

Abstract
Inflammation causes hyperalgesia, an enhanced sensitivity to noxious stimuli. Transient receptor potential vanilloid 1 (TRPV1), a thermo-TRP ion channel activated by painful levels of heat, is an important contributor because hyperalgesia is reduced when TRPV1 is either genetically deleted or pharmacologically blocked. Inflammatory mediators such as prostaglandin-E2 or bradykinin cause hyperalgesia by activating cellular kinases that phosphorylate TRPV1, a process that has recently been shown to rely on a scaffolding protein, AKAP79, to target the kinases to TRPV1. Here we use Förster resonance energy transfer, immunoprecipitation, and TRPV1 membrane trafficking experiments to identify a key region on AKAP79, between amino acids 326–336, which is responsible for its interaction with TRPV1. A peptide identical to this domain inhibited sensitization of TRPV1 in vitro, and when covalently linked to a TAT peptide to promote uptake across the cell membrane the peptide inhibited in vivo inflammatory hyperalgesia in mice. Critically, it did so without affecting pain thresholds in the absence of inflammation. These results suggest that antagonizing the TRPV1–AKAP79 interaction will be a useful strategy for inhibiting inflammatory hyperalgesia.
Introduction
The transient receptor potential vanilloid 1 ion channel (TRPV1), a member of the thermo-TRP channel family, is an important mechanism by which pain-sensitive neurons, or nociceptors, detect heat (Cesare and McNaughton, 1996; Caterina et al., 1997). TRPV1 is also activated by other stimuli such as capsaicin, anandamide, and protons, and consequently acts as a multimodal detector of potentially injurious events (Caterina and Julius, 2001; Vellani et al., 2001). The response of TRPV1 ion channels to heat in vitro is enhanced (sensitized) by inflammatory mediators, including bradykinin, prostaglandin-E2 and nerve growth factor, which are released following tissue damage (Huang et al., 2006a). Consistent with this, inflammatory heat hyperalgesia in vivo is significantly reduced when TRPV1 is either genetically deleted or blocked by specific antagonists (Caterina et al., 2000; Davis et al., 2000; Pomonis et al., 2003; Gavva et al., 2007, 2008; Steiner et al., 2007). A more surprising result, because TRPV1 is not thought to be directly sensitive to mechanical stimuli, is that the mechanical hyperalgesia caused by inflammation is also alleviated by blocking TRPV1 (Pomonis et al., 2003; Walker et al., 2003). TRPV1 is therefore an attractive pharmacological target, but TRPV1 antagonists developed to date as potential analgesics have two critical drawbacks: the threshold for detection of harmful heat is elevated, which could lead to accidental burns, and the core body temperature is increased (Vay et al., 2012).
Many inflammatory mediators activate the downstream kinases protein kinase A (PKA) and protein kinase C (PKC), which in turn phosphorylate TRPV1 and thus sensitize the channel to stimuli (Huang et al., 2006b). A scaffolding protein, A-Kinase Anchoring Protein 79 (AKAP79), has binding sites for these kinases and also binds to TRPV1, thus forming a signaling complex that promotes rapid and specific phosphorylation of critical sites on TRPV1 (Zhang et al., 2008). Genetic deletion or knockdown of AKAP150, the murine homolog of AKAP79, reduces PKC-mediated hyperalgesia in mice (Jeske et al., 2009). More recently, a peptide mimicking the TRPV1 binding site for AKAP79 has been shown to block TRPV1 sensitization in vitro and inflammatory pain in vivo (Fischer et al., 2013). The interaction domain between TRPV1 and AKAP79 is therefore a potential target for the development of novel analgesics that may lack the side effects observed with direct TRPV1 block.
Several ion channels, including NMDA glutamate receptors and L-type Ca2+ channels, are known to bind AKAP79, some directly and others via intermediate proteins such as MAGUKs (Sanderson and Dell'Acqua, 2011). Is the TRPV1 binding site on AKAP79 shared with other important effectors, or is it unique and therefore may offer possibilities for selective intervention? To answer this question we set out to localize the binding site of TRPV1 on AKAP79. We used Förster resonance energy transfer (FRET), coimmunoprecipitation, and TRPV1 trafficking to the membrane as indices of TRPV1–AKAP79 interaction, and we show that TRPV1 binds at a unique site within amino acids 326–336 on AKAP79. A peptide with sequence identical to the AKAP79 binding site blocks sensitization of TRPV1 in vitro, and, moreover, when the peptide is made cell permeable by addition of a TAT peptide, the peptide has a potent analgesic action in several mouse models of inflammatory pain in vivo.
Materials and Methods
Cloning and construction of cDNA plasmids.
Human AKAP79 cDNA was obtained from John Scott, University of Washington. AKAP79-YFP containing AKAP79 in a pEYFP-N1 vector was obtained from Mark Dell'Acqua, University of Colorado (Dell'Acqua et al., 1998, 2002). Human TRPV1 was subcloned into the pcDNA3-V5-His-TOPO vector and into the pEYFP-N1 vector as described previously (Zhang et al., 2008). AKAP79 truncations and the Δ326–336 deletion were generated without a tag, with a V5–6His-tag or with a fluorophore (CyPet or YFP) fused to the C terminus. CyPet was PCR amplified from pcEP4-CyPet and inserted via restriction sites BamHI and NotI instead of YFP in the AKAP79-YFP and TRPV1-YFP plasmids. Wild-type (WT) AKAP79 and truncations 1–235, 1–315, 1–325, 1–336, 1–360, and 1–384 were developed by PCR amplification and inserted into vectors: pEYFP-N1 with C terminus YFP or CyPet using 5′ HindIII and 3′ BamHI and pcDNA-V5-His-TOPO with C terminus V5-His using 5′ HindIII and 3′ NotI sites, respectively. AKAP79 truncation 32–427 and AKAP79 Δ326–336 were developed by site-directed mutagenesis using the QuikChange II XL Kit (Stratagene) following the manufacturer's protocol. Primers for the mutagenesis were developed as described previously (Liu and Naismith, 2008). All constructs were verified by DNA sequencing.
