were then added and incubated for 5 hours at the indicated effector:target (E:T) ratios. The differences between the killing of the RKO cells expressing miR-control and those expressing hcmv-miR-UL112 in the presence of the isotype-matched control mAb were significant (P < 0.01, t test). Error bars represent standard deviation of replicates.
To assess the function of hcmv-miR-UL112, we expressed this miRNA in various human tumor cell lines that endogenously express MICA and MICB with the use of recombinant lentiviral vectors: hcmv-miR-UL112 and two control vectors, a non-miRNA sequence (miR-control) and hcmv-miR-US5-1. The expression of hcmv-miR-UL112 was confirmed by quantative real-time polymerase chain reaction (qPCR) (fig. S1). The vectors contained green fluorescent protein (GFP) for monitoring the infection efficiency (7). No difference in the transduction efficiency of the different lentiviral vectors was measured (fig. S2). Analysis of the various tumor cells transduced with hcmv-miR-UL112 revealed a specific and extensive reduction of MICB and little or no reduction of MICA (Fig. 1B). The down-regulation was specific to MICB and to hcmv-miR-UL112, because no change in the level of major histocompatibility complex (MHC) class I was observed (Fig. 1B) and transduction with hcmv-miR-US5-1 had no effect (Fig. 1C).
To study whether the observed changes in MICB protein levels affected its interaction with NK cell activating receptor NKG2D, we stained RKO cells expressing either miR-control, hcmv-miR-US5-1, or hcmv-miR-UL112 with NKG2D fused to immunoglobulin G1 (IgG1), as previously described (11). Fluorescence-activated cell sorting (FACS) analysis revealed a measurable decrease in NKG2D-Ig staining of cells expressing hcmv-miR-UL112 compared with those expressing hcmv-miR-US5-1 or miR-control (Fig. 1D). The reduction in NKG2D binding was specifically due to the reduced levels of MICB, because the expression of other NKG2D ligands was not affected by hcmv-miR-UL112 (Fig. 1E).
The functional implication of the hcmv-miR-UL112–mediated reduction in NKG2D binding was demonstrated by measuring NK lysis of RKO cells expressing hcmv-miR-UL112 or miR-control. Cells expressing hcmv-miR-UL112 were killed less efficiently than cells expressing miR-control (Fig. 1F). When NKG2D interactions were blocked with a monoclonal antibody (mAb) against NKG2D (anti-NKG2D), killing levels of both cells were similar (Fig. 1F), indicating that this reduced killing was explicitly due to reduced NKG2D recognition.
Direct binding of hcmv-miR-UL112 to the 3′UTR of MICA and MICB was studied by luciferase reporter assays in HeLa cells ectopically expressing either hcmv-miR-UL112 or miR-control. In agreement with the staining results (Fig. 1B), a measurable decrease in the activity of the luciferase reporter gene was observed only when it was fused to the 3′UTR of MICB and only in cells expressing hcmv-miRUL112 (Fig. 2A). To demonstrate that the downregulation of MICB by hcmv-miR-UL112 is mediated through the predicted binding site, we generated two double substitution mutations, disrupting the predicted pairing between hcmv-miR-UL112 and the 3′UTR of MICB (Fig. 2B). Both mutations abolished the repression mediated by hcmv-miR-UL112 (Fig. 2A). The binding site in the 3′UTR of MICB has one additional putative paired nucleotide in comparison to MICA (Figs. 1A and 2B). To test whether the difference observed between MICA and MICB was due to the change in this single nucleotide, we generated a point substitution that changed the MICA binding site to that of MICB (MICA to B, Fig. 2B). This mutation indeed caused a reduction in luciferase activity but not as substantial as that with the 3′UTR of MICB, supporting previous suggestions that additional factors are involved in the determination of a functional binding site (13).
Fig. 2.

hcmv-miR-UL112 specifically binds to MICB-3′UTR and inhibits its translation. For all panels, one representative experiment is shown out of two performed. (A) hcmv-miR-UL112–mediated repression of luciferase reporter gene activity. The 3′UTR of MICA (303 nt) and a 350-nt segment of the 3′ UTR of MICB (including the predicted hcmv-miR-UL112 binding site) were inserted downstream of a firefly luciferase open reading frame. The figure demonstrates luciferase activity after the indicated reporter plasmids were transfected into HeLa cells expressing either hcmv-miR-UL112 (gray) or miR-control (white). Firefly luciferase activity was normalized to Renilla luciferase activity and then normalized to the average activity of the control reporter. Values are mean ± SD for triplicate samples. *Statistically significant difference between cells expressing miR-control and those expressing hcmv-miR-UL112 (P < 0.005 by t test). (B) Schematic representation of the mutations made in MICB and MICA 3′UTRs (blue) and their base-pairings with hcmv-miR-UL112 (red). Mutated positions are underlined. (C) qPCR analysis of MICB. Experiments were performed with RKO cells expressing either miR-control or hcmv-miR-UL112. The levels of 18S ribosomal RNA were used as internal standard control. Values are mean ±SD for triplicate samples.
