

ABSTRACT
The recent epidemic history of hepatitis B virus (HBV) infections in the United States is complex, as indicated by current disparity in HBV genotype distribution between acute and chronic hepatitis B cases and the rapid decline in hepatitis B incidence since the 1990s. We report temporal changes in the genetic composition of the HBV population using whole-genome sequences (n = 179) from acute hepatitis B cases (n = 1,206) identified through the Sentinel County Surveillance for Acute Hepatitis (1998 to 2006). HBV belonged mainly to subtypes A2 (75%) and D3 (18%), with times of their most recent common ancestors being 1979 and 1987, respectively. A2 underwent rapid population expansions in ca. 1995 and ca. 2002, coinciding with transient rises in acute hepatitis B notification rates among adults; D3 underwent expansion in ca. 1998. A2 strains from cases identified after 2002, compared to those before 2002, tended to cluster phylogenetically, indicating selective expansion of specific strains, and were significantly reduced in genetic diversity (P = 0.001) and frequency of drug resistance mutations (P = 0.001). The expansion of genetically close HBV A2 strains was associated with risk of infection among male homosexuals (P = 0.03). Incident HBV strains circulating in the United States were recent in origin and restricted in genetic diversity. Disparate transmission dynamics among phylogenetic lineages affected the genetic composition of HBV populations and their capacity to maintain drug resistance mutations. The tendency of selectively expanding HBV strains to be transmitted among male homosexuals highlights the need to improve hepatitis B vaccination coverage among at-risk adults.
IMPORTANCE Hepatitis B virus (HBV) remains an important cause of acute and chronic liver disease globally and in the United States. Genetic analysis of HBV whole genomes from cases of acute hepatitis B identified from 1998 to 2006 in the United States showed dominance of genotype A2 (75%), followed by D3 (18%). Strains of both subtypes were recent in origin and underwent rapid population expansions from 1995 to 2000, indicating increase in transmission rate for certain HBV strains during a period of decline in the reported incidence of acute hepatitis B in the United States. HBV A2 strains from a particular cluster that experienced the most recent population expansion were more commonly detected among men who have sex with men. Vaccination needs to be stepped up to protect persons who remain at risk of HBV infection.
INTRODUCTION
Hepatitis B virus (HBV) is one of the major causative agents of liver disease, including acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Infection with HBV is common, with an estimated prevalence of 240 million infected persons globally (1). In the United States, it is estimated that 1.4 million people are infected with HBV (2), and 43,000 new infections occur annually (3). The estimated incidence rate of acute HBV infection has declined from 13.5 cases per 100,000 population in 1987 to 2.8 cases per 100,000 population in 2002 (4, 5), which is attributable in part to nationwide implementation of vaccination against hepatitis B. However, this decline was not uniform: between 1999 and 2002, incidences were observed to increase transiently among adults in certain age groups (5). Despite the availability of hepatitis B vaccine and the implementation of comprehensive national guidelines, immunization coverage remains suboptimal in certain demographic and risk groups (6–9).
HBV is genetically diverse, and HBV strains are classified into eight major genotypes (A to H) and numerous subtypes (10). The Sentinel Counties Study of Acute Viral Hepatitis was a population-based study that enrolled acute viral hepatitis patients from six city/county health departments in the United States from 1982 through 2006 (11, 12). We recently showed that 75% of acute hepatitis B cases from 1998 to 2006 were infected by HBV genotype A2 and 17% were infected by genotype D (13). This distribution contrasts with the findings from a study of persons in the United States with chronic hepatitis B showing that only 37.4% were infected by genotype A2 and 10.4% were infected by genotype D (14). Strains identified among the acute cases appeared to be of limited genetic diversity, with many strains sharing identical sequences (13). These observations might, however, have reflected usage of the relatively conserved S gene as basis of HBV strain comparison. We and others have shown that analysis of whole-genome (WG) sequences of HBV confers significantly finer resolution to the study of viral diversity and better assessments of HBV evolution during natural infection and under selection pressure from antiviral drugs (15–17).
The disparity of genotype distribution between acute (13) and chronic (14) cases of HBV infection reflects complexity of epidemiological factors associated with patterns of HBV introduction, transmission, and maintenance in the United States. The disproportionately high representation of genotypes A2 and D in incident HBV infections imply greater efficacy of transmission from chronic infections with these genotypes than with other HBV genotypes or more frequent transmission of these two genotypes from acutely infected persons. Taken together with the observation of a substantial decline in incident HBV infection, the genotypic disparity between acute and chronic hepatitis B indicates a dynamic epidemic history of HBV infections over the last two decades. To gain insight into the evolutionary history and molecular epidemiology of incident HBV infection in the United States, we characterized HBV WG sequences from cases of acute HBV infection.
MATERIALS AND METHODS
In the Sentinel Counties Study (11), a case of acute hepatitis B was defined as having history of sudden onset of signs and symptoms consistent with hepatitis, together with serological detection of immunoglobulin M antibody to hepatitis B core antigen, or newly detected hepatitis B surface antigen within the window period of 60 days. The cases were treatment naive at the time of sampling. The six Sentinel sites were Jefferson County, Alabama; Pinellas County, Florida; Pierce County, Tacoma, Washington; Multnomah County, Oregon; San Francisco, California; and Denver County, Denver, Colorado. The study was approved by institutional review boards of the Centers for Disease Control and Prevention (CDC). For the present study, only sera originating from cases enrolled between 1998 through to 2006 that contained sufficient HBV titer to allow for WG amplification and sequencing were included.
