

SUMMARY
Bone marrow transplantation is an effective cell therapy but requires myeloablation, which increases infection-risk and mortality. Recent lineage-tracing studies documenting that resident macrophage populations self-maintain independent of hematologic progenitors prompted us to consider organ-targeted, cell-specific therapy. Here, using GM-CSF receptor-β deficient (Csf2rb−/−) mice that develop a myeloid cell disorder identical to hereditary pulmonary alveolar proteinosis (hPAP) in children with CSF2RA/CSF2RB mutations, we show that pulmonary macrophage transplantation (PMT) of either wild-type or Csf2rb-gene-corrected macrophages without myeloablation was safe, well-tolerated, and that one administration corrected the lung disease, secondary systemic manifestations, normalized disease-related biomarkers, and prevented disease-specific mortality. PMT-derived alveolar macrophages persisted for at least one year as did therapeutic effects. Results identify mechanisms regulating alveolar macrophage population size in health and disease, indicate that GM-CSF is required for phenotypic determination of alveolar macrophages, and support translation of PMT as the first specific therapy for children with hPAP.
Keywords: GM-CSF, M-CSF, Alveolar Macrophage, Surfactant Homeostasis, Myeloid Cell Disorder, Gene Therapy, Genetic Disease
Mutations in CSF2RA or CSF2RB, encoding GM-CSF receptor α or β, respectively, cause hereditary pulmonary alveolar proteinosis (hPAP) by impairing GM-CSF-dependent surfactant clearance by alveolar macrophages resulting in progressive surfactant accumulation in alveoli and hypoxemic respiratory failure1-5. Surfactant normally comprises a thin phospholipid/protein layer reducing tension on the alveolar surface6 that is maintained by balanced secretion by alveolar type II epithelial cells and clearance by these cells and alveolar macrophages7,8. PAP also occurs in people with GM-CSF-autoantibodies (~85-90% of all patients with PAP)9,10 and mice with disruption of GM-CSF/Csf211,12 or the GM-CSF receptor β subunit/Csf2rb13,14 (Csf2−/− or Csf2rb−/− mice, respectively). Characteristics of PAP caused by disruption of GM-CSF signaling include typical lung histopathology (well-preserved alveoli filled with surfactant and ‘foamy’ macrophages staining positive with periodic acid-Schiff (PAS) oroil-red-O); turbid, ‘milky’-appearing bronchoalveolar lavage (BAL) caused by accumulated surfactant and cell debris; and a disease-specific pattern of biomarkers (increased GM-CSF (hPAP), M-CSF/CSF1, and MCP-1 in BAL fluid and reduced mRNA for PU.1, PPARγ, and ABCG1 in alveolar macrophages)1-5,15-20.
Currently, no pharmacologic therapy of hPAP exists and surfactant must be removed by whole lung lavage, an inefficient, invasive procedure to physically remove excess surfactant2-4. In Csf2rb−/− (KO hereafter) mice, PAP was corrected by bone marrow transplantation (BMT) of wild type (WT)21 or Csf2rb gene-corrected KO hematopoietic stem/progenitor cells (HSPC)22. However, in humans, this approach resulted in death from infection before engraftment2, likely from required myeloablation/immunosuppressive therapy. Since pulmonary GM-CSF is increased in hPAP1-5 we hypothesized that macrophages administered directly into the lungs (pulmonary macrophage transplantation or PMT) without myeloablation would engraft and reverse the manifestations of hPAP.
RESULTS
We first validated KO mice as a model of human hPAP by demonstrating they had the same clinical, physiological, histopathological, and biochemical abnormalities, disease biomarkers, natural history (Fig. 1, Extended Data Fig. 1) as children with hPAP3.
Figure 1.
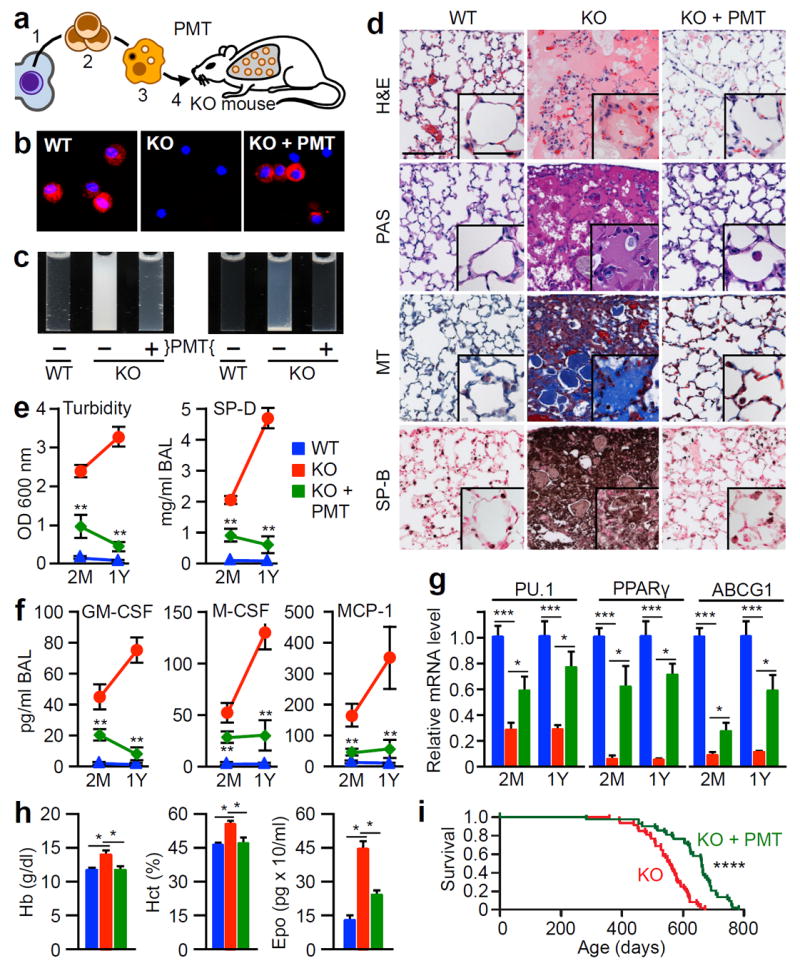
Therapeutic efficacy of PMT in Csf2rb−/− (KO) mice. (a) Schematic of the method used. WT HSPCs (1) were isolated, expanded (2), differentiated into macrophages (3), and administered by endotracheal instillation into 2 month-old KO mice (4) and evaluated after two months (2M) (e-g) or one year (1Y) (b-h) with age-matched, untreated WT or KO mice (KO+PMT, WT or KO, respectively). (b) CD131-immunostained BAL cells.(c) Appearance of BAL fluid (left) or sediment (right). (d) Lung histology after staining with H&E, PAS, Masson’s trichrome (MT), or surfactant protein B (SP-B). Scale bar, 100μm; inset, 50μm. (e) BAL turbidity and SP-D concentration. (f) BAL biomarkers. (g) Alveolar macrophage biomarkers. (h) Effects of PMT on blood hemoglobin (Hb), hematocrit (Hct), serum erythropoietin (Epo). (i) Kaplan-Meier analysis of PMT-treated (n=43) and untreated KO mice (n=48). Images are representative of 6 mice/group (b-d). Numeric data are Mean ± SEM of 7 (2M) or 6 (1Y) mice/group. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Characterization of macrophages before PMT
Bone marrow derived macrophages (BMDMs) from WT mice had morphology and phenotypic markers (F4/80+, CD11bHi, CD11c+, CD14+, CD16/32+, CD64+, CD68+, CD115+, CD131+, SiglecFLo, MerTK+, MHC class II+, Ly6G−, CD3−, CD19−) of macrophages (Extended Data Fig. 2a-c) and contained <0.0125% lineage negative (Lin−) Sca1+cKit+ (LSK) cells. Clonogenic analysis indicated <0.005% CFU-GM and no BFU-E, or CFU-GEMM progenitors (Extended Data Fig.2d-e). Functional evaluation23 showed they could clear surfactant (Extended Data Fig. 2f-g). These results demonstrated the cells used for PMT were highly purified, mature macrophages capable of surfactant clearance.
Efficacy of PMT of WT macrophages
To determine the therapeutic potential of PMT, KO mice received WT (Csf2rb+/+) BMDMs by PMT once (Fig. 1a). One year later, PMT-derived CD131+ BAL cells were present (Fig. 1b), alveolar macrophages expressed Csf2rb (Extended Data Fig. 3a), BAL was markedly improved with respect to opacification (Fig. 1c), sediment (Fig. 1c), and microscopic cytopathology (Extended Data Fig. 3b). Importantly, PMT nearly completely resolved the abnormal pulmonary histopathology (Fig. 1d, Extended Data Fig. 3c). Measurement of BAL turbidity and SP-D content (Fig. 1e), which reflect the extent of surfactant accumulation across the entire lung surface, confirmed the improvement in hPAP. BAL fluid biomarkers of hPAP were also improved (Fig. 1f). The effects of PMT were evident early as demonstrated by detection of CD131+ alveolar macrophages with Csf2rb mRNA and protein (not shown), reduced BAL opacification and cytopathology (not shown), BAL turbidity (Fig. 1e), SP-D (Fig. 1e), and BAL fluid biomarkers (Fig. 1f) two months after PMT, and reduced lung histopathology 4 months after PMT (not shown). In contrast, PMT of KO BMDMs had no effect on BAL turbidity, SP-D content, or BAL fluid biomarkers (not shown) demonstrating the importance of GM-CSF receptors on transplanted macrophages to the therapeutic effects.
To evaluate the effects of PMT on the alveolar macrophage population, we measured cellular biomarkers after PMT. Results showed alveolar macrophages from PMT-treated KO mice had increased mRNA for PU.1, PPARγ, and ABCG1, improvement was significant by two months, and the effects persisted one year after PMT (Fig. 1g).
Since KO mice develop polycythemia, a secondary consequence of hypoxemiain chronic lung diseases24, the effects of PMT on this systemic clinical manifestation were evaluated. Importantly, PMT corrected polycythemia in KO mice (Fig. 1h).
Finally, the effects of PMT on hPAP-associated mortality were evaluated by comparing the survival of PMT-treated and untreated KO mice. PMT increased the lifespan of KO mice by 107 days, from (median [interquartile range]) 555 [507-592] to 662 [604-692] days (Fig. 1i). In separate studies of KO mice surviving to 617 [604-631] days after PMT of WT BMDMs (561 [548-575] days after PMT), CD131+ alveolar macrophages were still present and BAL turbidity remained low compared to untreated KO mice that survived to 631 [631-631] days (OD6000.75±0.17 versus 2.63±0.44; n=8, 4, respectively; P<0.001). However, such long-term evaluation of laboratory abnormalities are obfuscated by reduced survival of untreated KO mice.
These results demonstrate PMT had a highly efficacious and durable therapeutic effect on the primary pulmonary and secondary systemic manifestations of hPAP in KO mice.
Macrophage engraftment efficiency
The effects of cell dose (0.5, 1, 2 and 4 million) and repeated administration (one versus four monthly transplantations) on PMT efficacy were evaluated (Extended Data Table 2 and 3, respectively). Neither significantly affected efficacy in the range evaluated and one dose of 2 million cells was used for PMT in the remaining studies.
To determine if WT macrophages had a survival advantage over KO macrophages, we measured GM-CSF bioactivity in BAL fluid and found it was detectable in KO but not WT mice (Extended Data Fig. 1h). WT macrophages had increased survival/proliferation compared to KO macrophages in vitro (Fig. 2a) and accumulated to greater numbers after PMT in KO mice than in WT mice (Fig. 2b and Extended Data Fig. 3d). PMT of WT Lys-MGFP knock-in mouse25 BMDMs into KO mice followed by Ki67 immunostaining revealed PMT-derived cells replicated in vivo (Extended Data Fig. 3e-g). The percentage of Ki67+ PMT-derived alveolar macrophages was 32.2 ± 6.05% one month after PMT and declined to 11.29 ± 2.2% by one year (Fig. 2c) similar to baseline Ki67+ immunostaining of alveolar macrophages in age-matched, normal WT mice (Extended Data Fig. 3f). To further define this survival advantage, we evaluated the engraftment kinetics after one PMT of WT BMDMs in KO mice. CD131+ cells increased steadily from zero to 69.0±2.5% of BAL cells (Fig. 2d) synchronous with a smooth decline in pulmonary GM-CSF to near normal (Fig. 2e). Similarly, BAL turbidity declined with the increase in CD131+ alveolar macrophages (Fig. 2f). One year after PMT, CD131+ cells were present (Fig. 1b), Csf2rb protein was detectable in alveolar macrophages (Extended Data Fig. 3a), and Csf2rb mRNA in BAL cells from PMT-treated KO mice was only slightly less than in WT and undetectable in untreated KO BAL cells (Fig. 2g). Importantly, numbers of CD131+ alveolar macrophages in PMT-treated KO and untreated WT mice were similar one year after PMT (Fig. 2h). These results demonstrate that WT macrophages 1) had a selective survival advantage over KO macrophages, and that after PMT into KO mice they 2) proliferated in vivo at a rate that slowed over time synchronous with reduction in pulmonary GM-CSF, 3) replaced dysfunctional KO alveolar macrophages, and 4) resulted in numbers of CD131+, GM-CSF-responsive alveolar macrophages similar to WT mice.
