

Summary
Tumor suppressors with widespread impact on carcinogenesis control broad spectra of oncogenic pathways. Protein degradation is an emerging mechanism by which tumor suppressors regulate a diversity of pathways and is exemplified by the SCFFbw7 ubiquitin ligase. Rapidly accumulating data indicate that SCFFbw7 regulates a network of crucial oncoproteins. Importantly, the FBXW7 gene, which encodes Fbw7, is one of the most frequently mutated genes in human cancers. These studies are yielding important new insights into tumorigenesis and may soon enable therapies targeting the Fbw7 pathway. Here, we focus on the mechanisms and consequences of Fbw7 deregulation in cancers and discuss possible therapeutic approaches.
Keywords: Fbw7, SCF, cancer, ubiquitin, phosphodegron, Fbxw7, hCDC4, tumor suppressor, proteasome, F-box protein
Protein degradation by the ubiquitin-proteasome system (UPS) controls a broad array of cellular processes (Hershko and Ciechanover, 1998). Ubiquitin-mediated proteolysis is regulated, rapid, and irreversible, and has important roles in cell division, growth, and differentiation. Cancers often contain mutations targeting the UPS that drive tumorigenesis (Nakayama and Nakayama, 2006). Indeed, the fundamental importance of the UPS in tumor cell homeostasis is highlighted by the emergence of pharmacologic UPS inhibitors as promising cancer therapies.
Perhaps the most commonly deregulated UPS protein in human cancers is the ubiquitin ligase component Fbw7, which targets a network of substrates for degradation, including some key human oncoproteins (Crusio et al., 2010; Welcker and Clurman, 2008). Most of these substrates are master transcription factors (TFs) allowing Fbw7 to regulate diverse pathways with oncogenic potential. Recent progress in many disciplines has illuminated Fbw7's central role in tumorigenesis. Large-scale cancer genome studies have shown that Fbw7 is among the most mutated cancer genes, mouse models have demonstrated its potent tumor suppressor functions, and new substrates and mutational mechanisms have been discovered that may drive Fbw7-associated cancer. These advances have not only increased our understanding of Fbw7's roles in tumorigenesis, but may also soon enable the development of Fbw7-targeted therapies.
Fbw7 is a ubiquitin ligase substrate receptor
Ubiquitin-mediated proteolysis is instigated by the attachment of K48 poly-ubiquitin chains to substrates, which provides a signal for recognition and degradation by the proteasome (Finley, 2009). In most cases, E3 ubiquitin ligases are needed to recognize substrates and facilitate their ubiquitylation. SCFs (Skp1, Cullin-1, F-box protein) are a class of E3s that use Cullin-1 as a scaffold and F-box proteins as substrate receptors, and have important roles in cancer biology (Fig. 1A) (Deshaies and Joazeiro, 2009; Lee and Diehl, 2014; Skaar et al., 2013). F-box proteins are thus adaptors that bring substrates into proximity with ubiquitylation enzymes.
Figure 1. Dimeric SCFFbw7 regulates a broad network of substrates.
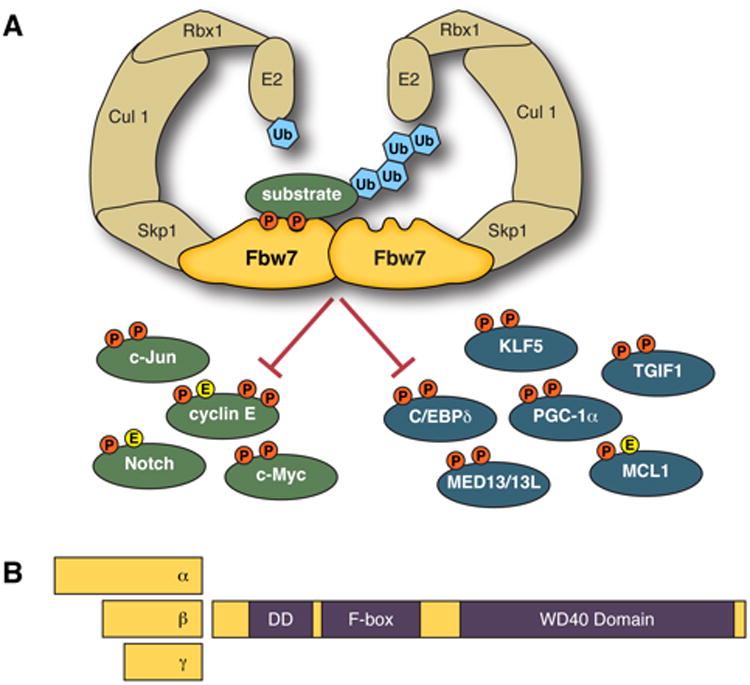
(A) Fbw7 binds to both phosphorylated substrates and the rest of the SCF complex (comprised of Skp1, Cullin-1, Rbx1, and an E2 enzyme), resulting in substrate poly-ubiquitylation and degradation by the proteasome. The network of Fbw7 substrates contains proteins with clear roles in carcinogenesis (shown in green) and others with emerging roles in Fbw7-associated tumors (shown in blue) (see text). Optimal (high-affinity) substrates have recognition signals termed CPDs that contain two phosphorylated residues (orange “P”); other, lower-affinity, CPDs contain a negatively charged amino acid (yellow “E”) in place of the second phosphate. (B) Fbw7 exists as three protein isoforms (α, β, and γ) that differ only by their N-terminal exons. All isoforms share three functional domains that are critical to their function as ubiquitin ligases: (1) the dimerization domain (“DD”) mediates Fbw7 dimerization, (2) the F-box binds to the rest of the SCF complex via Skp1 (Fig. 1A), and (3) the WD40 domain binds to phosphorylated substrates, and includes the three arginine residues that are mutational hotspots in cancers.
Fbw7 is an evolutionary conserved F-box protein, and although beyond the scope of this review, studies of its orthologues (Cdc4, S. cerevisiae; sel-10, C. elegans; Archipelago, Drosophila) have yielded fundamental insights into SCFFbw7 function. The human FBXW7 gene resides on chromosome 4q32, a region commonly deleted in cancers, and produces three mRNAs, each under their own transcriptional control (Spruck et al., 2002). The three Fbw7 mRNAs encode three protein isoforms that differ only by isoform-specific N-terminal exons that specify subcellular localization: Fbw7α is nucleoplasmic, Fbw7β is cytoplasmic, and Fbw7γ is nucleolar. All isoforms share three important functional domains: 1) the D-domain mediates Fbw7 dimerization, which regulates substrate binding modes and ubiquitylation, 2) the F-box binds Skp1 and links Fbw7 to the SCF complex, and 3) the WD40 domain forms a β-propeller that binds phosphorylated substrates (Fig. 1B) (Hao et al., 2007; Orlicky et al., 2003; Tang et al., 2007; Welcker and Clurman, 2007; Welcker et al., 2013; Zhang and Koepp, 2006). Fbw7α is thought to perform most Fbw7 functions, although specific roles for the other isoforms have also been described (Bonetti et al., 2008; Ekholm-Reed et al., 2013; Grim et al., 2008; Matsumoto et al., 2011; van Drogen et al., 2006; Welcker et al., 2004a).