HBV WG amplification, sequencing, and analyses.
HBV WG sequences were amplified using two rounds of PCR and sequenced, as previously described (15). The detection sensitivity of this approach is 5 × 102 IU/ml, using the third World Health Organization International standard for HBV DNA. HBV genotyping was performed by nucleotide (nt) sequencing of a 435-bp DNA segment amplified from the HBV S gene (from nt 222 to nt 656 of the HBV genome). Phylogenetic trees were constructed using a maximum-likelihood algorithm. Nucleotide diversity and the distribution of nucleotide distances were evaluated among A2 and D3 sequences. Analysis of molecular variance (AMOVA) was conducted to measure the fraction of heterogeneity in genetic distances among the A2 sequences that arose due to differences between the A2 time clusters.
Estimation of evolutionary dates and demographic history.
The time to the most recent common ancestor (tMRCA) for each genotype was calculated using BEAST (v1.7.1) (18). Estimates were calculated by using the HKY substitution model with four gamma rate categories and invariant sites. The calculations used a coalescent constant size tree prior and an uncorrelated lognormal molecular clock with an initial substitution rate estimate of 5 × 10−3 substitutions per site per year. The nucleotide mean was estimated with a uniform prior distribution. Bayesian skyline plot analysis was performed separately for genotypes A2 and D3 using mean substitution rate estimates from the tMRCA estimates and a constant rate uncorrelated lognormal molecular clock with a coalescent Bayesian skyline prior and 10 groups in a piecewise-constant skyline model. All calculations were run until the effective sample size was greater than 200.
Statistical analysis.
Risk ratio estimates were calculated to determine the association between patient characteristics and phylogenetic distribution of WG sequences. We used univariate and multivariate analyses to determine factors associated with infection by HBV genotypes. We also compared genetic distances among HBV strains by geographic or temporal distributions. SAS for Windows v9.3 was used for statistical analysis. Differences in strain mutations were tested by the Fisher exact test.
Nucleotide sequence accession numbers.
WG sequences of the HBV isolates have been deposited in the National Center for Biotechnology Information GenBank database (accession numbers KF779209 to KF779386).
RESULTS
HBV genetic diversity.
From 1998 through 2006, 1,206 acute hepatitis B cases were reported from the six survey sites. The HBV S gene could be sequenced from 614 cases (13). As we have shown earlier (13), its diversity was restricted: among the A2 strains (75%), 47% shared three sequences, with 32% sharing a single sequence, and among the D strains (18%), 41% shared a single sequence (Fig. 1a). WG sequences were obtained from 179 HBV strains, of which 134 (75%) belonged to A2, 32 (18%) belonged to D3, and the remaining 13 (8%) belonged to genotypes B, C, E, F, G, and H. There are some unavoidable limitations to our study. We did not have sufficient serum volume or titers for HBV WG genome sequencing of all incident cases genotyped using the S gene amplicon. However, the percent distribution of genotypes A, D, and the others was similar among HBV strains, from which WG or only S gene sequences were obtained, confirming a fair representation of the sampled population by the 179 WG sequences. The WG sequences were unique (Fig. 1b), except for those from two A2 and two D3 strains; the identical A2 strains were from cases in the same county and identified from the same year, and the identical D3 strains were from different counties but identified in the same year. Five strains were recombinant: two between A1 and A2 and three between D3 and A2. Cases infected by these recombinants were excluded from the analysis.
FIG 1.

Phylogenetic maximum-likelihood tree constructed using HBV S gene sequences (378 bp) (a) and WG sequences (3.2 kb) (b) from genotypes A and D. The frequencies of the S gene sequences are color-coded, with the color scale shown in the vertical bar for each phylogenetic tree. Red circles in panel b represent sequences that have the most common variant of the S gene found in 76 WGs. Each white circle represents a WG sequence in which S gene is unique.
The WG sequences were tightly clustered within A2 and D3 subtypes, producing a star-like phylogeny, indicating close genetic relatedness within each subtype (Fig. 1b) and recent selective sweep or population expansion for each subtype. Genetic relatedness among A2 strains varied from 97.4 to 100% and among D3 strains from 97.6 to 100% (see Fig. 5b). Among A2 strains from any county, the genetic relatedness ranged between 97.6 and 100%. The proportions of pairs of sequences differing at >3 nt positions were 99.8% for A2 strains and 97.5% for D3 strains.
FIG 5.

(a) Nucleotide diversity of the entire HBV genome, color-coded based according to the year of diagnosis. Each point is an average over a window of 301 nt, with a step of 1. The diagram at the bottom of the figure depicts the HBV genetic map in alignment with the nucleotide numbering on the x axis. (b) Frequency distribution of nucleotide distances among WG from A2 and D3 isolates, color-coded according to the year of diagnosis.
Geographic and temporal distributions.
No correlation between genetic distance and geographic distance for either subtype A2 or D3 was observed, although there was some degree of clustering of A2 and D3 strains from Pinellas County, FL, and Multnomah County, OR, in the phylogenetic tree (sequences highlighted in yellow, Fig. 2a). No correlation was observed between genetic distances among HBV strains and the year of diagnosis. For example, the eight sequences (highlighted in yellow, Fig. 2b) that formed a tight cluster in the tree were diagnosed 1 to 4 years apart.
