

SUMMARY
The receptor for advanced glycation end products (RAGE) is a pattern recognition receptor involved in inflammatory processes and is associated with diabetic complications, tumor outgrowth, and neurodegenerative disorders. RAGE induces cellular signaling events upon binding of a variety of ligands, such as glycated proteins, amyloid-β, HMGB1, and S100 proteins. The X-ray crystal structure of the VC1 ligand-binding region of the human RAGE ectodomain was determined at 1.85 Å resolution. The VC1 ligand-binding surface was mapped onto the structure from titrations with S100B monitored by heteronuclear NMR spectroscopy. These NMR chemical shift perturbations were used as input for restrained docking calculations to generate a model for the VC1-S100B complex. Together, the arrangement of VC1 molecules in the crystal and complementary biochemical studies suggest a role for self-association in RAGE function. Our results enhance understanding of the functional outcomes of S100 protein binding to RAGE and provide insight into mechanistic models for how the receptor is activated.
INTRODUCTION
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin (Ig) superfamily of cell surface receptors (Neeper et al., 1992; Schmidt et al., 1992). RAGE signaling plays a central role in the inflammatory response (Hofmann et al., 1999), mediating aspects of innate immunity (Tian et al., 2007), acute and chronic inflammatory disorders (Orlova et al., 2007), and certain cancers (Taguchi et al., 2000). Ligand binding to the ectodomain of RAGE is tightly coupled to the recruitment of intracellular factors such as Diaphanous-1 to the cytoplasmic domain, ultimately leading to the proliferation of intracellular signals (Hudson et al., 2008b). Activated RAGE is known to recruit extracellular signal–regulated kinase–1 and –2 (ERK-1/2) (Ishihara et al., 2003), resulting in downstream activation of NF-kκB via the MAP kinase pathway.
RAGE mediates physiological and pathological effects through interaction with a diverse set of ligands, which remarkably, are each associated with a specific disease. The first identified RAGE ligand was advanced glycation end products (AGEs), which form by nonenzymatic glycation of proteins and lipids and accumulate as a result of normal aging and inflammatory processes, particularly in diabetes (Yamagishi et al., 2003). RAGE is up-regulated in Alzheimer disease, and soluble amyloid-β has been shown to bind to the receptor inducing oxidative stress in neurons and the production of proinflammatory cytokines in microglia (Yan et al., 2003). RAGE in the endothelium mediates amyloid-β transport across the blood brain barrier into the central nervous system, promoting amyloid-β plaque formation (Deane et al., 2003). Activation of RAGE on neurons by high mobility group protein 1 (HMGB1) induces reorganization of the cytoskeleton and neurite extension (Hori et al., 1995). However, in cancer cells, RAGE-HMGB1 interaction stimulates tumor invasiveness and growth (Taguchi et al., 2000). Moreover, HMGB1-DNA complexes trigger the association of RAGE with Toll-like receptor 9, which is essential for the immune response to pathogens (Tian et al., 2007). Several S100 proteins bind and activate RAGE. In fact, the first native RAGE ligand discovered was EN-RAGE (S100A12), which initiates and sustains the inflammatory response (Hofmann et al., 1999). S100B binding to RAGE is among the best characterized, having been shown to induce multimerization of the receptor and promote neurite extension and neuronal survival (Huttunen et al., 2000; Leclerc et al., 2007; Ostendorp et al., 2007).
The specific association of RAGE with disease pathogenesis has resulted in a growing interest in RAGE as a therapeutic target. The initial steps toward validating RAGE as a target involved the use of the extracellular ligand-binding region of RAGE (sRAGE), which effectively competes with the receptor for ligands. For example, sRAGE blockage of receptor activation in cell culture has been useful for studies in mouse models for various diseases (Deane et al., 2003; Park et al., 1998; Yan et al., 2003). A model for advanced atherosclerosis shows severe plaque development in the aorta, the formation of which is suppressed in mice treated with exogenous sRAGE (Park et al., 1998). In a mouse model for enterocolitis, administration of sRAGE prevented immune cell infiltration into the colon and sepsis, presumably by blocking the S100A12-RAGE interaction and consequent signaling (Hofmann et al., 1999). Alternative splicing of RAGE mRNA results in an endogenous secretory form of RAGE (esRAGE). Levels of esRAGE are largely decreased in inflammatory and neurodegenerative disorders (Emanuele et al., 2005; Ghidoni et al., 2008; Sternberg et al., 2008; Wittkowski et al., 2007), implying a regulatory function for esRAGE.
In order to better understand the molecular basis for RAGE signaling and its role in cellular physiology, and to lay a foundation for exploiting RAGE as a therapeutic target, we have undertaken a comprehensive analysis of the structure and ligand-binding properties of human sRAGE. sRAGE is composed of a V-type immunoglobulin (Ig) domain and two C-type Ig domains (C1 and C2) (Schmidt et al., 1992). Previous characterization of sRAGE revealed the C-terminal Ig domain (C2) to be structurally independent of the first N-terminal two Ig domains (V and C1), which together form an integrated structural unit, VC1 (Dattilo et al., 2007). VC1 has been shown to bind AGE-modified BSA and S100B (Allmen et al., 2008; Dattilo et al., 2007). Here, we report a high-resolution crystal structure of the tandem VC1 domain pair and link its unique structural features to its binding properties. Heteronuclear NMR spectroscopy is used to map the VC1-binding site for S100B and the S100B-binding site for VC1. The NMR data are then used in combination with molecular docking calculations to generate a model of the complex. These results shed new light on models for RAGE activation.