Figure 2.
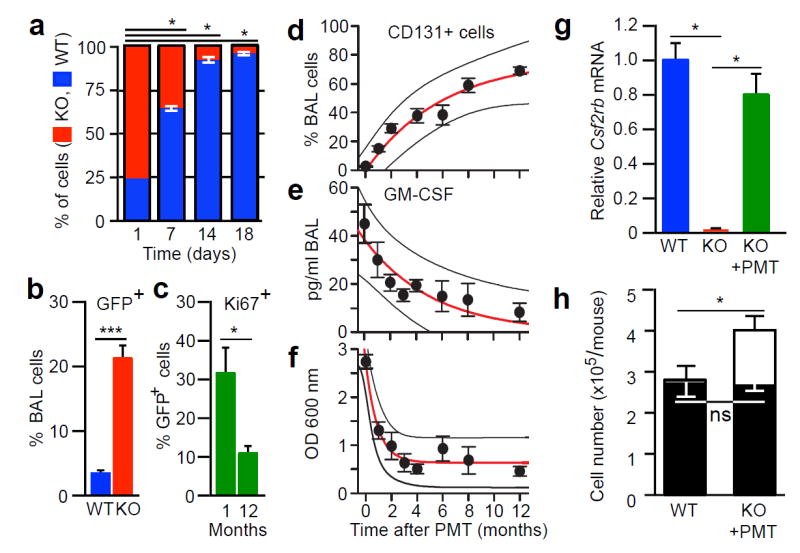
Pharmacokinetics and pharmacodynamics of PMT in KO mice. (a) Competitive proliferation of WT and KO BMDMs co-cultured with GM-CSF and M-CSF (n=3 plates/point).
(b) Quantification of GFP+ BAL cells 2 months after PMT of Lys-MGFP BMDMs into WT (n=3) or KO (n=6) mice. (c) Quantitation of Ki67+ Lys-MGFP cells in KO mice (n=3) one or twelve months after PMT. (d-f) KO mice received PMT of WT BMDMs and were evaluated at the indicated times to quantify CD131+BAL cells (d), BAL GM-CSF concentration (e), and BAL turbidity (f). Exponential regression (± prediction bands), R2=0.943 (d), R2=0.819 (e), R2=0.958 (f). Data are mean ± SEM for 3-7 mice/group. (g) Csf2rb mRNA in BAL cells from KO mice one year after PMT, or untreated, age-matched control mice (n=6). (h) Number of BAL cells (open bars) or CD131+ alveolar macrophages (closed bars) in KO mice one year after PMT (n = 5) or untreated WT mice (n=10). Data are mean ± SEM. *p < 0.05, ***P<0.001, not significant (ns).
Macrophage characterization after PMT
The fate of macrophages following PMT was evaluated to determine their spatial distribution, phenotype, and gene expression profile. Intra-pulmonary localization was evaluated one year after PMT of WT Lys-MGFP BMDMs by fluorescence microscopy to identify CD68+GFP+ (i.e., PMT-derived macrophages), which revealed 88.9 ± 0.87% were intra-alveolar and 11.1 ± 0.87% were interstitial (Extended Data Fig. 3h). GFP-immunohistochemical staining was done to eliminate potential interference from autofluorescence and confirmed these results; 90.5 ± 1.1% PMT-derived macrophages were intra-alveolar and 9.4 ± 1.1% were interstitial (Fig. 3a-b and Extended Data Fig. 3i). Localization was done in similarly-treated mice by flow cytometry to detect GFP+ cells two months (not shown) or one year after PMT (Extended Data Fig. 4a-b) and by PCR amplification of Lys-MGFP transgene-specific DNA (Extended Data Fig. 4c), all of which showed PMT-derived cells were present in the lungs but not detected in blood, bone marrow, or spleen. One year after PMT of CD45.1+ WT BMDMs into CD45.2+ KO mice, flow cytometric detection of CD45.1+ cells confirmed these findings (Extended Data Fig. 4e-g). Results show that the transplanted macrophages remained in the lungs, primarily within the intra-alveolar space.
Figure 3.
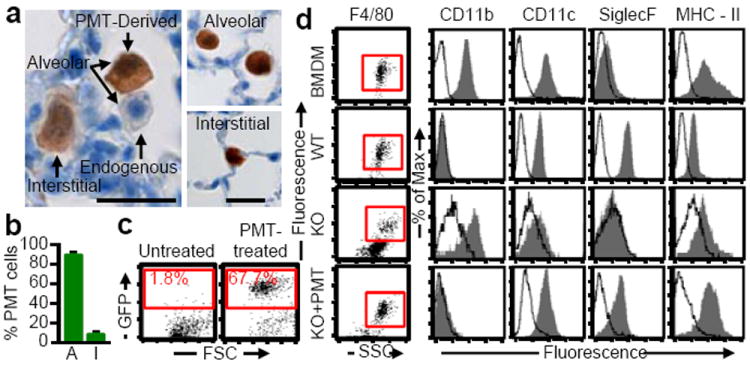
Localization and phenotype of transplanted macrophages. Lys-MGFP BMDMs were transplanted into KO mice and evaluated after one year. (a) Immunostained lung showing GFP+ cells (Scale bar, 200 μm; inset, 20 μm). (b) Localization of GFP+ macrophages to intra-alveolar (A) and interstitial (I) spaces (n=6). (c) GFP+ BAL cells identified by flow cytometry. (d) Phenotypic analysis of F4/80+ BMDMs before PMT, and alveolar macrophages from PMT-treated KO mice, or untreated, age-matched WT or KO mice (n=6/group). Data are mean±SEM.
The effects of the lung milieu on the phenotype of transplanted macrophages were evaluated by measuring cell-surface markers. One year after PMT of WT Lys-MGFP BMDMs into KO mice, PMT-derived alveolar macrophages comprised 68.7 ± 6.5% of BAL cells and had converted from CD11bHiSiglecFLo to CD11bLoSiglecFHi, similar to the phenotype of WT alveolar macrophages and different from recipient KO mice (CD11bHiSiglecFLo) (Fig. 3c-d). Similarly, one year after PMT of WT CD45.1+ BMDMs into CD45.2+ KO mice, CD45.1+ alveolar macrophages comprised 63.6±12.1% of BAL cells and had undergone the same phenotypic conversion (Extended Data Fig. 4h).
To determine the effects on gene expression, genome-wide expression profiling was performed on alveolar macrophages from KO mice one year after PMT of WT BMDMs and compared to results for untreated, age-matched WT or KO mice. Unsupervised analysis indicated marked co-clustering between PMT-treated KO and WT mice while KO mice clustered separately (Fig. 4). Expression of genes regulated by GM-CSF was reduced in KO mice and restored by PMT (Fig. 4, Extended Data Fig. 5a). Of 776 genes for which expression was disrupted in KO mice, PMT normalized expression of 600 including 80% of genes up-regulated and 76% of genes down-regulated in KO compared to WT mice (Extended Data Fig. 5b). Supervised Gene Ontology (GO) and detailed KEGG pathway analysis revealed that genes in multiple pathways involved in lipid metabolism, cellular proliferation, apoptosis, and host defense were coordinately down-regulated in KO mice, and normalized by PMT (Extended Data Fig. 5c,d). Results for multiple genes important in lipid metabolism (Abcg1, Nr1h3, Olr1, Lepr, Fabp1, Lipf, Abca1, Apoe, Apoc2, Pla2g7) were validated using separate samples (Extended Data Fig. 5e).
Figure 4.
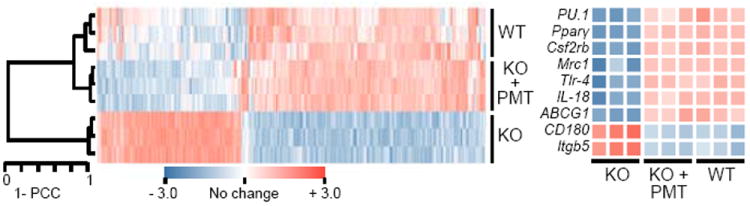
Microarray analysis of alveolar macrophages one year after PMT. Unsupervised hierarchical clustering dendrogram and heat-map of selected GM-CSF-regulated genes in PMT-treated KO mice or untreated, age-matched WT or KO mice (3/group). Pearson correlation coefficient (PCC).
Efficacy of gene therapy by PMT
Since PMT in humans would likely employ autologous, gene-corrected HSPC-derived macrophages, we evaluated PMT of KO macrophages derived from Lin−Sca-1+c-Kit+ (LSK) cells after lentiviral vector (LV)-mediated Csf2rb cDNA expression (Fig. 5a). Csf2rb gene-corrected (GM-R-LV-transduced) or sham-treated (GFP-LV-transduced) KO and non-transduced WT LSK-derived cells all had macrophage morphology, expressed CD68 (Extended Data Fig. 6a) and were F4/80+CD11bHiCD11c+ (not shown). In contrast, only WT and GM-R-LV-transduced KO cells were CD131+ and only LV transduced cells were GFP+ (Extended Data Fig. 6a). GM-R-LV restored GM-CSF signaling in KO macrophages (Fig. 5b and Extended Data Fig. 6b). Two months after PMT into KO mice, GM-CSF receptor-β was detected on alveolar macrophages only from mice receiving gene-corrected KO or WT macrophages (Extended Data Fig. 6c). The efficacy using gene-corrected KO or WT cells was equivalent as demonstrated by a similar degree in improvement in BAL appearance (Extended Data Fig. 6d), BAL turbidity, SP-D, and biomarkers of hPAP (Fig. 5c-d). Further, gene-corrected BMDMs localized to the lung (Extended Data Fig. 4d and Fig. 6e) and underwent phenotypic conversion to CD11bLo (Extended Data Fig. 6f). The long-term efficacy of gene-corrected macrophages one year after PMT was demonstrated by marked reduction in BAL turbidity, SP-D, and BAL fluid biomarkers of hPAP (Fig. 5c-d). These results demonstrate that PMT of gene-corrected macrophages had a therapeutic effect on hPAP in KO mice equivalent to that of WT macrophages and was durable, lasting at least one year.
Figure 5.
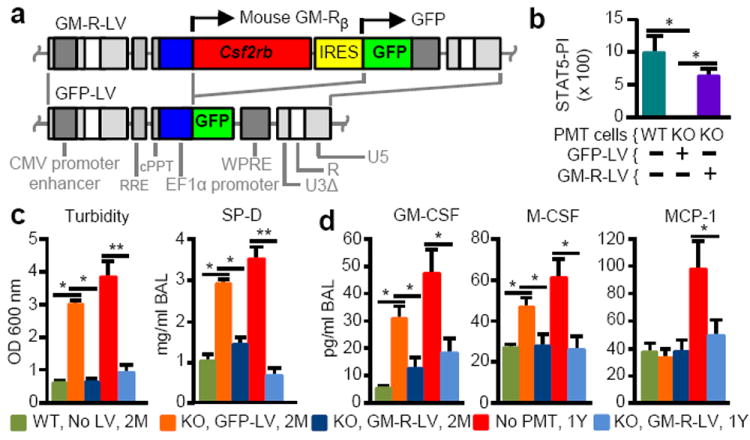
Effects of PMT of gene-corrected macrophages on hPAP severity and biomarkers. KO mice received PMT of non-transduced WT or LV-transduced KO macrophages and were evaluated after two month (2M) or one year (1Y) (with untreated, age-matched KO mice). Key indicates PMT cells used, prior LV treatment, and time after PMT analysis was performed. (a) LV schematics. (b) GM-CSF signaling measured by the STAT5 phosphorylation index (STAT5-PI) in the indicated cells before PMT. (c) BAL turbidity and SP-D concentration. (d) BAL biomarkers. Mean ± SEM of n=3 (b) or 5-10 (c-g) mice/group. *p < 0.05, **p < 0.01.
Figure 6.
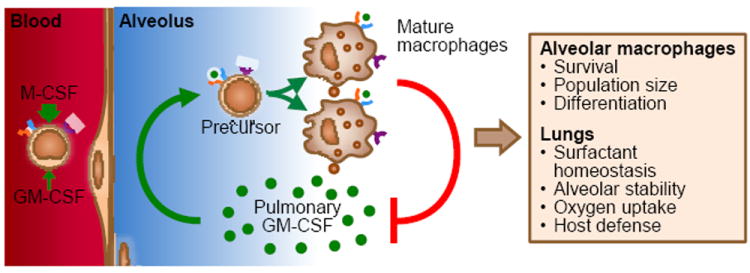
Proposed homeostatic reciprocal feedback mechanism by which pulmonary GM-CSF regulates alveolar macrophage population size in vivo.