FIG 2.
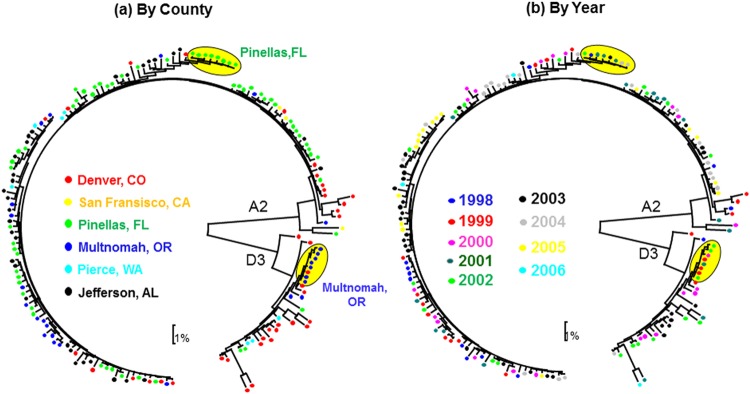
Phylogenetic analysis of WG sequences belonging to genotypes A and D, color-coded according to county and year of sampling. The sentinel Counties are Jefferson County, AL; Pinellas County, FL; Pierce County, WA; Multnomah County, OR; San Francisco County, CA; and Denver County, CO.
Recent origin of A2 and D3 strains.
The tight sequence clustering (Fig. 1) and the close genetic relatedness (Fig. 5b) among HBV strains of same subtypes indicate their recent origins. Bayesian analysis (Fig. 3) showed that the MRCA for A2 existed in ca. 1979 (range, 1961 to 1989) and for D3 in ca. 1987 (range, 1971 to 1998).
FIG 3.

Bayesian skyline plot showing HBV epidemic history in the United States. Middle line represents mean estimate of effective population size, plotted as the log Neτ, and blue lines shows the 95% highest posterior density (HPD) intervals for this estimate. The time scale encompasses a lower limit of 95% HPD for time of the most recent common ancestor (tMRCA) to the time of collection for the most recent specimens.
A2 and D3 population dynamics.
Skyline plot analysis of all HBV A2 sequences indicated expansion of the A2 strains between 1994 and 1996, followed by a transient decline, and a second expansion during 2002. For D3 strains, there was a trend toward population expansion between 1998 and 2000 (Fig. 3).
Differential A2 expansion.
Inspection of the A2 phylogenetic tree identified two rapidly evolving viral lineages that together form a cluster (designated cluster 1, indicated by a yellow circle in Fig. 4a). Although cluster 1 contained HBV strains sampled from 1998 to 2006, ∼80% of strains originated from cases diagnosed after 2002. A skyline plot constructed using sequences from cluster 1 showed a sharp population expansion during 2002 (Fig. 4b). The other A2 sequences (designated cluster 2, Fig. 4a) were mainly from samples collected before 2002. Analysis of these sequences showed an earlier expansion between 1994 and 1996 (Fig. 4c). These observations indicate that the HBV A2 strains experienced two recent expansions (Fig. 3a). The first expansion, mainly of cluster 2 strains, occurred during the mid-1990s. The second, of cluster 1 strains, occurred in 2002 and was accompanied by a population decline among the cluster 2 strains.
FIG 4.

(a) Phylogenetic analysis of WG (genotype A2) color-coded according to year of diagnosis. The bootstrap values are marked on the figure. Cluster 1 sequences (highlighted in yellow) represent most heterogeneous lineage of sequences compared to cluster 2 (not highlighted). (b) Skyline plot for the A2 cluster 1. The middle black line represents the estimated mean of the effective population size (logarithmic scale). The blue lines show the limits of the 95% HPD for this estimate. The vertical dotted line represents the lower 95% HPD for tMRCA. (c) Skyline plot for A2 cluster 2.
Changes in A2 and D3 heterogeneity.
Differential expansion of specific phylogenetic lineages should have a significant effect on the genetic heterogeneity of the HBV population over time. Indeed, the mean nucleotide diversity across all positions of the HBV genome was significantly greater (P = 0.001) in the A2 sequences of the 1998-2001 cases than in the 2002-2006 cases (Fig. 5). However, the genetic differences were not distributed uniformly along the HBV genome, with regions at positions 1 to 400, 600 to 1000, 1500 to 1900, and 2200 to 2700, showing more diversity for A2 strains sampled between 1998 and 2001 than between 2002 and 2006 (Fig. 5a). This observation suggests that these genomic regions contribute more than others to differentiation among HBV A2 strains sampled here. Comparison of sequences within and between clusters 1 and 2 by AMOVA showed that 91.4% of all genetic variations occur within each cluster (P = 0.0001) and only 8.7% of such variations occur between clusters (Fig. 5).
WG sequences were inspected for the presence of clinically important nucleotide changes (see Table 2). None of the substitutions known to confer vaccine escape were identified. The A1896 (stop codon at codon 28) precore mutation was found in three D3 strains and in no A2 strains. There were 18 strains carrying amino acid substitutions known to be associated with drug resistance: rtA194 (to adefovir/tenofovir; n = 17), rtM204 and rtV207 (to lamivudine; n = 16), and rtS202 and rtM250 (to entecavir; n = 3). These drug resistance-associated changes were observed in 38% of the D3 strains, 28% of the A2 cluster 2 strains, and in none of the cluster 1 strains (P < 0.0001) (see Table 2). Analysis of mutations within the HBsAg “a” determinant at amino acid positions 120 to 165 identified a single HBV A2 strain with the known vaccine escape mutation T126I (see Table 2).