RESULTS
VC1 Forms an Integrated Structural Unit
The tandem RAGE VC1 domains were crystallized and the X-ray structure was refined at 1.85Å resolution to a Rcryst of 20.9% and Rfree of 24.0% (Table 1). VC1 is found to form a bent elongated structure with an angle of 145° between the two Ig domains (Figure 1A). A specific interface between the domains is observed, which includes several interdomain hydrogen bonds and hydrophobic interactions (Figure 1A). Among these H-bonds, Gln119 in V engages in a pseudo β sheet hydrogen-bonding pattern with Tyr150 in C1, and the carboxylate oxygen atom of Glu94 in V forms an H-bond with the hydroxyl hydrogen of Tyr150 in C1. In addition, a water molecule bridges the amide nitrogen of Ile120 in C1 domain with the side chains of Gln119 and Arg29 in V. Hydrophobic contacts at the interface are mediated between side chain atoms of Pro215 from the C1 F-G loop and the side chain of Tyr118, as well as between the side chain of Tyr150 from the C1 B-C loop and Ile91. Notably, all residues at the VC1 interface are strictly conserved (see Figure S1 available online). These characteristics confirm that the two domains form an integrated structural unit (Dattilo et al., 2007).
Table 1.
Crystallographic Data and Refinement Statistics
| Native | Peak | Zn-MAD Inflection | Remote | |
|---|---|---|---|---|
| Data collection | ||||
|
| ||||
| Wavelength | 1.0082 | 1.2820 | 1.2825 | 1.0082 |
| Resolution (Å) | 47–1.85 (2.0–1.85)a | 47–2.6 (2.7–2.6)a | 47–2.6 (2.7–2.6)a | 47–2.4 (2.1–2.0)a |
| Space group | P21212 | P21212 | P21212 | P21212 |
| Cell dimensions (Å) | 74.85, 119.96, 28.89 | 74.76, 119.90, 28.87 | 74.76, 119.90, 28.87 | 74.76, 119.90, 28.87 |
| Total observations | 153,044 (23,037) | 57,942 (15,395) | 57,858 (15,401) | 124,565 (33,842) |
| Unique reflections | 28,559 (4,704) | 6,158 (1,619) | 6,138 (1,622) | 15,819 (4,599) |
| Completeness (%) | 99.7(99.8)a | 99.9 (100.0)a | 99.9 (100.0)a | 99.7 (99.7)a |
| Redundancy | 6.6 (6.0)a | 3.7 (3.8)a | 3.7 (3.8)a | 3.7 (3.4)a |
| Rsym b (%) | 8.2 (57.1)a | 6.0 (19.1)a | 5.5 (16.5)a | 7.6 (32.9)a |
| I / σI | 12.2 (2.6)a | 16.8 (6.7)a | 17.6 (7.4)a | 11.3 (3.6)a |
| Phasing power acentric | 1.391 | 1.382 | 0.419 | |
| FOM acentric/centric | 0.63279/0.44639 | |||
|
| ||||
| Refinement | ||||
|
| ||||
| Resolution (Å) | 1.85 | 2.0 | 2.0 | 2.0 |
| Rcryst c / Rfree d (%) | 20.9/24.0 (24.7/26.0)a | |||
| No. atoms | ||||
| Protein | 1938 | |||
| Ligand/ion | 4 | |||
| Water | 229 | |||
| B-factors | ||||
| Protein | 25.6 | |||
| Ligand/ion | 44.8 | |||
| Water | 47.0 | |||
| R.m.s deviations | ||||
| Bond angles (°) | 1.99 | |||
| Bond lengths (Å) | 0.018 | |||
|
| ||||
| Ramachandran plot | ||||
|
| ||||
| Most favored (%) | 87.8 | |||
| Additionally allowed (%) | 11.2 | |||
| Generously allowed (%) | 1.2 | |||
Numbers in parentheses apply to the highest-resolution shell.
Rsym = Σhkl Σj∣Ij – < I > ∣ Σhkl Σj ∣ Ij ∣.
Rcryst = Σhkl∣ Fobs – Fc ∣/ΣhklFobs, where Fobs and Fc are observed and calculated structure factors, respectively.
Five percent randomly selected reflections were excluded from refinement and used for the calculation of Rfree.
Figure 1. Structure of the Tandem VC1 Domains of RAGE.

(A) Stereo ribbon diagram VC1 with the V domain in green and the C1 domain in magenta. The two Ig domains adopt a fixed orientation that is stabilized by hydrogen bonds and hydrophobic contacts.
(B) Topology diagram of VC1 with the strands colored as in (A). The structure of the V domain shows it belongs to the I-set of Ig domains, whereas C1 remains in the C1 set. Note that a unique parallel β sheet is formed by strands A’ and G’, which stabilizes the C terminus of the C1 domain.
See also Figure S1.
On the basis of sequence alignments, the N-terminal domain of RAGE (V) had been assigned to the V-set type of Ig molecules. However, once the structure was determined, it revealed features typical of I-set topology (Harpaz and Chothia, 1994), which is characteristic for cell adhesion molecules (Casasnovas et al., 1997; Feinberg et al., 2001; Freigang et al., 2000; Kasper et al., 2000; Su et al., 1998) or muscle proteins (Holden et al., 1992; Zou et al., 2006). Strands A, C, C’, F, and G form one sheet, whereas strands B, D, and E form the second sheet (Figure 1). The region in the V domain corresponding to C” strand in V-set Ig domains instead forms an extended loop encompassing three glycine residues (Gly68, Gly69, and Gly70) and two proline residues (Pro66 and Pro71). The frequency of glycine and proline residues is inconsistent with formation of a β strand. High B-factors in this region suggest that the loop might be adopting variable conformations, which is not unusual for a short Gly and Pro-rich sequence. Although more correctly classified as an I domain, the V nomenclature will be retained here in order to maintain consistency with previous literature.