Safety of PMT Therapy in KO Mice
PMT was well-tolerated and without adverse effects. One year after PMT, there were no hematologic abnormalities (Extended Data Table 4), cellular inflammation or pulmonary fibrosis in mice receiving PMT of WT (Fig. 1d) or gene-corrected macrophages (not shown). KO mice had trivial elevations of IL-6 and TNFα in BAL that were reduced by PMT of WT macrophages (Extended Data Table 4). These data identify no safety concerns for PMT therapy of hPAP in KO mice.
DISCUSSION
Multiple lines of evidence indicate the high efficacy of PMT therapy of hPAP in KO mice was attributable to a selective survival advantage conferred by increased pulmonary GM-CSF to alveolar macrophages bearing functional GM-CSF receptors. However, pulmonary surfactant remained slightly increased one year after a single PMT. This could be because the treatment time was too short or exceeded the durability of the clinical benefit, or due to the continued presence of KO alveolar macrophages despite engraftment of GM-CSF-responsive macrophages. The latter is likely due to ongoing KO myelopoiesis, pulmonary recruitment of monocytes and local proliferation, and GM-CSF-independent survival as occurs in untreated KO mice. KO macrophages may provide a ‘protected intracellular niche’ for surfactant accumulation since without GM-CSF, alveolar macrophages internalize but cannot clear surfactant.26 Another factor may be reduction of the survival advantage over time, i.e., reduced pulmonary GM-CSF (driving WT cell proliferation) and reduced surfactant burden (driving surfactant-engorgement-related KO cell death). Notwithstanding, a single PMT of GM-CSF responsive cells cleared ~90% of the abnormal surfactant accumulation for at least one year.
The feasibility of translating PMT therapy to humans with hPAP is supported by the safety and efficacy of PMT in KO mice and the striking similarity of hPAP in mice and humans. Macrophages could be delivered by bronchoscopic instillation without endotracheal intubation, general anesthesia, or mechanical ventilation, which are required for whole lung lavage and increase risk. Preparative myeloablation would be unnecessary and use of autologous, gene-corrected cells would eliminate the need for immunosuppression, which are required for BMT. PMT may also be possible with gene-corrected, inducible pluripotent cell-derived macrophages recently prepared from children with hPAP23,27. However, formal preclinical toxicology studies related to PMT and to gene-transfer will be needed before this approach can be tested in humans. Since pulmonary GM-CSF is critical to lung host defense and clearance of a broad range of microorganisms28, PMT may also be useful in treating serious lung infections. Indeed, pulmonary administration of macrophages constitutively expressing IFN-γ improved host defense in SCID mice29. In such applications, inhaled GM-CSF could be used to promote survival of transplanted macrophages30.
Identification of a homeostatic mechanism by which pulmonary GM-CSF regulates alveolar macrophage population size (Fig. 6) was an unexpected but important finding. Its existence is supported by recent fate-mapping studies indicating that tissue-resident alveolar macrophages derive prior to birth and self-maintain by local replication independent of circulating monocytes at steady state30-32,33.
The concept of alveolar macrophages (and other tissue-resident macrophages) as short-lived, terminally-differentiated, non-dividing, representatives of a unified mononuclear phagocyte system replenished via monocyte intermediates has evolved considerably since its inception34. Alveolar macrophage half-life was initially estimated at 2 weeks based on studies of repopulation after lethal irradiation and allogeneic BMT35. Improved detection methods using GFP+ cells increased the estimate to 30 days36. Shielding the thorax during irradiation increased it further to 8 months37. Our data, obtained without irradiation or myeloablation, show that macrophages transplanted directly into the respiratory tract persisted for one and a half years. A caveat of such estimates is their inability to discern if persistence is due to prolonged survival or replication.
Normally, alveolar macrophages are phenotypically CD11bLoSiglecFHi while other macrophage populations are CD11bHiSiglecFLo. Surprisingly, WT BMDMs cultured in GM-CSF and M-CSF were CD11bHiSiglecFLo in vitro but converted to CD11bLoSiglecFHi after PMT. In contrast, BMDMs instilled in the peritoneum adopt the CD11bHi phenotype of peritoneal macrophages38. These changes agree with gene-expression profiling studies39 and indicate local microenvironments provide critical ‘phenotypically instructive’ cues that direct development of tissue-resident macrophage populations. Our results show for alveolar macrophages that GM-CSF provides one such phenotypic cue while the lung milieu provides another critical, albeit unidentified, cue.
Study limitations include that it did not establish a minimum effective dose, maximum tolerated dose, or a significant dose-response relationship. BMDMs were capable of clearing surfactant before transplantation but results did not determine if‘lung-conditioning’ further increased their clearance capacity. While the macrophages used for PMT contained very few progenitors, it is theoretically possible that clonal expansion of a progenitor subpopulation may have contributed to therapeutic efficacy and, if so, potential clonal shrinkage may have contributed to loss of benefit at later times. Thus, additional studies are needed to further confirm the identity of effect or cells and precise pharmacokinetics and durability of the therapeutic benefit.
METHODS
Mice
All mice were bred, housed, and studied in the Cincinnati Children’s Research Foundation Vivarium using protocols approved by the Institutional Animal Care and Use Committee. Csf2rb gene-targeted (Csf2rb−/− or KO) mice13, and mice expressing EGFP knocked-into the lysozyme M gene (Lys-MGFP mice)25 were all generated previously and backcrossed onto the C57Bl/6 background. C57Bl/6 mice (referred to as wild type or WT mice) were purchased from Charles River. B6.SJL-PtprcaPepcb/Boy J (CD45.1+) mice were from Jackson Laboratory.
Lung histology and immunohistochemistry
Animals were sacrificed by intraperitoneal pentobarbital administration and exsanguination by aortic transection. The trachea was exposed by a vertical midline skin incision, cannulated through a small transverse incision in its ventral surface away from the thoracic inlet, inflated with fixative (PBS, pH = 7.4, containing 4% paraformaldehyde) under a hydrostatic head of 25cm and ligated with suture while retracting the cannula to seal the lung under pressure. The sternum and diaphragm were transected sagittally, retracted laterally, and the lungs and heart separated from the chest wall by blunt dissection to avoid puncturing the mediastinal pleura and removed from the chest. The intact tissue block containing the heart, lungs, and ligated trachea was submerged in fixative and kept at 4°C for 24 hours. After fixation, the lung lobes were divided, removed from the tissue block, cut into ~2 mm thick slices along the long axis, washed in cold PBS, dehydrated, embedded in paraffin, and 5 μm thick sections were cut and stained with hematoxylin and eosin (H&E), periodic acid-Schiff reagent (PAS), or Masson’s trichrome as previously described40. Immunostaining for surfactant protein B (SP-B) was done by incubating slides with rabbit anti-SP-B polyclonal antibody (diluted 1:500, Seven Hills Bioreagents, Cincinnati, OH) and Vectastain ABC anti–rabbit immunohistochemical horseradish peroxidase kit (Vector Labs, Inc., Burlingame, CA) and counterstaining with hematoxylin as described41. To prepare frozen lung sections, the lungs were inflation fixed in-situ as described above and then the heart and lungs were removed enbloc and cryoprotected by sequential immersion in PBS containing increasing sucrose concentrations (10%, 15% and 20%; 8-12 hours, 4 °C, at each concentration). The lungs were then embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA), frozen and stored at -80 °C until use. Serial 6 μm sections were prepared for immunostaining or evaluation of GFP+ cells. Lung sections and sedimented lung cells were examined by light microscopy using a Zeiss Axioplan 2 microscope (Zeiss) equipped with AxioVision software (Zeiss).
Collection, handling, and evaluation of bronchoalveolar lavage fluid and cells
Epithelial lining fluid and non-adherent cells were collected from lung surface of mice by bronchoalveolar lavage (BAL) as described42 and processed immediately. Briefly, five 1 ml aliquots were instilled and immediately recovered per mouse and combined resulting in a BAL recovery of 93.9 ± 1.2% per mouse (BAL recovery data for 10 mice evaluated randomly). The photographs of fresh BAL specimens and the specimen after allowing sediment to be formed by overnight incubation at 4 °C were taken. The turbidity of BAL was determined as described41. Briefly, after gently mixing to ensure a homogeneous suspension of BAL, a 250 μl aliquot was diluted into 750 μl PBS and the optical density was measured at a wavelength of 600 nm and multiplying the result by the dilution factor. The total number of BAL cells recovered from each mouse was determined by counting cells in an aliquot of known volume using a hemocytometer and multiplying the result by the total volume of BAL and dividing by the volume of the aliquot used for counting. BAL cytology was evaluated in aliquots (~50,000 cells) after sedimentation (Cytospin, Shandon, Inc.; 500 rpm, 7 min, room temperature) onto glass slides and staining with DiffQuick, PAS, or oil-red-O (all from Fisher Scientific) as described41. The cell differential was determined by microscopic examination of DiffQuick stained cells and the total number of alveolar macrophages per mouse was determined by multiplying the percentage of alveolar macrophages in BAL cells by the total number of BAL cells recovered43. BAL fluid and cells were separated by low speed centrifugation (285 × G, 10 min, room temp) and stored at -80 °C until use (BAL fluid) or immediately evaluated (referred to as BAL cells) or used to isolate alveolar macrophages (see below). Primary alveolar macrophages were purified by brief adherence of BAL cells to plastic as described18. Viability was evaluated by Trypan blue exclusion and was ≥ 95%.
ELISA
The concentration of surfactant protein D (SP-D) in BAL fluid was measured by enzyme-linked immunosorbent assay (ELISA) as we described41. The concentration of several cytokines (GM-CSF, M-CSF, MCP-1, IL-1β, IL-6, TNFα) in BAL fluid and erythropoietin in serum was measured by ELISA (Mouse Quantikine Kits, R&D Systems) as described1.
Quantitative RT-PCR
Total RNA was isolated from alveolar macrophages using TRIzol Reagent (Life Technologies, Carlsbad, CA) and then used to purify mRNA using RNeasy (Qiagen, Valencia, CA), both as directed by the manufacturers. Purified mRNA was used to synthesize cDNA using the Invitrogen SuperScript III First-Strand Synthesis System (Life Technologies). Standard quantitative RT-PCR (qRT-PCR) was performed as previously described1 on an Applied Biosystems 7300 Real-Time PCR System (Life Technologies) to measure transcript abundance using TaqMan® oligonucleotide primer sets (all from Life Technologies) (Extended Data Table 1). Expression of target genes was normalized to the expression of 18s RNA. Data for each gene were shown as the fold change of the mean of results for wild type mice.
Bone marrow derived macrophages (BMDMs)
Bone marrow cells were obtained from 6 – 8 week-old WT, KO, or Lys-MGFP mice by isolating and flushing tibias and femurs with DMEM (Life Technologies) containing 10% heat-inactivated FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. After red blood cells were removed with BD Pharm Lyse (BD Biosciences), mononuclear cells were isolated by centrifugation on Ficoll-Paque (GE Healthcare) at room temperature for 30 minutes, washed, re-suspended in DMEM containing 10% heat-inactivated FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, 10ng/ml GM-CSF and 5 ng/ml M-CSF (both from R&D Systems), seeded into plastic dishes (Falcon) at a density of ~27 × 106 cells/10 cm dish (1/mouse) and cultured overnight at 37°C in a humidified environment containing 5% CO2. The next day, non- or weakly-adherent cells were recovered, transferred to a new dish and cultured under the conditions just described to permit differentiation and expansion of macrophages; firmly adherent cells were discarded. After 2 days the culture medium was changed and after 5 days from seeding, adherent bone marrow-derived macrophage were gently washed with PBS, harvested by brief exposure to trypsin-EDTA (Life Technologies), washed, and used for experiments. The cell purity was high as indicated by the percentage of CD68+ and F4/80+ cells (96.6 ± 0.3%, 95.4 ± 1.3%, respectively, not shown).
Some experiments used lineage-negative (Lin−) c-kit+Sca-1+ (LSK cells) which were obtained from mouse bone marrow as described44. Briefly, bone marrow from 6-8 week-old WT or KO mice was collected as above and lineage depleted with biotinylated lineage antibodies CD5 (53-7.3), CD8a (53-6.7), CD45R/B220 (RA3-6B2), CD11b (M1/70), Gr-1 (RB6-8C5), and TER-119 (TER-119) (BD Bioscience), and magnetic beads (Dynabeads sheep anti-rat IgG) (Life-Technologies). After removing lineage-positive cells, the remaining cells were stained with 7-ADD, FITC-Streptavidin (BD Biosciences) and antibodies to Sca-1 (D7) and c-kit (2B8) (BD Biosciences). Then, Lin−c-kit+Sca-1+7-ADD− cells were isolated by cell sorting on a FACSAria (BD Biosciences) and used immediately in experiments. Cell morphology was confirmed by Diff-Quick Staining of sedimented cells (Cytospin, Shandon) and viability was measured by Trypan blue exclusion as described18 and found to be ≥95%. In some experiments, cells were immunostained for CD68 (FA-11) (AbD Serotec), counter stained with DAPI as described41, and examined by light microscopy using a Zeiss Axioplan 2 microscope (Zeiss) equipped with AxioVision software (Zeiss).