Substrate phosphorylation stimulates Fbw7 binding
Substrate phosphorylation instigates Fbw7 binding to a conserved Cdc4 phosphodegron (CPD) motif (Koepp et al., 2001; Nash et al., 2001; Strohmaier et al., 2001; Welcker et al., 2003). Mutational and structural studies have provided insights into the interactions between CPDs and Fbw7's β-propeller (Hao et al., 2007; Orlicky et al., 2003; Tang et al., 2007). CPDs contain multiple residues that contact Fbw7, and typically include phosphorylated threonine/serine residues in the “0” and “+4” positions that interact with Fbw7 phosphate-binding pockets (Welcker and Clurman, 2008). CPD affinity varies among substrates; high-affinity CPDs contain two phosphorylations and other optimal residues, whereas low-affinity substrates may contain a negatively charged amino acid in lieu of a second phosphate or other unfavorable residues (Fig. 1A) (Hao et al., 2007; Nash et al., 2001; Welcker et al., 2013). Importantly, three arginine residues in Fbw7's WD40 domain interact with CPD phosphates and are mutational hotspots in cancers (see below).
Ultimately, the signaling pathways that mediate CPD phosphorylation regulate substrate degradation. The presence of two phosphorylation sites in most CPDs provides a mechanism through which multiple signals can control substrate degradation. Most substrates have multiple turnover pathways; therefore the regulation of CPD phosphorylation determines the contexts of their degradation by SCFFbw7. Glycogen synthase kinase 3 (GSK3) phosphorylates the central position of many CPDs, and this is opposed by mitogen-stimulation of the PI3K-AKT pathway, which inactivates GSK3 (Crusio et al., 2010; Welcker and Clurman, 2008). It is likely that by coupling Fbw7-mediated degradation with GSK3 activity, mitogenic signaling can coordinately stabilize substrates with key roles in cell proliferation (e.g. Myc, cyclin E, and Jun). The abnormal AKT and PTEN activity commonly found in cancers might similarly allow oncogenic Fbw7 substrates to accumulate.
Fbw7 dimerization provides another important level of regulation. Because each protomer of an Fbw7 dimer contains a substrate-binding domain, dimers can simultaneously bind two CPDs, which is particularly important for substrates with degrons that are too weak to drive Fbw7 binding alone (Figs. 1-2) (Welcker et al., 2013). The combinatorial impact of multiple CPDs allows highly specific control of substrate ubiquitylation. Additionally, dimerization promotes substrate degradation by expanding the number of substrate lysine residues that are accessible for ubiquitin conjugation (Hao et al., 2007; Tang et al., 2007; Welcker et al., 2013).
Figure 2. Possible mechanisms of Fbw7ARG missense mutations in cancers.

(A) Normal interactions of Fbw7 dimers with monomeric (left) or dimeric (right) substrates. Each protomer within an Fbw7 dimer can interact with a separate substrate CPD, leading to greatly increased binding affinity between substrates and Fbw7. The two substrate CPDs can be present within a monomeric substrate (e.g. cyclin E, left), or separated onto two interacting proteins (e.g. SREBP, right). The reduced number of contacts made by suboptimal degrons is indicated by a dashed line. (B) Model of heterozygous Fbw7ARG dominant-negative activity in cancers resulting from the formation of impaired Fbw7WT-Fbw7ARG heterodimers. We speculate that Fbw7WT-Fbw7ARG heterodimers differentially affect substrates, depending on CPD affinity. The degradation of high-affinity substrates may still be driven by the normal protomer of an Fbw7WT-Fbw7ARG heterodimer (left), whereas suboptimal substrates (depicted by glutamate instead of a second CPD phosphate) rely on the concerted biding of two CPDs to an Fbw7 dimer, and will not be ubiquitylated by Fbw7WT-Fbw7ARG heterodimers (right). (C) Fbw7 truncation mutants may also generate heterodimers with Fbw7WT, but are not nearly as frequent in tumors as Fbw7ARG. We therefore speculate that a full length Fbw7 protein is critical for the dominant-negative effect of Fbw7ARG, perhaps by retaining sufficient residual binding affinity (depicted by dashed line) for substrates with intermediate affinity.
Oncogenic Fbw7 substrates: the drivers of Fbw7-associated cancers
SCFFbw7 targets approximately two-dozen proteins with key roles in proliferation, differentiation, apoptosis, and metabolism (Fig. 1A and not shown). With few exceptions (e.g. cyclin E and MCL1), Fbw7 substrates are transcriptional regulators that control complex gene-expression programs; this extends Fbw7's impact far beyond its direct substrates. Below, we highlight only those substrates that are either known oncoproteins or have emerging roles in tumorigenesis.
Critical oncogenic Fbw7 substrates
Four Fbw7 substrates stand apart by virtue of their frequent mutation in many human cancers. c-Myc (hereafter called Myc) sustains gain-of function mutations, including amplifications in solid tumors and translocations in hematologic malignancies. Myc deregulation promotes tumorigenesis largely through its transcriptional regulation of proliferation, protein synthesis, apoptosis, metabolism, and differentiation. Fbw7α mediates Myc ubiquitylation in the nucleoplasm, whereas Fbw7γ ubiquitylates Myc in the nucleolus, which inhibits Myc's ability to promote cell growth (Bonetti et al., 2008; Grim et al., 2008; Welcker et al., 2004a; Welcker et al., 2004b; Yada et al., 2004). Early work found that phosphorylation of the threonine 58 (T58) CPD stimulates Myc degradation (Gregory et al., 2003; Sears et al., 2000) and this is now known to be mediated by Fbw7. The T58 region is targeted by missense mutations in lymphomas, suggesting a crucial role for impaired Myc degradation by Fbw7 (Bahram et al., 2000; Bhatia et al., 1993). This has been confirmed in mouse models (see below). N-Myc also contains the T58 CPD and is commonly amplified in cancers (e.g. neuroblastoma), but the significance of its degradation by Fbw7 is less well understood.