The C1-domain fits to the C1-set of Ig molecules, with strands A, B, D, and E forming the back β sheet and strands C, C’, F, and G forming the front sheet of the β sandwich. However, the RAGE C1-domain has a unique topology distinct from other Ig fold sets: it contains two additional β strands, A’ and G’, which belong neither to the front nor to the back sheet. Both strands form an additional parallel β sheet stabilizing the C terminus of the domain (Figures 1A and 1B). Further variation is observed for strands D and E. In comparison to most C1-type Ig domains, strand D is elongated by two residues encompassing Arg178 and Arg179, as well as the D-E loop, which is slightly longer through the inclusion of Pro181-Gly184. The E-F loop, which includes Thr195-Thr205, is also elongated by residues Arg198-Asp201. This loop has a unique interaction that stabilizes the C terminus of C1 involving a hydrogen bond between the Arg198 side chain and the Pro232 main chain carbonyl oxygen.
A well-ordered 10-residue C-terminal extension from C1 (Glu231-Leu240) is observed in the high-resolution structure (Figures 1A and 1B). This extension constitutes at least in part the linker to the C2 domains, which NMR studies have shown is flexible (Dattilo et al., 2007). A solution NMR structure of the isolated C2 domain has been determined (PDB 2ENS), but the N terminus is not well defined. In our structure, the extension is stabilized by a contact between Leu240 and the C1-domain of a neighboring molecule in the crystal. Hence, some uncertainty remains over the exact length of the linker between the C1 and C2 domains.
VC1 Contains a Highly Basic Surface
One of the most unusual properties of RAGE is its ability to engage very diverse ligands of different size, fold, or symmetry. The structure of the VC1 ligand-binding domain provides a means to assess what specific surface properties enable this unique characteristic. Our analysis revealed large positively charged patches on the V domain (Figure 2), which are formed by highly conserved Arg and Lys residues. A patch of positively charged electrostatic surface was also identified on the C1 domain. Thus, the entire V domain and a major part of C1 form a large area of electropositive surface (Figure 2) that fits well to the acidic (negative) character of the diverse RAGE ligands, including AGE-modified proteins, amyloid-β, and S100 proteins. Comparison of molecular surfaces displaying the isoelectrostatic field potential of structural homologs neural adhesion molecules TAG-1/axonin-1 (Freigang et al., 2000) and N-CAM (Kasper et al., 2000), as well as a structural model of the closest homolog ALCAM, reveals that the electrostatic potential of VC1 is significantly stronger (Figure 2), which suggests RAGE contains an electrostatic trap for negatively charged ligands.
Figure 2. Surface Analysis of VC1.
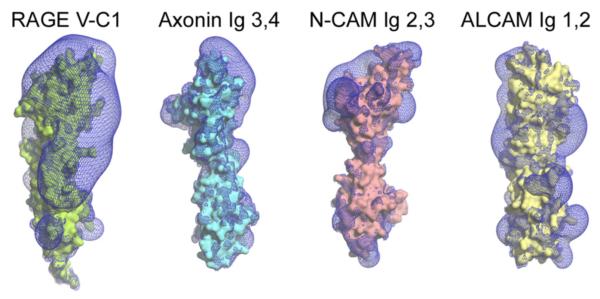
Surface representation of the tandem VC1 domains and its closest structural homologs TAG 1/axonin-1 (Ig domains 3,4), neuronal cell adhesion molecule (N-CAM Ig domains 2,3), and activated leukocyte adhesion molecule (ALCAM Ig domains 1,2). Positive electrostatic isopotential surfaces at 0.8 kT/e are shown in blue. RAGE VC1 exhibits an unusually large positive electrostatic potential. The isosurface shows a coherent potential across both Ig domains extending far into space. The figure was prepared using PyMOL (http://www.pymol.org) and APBS (Baker et al., 2001).
See also Figure S2.
Self-Association of RAGE
Recently, it was shown that RAGE forms oligomers on the plasma membrane (Xie et al., 2008), and it was suggested that oligomerization is mediated by the C1 domain (Xie et al., 2007). Indeed, in the crystal structure, there are side-by-side contacts between VC1 molecules mainly involving residues in the C1 domain. The solvent accessible surface area buried by this interface is 980 Å2 (Figure 3A). The contact surface contains several charged residues and is connected by a tight network of hydrogen bonds and polar interactions (Figures 3B and 3C). The guanidine of Arg178 side chain forms a salt bridge with the carboxylate group of Glu168′ and a hydrogen bond to the carbonyl oxygen of Pro163′ of the neighboring molecule (Figure 3C). The Gln176 side chain oxygen forms two hydrogen bonds to the Nε of Arg178, which positions the guanidine group for optimal interaction with the neighboring molecule (Figure 3C; Figure S2). These interactions are reminiscent of the critical salt bridge formed between Arg and Glu/Asp residues at the interface between Ig domains in the active dimer of the receptor tyrosine kinase KIT (Yuzawa et al., 2007) or between the membrane-proximal Ig-domains of the VEGF receptor (Yang et al., 2010). The extensive set of stabilizing interactions between C1 domains in adjacent molecules in the crystal structure is consistent with the proposal of RAGE aggregation in the plasma membrane. In addition to H-bonds and salt bridges, a Zn2+ ion is found at the intermolecular interface. The Zn2+ is coordinated by residues His180, Glu182, and His158′ from the adjacent VC1 molecule (Figure 3B). These residues, like most involved in at the C1-C1 interface, are conserved across species (Figure S1), which implies a role for Zn2+ in receptor multimerization. We note that Zn2+ can induce dimerization of the class II major histocompatibility complex, leading to cooperative binding of a superantigen ligand (Li et al., 2007). Moreover, homophilic interactions of receptor Ig-type ectodomains play a central role in ligand-binding and cellular activation (Stuttfeld and Ballmer-Hofer, 2009; Xu and Jin, 2010; Yang et al., 2010).