Colony forming cell (CFC) assay
BMDMs or Lin−bone marrow cells were evaluated for the presence of hematopoietic progenitors capable of forming colonies in semisolid medium in response to cytokine stimulation as previously described.45 Briefly, fresh Lin− bone marrow cells or BMDMs after induced differentiation into macrophages for five days were seeded into standard mouse methylcellulose media supplemented with insulin, transferrin, SCF, IL-3, IL-6, and erythropoietin (HSC007, R&D Systems, Minneapolis, MN). After seven days in culture, colonies of ≥50 cells were visible and were examined morphologically using whole plate stack images acquired using an AXIO-Z1 microscope and AXIO-vision software (Zeiss, Jena, Germany) to identify and enumerate burst-forming erythroid progenitors (BFU-E), colony-forming myeloid progenitors (CFU-GM), and the multi-potential progenitors (CFU-GEMM).
Surfactant Clearance Assay
BMDMs were evaluated functionally to demonstrate their ability to clear human surfactant as we previously reported41. Briefly, BMDMs from either WT or KO mice were seeded into 12-well plates (4 × 105 cells/well) in DMEM, 10% FBS, 10ng/ml GM-CSF, 5 ng/ml M-CSF. Human surfactant recovered by lavage of a patient with PAP was added to the media and cells were incubated for 24 hours to permit surfactant uptake into cells and then washed to remove extracellular surfactant. Cells were incubated for 24 hours to permit macrophages to clear internalized surfactant. Cells collected before, immediately after surfactant exposure or 24 hours after the completion of surfactant exposure were sedimented onto slides by cytocentrifugation (Shandon), stained with oil-red-O, and counterstained with hematoxylin. Oil-red-O staining was evaluated in ≥10 random 20x microscopic fields for each sample as described41.
Pulmonary macrophage transplantation (PMT)
BMDMs or LSK cell-derived macrophages were administered directly into the lungs of 8 week-old mice using a relatively non-invasive endotracheal instillation method described previously46. Briefly, mice received light anesthesia by isoflurane inhalation and were suspended on a flat board by a rubber band across the upper incisors and placed in a semi-recumbent (45-degree) position with the ventral surface and rostrum facing upwards. Using a curved blade Kelly forceps, the tongue was gently and partially retracted rostrally, and 50μl of PBS containing the macrophages to be administered was placed in the back of the oral cavity using a micropipette. The PBS and cells were inhaled into the lungs by subsequent respiratory efforts under direct visualization. Mice were then observed while recovering from anesthesia to ensure continued retention of the administered fluid and cells and then returned to their cages for routine care and handling. Because the efficacy of PMT at a dose of two million macrophages was optimal, this dose given as one administration was used throughout the study except where noted (Extended Data Table 2-3). Age-matched mice were used in all experiments to control for the degree of lung disease severity.
Flow cytometry
BAL cells were purified by centrifugation on Percoll to remove surfactant and debris 31. BAL cells or BMDMs were immunostained to detect CD115 (AFS98), F4/80 (BM8) (eBioscience), CD3 (145-2C11), CD11b (M1/70), CD11c HL3), CD16/32 (2.4G2), CD19 (1D3), CD64 (X54-5/7.1.1), CD131 (JORO50), Ly6G (1A8), CD45.1 (A20), CD45.2 (104), MHC class II (I-A/I-E) (M5/114.15.2), SiglecF (E50-2440) (BD Biosciences), CD14 (Sa14-2) (BioLegend), CD68 (FA-11) (AbD Serotec), and MerTK (108928) (R&D Systems), as previously described1, evaluated by flow cytometry using a FACSCaliber or FACSCanto flow cytometers (both from BD Biosciences, San Jose, CA), and the results were analyzed using CellQuest, FACSDiva (BD Biosciences), or FlowJo software (Tree Star). For intracellular staining of CD68, Leucoperm (AbDSerotec) was used as directed by the manufacturer.
Quantification of CD131+ alveolar macrophages
The percentage of BAL cells expressing the GM-CSF receptor β subunit was determined by immunostaining. Briefly, aliquots of BAL cells were sedimented onto glass slides and incubated (10 minutes, room temperature) in fixative (PBS containing 4% paraformaldehyde), washed with PBS and incubated (4°C, overnight) with anti-mouse GM-CSF receptor-β (CD131) antibody (sc-678) (Santa Cruz) diluted 1:400 in PBST (PBS containing 2.5% (w/v) Triton X-100 and 5% (v/v) goat serum). After incubation, slides were rinsed 5 times in PBST and incubated (room temperature, 1 hour) with the secondary detection antibody (Alexa Fluor 594-conjugated, anti-rabbit IgG (Life Technologies)) and counter stained with 4’,6-diamidino-2-phenylindoledihydrochloride (DAPI) (Vector Labs, Burlington, CA). Cells were examined using a Zeiss Axioplan 2 microscope (Zeiss) equipped with AxioVision software (Zeiss). The percentage of CD131+ BAL cells was determined by first counting the CD131+ and DAPI+ cells in 5 (or more) random 20x microscopic fields for each BAL sample. Then, the number of CD131+ cells in each field was divided by the number of DAPI+ cells in the same field and results for all fields examined were averaged and multiplied by 100. The total number of CD131+ cells per mouse was calculated by multiplying the percentage of CD131+ cells by the total number of BAL cells recovered from each mouse.
STAT5 phosphorylation index assay
GM-CSF bioactivity in BAL fluid and GM-CSF receptor function in transduced or WT macrophages was evaluated by measuring GM-CSF-stimulated phosphorylation of STAT5 in BMDMs or LSK cell-derived macrophages using anti-phospho STAT5 antibody (47/Stat5(pY694)) (BD Biosciences) by flow cytometry as previously reported1. The STAT5 phosphorylation index (STAT5-PI) was calculated as the mean fluorescence intensity of phosphorylated STAT5 staining in GM-CSF-stimulated cells minus that of non-stimulated cells, divided by that of non-stimulated cells, and multiplied by 100. In experiments to quantify GM-CSF bioactivity, WT BMDMs were incubated in BAL fluid containing anti-GM-CSF (22E9, eBioscience) or isotype control antibody (10 μg/ml) for 30 minutes and then evaluated.
Evaluation of macrophage proliferation
In vitro mixed-cell proliferation assay
CD45.1+ WT LSK-derived cells and CD45.2+ KO LSK-derived cells were isolated, seeded into dishes at an initial ratio of 1:3, respectively, and cultured in DMEM containing 10% bovine calf serum, 1% penicillin/streptomycin, GM-CSF (10 ng/ml) and M-CSF (5 ng/ml). Cells were collected at 1, 7, 14, and 18 days, immunostained with anti-murine CD45.1, anti-CD45.2 and evaluated by flow cytometry to determine the percentage of each cell type at these times.
In vivo evaluation of transplanted macrophage proliferation
Frozen lung tissue sections were immunostained with anti-Ki67 antibody (Roche) and examined using a Zeiss Axioplan 2 microscope (Zeiss). The percentage of proliferating PMT-derived cells was determined by enumerating GFP+ Ki67+ cells among total GFP+ cells in ≥7 random 20x microscopic fields for each sample. To confirm the specificity of Ki67 immunostaining, paraffin embedded sections or WT alveolar macrophages isolated by BAL and adherence were also stained with Ki67 and examined by light microscopy.
Western blotting
Detection of GM-CSF receptor β and actin by Western blotting was done as previously described1 with the following modifications. Briefly, primary alveolar macrophages (0.5 × 106/condition) or cultured BMDMs (1 × 106/condition, after culture with either GM-CSF or M-CSF at 10 ng/ml for 30 min) were collected by low speed centrifugation (285 × G, 4 °C, 10 min) and the pellets incubated on ice for 30 min in 200 μl of lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 % (v/v) nonidet p-40, 0.5 % (w/v) sodium deoxycholate, 0.1 % (w/v) sodium dodecyl sulfate (SDS), 0.004 % (w/v) sodium azide) containing 2 % (v/v) proteinase inhibitor cocktail (phenyl-methyl-sulfonyl-fluoride and sodium orthovanadate; Santa Cruz). Insoluble debris was removed by centrifugation 10,000 × G, 4 °C, 15 min and the supernatant transferred to a clean polypropylene tube. An equal volume of Laemmli sample loading buffer (Bio-Rad, CA) was added and the tubes were capped tightly, vortexed briefly, boiled for 5 min, and separated by electrophoresis on SDS- polyacrylamide gradient (4-12%) gels (Invitrogen) under reducing conditions. Separated proteins were transferred to PVDF membranes by electro-blotting, incubated in blotting solution (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 % (w/v) Non-fat dry milk (Kroger, Cincinnati, OH), 0.1 % (v/v) Tween 20; 4°C, overnight) to block non-specific binding. Diluted primary detection antibody (see below) was added and the membranes were incubated for 2 hours at room temperature and then washed in TBST (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1 % [v/v] Tween 20). Membranes were then incubated with the secondary HRP-conjugated detection antibody in blotting solution for 1 hour at room temperature and then washed as above and then incubated with ECL-Plus (GE Healthcare) as directed by the manufacturer. Anti-mouse GM-CSF receptor β antibody (sc-678) (Santa Cruz) diluted 1:500 and anti-actin (sc-1616) (Santa Cruz) diluted 1:1000 were used for primary antibodies.
Hematologic analysis
Blood was obtained from the superior vena cava from mice and 20 μl was used to measure complete blood counts on a fully automated Hemavet 850 (Drew Scientific). Data for the precision and linearity of measurements made with the Hemavet850 can be found online at www.drew-scientific.com/product_hemavet850.htm.
Microarray analysis
Alveolar macrophages were obtained from age-matched mice (3 per condition) and analyzed individually as follows. Total RNA was isolated as described above and microarray analysis was performed using the Mouse Gene 1.0 ST Array (Affymetrix, Santa Clara, CA) in the CCHMC Affymetrix Core using standard procedures as described47. Data (available at www.ncbi.nlm.nih.gov/geo/GSE60528) were analyzed using the Affymetrix package in the R statistical programming language (Bioconductor; www.bioconductor.org). Probes were corrected for background using the Microarray Analysis Suite algorithm, quantile normalized, and probe sets were summarized using the average difference of perfect matches only. Differential expression tests were performed using significance analysis of microarrays48 with Benjamini-Hochberg correction for multiple testing 49. Significant gene lists were selected with a Δ that constrained the false discovery rate to less than 10%. Cluster dendrogram was generated from unsupervised hierarchical clustering analysis of microarray data from probes for all 28,853 genes represented on the chip (Spearman correlation; 3 mice/group). In Venn diagrams, numbers of genes whose expression was altered in alveolar macrophages from KO compared to WT mice (WT→KO) or PMT-treated compared to untreated KO mice (KO→KO+PMT) were shown. Only genes with statistically significant changes (false detection rate < 10%) of at least two-fold were marked as increased (up arrows) or decreased (down arrows). The numbers of genes for which expression was disrupted in KO mice and normalized by PMT (or unchanged in both comparisons) is shown in the overlap regions. In gene ontology analysis, data show the coordinate increases (red) or decreases (blue) in expression of genes in all gene sets significant at or below a false detection rate of 10% calculated by the Gene Set Test with correction for multiple testing.
Lentiviral vectors, LSK-cell transduction, and differentiation and expansion of transduced macrophages
Gene transfer vectors were constructed using routine methods44 from the vector backbone of (Ery-GFP), a human immunodeficiency virus-based, self-inactivating (SIN) lentiviral vector (LV) harboring a 398-bp U3 deletion eliminating the strong viral promoter/enhancer element50. GM-R-LV contains a chimeric transgene comprised of the human elongation factor 1-alpha (ELF-1α) promoter (a 1189 bp fragment containing intron 1 ending 20 bp upstream of the ATG codonisolated from the pEF-BOS plasmid51) followed by the mouse Csf2rb cDNA (nucleotides -80 to 2691, GenBank accession no. M34397.1) located 3’ of the lentiviral central poly-purine tract and followed by an internal ribosome binding site (IRES) and then an enhanced green fluorescent protein (GFP) transgene (Figure 5A). GFP-LV is a LV of similar design except that the Csf2rb and IRES were omitted and the GFP transgene is driven from the ELF1α promoter (Figure 5A). Both vectors contain a viral splice donor site, packaging sequence, splice acceptor site, and central polypurine tract (cPPT) 5’ of the ELF1α promoter and a woodchuck hepatitis post-transcriptional regulatory element (WPRE) (nucleotides 1093 to 1684; GenBank accession no. J04514)52 located 3’ of the GFP stop codon as described50. LVs were produced by transient transfection as vesicular stomatitis virus-G (VSVG) virions, concentrated, and tittered as described44. KO LSK cells were isolated, transduced and expanded as described53 except that transductions were done at a multiplicity of infection (MOI) of 20 for two 12 hour periods, IL-11 was omitted, GM-CSF (10 ng/ml) and M-CSF (5 ng/ml) were included, SCF and Flt-3 ligand were sequentially reduced (50, 1, 0 ng/ml), and IL-3 was present early.