TABLE 2.
Distribution of mutationsa
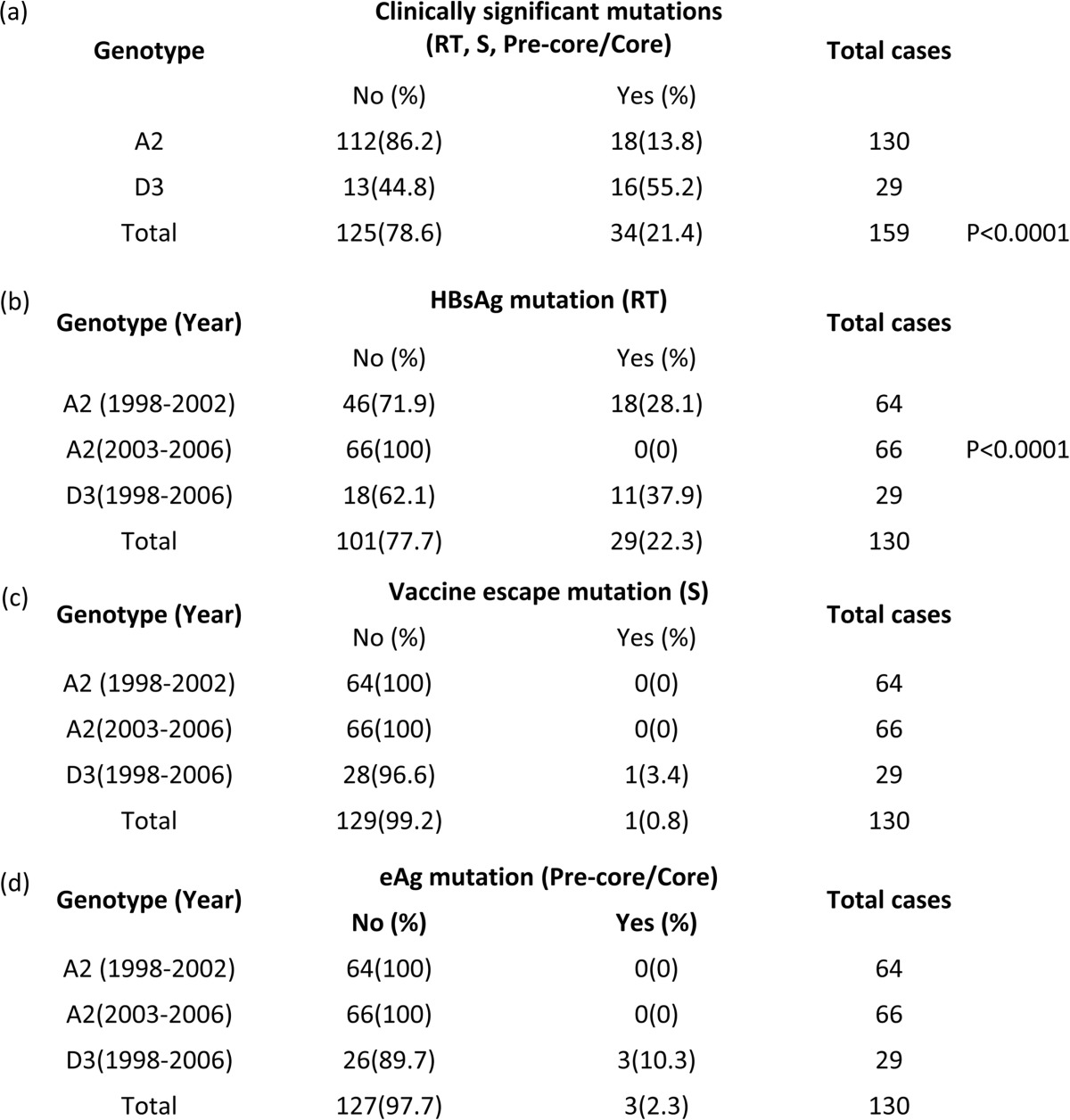
Lettered sections: a, any clinical significant nucleotide changes (in the precore/core, S, and polymerase regions) in genotypes A2 and D3 (the P values are based on a comparison between the percentages of the two subtypes); b, the drug resistance mutations rtA194 (to adefovir/tenofovir; n = 17), rtM204 and rtV207 (to lamivudine, n = 16), and rtS202 and rtM250 (to entecavir, n = 3) (the P value is based on comparison between the percentages of the two A2 clusters); c, S gene mutations (S120 to S165); d, precore (A1896, stop codon 28) mutation.
Factors associated with selective A2 expansion.