Notch proteins are transcriptional regulators of cell fate and differentiation that are broadly implicated in human cancers; they are typically dominant oncogenes, although they can be tumor suppressive in some cancers. All four Notch paralogs contain motifs homologous with the Notch1 CPD, but most studies have focused on Notch1 (Fryer et al., 2004; Gupta-Rossi et al., 2001; O'Neil et al., 2007; Oberg et al., 2001; Thompson et al., 2007; Wu et al., 2001). Notch1 is processed by a series of proteolytic cleavages, and the transcriptionally active Notch1 intracellular domain (NICD) is ubiquitylated by SCFFbw7. Activating Notch1 mutations occur in approximately 50% of T-cell acute lymphoblastic lymphomas (T-ALL) and often target the PEST domain, which contains the Fbw7 CPD (Weng et al., 2004). Fbw7 mutations are also common in T-ALL and mutually exclusive with Notch PEST mutations, underscoring the importance of Fbw7-dependent Notch degradation in this disease (Malyukova et al., 2007; Maser et al., 2007; O'Neil et al., 2007; Thompson et al., 2007). Furthermore, Fbw7 mutations in T-ALL may confer resistance to Notch inhibition by γ-secretase inhibitors, which prevent Notch processing (O'Neil et al., 2007).
Cyclin E, in conjunction with its catalytic partner cyclin-dependent kinase 2 (CDK2), regulates cell cycle entry and progression. Cyclin E has long been implicated in carcinogenesis and The Cancer Genome Atlas (TCGA) studies demonstrating frequent cyclin E amplifications in solid tumors (e.g. ovarian and breast) confirm its role as an oncogenic driver. Multi-site phosphorylation of two well-defined CPDs regulates cyclin E stability and periodicity (Clurman et al., 1996; Grim et al., 2008; Koepp et al., 2001; Minella et al., 2008; Spruck et al., 2002; Strohmaier et al., 2001; Welcker et al., 2003; Won and Reed, 1996). Genome instability is a critical consequence of constitutive cyclin E-CDK2 activity during the cell cycle caused by its impaired degradation (Ekholm-Reed et al., 2004; Keck et al., 2007; Loeb et al., 2005; Minella et al., 2007; Minella et al., 2002; Rajagopalan et al., 2004; Spruck et al., 1999). This appears to be a central mechanism through which cyclin E drives carcinogenesis, and is opposed by p53 activation.
c-Jun (hereafter called Jun) is a component of the AP-1 transcription factor and has essential roles in mitogen-stimulated cell proliferation. Jun overexpression is common in cancers and thought to drive oncogenesis; this is supported by the finding of Jun amplifications in some human cancers, such as liposarcomas. Two different Jun CPDs have been described, one of which is mutated in the v-Jun retroviral oncogene (Nateri et al., 2004; Wei et al., 2005).
Other oncogenic Fbw7 substrates
Many Fbw7 substrates have emerging roles in tumorigenesis, but uncertain contributions to Fbw7-associated cancers. Myeloid Cell Leukemia 1 (MCL1) is an anti-apoptotic protein that is overexpressed in cancers, and its stabilization in tumors with Fbw7 mutations causes resistance to chemotherapy (Inuzuka et al., 2011; Wertz et al., 2011). MED13/13L is the component of the Mediator transcriptional co-activator complex that recruits CDK8, an oncogene amplified in colorectal cancer (Davis et al., 2013). Because Mediator is required for all transcription and CDK8 regulates specific oncogenic transcriptional programs, MED13 degradation may greatly expand Fbw7's role in global transcriptional control. PPARγ Coactivator-1α (PGC-1α) is a transcriptional co-activator that coordinates mitochondrial biogenesis and cellular energetics, and has an expanding role in tumorigenesis (Olson et al., 2008). Similar to Notch, Kruppel-Like Factor 5 (KLF5) and CCAAT/Enhancer Binding Protein Delta (C/EBPδ) have either oncogenic or tumor suppressive functions in different cancers (Balamurugan et al., 2013; Liu et al., 2010; Zhao et al., 2010). TGIF1 is a transcriptional repressor that inhibits transforming growth factor β signaling, a crucial oncogenic pathway (Bengoechea-Alonso and Ericsson, 2010a). Additional oncoproteins have been reported to be Fbw7 substrates, but either lack consensus CPDs, or the contexts of their regulation by Fbw7 requires further confirmation (e.g. Hif1α, mTOR, Aurora kinase A, Myb) (Wang et al., 2012).
Fbw7 mutations in human cancers
Because many of Fbw7's first reported substrates were potent oncoproteins, its role as a tumor suppressor was quickly evaluated and confirmed. Early studies showed that 6-10% of colorectal carcinomas contain Fbw7 mutations, and subsequent work revealed Fbw7 mutations in a wide range of organ sites, including a high prevalence in T-ALL and cholangiocarcinoma (Akhoondi et al., 2007; Welcker and Clurman, 2008). These studies revealed a high frequency of heterozygous missense mutations of the three Fbw7 arginine residues that bind CPD phosphates (R465, R479, R505), hereafter called Fbw7ARG. Because these residues make the most critical contacts with the CPD phosphates and are absolutely required for high-affinity Fbw7-substrate interactions (Hao et al., 2007; Orlicky et al., 2003; Tang et al., 2007), Fbw7ARG mutations have a uniquely profound impact on substrate-binding affinity compared with other missense mutations. Moreover, Fbw7 dimers, which have increased substrate affinity, tolerate many missense mutations that disable monomers. Fbw7 dimerization thus greatly restricts the repertoire of deleterious Fbw7 missense mutations to only these most stringent positions (Welcker et al., 2013). There are many possible mutations that could produce either Fbw7 null alleles or truncated proteins, but they occur much less commonly than Fbw7ARG mutations. The strong biologic selection of Fbw7ARG mutations suggests that they are not simple loss-of-function alleles and is most consistent with dominant negative alleles, which has been confirmed in mouse models (see below).
TCGA studies have provided a wealth of mutational and expression data for different cancers (Cancer Genome Atlas, 2012; Cancer Genome Atlas Research et al., 2013; Kandoth et al., 2013). Although large-scale studies of T-ALL are still in progress, previous work indicates that T-ALL represents a special example of Fbw7-associated cancer with mutations in up to 30% of cases (Table 1) (Malyukova et al., 2007; Maser et al., 2007; O'Neil et al., 2007; Thompson et al., 2007). Fbw7 is significantly mutated (>10% of samples) in at least five tumor types: T-ALL, colorectal adenocarcinoma, uterine carcinosarcoma, uterine endometrial carcinoma, and bladder carcinoma. Other cancers with somewhat less frequent Fbw7 mutations include stomach adenocarcinoma, lung squamous cell carcinoma, cervical squamous cell carcinoma, and head and neck squamous cell carcinoma (Table 2). In contrast, Fbw7 mutations are not found in some cancers, such as acute myeloid leukemia (AML) and multiple myeloma. One possibility is that Fbw7 substrate stabilization is detrimental in these neoplasms. For example, the Fbw7 substrate C/EBPα suppresses AML (Bengoechea-Alonso and Ericsson, 2010b) and multiple myelomas require constitutive NF-κB signaling, therefore disruption of Fbw7-mediated Nκ-kB2 ubiquitylation in these tumors results in cell death (Busino et al., 2012). Despite extensive studies, the prognostic significance of Fbw7 mutations in cancer remains uncertain. For example, whereas TCGA Pan-Cancer analyses found Fbw7 mutations to be detrimental across multiple tissue types (Kandoth et al., 2013), the colorectal-specific TCGA study found no overlap between Fbw7 mutations and distant metastases, suggesting that mutations could be beneficial (Cancer Genome Atlas, 2012). Analogous studies of Fbw7 mutations in T-ALL have similarly yielded conflicting results.