Figure 3. Packing of VC1 in Crystal Lattice.

(A) Ribbon diagram of the VC1 assembly observed in the crystal. The molecules interact mainly via the C1 domain. (B and C) Contact between molecules involve a bridging Zn2+ ion (B) and a salt bridge and hydrogen bonds (C). Stereo view around the bridging Zn2+ ion (B) depicts a Fo-Fc map calculated after refinement of a coordinate file missing the Zn2+ and the side chains of the coordinating residues; the map is shown in purple at 3.0 σ in and in cyan at 12.0 σ, respectively.
See also Figure S1.
In order to further investigate self-association of VC1 and a potential role for Zn2+, we performed dynamic light scattering experiments. The data show that VC1 is monomeric in the absence of Zn2+ and at low pH. The tendency for VC1 to self-associate is heavily influenced by Zn2+ and increased pH into the physiological range (Figures 4A–4C). The degree of self-association of VC1 is concentration dependent (Figure 4D), which is consistent with self-association being a specific binding phenomenon. The concentration dependence of self-association is promoted by the presence of Zn2+ and shows a saturation behavior that could be fit by a hyperbolic function yielding a Kd app of 3.8 ± 1.3 μM.
Figure 4. Self Association of RAGE VC1 Monitored by Dynamic Light Scattering.

(A) VC1 exists as a monomer at pH 5.2.
(B and C) Increase of pH to 6.5 and 7.5 as well as the addition of Zn2+ leads to a shift toward oligomeric states.
(D) VC1 oligomer formation at pH 7.6 was dependent on protein concentration (− ) and is strongly increased by the presence of Zn2+ (C ). The Zn2+ and concentration-dependent oligomerization was fit to a hyperbolic function yielding an apparent dissociation constant (Kdapp) of 3.8 ± C1.3 μM.
See also Figure S2.
An NMR-Based Model for the Complex of VC1 with the RAGE Ligand S100B
Structural details about ligand binding to RAGE have been limited to a single study that highlighted severalr esidues in the V domain that are important for binding of AGEs (Matsumoto et al., 2008). We have investigated the structural basis for binding of the ligand S100B, combining two-dimensional heteronuclear NMR experiments to identify the binding interface on both VC1 and S100B and HADDOCK-based docking calculations to generate a model of the complex.
To map the S100B-binding site on RAGE, a series of 2D 15N-1H HSQC NMR experiments were acquired as Ca2+-S100B was titrated into a solution of 15N-enriched V domain (Figure 5B). The isolated V domain was used in NMR experiments rather than sRAGE or VC1 because, first, the majority of the S100B binding affinity has been shown to be localized to this domain (Dattilo et al., 2007), second, S100B is a homodimer and working with the smallest complex was important to facilitate the NMR analysis, and third, this enables direct comparison to a published study with AGE ligands (Matsumoto et al., 2008). Broadening of all NMR signals over the course of the titration was observed as expected for formation of the V-S100B complex. However, careful analysis of the data revealed differential line broadening effects. As demonstrated elsewhere (Takeuchi and Wagner, 2006), resonances exhibiting enhanced line broadening correspond to residues experiencing the largest perturbation upon binding of the ligand, and these can be correlated to the ligand-binding sites. The binding site was mapped by tracking 52 resonances distributed throughout the V domain that could be directly assigned from the published chemical shift assignments (Matsumoto et al., 2008). Quantification of normalized intensities for these resonances identified a subset of residues that are selectively broadened upon addition of Ca2+-S100B (Figure 5B).
Figure 5. Structural Model of the S100B-VC1 Complex.

(A) Surface representation of VC1 with the electrostatic potential mapped on the surface. Blue represents positively charged areas, and red represents negative charge.
(B) Overlay of the 600 MHz 15N-1H HSQC NMR spectrum of 15N-enriched V domain in the absence (black) and presence (red) of Ca2+-S100B.
(C) VC1 residues identified in (B) mapped on the VC1 structure. The V domain is colored in green, the C1 domain in light blue, and residues perturbed in the NMR experiments are colored yellow.
(D) Docking of S100B on VC1 based on the chemical shift perturbations identified in (B).
(E) Surface representations of the structural model of the S100B-VC1 complex demonstrating the contributions of charge complementarity to binding. The left-hand panel shows S100B engaged with VC1. The right-hand panel pulls apart the complex to reveal the intense negative potential of the binding face of S100B and the intense positive potential of the binding face of VC1.
(F) Structural model of the S100B-VC1 complex with the S100B ribbon and VC1 displayed with the electrostatic potential surface.
See also Figure S3.
When these changes are mapped onto the VC1 structure, they reveal that binding of Ca2+-S100B occurs to one face of the molecule (Figure 5C). Interestingly, these residues map to the highly basic surface of VC1 (Figure 5A), which is complementary to the acidic nature of S100B (Figure 5E). Notably, electrostatic forces have been shown to modulate ligand interactions with other Ig-like domains, such as in the CD2-CD58 protein complex (Wang et al., 1999). Moreover, the S100B-binding site appears to correspond to the site used by RAGE for binding AGE-BSA (Matsumoto et al., 2008). Key residues in AGE binding include Arg48, Arg98, and a patch around Arg104, all of which are perturbed upon binding of Ca2+-S100B. Additional residues include Val35, Lys39, Val89, Glu94, Phe97, Ala101, Asn105, Glu108, and Thr109 (Figure 5C). Mutagenesis experiments on V residues serve to validate this as the ligand-binding surface (Matsumoto et al., 2008). The similarity of binding sites for different RAGE ligands has been inferred previously (Matsumoto et al., 2008), but this work combined with the previous NMR studies on binding of AGE ligands, provides the first direct evidence in support of this hypothesis.