Analysis of demographic data of acute hepatitis B cases (Table 1) showed that HBV A2 strains comprising cluster 1 (Fig. 4a) tended to be carried by (i) persons of African-American origin rather than whites (OR, 3.22; 95% CI, 1.44 to 7.2; P = 0.005); individuals from Jefferson County, AL, rather than from Denver, CO (OR, 5.85; 95% CI, 1.37 to 24.89; P = 0.019); and (iii) persons who were diagnosed after 2002 compared to those diagnosed earlier than 2002 (OR, 4.47; 95% CI 95%, 2 to 9.98; P = 0.001). Among the risk factors examined, cluster 1 strains were more common among cases who were men who have sex with men (MSM) compared to those who were not (OR, 2.05; 95% CI, 1.03 to 4.05; P = 0.03). Multivariate analysis was conducted but, owing to small sample sizes, no statistically significant conclusions could be made.
TABLE 1.
Demographic and risk characteristics of A2-infected cases stratified according to infection by cluster 1 and 2 strains
| Demographic parameter | Genotype A2 |
OR (95% CI)a | Total | |||
|---|---|---|---|---|---|---|
| Cluster 1 |
Cluster 2 |
|||||
| n | % | n | % | |||
| Race/ethnicity | ||||||
| White | 21 | 31.3 | 46 | 68.7 | Referent | 67 |
| Black | 25 | 59.5 | 17 | 40.5 | 3.22 (1.44–7.20) | 42 |
| Hispanic | 4 | 30.8 | 9 | 69.2 | 0.97 (0.27–3.52) | 13 |
| County, state | ||||||
| Denver, CO | 3 | 20.0 | 12 | 80.0 | Referent | 15 |
| Jefferson, AL | 19 | 59.4 | 13 | 40.6 | 5.85 (1.37–24.89) | 32 |
| Multnomah. OR | 5 | 31.3 | 11 | 68.7 | 1.82 (0.35–9.46) | 16 |
| Pinellas, FL | 24 | 40.7 | 35 | 59.3 | 2.74 (0.70–10.77) | 59 |
| Pierce, WA | 2 | 50.0 | 2 | 50.0 | 4.00 (0.39–41.23) | 4 |
| San Francisco, CA | 0 | 0.0 | 3 | 100.0 | NA | 3 |
| Yr of collection | ||||||
| 1998-2001 | 11 | 21.2 | 41 | 78.8 | Referent | 52 |
| 2002-2006 | 42 | 54.5 | 35 | 45.5 | 4.47 (2.00–9.98) | 77 |
| Behavioral risk | ||||||
| MSM activity | ||||||
| No | 33 | 52.4 | 30 | 47.6 | Referent | 63 |
| Yes | 25 | 75.8 | 8 | 24.2 | 2.05 (1.03–4.05) | 33 |
| Injecting drug use | ||||||
| No | 45 | 39.8 | 68 | 60.2 | Referent | 113 |
| Yes | 6 | 54.5 | 5 | 45.5 | 0.55 (0.16–1.92) | 11 |
OR, exact odds ratio; CI, confidence interval; referent, the group with which another group is compared; NA, not applicable. Boldface indicates that P is >0.05.
DISCUSSION
Genetic analysis of HBV WG sequences confirmed significant prevalence of genotypes A and D among acute hepatitis B cases in the United States (1998 to 2006). All HBV genotype A variants belonged to subtype A2. Although the S gene sequences used in the previous study did not allow for a confident subtyping of the genotype D variants (13), the phylogenetic analysis of the WG sequences conducted here showed for the first time that all HBV genotype D variants belonged to subtype D3. The high incidence of A2 in acute cases in the United States parallels other global reports. In Europe, the majority of incident HBV strains belong to A2 (19, 20), with specific strains identified in English prisons (21), in outbreaks of nosocomial HBV transmissions in Germany (22) and Belgium (23), and among MSM in The Netherlands (24). In Japan, HBV A2 has been observed to be spreading not only within the MSM community (25) but also from there to the general population (26). The National Heart, Lung, and Blood Institute Retrovirus Epidemiology Donor Study-II (REDS-II) of 34 million U.S. blood donations between 2006 and 2009 reported that the majority of 193 donor HBV strains (37%) consisted of A2, with incident donors carrying higher frequencies of A2 (67%) compared to those in prevalent donors (27%) (27). A similar difference in genotype distribution between acute hepatitis and chronic hepatitis has been reported in Japan (28). There is preliminary evidence suggesting that the apparent high transmission rate of A2 may reflect the longer duration of HBV viremia in infected patients (29, 30).
Almost all HBV WG sequences were unique, with only 2 A2 and 2 D3 variants being identical. It is important to note that the observations made in the present study show that sharing the S gene sequence (13) is not an indication of the close genetic relatedness among HBV A2 or D3 variants, since HBV variants carrying identical S gene sequences were scattered without clustering within the subtype-specific branches in the phylogenetic tree constructed using WG sequences (Fig. 1). The identical S gene sequences shown as a single red node in Fig. 1a (32% of total sequences) are found in many different WG sequences in Fig. 1b. Thus, the extent of HBV genetic relatedness estimated using the S gene sequences should be interpreted with caution and limited to genotype assignment. Comprehensive analysis using WG rather than the S gene of HBV should lend confidence to establishing genetic relatedness of incident HBV and also to the detection of recombinants and clinically significant mutations.