Table 1. Fbw7 mutational frequency in T-ALL cell lines and patient samples.
| Study Reference | No. Samples | Source of sample | Fbw7 point mutations | Arginine hotspot mutations/total point mutations |
|---|---|---|---|---|
| O'Neil et al., 2007 | 20 | Cell lines | 35% | 86% |
| 81 | Primary samples | 9% | 100% | |
|
| ||||
| Maser et al., 2007 | 23 | Cell lines | 43% | 90% |
| 38 | Primary samples | 29% | 73% | |
|
| ||||
| Thompson et al., 2007 | 89 | Primary samples | 17% | 100% |
|
| ||||
| Malyukova et al., 2007 | 15 | Cell lines | 33% | 100% |
| 26 | Primary samples | 31% | 88% | |
TCGA studies for T-ALL have not been performed, therefore these data were collected from the four indicated studies.
Table 2. Fbw7 mutation and homozygous deletion frequency in selected cancers.
| Tumor Type | No. samples | Arginine hotspot mutations | Nonsense mutations | Other missense mutations | All point mutations | Arginine hotspot mutations/total point mutations | Homozygous deletions (%) |
|---|---|---|---|---|---|---|---|
| Uterine Carcinosarcoma | 56 | 23% | 4% | 14% | 39% | 59% | 0% |
| Colon and rectal adenocarcinoma | 212 | 8% | 3% | 6% | 17% | 49% | 0% |
| Uterine corpus endometrial carcinoma | 240 | 6% | 3% | 8% | 16% | 37% | 0% |
| Stomach adenocarcinoma | 219 | 5% | 2% | 2% | 9% | 59% | <1% |
| Urothelial bladder carcinoma | 127 | 3% | 5% | 4% | 9% | 33% | 2% |
| Lung squamous cell carcinoma | 178 | 2% | 2% | 2% | 6% | 36% | <1% |
| Head and neck squamous cell carcinoma | 302 | 2% | <1% | 3% | 5% | 33% | 1% |
| Cutaneous melanoma | 262 | <1% | 1% | 3% | 4% | 9% | <1% |
| Breast cancer | 962 | <1% | <1% | 1% | 2% | 20% | <1% |
| Cervical squamous adenocarcinoma | 36 | 0% | 3% | 3% | 6% | 0% | 0% |
| Lung adenocarcinoma | 230 | 0% | 1% | <1% | 2% | 0% | 0% |
| Glioblastoma multiforme | 281 | 0% | <1% | 0% | <1% | 0% | <1% |
Mutation and copy number alteration data were derived from analyses of TCGA data using cBioPortal (Cerami et al., 2012; Gao et al., 2013).
We performed a meta-analysis of TCGA data using the cBioPortal for Cancer Genomics online interface to compare and contrast Fbw7 mutations across cancer types (Table 2) (Cerami et al., 2012; Gao et al., 2013). These results are based upon data generated in whole by the TCGA Research Network (http://cancergenome.nih.gov/) and confirm the striking skewing towards Fbw7ARG mutations, particularly in organ sites where Fbw7 is most frequently mutated. Importantly, most tumors with Fbw7ARG mutations retain a normal second Fbw7 allele. This notably contrasts with the “two-hit” mutation pattern of some tumor suppressors. However, nonsense mutations (sometimes in combination with allelic loss) and homozygous null mutations are found in some tumors, thus Fbw7 exhibits classic tumor suppressor features in these cases. The frequency and types of Fbw7 mutations vary among organ sites. For example, colorectal cancers contain the entire spectrum of Fbw7 mutations (deletions and missense/nonsense mutations) while others, such as T-ALLs, exhibit nearly 100% heterozygous Fbw7ARG mutations (Tables 1 and 2). This suggests that distinct Fbw7 mutations produce unique biologic outcomes, presumably through differential effects on specific substrates (see below).
Instead of mutations in Fbw7 itself, some cancers contain substrate CPD mutations that prevent their ubiquitylation, including Myc CPD mutations in Burkitt's Lymphomas, Notch PEST mutations in T-ALL, and a KLF5 CPD mutation in colon cancer (Bahram et al., 2000; Bhatia et al., 1993; Bialkowska et al., 2014; Weng et al., 2004). This suggests particularly important roles for these substrates in specific tumors. However, the paucity of single substrate CPD mutations compared with Fbw7 mutations suggests that in most cases, Fbw7-associated tumorigenesis requires the concurrent deregulation of multiple oncoproteins.
Additional mechanisms of Fbw7 disruption in cancer
Mechanisms other than mutations and allelic loss also impair Fbw7 function in cancers. Fbw7 is targeted by oncogenic microRNAs such as miR-27, miR-92, and miR-223 in numerous cancers (Olive et al., 2013; Wang et al., 2014). Promoter hypermethylation downregulates Fbw7β expression in breast cancers and thymomas (Akhoondi et al., 2010; Gu et al., 2008b) and Fbw7 mRNA is also repressed in melanomas and gliomas, although the underlying mechanisms are not known (Cheng et al., 2013; Gu et al., 2008a). Fbw7β is a p53 target gene, therefore p53 mutations may reduce Fbw7 expression (Kimura et al., 2003). Two Fbw7 substrates feed back to control Fbw7 mRNA expression: C/EBPδ, which may contribute to mammary tumor metastasis (Balamurugan et al., 2013), and Hes5 (a Notch target gene) (Sancho et al., 2013). Many studies have examined low Fbw7 mRNA expression in tumors as a biomarker, which generally appears to be a high-risk feature.
Fbw7 protein stability is another regulatory mechanism that has been examined in tumors. One example is Fbw7β degradation by Parkin, which subsequently inhibits MCL-1 degradation (Ekholm-Reed et al., 2013). It has also been suggested that Pin1 overexpression in cancers destabilizes Fbw7 by directly generating Fbw7 monomers that are targeted for degradation through enhanced autoubiquitylation (Min et al., 2012). However, two other studies have disputed these findings (Kanatsu-Shinohara et al., 2014; Welcker et al., 2013).
Lessons learned from murine models of Fbw7-associated cancer models
Germline Fbw7 deletion in mice causes embryonic lethality (Tetzlaff et al., 2004; Tsunematsu et al., 2004). Therefore, conditional strains have been used to study Fbw7 in normal tissues and during tumorigenesis. Mice with knockin mutations that ablate the cyclin E degrons (hereafter termed cyclin EΔCPD) have also been used to specifically study impaired cyclin E degradation. These models have convincingly demonstrated that 1) FBXW7 is a bona fide tumor suppressor gene, 2) Fbw7ARG mutations have unique functional consequences, 3) specific Fbw7 substrates contribute to tumorigenesis, and 4) multiple oncogenic pathways cooperate with Fbw7 mutations.