Transduction, expansion, and differentiation of LSK cells into gene-corrected macrophages was done by adjusting the cytokine ‘cocktail’ mixture to optimize the culture conditions for each of four sequential stages, which included: 1) LSK transduction –murine SCF (R&D) 50ng/ml, mIL-3 (PeproTech) 10 ng/ml, hFlt3-L (PeproTech) 50 ng/ml and GM-CSF (R&D) 10 ng/ml), culture time – two 12 hour periods; 2) progenitor expansion - mSCF 50 ng/ml, hFlt3-L 50 ng/ml and GM-CSF 10 ng/ml, culture time - 4 days; 3) macrophage lineage commitment - mSCF 1 ng/ml, hFlt3-L 1 ng/ml, GM-CSF 10 ng/ml, M-CSF (R&D) 5 ng/ml, culture time - 3 days; and 4) macrophage differentiation - GM-CSF 10 ng/ml and M-CSF 5 ng/ml, culture - 4 days. StemSpan (STEMCELL Technologies) containing 2% FBS, 1% penicillin/streptomycin, 10 mM dNTP, and low density lipoprotein was used as the culture medium for the LSK transduction and DMEM with 10% FBS, 50 U/ml penicillin and 50 ug/ml streptomycin was used for all other stages. Phenotype markers (F4/80, CD11b, CD11c) were analyzed by flow cytometry at each stage to monitor macrophage differentiation. Only adherent macrophages at the end of this procedure were used for PMT.
Localization of PMT-derived cells after transplantation
Several approaches were utilized to identify and localize PMT-derived cells within the lung parenchyma and in different organs.
Intra-pulmonary localization of PMT-derived cells
CD131 immunostaining and fluorescence microscopy or flow cytometry was used to detect and quantify transplantation-derived donor macrophages among BAL cells from the lungs of KO mice that previously received PMT of WT (C57BL/6) BMDMs, Lys-MGFP BMDMs, CD45.1+ WT BMDMs, or GM-R-GFP-LV Csf2rb gene-corrected KO LSK-derived macrophages.
To localize PMT-derived macrophages to intra-alveolar space or interstitium of the lung, frozen lung sections from mice that received PMT of Lys-MGFP BMDMs one month or one year earlier were immunostained with CD68, counterstained with DAPI (Vector Labs) and evaluated by fluorescence microscopy to identify macrophages, PMT-derived cells, and nucleated cells, respectively. PMT-derived macrophages (i.e., GFP+ CD68+ cells) located within the intra-alveolar space or the interstitium were then enumerated. To eliminate the possibility of any interference from non-specific auto-fluorescence of alveolar macrophages, paraffin-embedded lung sections from these mice were immunostained with anti-GFP antibody (Life Technologies) and examined by light microscopy to enumerate immunohistochemically marked macrophages located within the intra-alveolar space or interstitium.
Organ-specific localization of PMT-derived cells
In one approach, KO mice received PMT of Lys-MGFP BMDMs and one year later, cells isolated from the BAL, blood, bone marrow, and spleen were evaluated by flow cytometry to detect GFP+ cells as a marker for PMT-derived cells.
In a second approach, CD45.2+ KO mice received PMT of CD45.1+ BMDMs and one year later, cells isolated from the BAL, blood, bone marrow, and spleen were evaluated by flow cytometry to detect CD45.1+ cells as a marker for PMT-derived cells.
In a third approach, KO mice received PMT of Lys-MGFP BMDMs and one year later, DNA was extracted from the BAL cells (lung), blood leukocytes, bone marrow cells, and spleen using a DNeasy Blood & Tissue Kit (Qiagen). Organ-specific DNA was subjected to PCR amplification using oligonucleotide primers (Extended Data Table 1) specific for the Lys-MGFP knock-in transgene or the unmodified Lysozyme M gene to detect PMT-derived and endogenous cells, respectively, as previously reported25.
A fourth approach was conducted using a specific operating procedure (TSL 6-13) and Good Laboratory Practice (GLP) conditions within the CCHMC Translational Core Laboratory. Here, DNA was extracted from the BAL cells (lung), blood leukocytes, bone marrow cells, and spleen of KO mice that had received PMT of GM-R-GFP-LV Csf2r gene-corrected, KO LSK-derived macrophages one year earlier and subjected to quantitative PCR amplification with oligonucleotide primers specific for the R-U5 of GM-R-GFP-LV using Applied Biosystems ABI7900HT Fast Real-Time PCR System (Life Technologies). The number of GM-R-GFP-LV vector copies per microgram of organ-specific DNA was quantified and normalized to the level of mouse apolipoprotein B gene as described previously54.
Statistical Analysis
Numeric data were evaluated for normality and variance using the Shapiro-Wilk and Levene median tests, respectively, and presented as mean± SEM (parametric data) or median and interquartile range (nonparametric data). Statistical comparisons were made with Student t test, one-way analysis of variance, or Kruskal-Wallis rank-sum test as appropriate; post-hoc pairwise multiple comparison procedures were done using the Student-Newman-Keuls or Dunn’s method as appropriate. P-values of ≤ 0.05 were considered to indicate statistical significance. Based on use of BAL turbidity (the primary outcome variable for efficacy) measured two months after PMT of WT BMDMs into KO mice and compared to age-matched, untreated KO mice, 6 mice per group had a power 0.8 to detect a difference of 1.4 O.D. 600 nm using a two-tailed Student’s t-test and a P value of 0.05. All studies used male and female mice by randomly assigning mice housed in the same cage to separate experimental groups but without formal randomization or blinding. Results from all mice were included in the final analysis without exclusion. Analyses, including Kaplan-Meyer survival analysis, were performed with SigmaPlot, Version 12.5 (Systat Software, San Jose, CA). All experiments were repeated at least twice, with similar results.
Extended Data
Extended Data Figure 1. Validation of Csf2rb-/- (KO) mice as an authentic model of human hPAP.
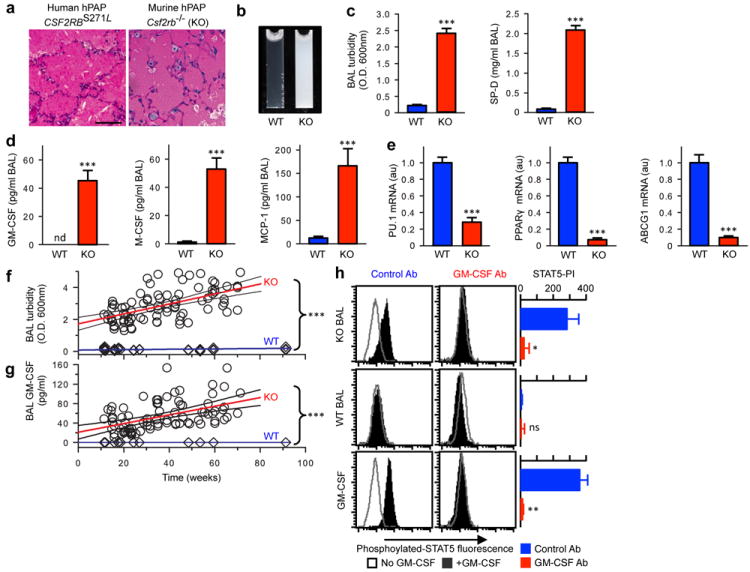
(a) Typical lung pathology showing surfactant-filled alveoli with well-preserved septa in a child homozygous for CSF2RBS271L mutations and identical pulmonary histopathology in a KO mouse. PAS stain. Scale bar, 100 μm. (b) Photographs of ‘milky’-appearing BAL from a 14 month-old KO mouse and normal-appearing BAL from an age-matched WT mouse (representative of n=6 mice/group). (c) Increased BAL turbidity and SP-D concentration in 4 month-old mice KO compared to age-matched WT mice. (d) BAL fluid biomarkers of hPAP (GM-CSF, M-CSF, and MCP-1) are reduced in 4 month-old KO mice compared to age-matched WT mice. (e) Alveolar macrophage biomarkers (PU.1, Pparg, Abcg1 mRNA) are reduced in 4 month-old KO compared to age-matched WT mice. (f) Progressive increase in BAL turbidity in KO mice but not age-matched WT mice (linear regression: KO, slope = 0.1271 ± 0.16 (r2, 0.311); WT, slope =0.031± 0.005). (g) Progressive increase in BAL fluid GM-CSF level in KO mice but not age-matched WT mice (linear regression: KO, slope =0.89±0.016 (r2, 0.249); WT, slope =0). Data are mean ± SEM of n=7 mice/group (c-j) or symbols representing individual WT (n=38) or KO (n=84) mice and the regression fit ± 95% CI lines. (h) GM-CSF bioactivity in BAL fluid from 10 month-old KO or WT mice (or 1 ng/ml murine GM-CSF) measured in the presence of anti-GM-CSF antibody (GM-CSF Ab) or isotype control (Control Ab) using the GM-CSF-stimulated STAT5 phosphorylation index (STAT5-PI) assay. Data are mean ± SEM of n=7 mice/group (c-e), n=4 (h) or symbols representing individual WT (n=38) or KO (n-84) mice and regression fit ± 95% CI (f-g). *p < 0.05, ***P<0.001, not significant (ns).
Extended Data Figure 2. Characterization of BMDMs before PMT.
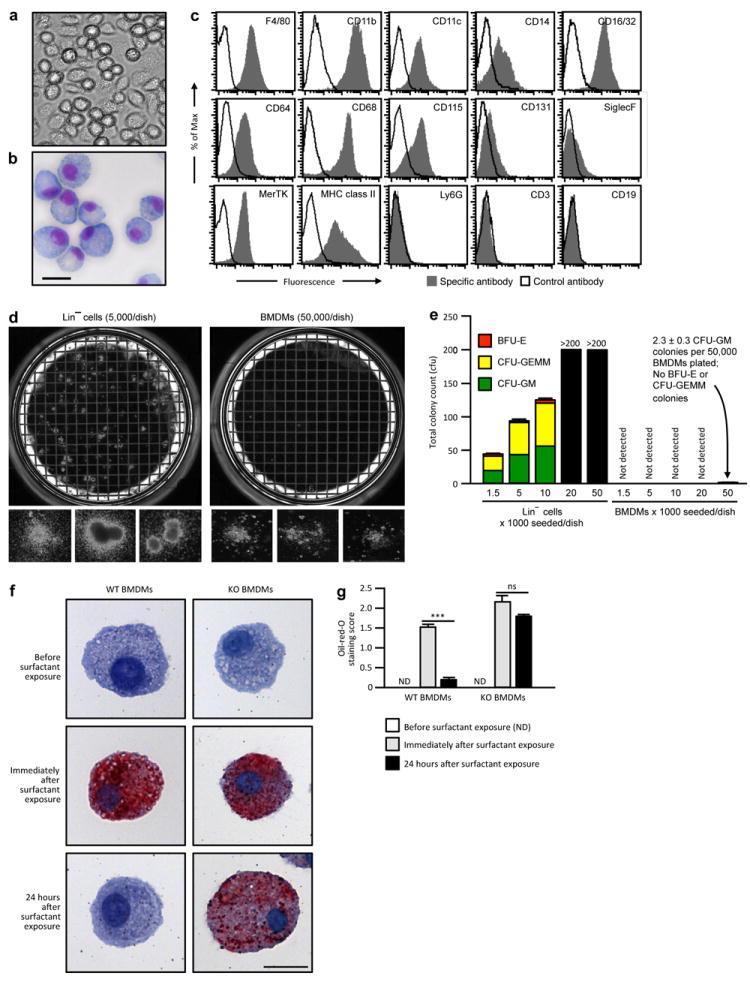
(a-b) Photomicrographs of WT BMDMs prior to transplantation phase-contrast (a) or Diff-Quick staining (b) (Representative of n=7 BMDM preparations). Scale bar, 20 μm. (c) Flow cytometry evaluation of cell-surface phenotypic markerson WT BMDMs before PMT. (d) Photographs of methylcellulose cultures of Lin− cells (5,000/dish) from bone marrow (left) and BMDMs (50,000/dish) prepared as described in the Methods (right) and typical colonies (below) (representative n=3 per condition). (e) Colony counts of BFU-E, CFU-GEMM and CFU-GM showing BMDMs contained <0.005% CFU-GM and no BFU-E or CFU-GEMM progenitors, corresponding to 93 CFU-GM per dose of BMDMs administered (n=3 determinations per condition). (f-g) Evaluation of surfactant clearance capacity. Representative photomicrographs of BMDMs from WT (left) or KO (right) were examined before (top) or immediately after incubation with surfactant for 24 hours (middle), or after exposure, removal of extracellular surfactant and culture for 24 hours in the absence of surfactant (lower) after oil-red-O staining (Representative of n=3 per condition). Scale bar, 20 μm. (g) Measurement of surfactant clearance by BMDMs after exposure as just described (Panel f) and quantified using a visual grading scale (the oil-red-O staining index) to measure the degree of staining. Bars represent the mean ± SEM (n=3/condition) of oil-red-O staining score for 10 high-power fields for each group. Not detected (ND).***P<0.001.
Extended Data Figure 3. Efficacy of PMT in KO mice and characterization of macrophages after PMT.