Both A2 and D3 strains are of relatively recent origin, with the MRCA for each subtype predicted to exist only 30 to 50 years ago, observations that also substantiate the close genetic relatedness observed within each subtype. Despite their short evolutionary histories, both genotypes experienced population expansions. The effective population size of D3 increased between 1998 and 2000. The A2 strains underwent two rounds of population expansion (Fig. 3 and 4). The first expansion, between 1994 and 1996, coincided with a transient rise in notifications of acute hepatitis B from 1994 to 1998 (4). The second expansion, during 2002, was observed for the two lineages that comprise cluster 1. It was preceded or accompanied by a modest decline in the effective population size for the other A2 strains (cluster 2, Fig. 4). The second A2 expansion coincided with increased acute hepatitis B notifications reported by the U.S. National Notifiable Disease Surveillance (NNDSS) between 1999 and 2002 among men >19 years old (5%) and among men and women >40 years old (20 and 31%, respectively) (5).
The dynamic nature of HBV's epidemic history during the study period has resulted in significant changes in genetic composition of the HBV A2 population carried by acute hepatitis B cases. The A2 strains sampled after 2002 were restricted in genetic heterogeneity, with a reduction in the number of polymorphic sites unevenly distributed across the HBV genome (Fig. 5a). No known drug resistance substitutions were found in these recently expanded strains, contrasting with almost a third of strains sampled before 2002 that carried such substitutions (Table 2). Consistent with the differential expansion of a single A2 cluster (Fig. 4a), the findings indicate that incident HBV populations circulating after 2002 were genetically distinct from those before 2002.
Reduction in genetic heterogeneity and the number of preexisting drug resistance mutations over time suggests the potentially changing capacity of HBV populations to respond to antiviral treatment. Predisposition to drug resistance is a convergent trait encoded in epistatic connectivity among HBV genomic sites (16, 31). The shift in the HBV population structure observed in the current study likely reflects changes in epistatic connectivity pertaining to the presentation of drug resistance mutations. In addition, the rapid population expansion of the closely related HBV strains is likely consequence of frequent opportunities for transmission from persons with primary infections, who tend to carry fewer intrahost HBV variants (32), than from persons with chronic infection. Thus, reduction in frequency of the detected drug resistance mutations after the second expansion period is likely associated with changing genetic structure and declining heterogeneity of the HBV population than with specific selection pressures related to therapeutic treatment.
Analysis of demographic characteristics of the cases showed that the cluster 1 HBV A2 strains tended to be detected among African-Americans, persons from Jefferson County in Alabama, those diagnosed after 2002, and MSM (Table 1). Multivariate analysis did not produce statistically significant results due to the relatively small sample size. Nonetheless, although each of the three factors was shown to be independently associated with infection by cluster 1 strains, MSM activity is the behavioral risk factor identified that would account for the observed increase in the rate of transmission of these genetically close strains. Although locally circulating HBV strains may be expected to be genetically related, geographical location or race by itself cannot explain the increase in the rate of transmission. Furthermore, race cannot be independently associated with genetic relatedness among incident HBV strains.
The demographic and risk associations to infection by cluster 1 HBV A2 strains are consistent with NNDSS data showing a rise in incidence rate of acute hepatitis B between 1999 and 2002, particularly in the southern United States, and among persons reporting engagement in high-risk practices, such as MSM activity and injecting drug use (4). Contemporaneous reports from other parts of the world have shown sharp increases of acute infections with genotype A2 strains linked to similar high-risk behaviors (20, 21). As noted, rapid transmissions among acutely infected MSM in the Netherlands and Japan (25, 26) were associated with genetically identical or closely related A2 strains. The data from Japan also indicate a greater propensity of primary infection with A2, compared to other genotypes, associated with persistent HBV infection (29, 30). Although there are currently no data indicating such propensity of the U.S. A2 strains that underwent selective transmission, it would be important to monitor the natural history of infection by these strains. Our results indicate the possibility of missed opportunities to vaccinate MSM. Gaps in implementing existing vaccination strategies must be addressed to increase hepatitis B vaccination coverage for MSM.
This study highlights that full-length genomes are critical for complete characterization of the biological properties of HBV variants and understanding the epidemiology of the disease. Despite a decline in the rate of incident HBV infection in the United States, HBV strains identified among acute hepatitis B cases have experienced three population expansions. The expansions altered significantly the viral genetic composition and affected the capacity of HBV populations to maintain resistance to antiviral treatment. A strong association between phylogeny and transmission rates reflected in selective viral expansion of specific HBV lineages, particularly of A2 strains, suggests biological differences among HBV variants affecting their dissemination or the existence of host contact networks linked to specific risks, such as among MSM. Integration of a hepatitis B vaccine into routine childhood vaccination schedules has dramatically increased immunization coverage among younger age groups, but similar gains in coverage have not been demonstrated among high-risk adults (33, 34), a population that accounts for an estimated 75 to 95% of all incident HBV infection cases in the United States (4, 33). Increased HBV transmission rate among MSM reflects inadequate hepatitis B vaccination coverage, highlighting the need to improve coverage of at-risk adults (9, 35, 36).
ACKNOWLEDGMENTS
We acknowledge the contributions of J. W. Ward, D. Holtzman, S. D. Holmberg, H. Roberts, J. Groeger, and V. Barry.
This information has not been formally disseminated by the Centers for Disease Control and Prevention/Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. No conflict of interest exists for any author or for financial support.