Fbw7 regulates stem cells, differentiation, and genome stability
Studies of normal tissues have provided valuable insights into pathways that are also important for tumorigenesis. Fbw7 loss profoundly affects differentiation and proliferation in stem and progenitor cell types, often through similar mechanisms. For example, in neural stem cells and intestinal crypt progenitor cells, loss of Fbw7 leads to increased proliferation and differentiation defects through the combined actions of Jun and Notch, respectively (Grim et al., 2012; Hoeck et al., 2010; Sancho et al., 2010). Similarly, aberrant Notch activity causes differentiation defects in hepatocytes (Onoyama et al., 2011). Novel TF substrates may mediate differentiation phenotypes in other tissues. For example, Fbw7 loss reprograms pancreatic ductal cells towards endocrine lineages by stabilizing Ngn3 (Sancho et al., 2014).
Fbw7 also controls hematopoietic stem cell (HSC) quiescence and self-renewal (Matsuoka et al., 2008; Thompson et al., 2008). HSCs are quickly exhausted following Fbw7 deletion, largely because of the detrimental effects of Myc overexpression on proliferation and apoptosis, although impaired cyclin E degradation also causes defective HSC self-renewal after hematologic stress (Reavie et al., 2010; Siu et al., 2014). Importantly, Fbw7ARG/+ HSCs have intermediate Myc abundance compared with Fbw7-/- and Fbw7+/+ HSCs, and do not exhibit these phenotypes—suggesting a unique role for heterozygous Fbw7ARG alleles and dose-dependent Myc effects in preserving HSC function (King et al., 2013).
These phenotypes have also been studied in cyclin EΔCPD mice to isolate cyclin E from other substrates affected by Fbw7 mutations. Impaired cyclin E degradation causes epithelial cell hyperproliferation, abnormal cell cycle control, impaired erythroid differentiation, and genome instability (Loeb et al., 2005; Minella et al., 2008).
Colorectal cancer (CRC)
Although Fbw7 mutations are found in early stage human colon adenomas (Rajagopalan et al., 2004), its deletion from the mouse gut is not sufficient to cause neoplasia. However, loss of Fbw7 in mice collaborates with other mutations commonly found in human CRC, including APCMin alleles (which mimic the WNT pathway activation seen in nearly all human CRC) and p53 loss. When combined with APCMin, both Fbw7-/- and Fbw7ARG/+ mutations decreased tumor latency and increased tumor burden (Babaei-Jadidi et al., 2011; Davis et al., 2014; Sancho et al., 2010). However, these neoplasms did not progress beyond the adenoma stage, and were neither invasive nor metastatic. Fbw7ARG/+ mice exhibited increased tumorigenesis compared with Fbw7+/- animals, confirming that Fbw7ARG is not simply a null allele (Davis et al., 2014). TGIF1 and KLF5 were elevated in Fbw7ARG/+ but not Fbw7+/- tumors (Davis et al., 2014), thus these proteins may similarly contribute to Fbw7-associated human CRC and could represent examples of substrate-specific consequences of Fbw7ARG/+ mutations. In Fbw7-/-tumors, Jun and Notch abundance was increased, and concurrent Jun deletion decreased tumor size (Sancho et al., 2010).
In a second approach, p53 and Fbw7 were co-deleted from the mouse gut, which caused advanced adenocarcinomas that were highly invasive and metastatic (Grim et al., 2012). In accordance with the findings that p53 suppresses cyclin E-induced genomic instability caused by Fbw7 loss, these adenocarcinomas exhibited a chromosomal instability (CIN) phenotype, which is the most common form of genome instability in human CRC, but rare in mouse tumors.
Hematologic cancers
Unlike CRC, Fbw7 deletion in either T-cells or HSCs is sufficient to cause T-ALL and this is accelerated by concurrent p53 loss, PTEN loss, or Notch activation (King et al., 2013; Kwon et al., 2012; Matsuoka et al., 2008; Onoyama et al., 2007; Thompson et al., 2008). The specific roles of Fbw7ARG alleles in leukemogenesis and leukemia-initiating cells (LICs), rare cells with stem cell-like properties (e.g. self-renewal, quiescence) and important roles in disease propagation and resistance to therapy, have also been studied (King et al., 2013). Fbw7ARG/+ mice did not develop spontaneous T-ALL, demonstrating another example of differences between Fbw7 missense and homozygous null mutations. However, the Fbw7ARG/+ mutation strongly cooperated with Notch deregulation to drive T-ALL, and resulted in LIC expansion that is largely attributable to increased Myc abundance. Genetic or pharmacologic Myc inhibition depleted LICs in this model, demonstrating an essential role for Myc, although this occurs independently of Fbw7 mutational status.
These data suggest that Myc abundance in Fbw7ARG/+ cells is finely tuned such that it does not affect HSC maintenance while still being sufficiently elevated to achieve tumorigenic effects in LICs. This may, in part, explain the high prevalence of Fbw7ARG mutations in T-ALL. While Myc and Notch are clearly key players in Fbw7-associated T-ALL, cyclin EΔCPD mice also developed T-cell malignancies that exhibited CIN (Siu et al., 2014), which is not seen in Fbw7ARG/+ T-ALL. Different lesions in the Fbw7 pathway may thus lead to T-ALL through different mechanisms. In addition to Fbw7-associated malignancies, mouse models have also shown that Fbw7 depletion induced apoptosis of chronic myeloid leukemia (CML) LICs and B-cell lymphomas (Olive et al., 2013; Reavie et al.; Takeishi et al., 2013).
Consequences of Fbw7ARG mutations in cancers
Fbw7's mutational spectrum strongly suggests that Fbw7ARG alleles have dominant negative activity. The mouse models described above confirm this idea, and demonstrate that heterozygous Fbw7ARG mutations impact Fbw7 function intermediate to that caused by single allele loss and homozygous null mutations. The concept that this intermediate Fbw7 inactivation favors tumorigenesis has been termed the “just-enough” hypothesis (Davis and Tomlinson, 2012), and the stabilization of Myc in Fbw7ARG/+ HSCs to levels that favor leukemogenesis, but not apoptosis, likely represents this mechanism (King et al., 2013).
Two important aspects of Fbw7ARG mutations remain poorly understood: 1) how do they produce dominant negative effects, and 2) why are they so frequently selected for in tumors compared with nonsense and homozygous null mutations? To understand these issues, we believe it is crucial to consider Fbw7's mode of action as a dimer, which enhances substrate-binding affinity by engaging multiple CPDs simultaneously (Fig. 2A) (Welcker et al., 2013). Because Fbw7ARG can dimerize with wild-type Fbw7 and potentially produce impaired Fbw7WT-Fbw7ARG heterodimers (that contain only a single intact WD40 domain), we suggest that Fbw7ARG proteins dominantly inhibit wild-type Fbw7 (Fig. 2B).