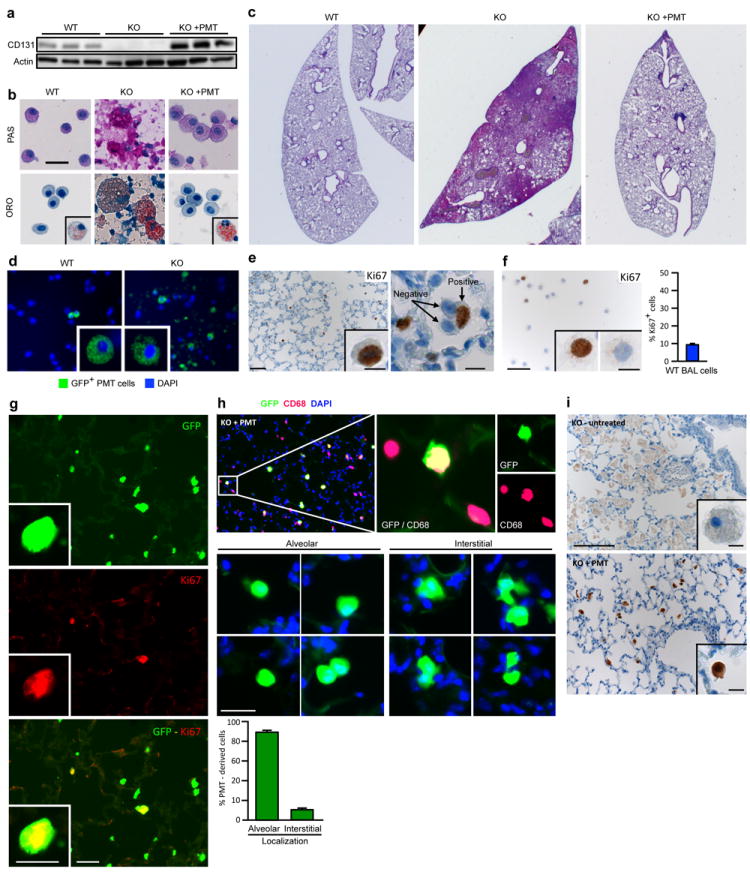
(a) Detection of CD131 (top) or actin (bottom) in BAL cells by western blotting one year after PMT (each lane represents one mouse of 6/group). (b) Representative cytology of BAL obtained one year after PMT after staining with PAS or oil-red-O (ORO) (6 mice/group). Scale bar, 25 μm. Oil-red-O positive cells were seen rarely in WT mice and occasionally in PMT-treated KO mice (insets). Cytological abnormalities in BAL from untreated KO mice including large, ‘foamy’, PAS- and oil-red-O-stained alveolar macrophages and PAS-stained cellular debris, were corrected by PMT. (c) Representative photomicrographs of PAS-stained whole-mount lung sections one year after PMT. Note that some residual disease remained at one year. (1×). (d) GFP+ cells in BAL cells from WT or KO mice 2 months after PMT of Lys-MGFP BMDMs. (e) Macrophage replication after PMT. KO mice received Lys-MGFP BMDMs by PMT and paraffin-embedded lung was immunostained for Ki67 one month or one year later. Scale bar, 50 μm; inset, 10μm. (f) Ki67 staining of BAL cells from an untreated WT mice (e). Inset shows positive (left) or negative (right) staining. Scale bar, 50 μm; inset 10 μm. Graph shows the percent Ki67+ BAL cells in age-matched WT mice (n = 5). (g) Representative immunofluorescence photomicrographs of frozen lung sections one year after PMT of Lys-MGFP into KO mice identifying GFP+ cells (top), Ki67+ cells (middle), and GFP+ Ki67+ (replicating, PMT-derived) cells (bottom). Scale bar, 20 μm; inset scale bar, 10μm. Quantitative summary data are shown in the manuscript (Fig. 2c). (h) Localization of macrophages within the lungs one year after PMT of Lys-MGFP BMDMs into KO mice and visualization in frozen lung sections after CD68 immunostaining, DAPI counter staining, and fluorescence microscopy to detect CD68+/GFP+ cells (i.e., PMT-derived macrophages) or CD68+/GFP− cells (i.e., non-PMT-derived endogenous macrophages). Graph shows quantitative data for n=6 mice. (i) Localization of macrophages in these same mice (Panel h) by detecting GFP by immunohistochemical staining of paraffin-embedded lung sections using light microscopy to eliminate potential interference from autofluorescence. Quantitative summary data are shown in the manuscript (Fig. 3b).
Extended Data Figure 4. Tissue distribution and characterization of transplanted cells one year after PMT.
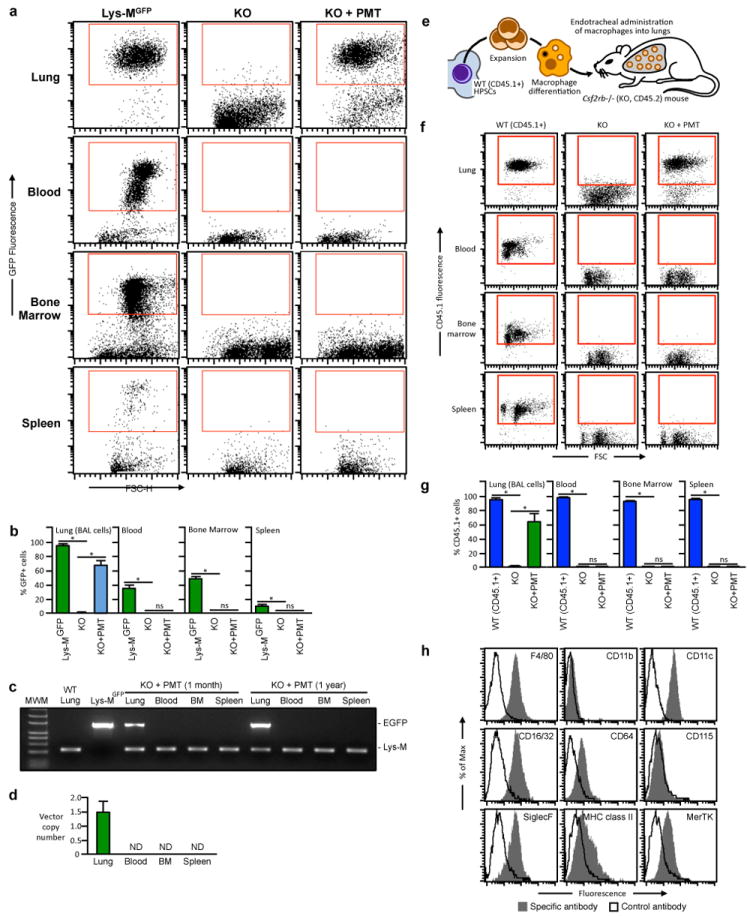
(a-d) Two month-old KO mice (4/group) received one PMT of Lys-MGFP BMDMs. Twelve months later, untreated, age-matched WT Lys-MGFP or KO mice and PMT-treated KO mice were evaluated using flow cytometry to detect GFP+ cells in the indicated organs. Representative data (a) and the percentage of GFP+ cells in the gated region are shown (b). Similar results were observed in KO mice two months after PMT of Lys-MGFP BMDMs (not shown). (c) Detection of Lys-MGFP PMT cells by PCR. PCR of genomic DNA from BAL cells (Lung), white blood cells (Blood), bone marrow (BM) cells and splenocytes (Spleen) of 1 month or 1 year after Lys-MGFP BMDMs PMT were performed to detect EGFP and Lysozyme M gene. BAL cells (Lung) from WT and Lys-MGFP were shown as negative and positive control for EGFP. EGFP was only detected in lung. (d) Vector copy number analysis after gene-corrected BMDMs PMT. Quantitative PCR with vector-specific primers (R-U5) was performed using genomic DNA from BAL cells (Lung), white blood cells (Blood), bone marrow (BM) cells and splenocytes (Spleen) obtained 1 year after PMT of gene-corrected macrophages. Note that the viral vector was only detected in lung. (e-h) CD45.2+ KO mice received one PMT of CD45.1+ BMDMs from congenic WT mice (e) and one year later, untreated, age-matched WT (CD45.1+) or KO (CD45.2+) mice and PMT-treated KO mice were evaluated by flow cytometry to detect CD45.1+ cells in the indicated organs. Representative data (f) and the percentage of CD45.1+cells in the gated regions are shown (g). Phenotypic characterization of PMT-derived (CD45.1+) cells (as shown in the gated region (f)). Results are similar to those for PMT of Lys-MGFP BMDMs (Fig 3d). Numeric data are mean ± SEM of n=4mice/group (a, d). Not detected (ND).*p < 0.05. Not significant (ns).
Extended Data Figure 5. Global gene expression analysis of alveolar macrophages from age-matched WT, KO, and KO mice one year after PMT of WT BMDMs.
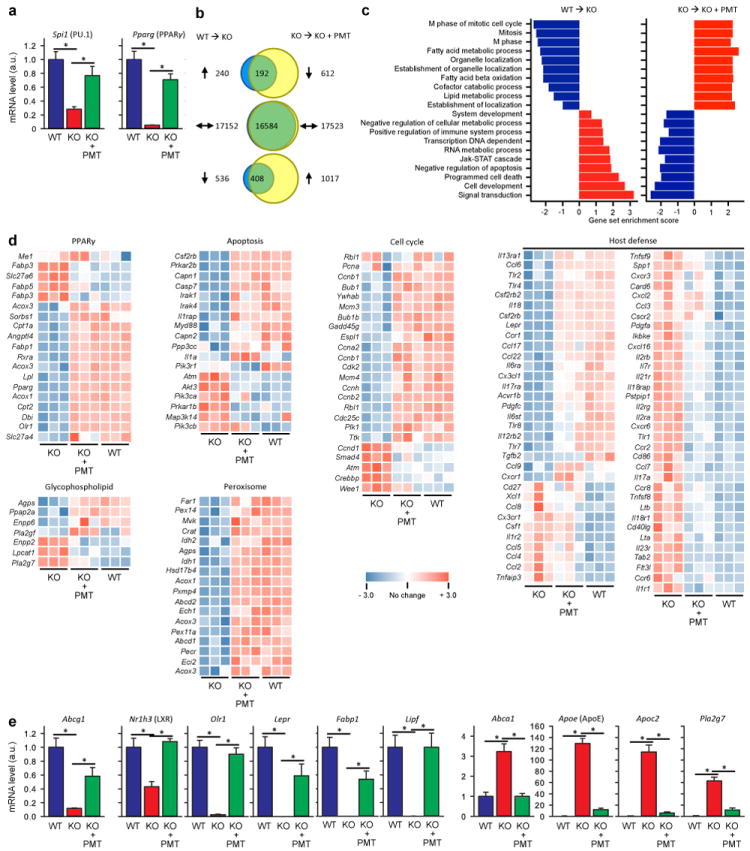
(a) Expression of Spi1 (PU.1) and Pparg (PPARγ) were confirmed by qRT-PCR using independent samples (6 mice/group). (b) Venn diagrams showing numbers of genes whose expression was altered in alveolar macrophages from KO compared to WT mice (WT→KO) or PMT-treated compared to untreated KO mice (KO→KO+PMT). Only genes with statistically significant changes (false detection rate < 10%) of at least two-fold were marked as increased (up arrows) or decreased (down arrows). The numbers of genes for which expression was disrupted in KO mice and normalized by PMT (or unchanged in both comparisons) is shown in the overlap regions. (c) Gene ontology analysis identifying pathways disrupted in KO mice and restored by PMT. Data show the coordinate increases (red) or decreases (blue) in expression of genes in all gene sets significant at or below a false detection rate of 10% calculated by the Gene Set Test with correction for multiple testing. (d) Heat maps showing differentially expressed genes in multiple KEGG pathways including PPARγ-regulated genes, glycophospholipid metabolism, peroxisome function apoptosis, cell cycle control, and immune host defense. Genes with increased or decreased transcript levels are shown by red and blue colors, respectively. (e) Confirmation by qRT-PCR for selected genes important in lipid metabolism. Data are mean ± SEM using independent samples (6 mice/group). *p < 0.05.
Extended Data Figure 6.