In order to map the VC1 binding surface on S100B, a series of 2D 15N-1H HSQC NMR experiments were acquired over the course of a reciprocal titration of RAGE VC1 into a solution of 15N-enriched Ca2+-S100B. The effects observed in these spectra were similar to those seen for the S100B titration into the V domain (i.e., differential line broadening for a specific set of S100B resonances). The residues affected include His42, Val52, Asn62, Asp69, Phe70, and Ala78, which cluster to a specific surface on S100B. We note that this surface, and in particular His42, Val52, and Ala78, were shown previously to be important for interactions with other S100B-specific ligands such as NDR kinase (Bhattacharya et al., 2003).
Because residues that are selectively perturbed upon formation of the complex could be identified for both RAGE-V and S100B, we proceeded to generate a model of the complex. The program HADDOCK (de Vries et al., 2007; Dominguez et al., 2003) was used for these calculations. A representative structure was selected on the basis of the lowest HADDOCK score and is shown in Figures 5D–5F. The models show S100B uses a concave, negatively charged surface to clamp around a complementary positively charged face of the RAGE V domain (Figure 5E). A surface area greater than 1400 Å2 is buried by formation of the complex. Analysis of the model revealed a number of possible polar hydrogen bonds and salt bridges in the interface. Thus, the model suggests that clustering of basic residues on the binding surface of VC1 and highly acidic surface of S100B are the major driving forces in formation of the complex. In order to validate the result obtained by HADDOCK, we performed a second set of calculations without NMR restraints using the computational docking program HEX (Ritchie and Kemp, 2000; Ritchie et al., 2008). The models obtained in this manner were remarkably similar to the models obtained using the NMR restraints in HADDOCK (Figure S3). Analysis of these models of the VC1-S100B complex reveals a strong electrostatic component to binding, consistent with the proposed electrostatic trap contributing significantly to RAGE ligand binding.
DISCUSSION
Initiation of signal cascades by ligand-induced receptor oligomerization has been proposed as a general mechanism of receptor activation. In some models, multimeric ligands activate their receptors by recruitment of receptor molecules. Hence, association of the extracellular ligand-binding domains drives the colocalization of the cytoplasmic domains, which is needed for signal transduction. In these models, the geometry of the ligand will control activity because the cytoplasmic domains need to be brought into close proximity in a specific orientation. This model has been invoked for receptor tyrosine kinases (Yuzawa et al., 2007). However, it has also been shown that many receptors, including TNFα-receptor (Chan, 2007; Chan et al., 2000), interleukin-receptor (Kramer et al., 2006), EPO receptor (Livnah et al., 1999), and EGF receptor (Stuttfeld and Ballmer-Hofer, 2009; Yang et al., 2010), preassemble in the absence of a ligand on the cell surface. In these cases, it is proposed that the clustering of receptor molecules increases the affinity for ligands and is a requirement for effective receptor signaling. It is notable that the distance of two adjacent VC1 molecules in the crystal structure is 20–25 Å, close to the distance between subunits in the receptor tyrosine kinase complex (Yuzawa et al., 2007) and predicted in the active EGF receptor complex (Burgess et al., 2003). Consistent with a recent FRET study (Xie et al., 2008), our data suggest a similar preassembly model for RAGE.
Preassembly has substantial implications for the mechanism of RAGE activation by its diverse ligands. The molecular basis for RAGE activation by its diverse set of ligands has remained enigmatic; RAGE ligands exhibit different structure, size, and symmetry or even no symmetry, as in the case of glycated proteins or amyloid-β. Clearly, single copies of dissimilar ligands cannot induce a regular arrangement of the intracellular receptor domains as required for initiation of a signal cascade. The common factor linking these ligands is their tendency to oligomerize. The structural and computational data provided here strongly imply a critical role for the positive electrostatic potential in ligand recognition and binding.
If RAGE assembles on the cell surface in the absence of a ligand, ligand binding can be viewed as shifting the equilibrium distribution between monomer and higher order oligomerization states (Figure 6A). Thus, receptor assemblies would be stabilized by the binding of multimeric ligands such as S100B, S100A12, or polymeric glycated proteins. This explains why RAGE activation by such ligands leads to a positive feedback cycle to sustain elevated RAGE expression. Moreover, because increased receptor levels at the cell surface will promote preassembly, this would explain the hyperactivation of the RAGE pathway in the development of chronic inflammatory and neurodegenerative disorders.
Figure 6. Models of RAGE Activation and Inhibition by sRAGE.

(A) Model in which RAGE preassembles in the plasma membrane. Ligand binding to RAGE stabilizes oligomers, which then can bind a signaling adaptor protein (gray sphere) to the cytoplasmic region of RAGE.
(B) Diagram showing the action of sRAGE, in which interaction with intact RAGE to form a hetero-oligomer limits binding of intracellular adaptors and blocks signal transduction.
The importance of RAGE oligomerization is underscored by a number of functional studies. A fusion of the RAGE cytoplasmic domains to the C terminus of the homodimeric glutathione S-transferase (GST) was able to bind the cytoplasmic RAGE mediators Diaphanous-1 (Hudson et al., 2008b) and ERK kinase (Ishihara et al., 2003) in vitro, implying that the GST-fusion protein mimics the active receptor. Because the C termini in the GST homodimer are located in the same orientation and at a distance of approximately 25 Å, these observations imply the cytoplasmic domains in the fusion are separated by the same distance as in the activated receptor.