Fbw7WT-Fbw7ARG heterodimers may also have special properties that produce substrate-specific effects (Welcker and Clurman, 2008). While the ubiquitylation of low-affinity substrates depends upon both substrate-binding domains of an Fbw7WT-Fbw7WT dimer, other substrates may have enough binding affinity to be ubiquitylated by Fbw7WT-Fbw7ARG heterodimers that contain only a single substrate-binding domain (Fig. 2B). Therefore, dimer-dependent substrate interactions might account for the selection of heterozygous Fbw7ARG mutations in cancers by allowing Fbw7WT-Fbw7ARG heterodimers to target substrates whose degradation is permissive or advantageous for tumorigenesis, while sparing the oncoproteins that drive tumor formation. Finally, although truncated Fbw7 proteins that can dimerize with Fbw7WT might similarly act as dominant negatives, these types of mutations are less common. We suggest that the selection for mutations that produce full-length Fbw7ARG protein reflects important, yet undefined, roles for continued interactions between substrates and a mutated, but full-length, Fbw7 protein (Fig. 2C).
Therapeutic strategies targeting the Fbw7 pathway in cancer
Given the high prevalence of Fbw7 mutations, the development of therapies targeting the Fbw7 pathway may have great potential. Figure 3 shows four possible therapeutic approaches; the first three specifically target tumors with Fbw7 mutations, whereas the fourth may represent a more general treatment strategy.
Figure 3. Potential therapeutic opportunities targeting Fbw7 pathway mutations in cancer.

(A) Tumors with heterozygous Fbw7ARG mutations could be treated with a small-molecule agonist (shown in purple) that restores the binding affinity between the mutated Fbw7ARG β–propeller and a phosphorylated substrate, leading to oncoprotein ubiquitylation and proteasomal destruction. (B) In cases where deregulation of particular Fbw7 substrates is necessary for tumor cell survival, inhibiting the functions of the relevant substrates and/or pathways, rather than targeting Fbw7 itself, may lead to anti-tumor activities. This approach would be applicable to tumors containing any type of Fbw7 mutation. (C) Synthetic lethality between decreased Fbw7 function and secondary pathways (“Pathway X”) could lead to selective killing of tumor cells, while sparing other cells that have normal Fbw7 function. Similar to (B), synthetic lethality could be applied to tumors containing any type of Fbw7 mutation. (D) Because leukemia-initiating cells are exquisitely sensitive to Myc abundance (Reavie et al., 2013; Takeishi et al., 2013), Fbw7 inhibition by small molecules (or other approaches) may sufficiently increase Myc abundance to cause LIC exhaustion and inhibit disease progression.
First, because Fbw7ARG proteins have decreased affinity for substrates, small molecule agonists might increase the Fbw7ARG-substrate binding affinity to levels that would restore substrate ubiquitylation (Fig. 3A). These agonists could act at the substrate-Fbw7 interface, similar to the plant hormone Auxin that functions as a “molecular glue“ between an F-box protein and its substrate (Tan et al., 2007). Alternatively, Fbw7ARG-substrate interactions might be bolstered by allosteric regulators, similar to a described inhibitor of the yeast Cdc4 protein (Orlicky et al., 2010).
Second, the downstream oncoproteins activated by Fbw7 mutations might be therapeutically targeted, rather than Fbw7 itself (Fig. 3B). Studies with a pharmacologic Myc inhibitor in mouse T-ALL suggest that this may be a viable strategy in some cases (King et al., 2013). A variation of this approach has been suggested for neuroblastoma, in which stable binding to AURKA prevents N-Myc degradation by Fbw7. Small molecules that dissociate N-Myc and AURKA allow the liberated N-Myc to be targeted for degradation by Fbw7, leading to anti-tumor effects (Brockmann et al., 2013).
Third, synthetic lethal strategies can be utilized to identify and pharmacologically target pathways that specifically inhibit cancers with Fbw7 pathway mutations, while sparing normal cells with intact Fbw7 function (Fig. 3C). This approach could be used against tumors with Fbw7 missense, nonsense, or null mutations, or in CPD-mutant tumors such as Burkitt's lymphoma.
Finally, studies in mice show that Fbw7 inhibition eliminates CML by increasing Myc abundance, thereby leading to LIC exhaustion (Reavie et al., 2013; Takeishi et al., 2013). This work raised the idea that Myc toxicity in cancer stem cells might provide the basis for using Fbw7 inhibition to eradicate LICs (Fig. 3D). However, inhibiting a potent tumor suppressor as a therapeutic strategy comes with obvious caveats, such as promoting cancers in other sites.
Conclusions
Our understanding of the Fbw7 pathway's role in tumorigenesis has increased enormously over the past 5 years; new substrates have deepened our understanding of the diverse oncogenic pathways that Fbw7 regulates and mouse models are providing insights into how Fbw7 mutations lead to cancer. The consequences of multiple oncoprotein deregulation by Fbw7 mutations are still incompletely understood, but some general themes are emerging. First, many Fbw7 substrates are broadly acting TFs that regulate processes fundamental to carcinogenesis, such as differentiation and proliferation. Therefore, Fbw7 mutant tumors exhibit alterations in many of these pathways. Second, Fbw7 has particularly important roles in normal and neoplastic stem cells, and this probably impacts both tumor development and therapeutic strategies. Third, genome instability is one mechanism that may drive tumor progression, and several Fbw7 substrates cause genomic instability when their activity is abnormally high. Fourth, the biologic selection for Fbw7ARG mutations likely reflects their unique functions and may provide important therapeutic opportunities. Finally, although identifying critical Fbw7 substrates is important for understanding tumorigenesis and discovering therapeutic targets, it should be stressed that Fbw7 mutations deregulate an entire network of oncoproteins, and tumorigenesis almost certainly results from their combined biologic output. The widely altered homeostasis caused by Fbw7 mutations may soon enable the development of novel therapies targeting the Fbw7 pathway in cancer.