Footnotes
Published ahead of print 3 September 2014
REFERENCES
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Weinbaum CM, Williams I, Mast EE, Wang SA, Finelli L, Wasley A, Neitzel SM, Ward JW. 2008. Recommendations for identification and public health management of persons with chronic hepatitis B virus infection. MMWR Morb. Mortal. Wkly. Rep. 57:1–20. [PubMed] [Google Scholar]
- 3.Lu PJ, Byrd KK, Murphy TV, Weinbaum C. 2011. Hepatitis B vaccination coverage among high-risk adults 18-49 years, U.S., 2009. Vaccine 29:7049–7057. 10.1016/j.vaccine.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein ST, Alter MJ, Williams IT, Moyer LA, Judson FN, Mottram K, Fleenor M, Ryder PL, Margolis HS. 2002. Incidence and risk factors for acute hepatitis B in the United States, 1982-1998: implications for vaccination programs. J. Infect. Dis. 185:713–719. 10.1086/339192. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2004. Incidence of acute hepatitis B–United States, 1990-2002. MMWR Morb. Mortal. Wkly. Rep. 52:1252–1254. [PubMed] [Google Scholar]
- 6.Ladak F, Gjelsvik A, Feller E, Rosenthal SR, Montague BT. 2012. Hepatitis B in the United States: ongoing missed opportunities for hepatitis B vaccination, evidence from the Behavioral Risk Factor Surveillance Survey, 2007. Infection 40:405–413. 10.1007/s15010-011-0241-2. [DOI] [PubMed] [Google Scholar]
- 7.Lum PJ, Hahn JA, Shafer KP, Evans JL, Davidson PJ, Stein E, Moss AR. 2008. Hepatitis B virus infection and immunization status in a new generation of injection drug users in San Francisco. J. Viral Hepat. 15:229–236. 10.1111/j.1365-2893.2007.00933.x. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. 2012. Adult vaccination coverage–United States, 2010. MMWR Morb. Mortal. Wkly. Rep. 61:66–72. [PubMed] [Google Scholar]
- 9.Pitasi MA, Bingham TA, Sey EK, Smith AJ, Teshale EH. 2014. Hepatitis B virus (HBV) infection, immunity and susceptibility among men who have sex with men (MSM), Los Angeles County, U.S.A. AIDS Behavior 18(Suppl 3):S248–S255. 10.1007/s10461-013-0670-2. [DOI] [PubMed] [Google Scholar]
- 10.Kramvis A. 2014. Genotypes and genetic variability of hepatitis B virus. Intervirology 57:141–150. 10.1159/000360947. [DOI] [PubMed] [Google Scholar]
- 11.Alter MJ, Hadler SC, Margolis HS, Alexander WJ, Hu PY, Judson FN, Mares A, Miller JK, Moyer LA. 1990. The changing epidemiology of hepatitis B in the United States. Need for alternative vaccination strategies. JAMA 263:1218–1222. [PubMed] [Google Scholar]
- 12.Nainan OV, Alter MJ, Kruszon-Moran D, Gao FX, Xia G, McQuillan G, Margolis HS. 2006. Hepatitis C virus genotypes and viral concentrations in participants of a general population survey in the United States. Gastroenterology 131:478–484. 10.1053/j.gastro.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Teshale EH, Ramachandran S, Xia GL, Roberts H, Groeger J, Barry V, Hu DJ, Holmberg SD, Holtzman D, Ward JW, Teo CG, Khudyakov Y. 2011. Genotypic distribution of hepatitis B virus (HBV) among acute cases of HBV infection, selected United States counties, 1999-2005. Clin. Infect. Dis. 53:751–756. 10.1093/cid/cir495. [DOI] [PubMed] [Google Scholar]
- 14.Chu CJ, Keeffe EB, Han SH, Perrillo RP, Min AD, Soldevila-Pico C, Carey W, Brown RS, Jr, Luketic VA, Terrault N, Lok AS. 2003. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 125:444–451. 10.1016/S0016-5085(03)00895-3. [DOI] [PubMed] [Google Scholar]
- 15.Ramachandran S, Zhai X, Thai H, Campo DS, Xia G, Ganova-Raeva LM, Drobeniuc J, Khudyakov YE. 2011. Evaluation of intra-host variants of the entire hepatitis B virus genome. PLoS One 6:e25232. 10.1371/journal.pone.0025232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thai H, Campo DS, Lara J, Dimitrova Z, Ramachandran S, Xia G, Ganova-Raeva L, Teo CG, Lok A, Khudyakov Y. 2012. Convergence and coevolution of hepatitis B virus drug resistance. Nat. Commun. 3:789. 10.1038/ncomms1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Utsumi T, Lusida MI, Yano Y, Nugrahaputra VE, Amin M, Juniastuti Soetjipto, Hayashi Y, Hotta H. 2009. Complete genome sequence and phylogenetic relatedness of hepatitis B virus isolates in Papua, Indonesia. J. Clin. Microbiol. 47:1842–1847. 10.1128/JCM.02328-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sloan RD, Strang AL, Ramsay ME, Teo CG. 2009. Genotyping of acute HBV isolates from England, 1997-2001. J. Clin. Virol. 44:157–160. 10.1016/j.jcv.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Laoi BN, Crowley B. 2008. Molecular characterization of hepatitis B virus (HBV) isolates, including identification of a novel recombinant, in patients with acute HBV infection attending an Irish hospital. J. Med. Virol. 80:1554–1564. 10.1002/jmv.21273. [DOI] [PubMed] [Google Scholar]