Although preassembly of RAGE might facilitate ligand binding and increase its efficiency in signaling, preassembled receptors could promote nonspecific activation with adverse consequences. Therefore, preassembly of RAGE must be tightly regulated. The major alternative splice product of RAGE comprises the extracellular region and occurs as soluble protein (sRAGE) (Hudson et al., 2008a), which represents an adaptable regulator of RAGE activation. It has been proposed that soluble alternative splice products of RAGE can act as decoy receptors, decreasing the concentration of available ligands. Our data suggest a different explanation for how sRAGE might interfere with RAGE activation: both sRAGE and the splice variant lacking only the intracellular domain (DN-RAGE) can form heterocomplexes with full-length RAGE, resulting in nonfunctional assemblies (Figure 6B). This notion is largely supported by the observation that coexpression of a RAGE isoform devoid of its intracellular domain has a dominant negative effect on RAGE signaling (Huttunen et al., 1999; Taguchi et al., 2000). Dominant inhibition of signaling by heterocomplex formation has also been observed for tumor necrosis factor–related apoptosis-inducing ligand receptor (Clancy et al., 2005) and Fas death receptor (Siegel et al., 2000), where coexpression of truncated forms of the receptors blocked signaling of full-length receptors by ligand-independent association.
RAGE is a critically important mediator of the inflammatory response and plays an important role in innate immunity. Therapeutic approaches to target RAGE will be valuable for the treatment of diabetic complications, chronic inflammations, and neurodegenerative disorders. The structure of VC1 and NMR-based models of the VC1-S100B complex provide detailed insights into the mechanism of ligand recognition and binding, as well as how RAGE can be activated by the diverse range of ligands. Both structural and biochemical data support the proposal that RAGE assembles in the cytoplasmic membrane without the presence of a ligand. These results define approaches to block ligand binding or reduce self-association, which represent a promising avenue for the pursuit of therapeutic strategies based on interfering with RAGE activation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification Protein encompassing the V and C1 domains of RAGE and S100 proteins were expressed in Escherichia coli and purified as described previously (Allmen et al., 2008; Dattilo et al., 2007; Ostendorp et al., 2005).
Crystallization, Data Collection, and Structure Determination
Crystals were grown by hanging drop vapor diffusion at 298 K by mixing 2 μl of protein with 2 μl of 0.1 M Na cacodylate, 0.2 M Zn acetate (pH 6.5), and 11% PEG 8000. Crystals of a size of 200 μm 3 100 μm 200 μm appeared after 10 to 20 days. The crystals were soaked for 1 min in three consecutive steps in mother liquor containing 4%, 8%, and 12% glycerol and were flash-frozen in liquid nitrogen. Crystals diffracted to a resolution of 1.85 Å. Because Zn2+ turned out to be essential for crystallization, we assumed that Zn2+ specifically interacts with RAGE VC1 and could, therefore, be used for experimental phasing. Multiple anomalous dispersion data were collected at a wavelength of 1.28 Å at the Swiss Light Source (Villigen, Switzerland) beamline X06SA at 100 K. An additional data set collected at a wavelength of 1.0 Å was used for refinement. The data sets were reduced and scaled with the XDS package (Kabsch, 1993). Crystals belonged to the space group P21212 with unit cell parameters of 74.85, 119.96, and 28.89, as determined by XPREP (2005 version, Bruker-AXS). The zinc substructure was determined using SHELXD (Schneider and Sheldrick, 2002). Refinement of the zinc sites and phase calculation was done with SHARP (Fortelle and Bricogne, 1997) followed by solvent flattening and initial fragment building using RESOLVE (Terwilliger, 2000). The final model was built in COOT (Emsley and Cowtan, 2004) and was refined using REFMAC5 (Murshudov et al., 1997; Winn et al., 2001) with final Rwork and Rfree of 20.9% and 24.0%, respectively (Table 1). Ninety-nine percent of the residues were located in the allowed regions, and 1% were located in the generously allowed regions of the Ramachandran plot (Table 1).
Solution NMR Mapping Studies with Ca2+-Loaded S100B
Substoichiometric additions of Ca2+-loaded S100B were made into uniformly labeled 15N-V in 10 mM Imidazole-d4 (pH 6.5), 100 mM NaCl, and 5 mM CaCl2. Similar additions of VC1 were made into a uniform solution of 15N-labeled S100B in 20 mM KCl (pH 7.1), 2.5 mM Ca2+, and 5% D2O. Perturbations in the NMR signals were monitored by 2D 15N-1H HSQC spectra using a Bruker DRX500 spectrometer. Data were processed using Topspin 2.0b (Bruker) and analyzed using NMRViewJ. The intensities (I) were extrapolated from peak heights and plotted as a ratio (I/Io) versus the initial intensity (Io).
Protein Oligomerization Assay
Protein oligomerization assays of VC1 were performed at 298 K. A stock solution of VC1 with 10 mg ml−1 in 10 mM sodium acetate buffer (pH 5.0) was diluted into 20 mM Tris and 150 mM NaCl (pH 7.6). Large oligomers formed at increasing VC1 concentrations and in the presence of Zn2+. The oligomers were pelleted by centrifugation (10 min at 12,000 g). The amount of oligomer formed was calculated by subtracting the amount of protein remaining in the supernatant from total protein. VC1 concentration in the supernatant was determined by the UV absorbance using an extinction coefficient of ε278nm = 32500 M−1 cm−1. In experiments monitoring the effect of Zn2+ ions, 200 μM ZnCl2 was present in the buffer.
Docking Calculations
Residues exhibiting significant NMR chemical shift perturbations were used as input for HADDOCK calculations (Dominguez et al., 2003) to generate an experimentally derived model of the S100B-VC1 complex. Specifically, those residues in S100B and in the V-domain of RAGE whose intensities were reduced by more than one standard deviation from the mean reduction in intensity were selected. The active and passive categories of restraint were assigned based on criteria outlined in HADDOCK2 (de Vries et al., 2007).