Acknowledgments
The authors apologize to their many colleagues whose work could not be cited due to space constraints. This work was supported by NIH grants CA084069 and CA102742 (BEC) and by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-0718124 and DGE-1256082 (RJD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhoondi S, Lindstrom L, Widschwendter M, Corcoran M, Bergh J, Spruck C, Grander D, Sangfelt O. Inactivation of FBXW7/hCDC4-beta expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010;12:R105. doi: 10.1186/bcr2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer research. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- Babaei-Jadidi R, Li N, Saadeddin A, Spencer-Dene B, Jandke A, Muhammad B, Ibrahim EE, Muraleedharan R, Abuzinadah M, Davis H, et al. FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. The Journal of experimental medicine. 2011;208:295–312. doi: 10.1084/jem.20100830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–2110. [PubMed] [Google Scholar]
- Balamurugan K, Sharan S, Klarmann KD, Zhang Y, Coppola V, Summers GH, Roger T, Morrison DK, Keller JR, Sterneck E. FBXW7alpha attenuates inflammatory signalling by downregulating C/EBPdelta and its target gene Tlr4. Nature communications. 2013;4:1662. doi: 10.1038/ncomms2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. Tumor suppressor Fbxw7 regulates TGFbeta signaling by targeting TGIF1 for degradation. Oncogene. 2010a;29:5322–5328. doi: 10.1038/onc.2010.278. [DOI] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. The ubiquitin ligase Fbxw7 controls adipocyte differentiation by targeting C/EBPalpha for degradation. Proceedings of the National Academy of Sciences of the United States of America. 2010b;107:11817–11822. doi: 10.1073/pnas.0913367107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt's lymphoma and mouse plasmacytomas. Nature genetics. 1993;5:56–61. doi: 10.1038/ng0993-56. [DOI] [PubMed] [Google Scholar]
- Bialkowska AB, Liu Y, Nandan MO, Yang VW. A colon cancer-derived mutant of Kruppel-like factor 5 (KLF5) is resistant to degradation by glycogen synthase kinase 3beta (GSK3beta) and the E3 ubiquitin ligase F-box and WD repeat domain-containing 7alpha (FBW7alpha) The Journal of biological chemistry. 2014;289:5997–6005. doi: 10.1074/jbc.M113.508549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti P, Davoli T, Sironi C, Amati B, Pelicci PG, Colombo E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. The Journal of cell biology. 2008;182:19–26. doi: 10.1083/jcb.200711040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer cell. 2013;24:75–89. doi: 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O'Connor O, Hoffmann A, Elenitoba-Johnson KS, Pagano M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nature cell biology. 2012;14:375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Chen G, Martinka M, Ho V, Li G. Prognostic significance of Fbw7 in human melanoma and its role in cell migration. The Journal of investigative dermatology. 2013;133:1794–1802. doi: 10.1038/jid.2013.58. [DOI] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes & development. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–4873. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H, Lewis A, Behrens A, Tomlinson I. Investigation of the atypical FBXW7 mutation spectrum in human tumours by conditional expression of a heterozygous propellor tip missense allele in the mouse intestines. Gut. 2014;63:792–799. doi: 10.1136/gutjnl-2013-304719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H, Tomlinson I. CDC4/FBXW7 and the ‘just enough’ model of tumourigenesis. The Journal of pathology. 2012;227:131–135. doi: 10.1002/path.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Larimore EA, Fissel BM, Swanger J, Taatjes DJ, Clurman BE. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes & development. 2013;27:151–156. doi: 10.1101/gad.207720.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annual review of biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Ekholm-Reed S, Goldberg MS, Schlossmacher MG, Reed SI. Parkin-dependent degradation of the F-box protein Fbw7beta promotes neuronal survival in response to oxidative stress by stabilizing Mcl-1. Molecular and cellular biology. 2013;33:3627–3643. doi: 10.1128/MCB.00535-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. The Journal of cell biology. 2004;165:789–800. doi: 10.1083/jcb.200404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annual review of biochemistry. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Molecular cell. 2004;16:509–520. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. The Journal of biological chemistry. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW, Clurman BE. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. The Journal of cell biology. 2008;181:913–920. doi: 10.1083/jcb.200802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim JE, Knoblaugh SE, Guthrie KA, Hagar A, Swanger J, Hespelt J, Delrow JJ, Small T, Grady WM, Nakayama KI, et al. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Molecular and cellular biology. 2012;32:2160–2167. doi: 10.1128/MCB.00305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Inomata K, Mitsui H, Horii A. Promoter hypermethylation is not the major mechanism for inactivation of the FBXW7 beta-form in human gliomas. Genes Genet Syst. 2008a;83:347–352. doi: 10.1266/ggs.83.347. [DOI] [PubMed] [Google Scholar]
- Gu Z, Mitsui H, Inomata K, Honda M, Endo C, Sakurada A, Sato M, Okada Y, Kondo T, Horii A. The methylation status of FBXW7 beta-form correlates with histological subtype in human thymoma. Biochem Biophys Res Commun. 2008b;377:685–688. doi: 10.1016/j.bbrc.2008.10.047. [DOI] [PubMed] [Google Scholar]
- Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israel A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. The Journal of biological chemistry. 2001;276:34371–34378. doi: 10.1074/jbc.M101343200. [DOI] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Molecular cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annual review of biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, Behrens A. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nature neuroscience. 2010;13:1365–1372. doi: 10.1038/nn.2644. [DOI] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Onoyama I, Nakayama KI, Shinohara T. Skp1-Cullin-F-box (SCF)-type ubiquitin ligase FBXW7 negatively regulates spermatogonial stem cell self-renewal. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:8826–8831. doi: 10.1073/pnas.1401837111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck JM, Summers MK, Tedesco D, Ekholm-Reed S, Chuang LC, Jackson PK, Reed SI. Cyclin E overexpression impairs progression through mitosis by inhibiting APC(Cdh1) The Journal of cell biology. 2007;178:371–385. doi: 10.1083/jcb.200703202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Gotoh M, Nakamura Y, Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer science. 2003;94:431–436. doi: 10.1111/j.1349-7006.2003.tb01460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A, Shi J, Vakoc C, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552–1566. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- Kwon YW, Kim IJ, Wu D, Lu J, Stock WA, Jr, Liu Y, Huang Y, Kang HC, DelRosario R, Jen KY, et al. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Molecular cancer research : MCR. 2012;10:834–844. doi: 10.1158/1541-7786.MCR-12-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Diehl JA. SCFs in the new millennium. Oncogene. 2014;33:2011–2018. doi: 10.1038/onc.2013.144. [DOI] [PubMed] [Google Scholar]