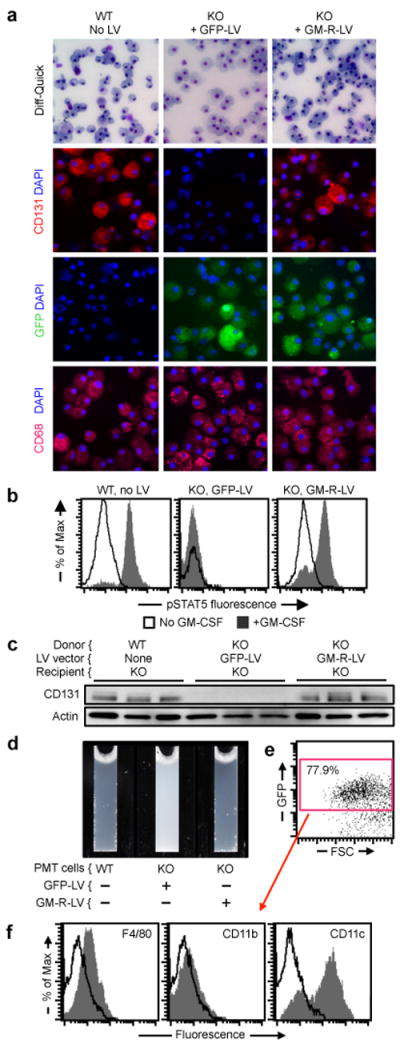
Effects of PMT of gene-corrected macrophages on hPAP. (a) Macrophages derived from KO LSK cells transduced with GM-R-LV or GFP-LV, or from non-transduced WT LSK cells (indicated) were examined by light microscopy after Diff-Quick staining (top), or by immunofluorescence microscopy after staining with anti-CD131 (GM-CSF-R-β) and DAPI (upper middle), DAPI alone (lower middle), or anti-CD68 and DAPI (bottom). Images are representative of 3 experiments per condition. (b) Evaluation of GM-CSF receptor signaling in the indicated cells (before PMT) by measurement of GM-CSF-stimulated STAT5 phosphorylation by flow cytometry. Representative of n=3 experiments per condition. Quantitative summary data are shown in the manuscript (Fig. 5b). (c) Western blotting to detect GM-CSF receptor-β (CD131) (top) or actin (bottom, as a loading control) in BAL cells from age-matched KO mice 2 months after PMT as indicated (each lane represents one mouse of n=10, 8, 10/group, respectively). (d) Appearance of BAL from age-matched KO mice 2 months after PMT as indicated (representative of n=10, 8, 10/group, respectively). (e-f) One year after PMT of GM-R-LV transduced KO LSK cell-derived macrophages in KO mice, GFP+ cells were identified (e) and evaluated for cell surface markers by flow cytometry (f) (representative of n=7 mice).
Extended Data Table 1.
Oligonucleotide primers used to quantify mRNA transcripts by qRT-PCR and Detection of PMT-derived cellular DNA by PCR.*
| TaqMan® Primers | |||
|---|---|---|---|
| Gene name | Accession no. | Product (bp) | Catalogue no. |
| Spi1(PU.1) | NM_011355.1 | 91 | Mm00488142_m1 |
| Pparg | NM_001127330.1 | 101 | Mm01184322_m1 |
| Abcg1 | NM_009593.2 | 65 | Mm00437390_m1 |
| Csf1 | NM_001113529.1 | 70 | Mm00432686_m1 |
| Csf2 | NM_009969.4 | 125 | Mm01290062_m1 |
| Csf2rb | NM_007780.4 | 60 | Mm00655745_m1 |
| Nr1h3 (LXR) | NM_001177730.1 | 57 | Mm00443451_m1 |
| Olr1 | NM_138648.2 | 64 | Mm00454586_m1 |
| Lepr | NM_001122899.1 | 97 | Mm00440181_m1 |
| Fabp1 | NM_007980.2 | 116 | Mm00433188_m1 |
| Lipf | NM_026334.3 | 87 | Mm00471152_m1 |
| Abca1 | NM_013454.3 | 55 | Mm00442646_m1 |
| Apoe (Apo E) | NM_009696.3 | 64 | Mm01307192_m1 |
| Apoc2 | NM_009695.3 | 60 | Mm00437571_m1 |
| Pla2g7 | NM_013737.5 | 111 | Mm00479105_m1 |
| Gapdh | NM_008084.2 | 107 | 4352932E |
| 18S RNA | X03205.1 | 187 | 4310893E |
| Custom Primers | |||
| Gene name | Accession no. | Product (bp) | Sequence (5’ → 3’) |
| Lys-MGFP | NA - transgene | 680 | aag ctg ttg gga aag gag gg gtc gcc gat ggg ggt gtt ct |
| Lysozyme-M | M21049 | 220 | aag ctg ttg gga aag gag gg tcg gcc agg ctg act cca ta |
Primer sets were purchased from Life Technologies.
Extended Data Table 2.
Effect of the number of macrophages transplanted on the efficacy of PMT therapy of hPAP in KO mice*
| Parameter | WT | KO | KO + PMT (Macrophages/dose × 106)
|
|||
|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | 4 | |||
|
| ||||||
| Turbidity | 0.0553 | 1.96 † | 0.765 ‡ | 0.685 ‡ | 0.38 ‡ | 0.536 ‡ |
| O.D. 600 nm | 0.023-0.21 | 1.85-2.74 | 0.599-0.823 | 0.472-0.997 | 0.283-0.685 | 0.301-0.732 |
|
| ||||||
| SP-D | 75.9 | 2105 † | 1475 ‡ | 1414 ‡ | 911 ‡ | 1299 ‡ |
| ug/ml BAL | 51-84 | 1739-2396 | 1367-2034 | 656-1951 | 660-1179 | 762-1634 |
|
| ||||||
| GM-CSF | 0 | 40.8 † | 28.1 ‡ | 17.2 ‡ | 13.8 ‡ | 14.8 ‡ |
| pg/ml BAL | 0-0 | 21.4-54.5 | 15.5-37.8 | 12.2-20.5 | 6.97-17.5 | 10.4-18.0 |
|
| ||||||
| M-CSF | 0 | 45.0 † | 30.4 § | 25.4 § | 21.7 § | 29.3 § |
| pg/ml BAL | 0-0 | 32.3-81.9 | 14.2-36.0 | 14.8-36.3 | 20.1-42.7 | 23.5-40.5 |
|
| ||||||
| MCP-1 | 0.88 | 135 † | 72.1 ‡ | 57.5 ‡ | 49.0 ‡ | 64.4 ‡ |
| pg/ml BAL | 0-25.1 | 123-163 | 36.9-121 | 45.9-80.7 | 26.1-63.0 | 28.2-109 |
|
| ||||||
| Csf2rb mRNA | 1.05 | 0 † | 0.108 ‡ | 0.167 ‡ | 0.265 ‡ | 0.447 ‡ |
| A.U. | 0.86-1.08 | 0-0 | 0.085-0.213 | 0.095-0.82 | 0.097-1.46 | 0.197-0.88 |
|
| ||||||
| Spi1 mRNA | 1.02 | 0.306 † | 0.468 ‡ | 0.474 ‡ | 0.475 ‡ | 0.480 ‡ |
| A.U. | 0.78-1.13 | 0.289-0.393 | 0.27-0.470 | 0.351-0.707 | 0.367-0.803 | 0.446-0.595 |
|
| ||||||
| Pparg mRNA | 0.929 | 0.052 † | 0.241 ‡ | 0.295 ‡ | 0.516 ‡ | 0.360 ‡ |
| A.U. | 0.902-1.13 | 0.0-0.106 | 0.196-0.362 | 0.267-0.607 | 0.327-0.923 | 0.318-0.603 |
|
| ||||||
| Abcg1 mRNA | 0.933 | 0.08 † | 0.148 ‡ | 0.232 ‡ | 0.179 ‡ | 0.220 ‡ |
| A.U. | 0.833-1.12 | 0.07-0.153 | 0.135-0.183 | 0.134-0.327 | 0.106-0.455 | 0.173-0.229 |
Definitions: A.U. – arbitrary units, BAL- bronchoalveolar lavage, hPAP – hereditary pulmonary alveolar proteinosis, KO – Csf2rb knock out mice, O.D. – optical density, PMT – pulmonary macrophage transplantation, WT – wild type.
Mice received the indicated numbers of WT BMDMs once by PMT. Three months later, BAL fluid and cells were obtained from PMT-treated KO mice, and age-matched, untreated WT or KO mice (7 mice per group for each condition evaluated). BAL turbidity, the concentration of SP-D, GM-CSF, M-CSF, and MCP-1 in BAL fluid, and the relative abundance of Csf2rb, Spi1 (PU.1), Pparg, and Abcg1 mRNA transcripts in alveolar macrophages were measured as described in the methods. All data are presented as median (interquartile range (IQR)) and between-group comparisons were done using non-parametric methods for consistency since results for some groups were either undetectable, not normally distributed or of unequal variance.
Result is significantly different compared to untreated WT mice (Mann-Whitney Rank Sum Test, P<0.001).
Result is significantly different compared to untreated KO mice (Kruskal-Wallis One Way Analysis of Variance on Ranks with Pairwise comparison to untreated KO mice by the Student-Neuman-Keuls method, P<0.05).
Result is not significantly different compared to untreated KO mice (Kruskal-Wallis One Way Analysis of Variance on Ranks, P=0.133).
Extended Data Table 3.
Comparison of the effects of single versus repeated macrophage administrations on the efficacy of PMT therapy of hPAP in KO mice*
| Parameter | Number of PMT Administrations | P-value † | |
|---|---|---|---|
| One | Four | ||
| Turbidity, O.D. 600 nm | 2.014 (1.77-2.53) | 1.68 (1.49-3.29) | 0.486 |
| SP-D, μg/ml BAL | 816 (750-996) | 772 (653-796) | 0.486 |
| GM-CSF, pg/ml BAL | 18.3 (15.3-35.6) | 14.3 (13.8-27.8) | 0.20 |
| M-CSF, pg/ml BAL | 55.5 (50.8-65.6) | 29.7 (28.1-49.8) | 0.114 |
| MCP-1, pg/ml BAL | 88.3 (74.0-118) | 53.6 (35.2-78.8) | 0.114 |
| Spi1 mRNA, A.U. | 0.377 (0.284-0.545) | 0.322 (0.268-0.362) | 0.686 |
| Pparg mRNA, A.U. | 0.201 (0.122-0.474) | 0.234 (0.169-0.303) | 1.0 |
| Abcg1 mRNA, A.U. | 0.116 (0.098-0.236) | 0.117 (0.083-0.131) | 0.886 |
Definitions: A.U. – arbitrary units, BAL- bronchoalveolar lavage, hPAP – hereditary pulmonary alveolar proteinosis, KO – Csf2rb knock out mice, O.D. – optical density, PMT – pulmonary macrophage transplantation.
KO mice received 2×106 macrophages by PMT either once or as four monthly doses. Four months after the initial PMT administration in both groups, BAL fluid and cells were obtained from all mice (6 per group for each condition evaluated). BAL turbidity, the concentration of SP-D, GM-CSF, M-CSF, and MCP-1 in BAL fluid, and the relative abundance of Spi1 (PU.1), Pparg, and Abcg1 mRNA transcripts in alveolar macrophages were measured as described in the methods. All data are presented as median (interquartile range (IQR)) and between-group comparisons were done using non-parametric methods for consistency since results for some groups were either not normally distributed or of unequal variance.
Mann-Whitney Rank Sum Test. P-values of ≤ 0.05 was considered to indicate statistical significance.
Extended Data Table 4.