The calculation proceeded in a similar manner to that reported by Nordquist et al. (2010). To improve the conformation sampling, twenty structures each were generated from the X-ray crystal structures of S100B (PDB accession number 2H61) and VC1 using room temperature MD simulations in AMBER 9 (Pearlman and Connelly, 1995). Using these starting structures, 1000 complexes were generated from rigid-body docking, of which 200 were selected for further refinement in explicit water. One hundred and four of the 200 structures fell into 11 clusters, of which the two with the lowest target energies were the most populated (cluster 1,25; cluster 2,16). Cluster 1 had the lowest average Haddockscore and highest average buried surface area, so the 20 structures with the lowest energies were selected as the representative ensemble for further analysis.
Free docking calculations of Ca2+-loaded S100B with VC1 were performed using HEX 5.0 (Mustard and Ritchie, 2005; Ritchie and Kemp, 2000) on a multiprocessor Linux workstation. HEX uses 3D parametric functions, which are used to encode both surface shape and electrostatic charge and potential distributions for docking calculations.
Modeling and Analysis
A homology model of the activated leukocyte adhesion molecule (ALCAM) Ig domains 1 and 2 was constructed using VC1 as a template in MODELLER (Eswar et al., 2007; Sali et al., 1995). Electrostatic potentials were calculated with APBS (Baker et al., 2001). All molecular figures were generated using PyMol (DeLano Scientific).
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff at the Swiss Light Source beamline X06SA for excellent assistance with data collection and Benjamin Chagot for technical expertise with HADDOCK2.0. Financial support was provided by the Deutsche Forschungsgemeinschaft (grants FR 1488/3-1 and FR 1488/5-1 to G.F.) and the National Institutes of Health (grant RO1 GM62112 to W.J.C.). S.C. was supported by a Canadian Institutes of Health and Research postdoctoral fellowship.
Footnotes
ACCESSION NUMBERS Coordinates and structure-factor amplitudes have been deposited in the Protein Data Bank with accession number 3CJJ.
SUPPLEMENTAL INFORMATION Supplemental Information includes three figures and can be found with this article online at doi:10.1016/j.str.2010.05.017.
REFERENCES
- Allmen EU, Koch M, Fritz G, Legler DF. V domain of RAGE interacts with AGEs on prostate carcinoma cells. Prostate. 2008;68:748–758. doi: 10.1002/pros.20736. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Large E, Heizmann CW, Hemmings B, Chazin WJ. Structure of the Ca2+/S100B/NDR kinase peptide complex: insights into S100 target specificity and activation of the kinase. Biochemistry. 2003;42:14416–14426. doi: 10.1021/bi035089a. [DOI] [PubMed] [Google Scholar]
- Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- Casasnovas JM, Springer TA, Liu JH, Harrison SC, Wang JH. Crystal structure of ICAM-2 reveals a distinctive integrin recognition surface. Nature. 1997;387:312–315. doi: 10.1038/387312a0. [DOI] [PubMed] [Google Scholar]
- Chan FK. Three is better than one: pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine. 2007;37:101–107. doi: 10.1016/j.cyto.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–2354. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- Clancy L, Mruk K, Archer K, Woelfel M, Mongkolsapaya J, Screaton G, Lenardo MJ, Chan FK. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc. Natl. Acad. Sci. USA. 2005;102:18099–18104. doi: 10.1073/pnas.0507329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dattilo BM, Fritz G, Leclerc E, Vander Kooi CW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry. 2007;46:6957–6970. doi: 10.1021/bi7003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Emanuele E, D’Angelo A, Tomaino C, Binetti G, Ghidoni R, Politi P, Bernardi L, Maletta R, Bruni AC, Geroldi D. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch. Neurol. 2005;62:1734–1736. doi: 10.1001/archneur.62.11.1734. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2 9. [DOI] [PubMed] [Google Scholar]
- Feinberg H, Mitchell DA, Drickamer K, Weis WI. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science. 2001;294:2163–2166. doi: 10.1126/science.1066371. [DOI] [PubMed] [Google Scholar]
- Fortelle E.l., Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwave-length anomalous diffraction methods. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- Freigang J, Proba K, Leder L, Diederichs K, Sonderegger P, Welte W. The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell. 2000;101:425–433. doi: 10.1016/s0092-8674(00)80852-1. [DOI] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Glionna M, Franzoni M, Geroldi D, Emanuele E, Binetti G. Decreased plasma levels of soluble receptor for advanced glycation end products in mild cognitive impairment. J. Neural Transm. 2008;115:1047–1050. doi: 10.1007/s00702-008-0069-9. [DOI] [PubMed] [Google Scholar]
- Harpaz Y, Chothia C. Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J. Mol. Biol. 1994;238:528–539. doi: 10.1006/jmbi.1994.1312. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Holden HM, Ito M, Hartshorne DJ, Rayment I. X-ray structure determination of telokin, the C-terminal domain of myosin light chain kinase, at 2.8 Å resolution. J. Mol. Biol. 1992;227:840–851. doi: 10.1016/0022-2836(92)90226-a. [DOI] [PubMed] [Google Scholar]
- Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008a;22:1572–1580. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008b;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003;550:107–113. doi: 10.1016/s0014-5793(03)00846-9. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- Kasper C, Rasmussen H, Kastrup JS, Ikemizu S, Jones EY, Berezin V, Bock E, Larsen IK. Structural basis of cell-cell adhesion by NCAM. Nat. Struct. Mol. Biol. 2000;7:389–393. doi: 10.1038/75165. [DOI] [PubMed] [Google Scholar]