- Liu N, Li H, Li S, Shen M, Xiao N, Chen Y, Wang Y, Wang W, Wang R, Wang Q, et al. The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. The Journal of biological chemistry. 2010;285:18858–18867. doi: 10.1074/jbc.M109.099440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb K, Kostner H, Firpo E, Norwood T, Tsuchiya K, Clurman BE, Roberts JM. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer cell. 2005;8:35–47. doi: 10.1016/j.ccr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Malyukova A, Dohda T, von der Lehr N, Akhoondi S, Corcoran M, Heyman M, Spruck C, Grander D, Lendahl U, Sangfelt O. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer research. 2007;67:5611–5616. doi: 10.1158/0008-5472.CAN-06-4381. [DOI] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O'Neil J, Gutierrez A, Ivanova E, Perna I, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Tateishi Y, Onoyama I, Okita Y, Nakayama K, Nakayama KI. Fbxw7beta resides in the endoplasmic reticulum membrane and protects cells from oxidative stress. Cancer science. 2011;102:749–755. doi: 10.1111/j.1349-7006.2011.01851.x. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes & development. 2008;22:986–991. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SH, Lau AW, Lee TH, Inuzuka H, Wei S, Huang P, Shaik S, Lee DY, Finn G, Balastik M, et al. Negative regulation of the stability and tumor suppressor function of Fbw7 by the Pin1 prolyl isomerase. Molecular cell. 2012;46:771–783. doi: 10.1016/j.molcel.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minella AC, Grim JE, Welcker M, Clurman BE. p53 and SCF(Fbw7) cooperatively restrain cyclin E-associated genome instability. Oncogene. 2007 doi: 10.1038/sj.onc.1210518. [DOI] [PubMed] [Google Scholar]
- Minella AC, Loeb KR, Knecht A, Welcker M, Varnum-Finney BJ, Bernstein ID, Roberts JM, Clurman BE. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes & development. 2008;22:1677–1689. doi: 10.1101/gad.1650208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol. 2002;12:1817–1827. doi: 10.1016/s0960-9822(02)01225-3. [DOI] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nature reviews Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. The Journal of experimental medicine. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. The Journal of biological chemistry. 2001;276:35847–35853. doi: 10.1074/jbc.M103992200. [DOI] [PubMed] [Google Scholar]
- Olive V, Sabio E, Bennett MJ, De Jong CS, Biton A, McGann JC, Greaney SK, Sodir NM, Zhou AY, Balakrishnan A, et al. A component of the mir-17-92 polycistronic oncomir promotes oncogene-dependent apoptosis. eLife. 2013;2:e00822. doi: 10.7554/eLife.00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson BL, Hock MB, Ekholm-Reed S, Wohlschlegel JA, Dev KK, Kralli A, Reed SI. SCFCdc4 acts antagonistically to the PGC-1{alpha} transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes & development. 2008;22:252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoyama I, Suzuki A, Matsumoto A, Tomita K, Katagiri H, Oike Y, Nakayama K, Nakayama KI. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. The Journal of clinical investigation. 2011;121:342–354. doi: 10.1172/JCI40725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, Nakayama KI. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. The Journal of experimental medicine. 2007;204:2875–2888. doi: 10.1084/jem.20062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Neduva V, Elowe N, Brown ED, Sicheri F, Tyers M. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nature biotechnology. 2010;28:733–737. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, Abdel-Wahab O, Ibrahim S, Nakayama KI, Aifantis I. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer cell. 2013;23:362–375. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reavie L, Della Gatta G, Crusio K, Aranda-Orgilles B, Buckley SM, Thompson B, Lee E, Gao J, Bredemeyer AL, Helmink BA, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol. 2010;11:207–215. doi: 10.1038/ni.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho R, Blake SM, Tendeng C, Clurman BE, Lewis J, Behrens A. Fbw7 repression by hes5 creates a feedback loop that modulates Notch-mediated intestinal and neural stem cell fate decisions. PLoS biology. 2013;11:e1001586. doi: 10.1371/journal.pbio.1001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho R, Gruber R, Gu G, Behrens A. Loss of Fbw7 Reprograms Adult Pancreatic Ductal Cells into alpha, delta, and beta Cells. Cell stem cell. 2014;15:139–153. doi: 10.1016/j.stem.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho R, Jandke A, Davis H, Diefenbacher ME, Tomlinson I, Behrens A. F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology. 2010;139:929–941. doi: 10.1053/j.gastro.2010.05.078. [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & development. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu KT, Xu Y, Swartz KL, Bhattacharyya M, Gurbuxani S, Hua Y, Minella AC. Chromosome instability underlies hematopoietic stem cell dysfunction and lymphoid neoplasia associated with impaired fbw7-mediated cyclin e regulation. Molecular and cellular biology. 2014;34:3244–3258. doi: 10.1128/MCB.01528-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nature reviews Molecular cell biology. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI. hCDC4 gene mutations in endometrial cancer. Cancer research. 2002;62:4535–4539. [PubMed] [Google Scholar]
- Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer cell. 2013;23:347–361. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- Tan X, Calderon-Villalobos LI, Sharon M, Zheng C, Robinson CV, Estelle M, Zheng N. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature. 2007;446:640–645. doi: 10.1038/nature05731. [DOI] [PubMed] [Google Scholar]
- Tang X, Orlicky S, Lin Z, Willems A, Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F, Tyers M. Suprafacial Orientation of the SCF(Cdc4) Dimer Accommodates Multiple Geometries for Substrate Ubiquitination. Cell. 2007;129:1165–1176. doi: 10.1016/j.cell.2007.04.042. [DOI] [PubMed] [Google Scholar]
- Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3338–3345. doi: 10.1073/pnas.0307875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. The Journal of experimental medicine. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. The Journal of experimental medicine. 2008;205:1395–1408. doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. The Journal of biological chemistry. 2004;279:9417–9423. doi: 10.1074/jbc.M312337200. [DOI] [PubMed] [Google Scholar]
- van Drogen F, Sangfelt O, Malyukova A, Matskova L, Yeh E, Means AR, Reed SI. Ubiquitylation of cyclin E requires the sequential function of SCF complexes containing distinct hCdc4 isoforms. Molecular cell. 2006;23:37–48. doi: 10.1016/j.molcel.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Wang L, Ye X, Liu Y, Wei W, Wang Z. Aberrant regulation of FBW7 in cancer. Oncotarget. 2014;5:2000–2015. doi: 10.18632/oncotarget.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, Wei W. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS letters. 2012;586:1409–1418. doi: 10.1016/j.febslet.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. Fbw7/hCDC4 dimerization regulates its substrate interactions. Cell Div. 2007;2:7. doi: 10.1186/1747-1028-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nature reviews Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- Welcker M, Larimore EA, Swanger J, Bengoechea-Alonso MT, Grim JE, Ericsson J, Zheng N, Clurman BE. Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes & development. 2013;27:2531–2536. doi: 10.1101/gad.229195.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004a;14:1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004b;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Molecular cell. 2003;12:381–392. doi: 10.1016/s1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. The EMBO journal. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies RJ, Kitajewski J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Molecular and cellular biology. 2001;21:7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Koepp DM. Fbw7 isoform interaction contributes to cyclin E proteolysis. Molecular cancer research : MCR. 2006;4:935–943. doi: 10.1158/1541-7786.MCR-06-0253. [DOI] [PubMed] [Google Scholar]
- Zhao D, Zheng HQ, Zhou Z, Chen C. The Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated degradation and suppresses breast cell proliferation. Cancer research. 2010;70:4728–4738. doi: 10.1158/0008-5472.CAN-10-0040. [DOI] [PubMed] [Google Scholar]