To determine whether hcmv-miR-UL112 reduced MICB expression by influencing its mRNA degradation, we tested MICB mRNA levels in RKO cells expressing either miR-control or hcmv-miR-UL112 by using qPCR. No significant change in MICB mRNA level was observed (Fig. 2C), suggesting that the down-regulation of MICB was due to translation inhibition.
We next explored the function of hcmv-miR-UL112 during authentic HCMV infection with use of a wild-type HCMV-AD169 strain and a HCMV-AD169 mutant (14). In this mutant the UL114 gene is deleted, and therefore the hcmv-miR-UL112 gene that resides on the complementary strand is also absent. HCMV-AD169 laboratory strain is adapted to infect primary fibroblasts and a limited number of cell lines. Expression of MICB protein was not observed in several primary fibroblasts and cell lines, before or after infection (fig. S3). We used this to our benefit and expressed MICB with its 3′UTR (MICB-3′UTR) or MICB without its 3′UTR (MICB) in primary human foreskin fibroblasts (HFF) cells. Ectopic expression of hcmv-miR-UL112 in these cells resulted in down-regulation of only MICB-3′ UTR protein (fig. S4), indicating that these cells are adequate for testing the effect of viral hcmv-miR-UL112 on MICB expression.
We used the HFF cells expressing either MICB-3′UTR or MICB and infected them with the AD169-wild-type or with the AD169-mutated virus. The protein expressed by MICB-3′UTR was almost completely down-regulated on day 3 after infection in the AD169-wild-type– infected cells but not in the cells infected with the AD169-mutated virus (Fig. 3A). This is consistent with the accumulation of hcmv-miR-UL112 3 days after infection (fig. S5) (15). In contrast, MICB down-regulation in the absence of the 3′UTR (which was mediated by the UL16 protein) was similar in cells infected with the AD169-wild-type and mutated viruses (Fig. 3A). The effect of hcmv-miR-UL112 on MICB-3′UTR expression was specific, because the level of MHC class I [known to be down-regulated by the virus during infection (16)] was similarly reduced by both viruses (Fig. 3B).
Fig. 3.
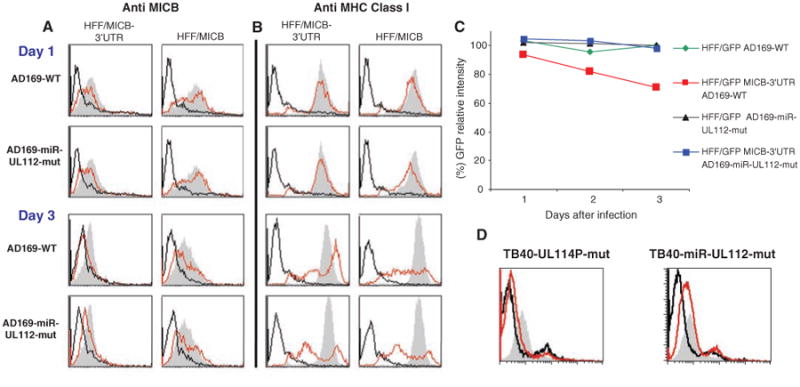
hcmv–miR-UL112–mediated down-regulation of MICB during authentic viral infection. (A and B) Time course expression of MICB (A) and MHC class I (B) on HFF cells expressing either MICB-3′UTR or only MICB. Cells were infected with either the AD169-wild-type virus (AD169-WT) or the AD169-miR-UL112 mutant virus (AD169-miR-UL112-mut). The expression levels of MICB (A) and MHC class I (B) were assessed by FACS staining (red histograms). The gray histograms represent staining of the corresponding uninfected cells. Background levels (black histogram) are the secondary fluorescein isothiocyanate (FITC)–conjugated Abs. (C) HFF cells expressing GFP or GFP fused to the 3′UTR of MICB were infected with either AD169-wild-type or with AD169-miR-UL112-mut viruses, and the levels of GFP expression were measured along the course of infection. The symbols represent the percentage of GFP compared to the corresponding uninfected cells. For (A) to (C), one representative experiment is shown out of three performed. (D) HUVECs were infected with either TB40 UL114-mutant virus (TB40-UL114P mut) or with the TB40 hcmv-miR-UL112 mutant virus (TB40-miR-UL112 mut), and the expression of MICB was measured (red histograms). The gray histograms represent the staining of the corresponding uninfected cells. Background levels (black histogram) are the secondary FITC-conjugated Abs. Shown is one representative experiment out of two performed.