- 21.Hallett RL, Ngui SL, Meigh RE, Mutton KJ, Boxall EH, Teo CG. 2004. Widespread dissemination in England of a stable and persistent hepatitis B virus variant. Clin. Infect. Dis. 39:945–952. 10.1086/423962. [DOI] [PubMed] [Google Scholar]
- 22.Dreesman JM, Baillot A, Hamschmidt L, Monazahian M, Wend UC, Gerlich WH. 2006. Outbreak of hepatitis B in a nursing home associated with capillary blood sampling. Epidemiol. Infect. 134:1102–1113. 10.1017/S0950268806005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pourkarim MR, Verbeeck J, Rahman M, Amini-Bavil-Olyaee S, Forier AM, Lemey P, Maes P, Van Ranst M. 2009. Phylogenetic analysis of hepatitis B virus full-length genomes reveals evidence for a large nosocomial outbreak in Belgium. J. Clin. Virol. 46:61–68. 10.1016/j.jcv.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 24.van Houdt R, Bruisten SM, Geskus RB, Bakker M, Wolthers KC, Prins M, Coutinho RA. 2010. Ongoing transmission of a single hepatitis B virus strain among men having sex with men in Amsterdam. J. Viral Hepat. 17:108–114. 10.1111/j.1365-2893.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- 25.Fujisaki S, Yokomaku Y, Shiino T, Koibuchi T, Hattori J, Ibe S, Iwatani Y, Iwamoto A, Shirasaka T, Hamaguchi M, Sugiura W. 2011. Outbreak of infections by hepatitis B virus genotype A and transmission of genetic drug resistance in patients coinfected with HIV-1 in Japan. J. Clin. Microbiol. 49:1017–1024. 10.1128/JCM.02149-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamada Y, Yatsuhashi H, Masaki N, Nakamuta M, Mita E, Komatsu T, Watanabe Y, Muro T, Shimada M, Hijioka T, Satoh T, Mano Y, Komeda T, Takahashi M, Kohno H, Ota H, Hayashi S, Miyakawa Y, Abiru S, Ishibashi H. 2012. Hepatitis B virus strains of subgenotype A2 with an identical sequence spreading rapidly from the capital region to all over Japan in patients with acute hepatitis B. Gut 61:765–773. 10.1136/gutjnl-2011-300832. [DOI] [PubMed] [Google Scholar]
- 27.Delwart E, Slikas E, Stramer SL, Kamel H, Kessler D, Krysztof D, Tobler LH, Carrick DM, Steele W, Todd D, Wright DJ, Kleinman SH, Busch MP, Group N-R-IS. 2012. Genetic diversity of recently acquired and prevalent HIV, hepatitis B virus, and hepatitis C virus infections in US blood donors. J. Infect. Dis. 205:875–885. 10.1093/infdis/jir862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda Y, Katano Y, Hayashi K, Honda T, Yokozaki S, Nakano I, Yano M, Yoshioka K, Toyoda H, Kumada T, Goto H. 2006. Difference of HBV genotype distribution between acute hepatitis and chronic hepatitis in Japan. Infection 34:201–207. 10.1007/s15010-006-5099-3. [DOI] [PubMed] [Google Scholar]
- 29.Yamada N, Yotsuyanagi H, Okuse C. 2010. Duration of HBs antigenemia in patients with acute hepatitis B. Kanzo 51:534–535. 10.2957/kanzo.51.534. [DOI] [Google Scholar]
- 30.Ishii K, Kogame M, Shiratori M. 2010. Clinical characteristics of patients with acute hepatitis B genotype A. Kanzo 51:397–399. 10.2957/kanzo.51.397. [DOI] [Google Scholar]
- 31.Khudyakov Y. 2010. Coevolution and HBV drug resistance. Antivir. Ther. 15:505–515. 10.3851/IMP1515. [DOI] [PubMed] [Google Scholar]
- 32.Osiowy C, Giles E, Tanaka Y, Mizokami M, Minuk GY. 2006. Molecular evolution of hepatitis B virus over 25 years. J. Virol. 80:10307–10314. 10.1128/JVI.00996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mast EE, Weinbaum CM, Fiore AE, Alter MJ, Bell BP, Finelli L, Rodewald LE, Douglas JM, Jr, Janssen RS, Ward JW, Advisory Committee on Immunization Practices, Centers for Disease Control and Prevention 2006. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). II. Immunization of adults. MMWR Morb. Mortal. Wkly. Rep. 55:1–34. [PubMed] [Google Scholar]
- 34.Matthews JE, Stephenson R, Sullivan PS. 2012. Factors associated with self-reported HBV vaccination among HIV-negative MSM participating in an online sexual health survey: a cross-sectional study. PLoS One 7:e30609. 10.1371/journal.pone.0030609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Centers for Disease Control and Prevention. 2012. Adult vaccination coverage: United States 2010. MMWR Recomm. Rep. 61:66–72. [PubMed] [Google Scholar]
- 36.Rijckevorsel GV, Whelan J, Kretzschmar M, Siedenburg E, Sonder G, Geskus R, Coutinho R, van den Hoek A. 2013. Targeted vaccination programme successful in reducing acute hepatitis B in men having sex with men in Amsterdam, The Netherlands. J. Hepatol. 59:1177–1183. 10.1016/j.jhep.2013.08.002. [DOI] [PubMed] [Google Scholar]