- Kramer JM, Yi L, Shen F, Maitra A, Jiao X, Jin T, Gaffen SL. Evidence for ligand-independent multimerization of the IL-17 receptor. J. Immunol. 2006;176:711–715. doi: 10.4049/jimmunol.176.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J. Biol. Chem. 2007;282:31317–31331. doi: 10.1074/jbc.M703951200. [DOI] [PubMed] [Google Scholar]
- Li H, Zhao Y, Guo Y, Li Z, Eisele L, Mourad W. Zinc induces dimerization of the class II major histocompatibility complex molecule that leads to cooperative binding to a superantigen. J. Biol. Chem. 2007;282:5991–6000. doi: 10.1074/jbc.M608482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–990. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Yoshida T, Murata H, Harada S, Fujita N, Nakamura S, Yamamoto Y, Watanabe T, Yonekura H, Yamamoto H, et al. Solution structure of the variable-type domain of the receptor for advanced glycation end products: new insight into AGE-RAGE interaction. Biochemistry. 2008;47:12299–12311. doi: 10.1021/bi800910v. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Mustard D, Ritchie DW. Docking essential dynamics eigenstructures. Proteins. 2005;60:269–274. doi: 10.1002/prot.20569. [DOI] [PubMed] [Google Scholar]
- Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- Nordquist KA, Dimitrova YN, Brzovic PS, Ridenour WB, Munro KA, Soss SE, Caprioli RM, Klevit RE, Chazin WJ. Structural and functional characterization of the monomeric U-box domain from E4B. Biochemistry. 2010;49:347–355. doi: 10.1021/bi901620v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostendorp T, Heizmann CW, Kroneck PM, Fritz G. Purification, crystallization and preliminary X-ray diffraction studies on human Ca2+-binding protein S100B. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005;61:673–675. doi: 10.1107/S1744309105018014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostendorp T, Leclerc E, Galichet A, Koch M, Demling N, Weigle B, Heizmann CW, Kroneck PM, Fritz G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007;26:3868–3878. doi: 10.1038/sj.emboj.7601805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr., Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- Pearlman DA, Connelly PR. Determination of the differential effects of hydrogen bonding and water release on the binding of FK506 to native and Tyr82/ Phe82 FKBP-12 proteins using free energy simulations. J. Mol. Biol. 1995;248:696–717. doi: 10.1006/jmbi.1995.0252. [DOI] [PubMed] [Google Scholar]
- Ritchie DW, Kemp GJ. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39:178–194. [PubMed] [Google Scholar]
- Ritchie DW, Kozakov D, Vajda S. Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions. Bioinformatics. 2008;24:1865–1873. doi: 10.1093/bioinformatics/btn334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. doi: 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]
- Sternberg Z, Weinstock-Guttman B, Hojnacki D, Zamboni P, Zivadinov R, Chadha K, Lieberman A, Kazim L, Drake A, Rocco P, et al. Soluble receptor for advanced glycation end products in multiple sclerosis: a potential marker of disease severity. Mult. Scler. 2008;14:759–763. doi: 10.1177/1352458507088105. [DOI] [PubMed] [Google Scholar]
- Stuttfeld E, Ballmer-Hofer K. Structure and function of VEGF receptors. IUBMB Life. 2009;61:915–922. doi: 10.1002/iub.234. [DOI] [PubMed] [Google Scholar]
- Su XD, Gastinel LN, Vaughn DE, Faye I, Poon P, Bjorkman PJ. Crystal structure of hemolin: a horseshoe shape with implications for homophilic adhesion. Science. 1998;281:991–995. doi: 10.1126/science.281.5379.991. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Wagner G. NMR studies of protein interactions. Curr. Opin. Struct. Biol. 2006;16:109–117. doi: 10.1016/j.sbi.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- Wang JH, Smolyar A, Tan K, Liu JH, Kim M, Sun ZY, Wagner G, Reinherz EL. Structure of a heterophilic adhesion complex between the human CD2 and CD58 (LFA-3) counterreceptors. Cell. 1999;97:791–803. doi: 10.1016/s0092-8674(00)80790-4. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Wittkowski H, Hirono K, Ichida F, Vogl T, Ye F, Yanlin X, Saito K, Uese K, Miyawaki T, Viemann D, et al. Acute Kawasaki disease is associated with reverse regulation of soluble receptor for advance glycation end products and its proinflammatory ligand S100A12. Arthritis Rheum. 2007;56:4174–4181. doi: 10.1002/art.23042. [DOI] [PubMed] [Google Scholar]
- Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J. Biol. Chem. 2007;282:4218–4231. doi: 10.1074/jbc.M608888200. [DOI] [PubMed] [Google Scholar]
- Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE) J. Biol. Chem. 2008;283:27255–27269. doi: 10.1074/jbc.M801622200. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jin B. A novel interface consisting of homologous immunoglobulin superfamily members with multiple functions. Cell. Mol. Immunol. 2010;7:11–19. doi: 10.1038/cmi.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi S, Takeuchi M, Inagaki Y, Nakamura K, Imaizumi T. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy. Int. J. Clin. Pharmacol. Res. 2003;23:129–134. [PubMed] [Google Scholar]
- Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, Schmidt AM, Brown C, Stern A, LaFaille J, et al. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat. Med. 2003;9:287–293. doi: 10.1038/nm831. [DOI] [PubMed] [Google Scholar]
- Yang Y, Xie P, Opatowsky Y, Schlessinger J. Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc. Natl. Acad. Sci. USA. 2010;107:1906–1911. doi: 10.1073/pnas.0914052107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130:323–334. doi: 10.1016/j.cell.2007.05.055. [DOI] [PubMed] [Google Scholar]
- Zou P, Pinotsis N, Lange S, Song YH, Popov A, Mavridis I, Mayans OM, Gautel M, Wilmanns M. Palindromic assembly of the giant muscle protein titin in the sarcomeric Z-disk. Nature. 2006;439:229–233. doi: 10.1038/nature04343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.