To segregate the miRNA posttranscriptional regulation of MICB from the posttranslational regulation mediated by HCMV UL16 protein, (12) we examined the effect of hcmv-miR-UL112 in HFF cells expressing either GFP, GFP fused to the 3′UTR of MICB, or GFP fused to the 3′UTR of MICA. Consistent with the observed MICB-3′UTR down-regulation (Fig. 3A) and the qPCR results (fig. S5), the most substantial reduction in GFP expression was observed on day 3, only when the cells were infected with the AD169-wild-type virus and only in those expressing the GFP fused to 3′UTR of MICB (Fig. 3C). No changes in the GFP levels were observed in cells carrying GFP alone or GFP fused to the 3′UTR of MICA (Fig. 3C and fig. S5).
To further demonstrate the biological relevance of hcmv-miR-UL112 and to exclude the possibility that the deletion of the UL114 protein-encoding gene affected MICB down-regulation, we constructed two new mutated viruses in the HCMV TB40 strain (17): a control virus in which UL114 is mutated but hcmv-miR-UL112 is intact (TB40-UL114P-mut) and a mutated UL114 virus in which hcmv-miR-UL112 was deleted (TB40-miR-UL112-mut). The HCMV TB40 strain is similar to clinical strains and infects endothelial cells, one of the natural targets of HCMV in vivo (17). Infection of human umbilical vein endothelial cells (HUVECs) that endogenously express MICB with the TB40-UL114P-mut resulted in an almost complete down-regulation of MICB expression after 3 days, whereas MICB expression was still evident in cells infected with the TB40-miR-UL112-mut virus (Fig. 3D). The level of MHC class I was not reduced during viral infection, because the TB40 viruses we used lack the viral US2-US6 genes that mediate MHC class I down-regulation (7). This result suggests that, during clinical viral infection, endogenous MICB is down-regulated by hcmv-miR-UL112.
We next performed killing assays in parallel with the staining presented in Fig. 3, A, B, and D. On day 3 of the infection, the cells expressing MICB-3′UTR and infected with the AD169-mutant virus were killed more efficiently than those infected with the AD169-wild-type virus (Fig. 4, A and B). Similarly, HUVECs infected with the TB40-miR-UL112-mut virus were killed more efficiently than those infected with the TB40-UL114P-mut virus (Fig. 4C). Addition of anti-NKG2D mAb abolished those differences (Fig. 4, B and C). The killing levels of the infected HUVECs were low, even at higher effector:target ratios, probably because of the high levels of MHC class I molecules, which inhibit NK cytotoxicity. Thus, during authentic viral infection, hcmv-miR-UL112 down-regulates MICB, perturbing its binding with NKG2D and consequently aiding in NK attack evasion.
Fig. 4.

hcmv-miR-UL112–mediated down-regulation of MICB during viral infection reduces NK cells cytotoxicity. Experiments were performed concomitantly with the FACS staining presented in Fig. 3. Error bars represent standard deviation of three replicates. Shown is one representative experiment out of two performed. (A) HFF cells expressing either MICB-3′UTR or MICB were infected with either AD169-WT (black) or with AD169-miR-UL112-mut viruses (gray) and incubated with bulk NK cells at the indicated effector:target (E:T) ratios. (B and C) Bulk NK cells were preincubated with either anti-NKG2D or with an isotype-match control mAb. In (B), HFF cells expressing either MICB-3′UTR or MICB that were infected for 3 days either with AD169-wild-type (white) or with AD169-miR-UL112-mut (gray) were then added at a final effector:target ratio of 20:1. In (C), HUVECs that were infected for 3 days either with TB40-UL114P mutant (white) or with TB40-miR-UL112 mutant (gray) were then added at a final effector:target ratio of 15:1.