Effect of PMT of WT or gene-corrected macrophages on hematologic indices and lung proinflammatory cytokine levels*
| Blood Safety Evaluation - PMT Using Wild-Type Macrophages | ||||
|---|---|---|---|---|
| Hematologic Parameter | Normal range | WT (n=6) | KO (n=6) | KO + PMT (n=6) |
| Hemoglobin, g/dL | 11.0 – 15.1 | 12.7 (12.2 – 13.2) | 14.5 (13.9 – 15.1) ‡ | 12.9 (12.2 – 13.4) ¶ |
| Hematocrit, % | 35.1 – 45.4 | 46.5 (44.5 – 47.3) | 53.0 (50.2 – 56.3) ‡ | 47.2 (43.0 – 50.5) ¶ |
| WBC, ×103/μl | 1.8 – 10.7 | 2.53 (1.53 – 5.35) | 5.08 (3.93 – 6.34) † | 3.27 (2.80 – 4.10) ¶ |
| Neutrophils, ×103/μl | 0.1 – 2.4 | 0.29 (0.107 – 0.67) | 0.89 (0.125 – 2.63) † | 0.670 (0.102 – 1. 41) § |
| Lymphocytes, ×103/μl | 0.9 – 9.3 | 2.18 (1.06 – 4.26) | 3.24 (1.60 – 5.79) † | 2.37 (1.76 – 2.94) § |
| Monocytes, ×103/μl | 0.0 – 0.4 | 0.15 (0.11 – 0.24) | 0.155 (0.12 – 0.32) † | 0.19 (0.15 – 0.25) § |
| Eosinophils, ×103/μl | 0.0 – 0.2 | 0.01 (0.01 – 0.04) | 0.02 (0.01 – 0.06) † | 0.015 (0.01 – 0.02) § |
| Basophils, ×103/μl | 0.0 – 0.2 | 0.0 (0.0 – 0.01) | 0.01 (0.01 – 0.01) † | 0.0 (0.0 – 0.003) ¶ |
| Platelets, ×103/μl | 592 – 2972 | 558 (463 – 735) | 1035 (830 – 1145) ‡ | 993 (838 – 1031) § |
| Blood Safety Evaluation - PMT Using Gene-Corrected Macrophage | ||||
| Hematologic Parameter | Normal range | WT (n=5) | KO (n=5) | KO + PMT (n=7) |
| Hemoglobin, g/dL | 11.0 – 15.1 | 10.2 (8.5 – 10.8) | 16.9 (13.8 – 18.5) ‡ | 12.2 (11.1 – 12.9) ¶ |
| Hematocrit, % | 35.1 – 45.4 | 37.5 (33.3 – 40.4) | 68.8 (55.4 – 80.3) ‡ | 49.4 (48.8 – 55.9) ¶ |
| WBC, ×103/μl | 1.8 – 10.7 | 1.90 (1.30 – 10.7) | 6.48 (5.11 – 8.57) † | 5.08 (2.32 – 5.88) § |
| Neutrophils, ×103/μl | 0.1 – 2.4 | 1.31 (0.67 – 8.01) | 2.61 (1.71 – 2.85) † | 1. 49 (0.96 – 2.89) § |
| Lymphocytes, ×103/μl | 0.9 – 9.3 | 0.65 (0.26 – 1.38) | 3.39 (2.70 – 5.15) ‡ | 2.03 (1.15 – 3.33) ¶ |
| Monocytes, ×103/μl | 0.0 – 0.4 | 0.25 (0.15 – 0.90) | 0.44 (0.29 – 0.87) † | 0.25 (0.15 – 0.40) § |
| Eosinophils, ×103/μl | 0.0 – 0.2 | 0.01 (0.01 – 0.43) | 0.02 (0.02 – 0.07) † | 0.08 (0.01 – 0.16) § |
| Basophils, ×103/μl | 0.0 – 0.2 | 0 (0.0 – 0.07) | 0.01 (0.00 – 0.04) † | 0.01 (0.00 – 0.03) § |
| Platelets, ×103/μl | 592 – 2972 | 619 (441 – 1478) | 1229 (1094 – 1460) † | 1381 (872 – 1614) § |
| Lung Safety Evaluation - PMT Using WT Macrophages | ||||
| Cytokine in BAL Fluid | WT (n=6) | KO (n=6) | KO + PMT (n=6) | |
| IL-6, pg/ml | 3.01 (1.35 – 3.89) | 164 (43.6 – 364) ‡ | 11.02 (4.87 – 60.1) ¶ | |
| IL-1β, pg/ml | 0 (0.0 – 0.0) | 3.49 (2.42 – 9.52) ‡ | 0.92 (0.04 – 1.02) ¶ | |
| TNFα, pg/ml | 0.52 (0.0 – 0.94) | 12.4 (7.22 – 17.9) ‡ | 2.75 (1.08 – 3.17) ¶ | |
| Lung Safety Evaluation - PMT Using Gene Corrected Macrophages | ||||
| Cytokine in BAL Fluid | WT (n=5) | KO (n=5) | KO + PMT (n=7) | |
| IL-6, pg/ml | 0.30 (0.0 – 2.43) | 16.2 (14.1 – 62.1) ‡ | 29.5 (13.7 – 42.5) § | |
| IL-1β, pg/ml | 0 (0.0 – 0.29) | 2.01 (0.47 – 8.1) † | 1.65 (0.94 – 3.08) § | |
| TNFα, pg/ml | 2.45 (0.46 – 2.76) | 6.14 (3.68 – 7.99) ‡ | 3.06 (2.45 – 7.98) § | |
Definitions: BAL – bronchoalveolar lavage, hPAP – hereditary pulmonary alveolar proteinosis, KO – Csf2rb knock out mice, O.D. – optical density, PMT – pulmonary macrophage transplantation, WT – wild type.
KO mice received WT or Csf2rb gene-corrected KO macrophages (2×106 cells/mouse) once by PMT and 12 months later, blood and BAL fluid were obtained from PMT-treated KO mice, or age-matched, untreated KO or WT mice and evaluated as described in the Methods. Number of mice/ group is indicated. All data are presented as median (interquartile range (IQR)) and between-group comparisons were done using non-parametric methods (Mann-Whitney Rank Sum Test) for consistency since results for some groups were either not normally distributed or of unequal variance. P-Values ≤ 0.05 were considered to be significant
Result is not significantly different compared to WT mice.
Result is significantly different compared to WT mice.
Result is not significantly different compared to untreated KO mice.
Result is significantly different compared to untreated KO mice.
Acknowledgments
Supported by grants from the NIH (R01 HL085453, R21 HL106134, R01HL118342, 8UL1TR000077-05, AR-47363, DK78392, DK90971), American Thoracic Society Foundation Unrestricted Research Grant, CCHMC Foundation Trustee Grant, Deutsche Forschungsgemeinschaft (DFG; Cluster of Excellence Rebirth; Exc 62/1), and the Else Kroner-Fresenius Stiflung, Eva-Luise Koehler Research for Rare Diseases 2013, and by the Pulmonary Biology Division, CCHMC. Flow cytometric data were acquired within the Research Flow Cytometry Core in the Division of Rheumatology, CCHMC. We thank our hPAP patients and their family members in the United States and internationally for their collaboration; Jeffrey Whitsett (CCHMC) and Frank McCormack (UCMC) for critical reading of the Manuscript; Jeff Krischer (U. of South Florida) for helpful discussions; Susan Wert for help with lung histology, and Diane Black, Kevin Link, and Catherine Fox (CCHMC), and Sebastian Brennig and Henning Kempf (Hannover Medical School) for their technical help.
Footnotes
Author Contributions
T.S., P.A., N.L., S.A., T.M., P.M., and B.C.T. designed research. T.S., P.A., T.S., N.L., C.C., A.S., S.A., B.C. and B.C.T. performed research. T.S., P.A., T.S., N.L., S.A., C.T., T.M., P.M., and B.C.T. analyzed data. T.S., P.A., N.L., P.M., C.L., R.E.W., and B.C.T. wrote the paper.
The authors declare no competing financial interests.
Online Content
Methods, Extended Data display items and Source Data are available in the online version of the paper: references unique to these sections appear only in the online paper.
References
- 1.Suzuki T, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. The Journal of experimental medicine. 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Moczygemba M, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFR alpha gene in the X chromosome pseudoautosomal region 1. The Journal of experimental medicine. 2008;205:2711–2716. doi: 10.1084/jem.20080759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki T, et al. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. American journal of respiratory and critical care medicine. 2010;182:1292–1304. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka T, et al. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J Med Genet. 2011;48:205–209. doi: 10.1136/jmg.2010.082586. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki T, et al. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. The European respiratory journal. 2011;37:201–204. doi: 10.1183/09031936.00090610. [DOI] [PubMed] [Google Scholar]
- 6.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annual review of medicine. 2010;61:105–119. doi: 10.1146/annurev.med.60.041807.123500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawgood S, Poulain FR. The pulmonary collectins and surfactant metabolism. Annual review of physiology. 2001;63:495–519. doi: 10.1146/annurev.physiol.63.1.495. [DOI] [PubMed] [Google Scholar]
- 8.Ikegami M, et al. Surfactant metabolism in transgenic mice after granulocyte macrophage-colony stimulating factor ablation. The American journal of physiology. 1996;270:L650–658. doi: 10.1152/ajplung.1996.270.4.L650. [DOI] [PubMed] [Google Scholar]
- 9.Kitamura T, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine. 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. The New England journal of medicine. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 11.Stanley E, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dranoff G, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 13.Robb L, et al. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9565–9569. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishinakamura R, et al. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor- deficient mice are normal. Immunity. 1995;2:211–222. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 15.Bonfield TL, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. American journal of respiratory cell and molecular biology. 2003;29:677–682. doi: 10.1165/rcmb.2003-0148OC. [DOI] [PubMed] [Google Scholar]
- 16.Bonfield TL, et al. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. American journal of physiology. Lung cellular and molecular physiology. 2003;285:L1132–1136. doi: 10.1152/ajplung.00216.2003. [DOI] [PubMed] [Google Scholar]
- 17.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. American Joural of Respiratory and Critical Care Medicine. 2002;166:215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 18.Shibata Y, et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 19.Thomassen MJ, et al. ABCG1 is deficient in alveolar macrophages of GM-CSF knockout mice and patients with pulmonary alveolar proteinosis. J Lipid Res. 2007;48:2762–2768. doi: 10.1194/jlr.P700022-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Iyonaga K, et al. Elevated bronchoalveolar concentrations of MCP-1 in patients with pulmonary alveolar proteinosis. The European respiratory journal. 1999;14:383–389. doi: 10.1034/j.1399-3003.1999.14b24.x. [DOI] [PubMed] [Google Scholar]
- 21.Nishinakamura R, et al. The pulmonary alveolar proteinosis in granulocyte macrophage colony-stimulating factor/interleukins 3/5 beta c receptor-deficient mice is reversed by bone marrow transplantation. The Journal of experimental medicine. 1996;183:2657–2662. doi: 10.1084/jem.183.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleff V, et al. Gene therapy of beta (c)-deficient pulmonary alveolar proteinosis (beta(c)-PAP): studies in a murine in vivo model. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:757–764. doi: 10.1038/mt.2008.7. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, et al. Use of Induced Pluripotent Stem Cells To Recapitulate Pulmonary Alveolar Proteinosis Pathogenesis. American journal of respiratory and critical care medicine. 2014;189:183–193. doi: 10.1164/rccm.201306-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stradling JR, Lane DJ. Development of secondary polycythaemia in chronic airways obstruction. Thorax. 1981;36:321–325. doi: 10.1136/thx.36.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- 26.Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF regulates surfacant Protein-A and lipid catabolism by alveolar macrohpages. American journal of physiology. Lung cellular and molecular physiology. 2001;280:L379–L386. doi: 10.1152/ajplung.2001.280.3.L379. [DOI] [PubMed] [Google Scholar]
- 27.Lachmann N, et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. American journal of respiratory and critical care medicine. 2014;189:167–182. doi: 10.1164/rccm.201306-1012OC. [DOI] [PubMed] [Google Scholar]
- 28.Seymour JF, et al. Mice lacking both granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF have impaired reproductive capacity, perturbed neonatal granulopoiesis, lung disease, amyloidosis, and reduced long-term survival. Blood. 1997;90:3037–3049. [PubMed] [Google Scholar]
- 29.Wu M, et al. Genetically engineered macrophages expressing IFN-gamma restore alveolar immune function in scid mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14589–14594. doi: 10.1073/pnas.251451498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guilliams M, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. The Journal of experimental medicine. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashimoto D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz C, et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 34.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. The Journal of experimental medicine. 1968;128:415–435. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Godleski JJ, Brain JD. The origin of alveolar macrophages in mouse radiation chimeras. The Journal of experimental medicine. 1972;136:630–643. doi: 10.1084/jem.136.3.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matute-Bello G, et al. Optimal timing to repopulation of resident alveolar macrophages with donor cells following total body irradiation and bone marrow transplantation in mice. Journal of immunological methods. 2004;292:25–34. doi: 10.1016/j.jim.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. American journal of respiratory cell and molecular biology. 2008;38:380–385. doi: 10.1165/rcmb.2007-0224RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guth AM, et al. Lung environment determines unique phenotype of alveolar macrophages. American journal of physiology. Lung cellular and molecular physiology. 2009;296:L936–946. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wert SE, et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5972–5977. doi: 10.1073/pnas.100448997. In Process Citation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakagami T, et al. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. American journal of respiratory and critical care medicine. 2010;182:49–61. doi: 10.1164/rccm.201001-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. The Journal of clinical investigation. 1999;103:563–569. doi: 10.1172/JCI5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berclaz PY, et al. GM-CSF regulates a PU.1-dependent transcriptional program determining the pulmonary response to LPS. American journal of respiratory cell and molecular biology. 2007;36:114–121. doi: 10.1165/rcmb.2006-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perumbeti A, et al. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood. 2009;114:1174–1185. doi: 10.1182/blood-2009-01-201863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10716–10721. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walters DM, Breysse PN, Wills-Karp M. Ambient urban Baltimore particulate – induced airway hyperresponsiveness and inflammation in mice. American journal of respiratory and critical care medicine. 2001;164:1438–1443. doi: 10.1164/ajrccm.164.8.2007121. [DOI] [PubMed] [Google Scholar]
- 47.Maeda Y, et al. Kras (G12D) and Nkx2-1 haploinsufficiency induce mucinous adenocarcinoma of the lung. The Journal of clinical investigation. 2012;122:4388–4400. doi: 10.1172/JCI64048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- 50.Richard E, et al. Gene therapy of a mouse model of protoporphyria with a self-inactivating erythroid-specific lentiviral vector without preselection. Molecular therapy : the journal of the American Society of Gene Therapy. 2001;4:331–338. doi: 10.1006/mthe.2001.0467. [DOI] [PubMed] [Google Scholar]
- 51.Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic acids research. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zufferey R, Donello JE, Trono D, Hope TJ. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. Journal of virology. 1999;73:2886–2892. doi: 10.1128/jvi.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arumugam PI, et al. The 3’ region of the chicken hypersensitive site-4 insulator has properties similar to its core and is required for full insulator activity. PloS one. 2009;4:e6995. doi: 10.1371/journal.pone.0006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arumugam PI, et al. Improved human beta-globin expression from self-inactivating lentiviral vectors carrying the chicken hypersensitive site-4 (cHS4) insulator element. Molecular therapy : the journal of the American Society of Gene Therapy. 2007;15:1863–1871. doi: 10.1038/sj.mt.6300259. [DOI] [PubMed] [Google Scholar]