The discovery of viral miRNAs has raised the intriguing possibility of their involvement in immune evasion (4). The first direct evidence for an miRNA-related immunoevasion mechanism was discovered in the SV40 virus, where a viral miRNA targets a viral gene, resulting in CTL evasion (18). Our results demonstrate a novel miRNA-based evasion strategy used by HCMV, in which a viral miRNA directly down-regulates a host immune defense gene. HCMV is known to rely on several functionally redundant immuno-evasive proteins that can cooperatively target the same process or even the same immune protein of the host (6). Our results expand this view, demonstrating a cooperative mechanism between a viral miRNA (hcmv-miR-UL112) and a viral protein [HCMV UL16 (12)], both targeting the host MICB protein. Down-regulation of host genes by interactions of their mRNAs with viral miRNAs has been also supported by recent findings in herpes simplex virus–1 (19). The advantages of a viral miRNA-based evasion mechanism are multiple. First, these molecules are small, nonimmunogenic, and specific (4). Second, from an evolutionary perspective it should be simpler to develop a regulatory antisense molecule rather than a regulatory protein. Lastly, the combination of protein-mediated and miRNA-mediated posttranscriptional regulation provides a tighter evasion strategy, which is more resistant to the host defense mechanisms because two immunomodulatory elements need to be impaired. The therapeutic implications of such a mechanism are intriguing, because targeting these viral miRNAs might constitute an antiviral therapy while mimicking their role could provide a means of immunosuppressive therapy.
Supplementary Material
Acknowledgments
We thank D. Cosman for the NKG2D-Ig and C. Sinzger for bacterial artificial chromosome plasmid pTB40 reagent; T. Tuschl, S. Altuvia, and D. M. Davis for useful discussions; R. Melnikov for technical support; and E. Akiva and G. Lithwick for their useful comments on the manuscript. This study was supported by grants from the U.S.–Israel Binational Science Foundation (H.M. and O.M.), the Israeli Cancer Research Foundation (H.M. and O.M.), the Israeli Science Foundation (O.M.), the European Consortium (grant nos. MRTN-CT-2005 and LSCH-CT-2005-518178, O.M.), the Hadassah Women's Health Fund (S.Y.), the Deutsche Forschungsgemeinschaft (grant HE 2526/7-1, H.H.), and the National Institute for Allergy and Infectious Disease (grant no. N01-30049, M.P.).
References and Notes
- 1.Bartel DP. Cell. 2004;116:281. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Pfeffer S, et al. Nat Methods. 2005;2:269. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan CS, Ganem D. Mol Cell. 2005;20:3. doi: 10.1016/j.molcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Cullen BR. Nat Genet. 2006;38(suppl):S25. doi: 10.1038/ng1793. [DOI] [PubMed] [Google Scholar]
- 5.French AR, Yokoyama WM. Curr Opin Immunol. 2003;15:45. doi: 10.1016/s095279150200002x. [DOI] [PubMed] [Google Scholar]
- 6.Mocarski ES., Jr Cell Microbiol. 2004;6:707. doi: 10.1111/j.1462-5822.2004.00425.x. [DOI] [PubMed] [Google Scholar]
- 7.Materials and methods are available as supporting material on Science Online.
- 8.Rajewsky N. Nat Genet. 2006;38(suppl. 1):S8. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 9.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. Nucleic Acids Res. 2006;34:D140. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer S, et al. Science. 1999;285:727. [Google Scholar]
- 11.Cosman D, et al. Immunity. 2001;14:123. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 12.Dunn C, et al. J Exp Med. 2003;197:1427. doi: 10.1084/jem.20022059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nilsen TW. Trends Genet. 2007;23:243. doi: 10.1016/j.tig.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Prichard MN, Duke GM, Mocarski ES. J Virol. 1996;70:3018. doi: 10.1128/jvi.70.5.3018-3025.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grey F, et al. J Virol. 2005;79:12095. doi: 10.1128/JVI.79.18.12095-12099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mocarski ES., Jr Trends Microbiol. 2002;10:332. doi: 10.1016/s0966-842x(02)02393-4. [DOI] [PubMed] [Google Scholar]
- 17.Sinzger C, et al. J Gen Virol. 1999;80:2867. doi: 10.1099/0022-1317-80-11-2867. [DOI] [PubMed] [Google Scholar]
- 18.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. Nature. 2005;435:682. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 19.Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Nature. 2006;442:82. doi: 10.1038/nature04836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.