Cell culture and transfection.
HEK293 and COS7 cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, 60 μg/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml glutamine (Invitrogen) at 37°C and 5% CO2. Cells were transfected with Metafectene (Biontex) in an antibiotic and serum-free medium for at least 6 h. For immunocytochemistry, HEK293 cells were reseeded 6–8 h after transfection onto poly-lysine (1 mg/ml)-coated glass coverslips. COS7 cells were plated and transfected (1:5 ratio DNA:Metafectene) on glass-bottomed dishes and FRET was measured 24 h after transfection. Cotransfections of cDNA were performed in a 1:1 mass ratio.
Dorsal root ganglion (DRG) neurons were harvested from 6- to 12-week-old C57B1/6 mice of either sex. Ganglia were collected in DMEM without supplements, then transferred to medium containing collagenase type IV 0.25% (Invitrogen) for 1 h at 37°C. DRG neurons were dissociated using pipette tips with decreasing diameter and then plated on dishes coated with poly-l-lysine and laminin (1 mg/ml; Sigma-Aldrich) immersed in DMEM supplemented as above and containing mouse nerve growth factor (NGF) 2.5S (100 ng/ml; Alomone Labs). NGF increases TRPV1 expression at the plasma membrane but does so by a mechanism that is independent of AKAP79 (Zhang et al., 2005, 2008). Patch-clamp and calcium imaging recordings were performed within 2 d of dissection.
FRET acceptor photobleach.
COS7 cells were transfected with YFP or CyPet fusion proteins of AKAP79 and TRPV1. We used CyPet in preference to CFP because of the improved stability of the fluorophore (Nguyen and Daugherty, 2005). CyPet is reported to have a higher FRET efficiency when paired with the YFP-derived YPet, which renders it more sensitive to observe dynamic changes of the protein–protein interactions (Nguyen and Daugherty, 2005). However, in pilot experiments using YPet as acceptor, photobleaching required ∼1 min (see Fig. 2A), and movement of live cells within this time compromised analysis of the cell membrane. We observed that YFP bleaches more rapidly than YPet (Fig. 2A) and therefore used YFP as acceptor.
Figure 2.
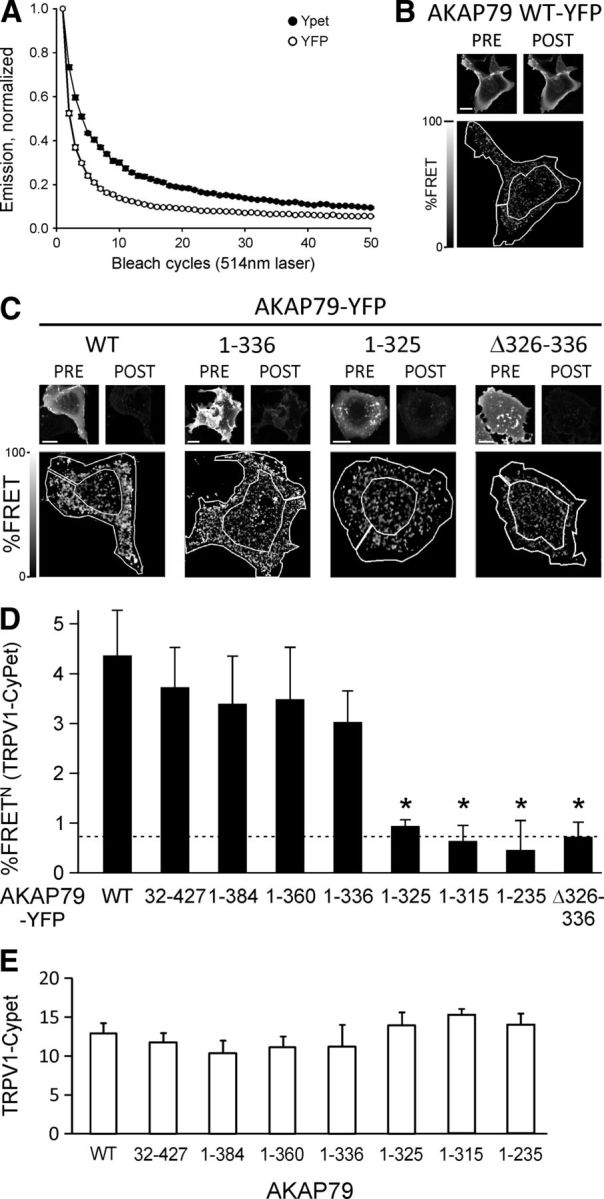
FRET between AKAP79-YFP and TRPV1-CyPet is disrupted when amino acids 326–336 of AKAP79 are removed. A, Fluorophore bleaching in HEK293 cells transfected with AKAP79-YFP or AKAP79-YPet. Live cells were bleached with an Argon Laser (514 nm, 40%) for 50 cycles within 61 s. Emission collected in the range 518–580 nm and normalized to the initial fluorescence intensity. YFP bleached faster than YPet (1/e in 3.2 ± 0.1 vs 6.9 ± 0.1 cycles, p < 0.001, n = 14 each, t test, independent samples). B, Control experiment following FRET acceptor photobleach protocol but with 0% laser power, i.e., no bleach. Representative images of AKAP79-YFP in live COS7 cell at pre-bleach and post-bleach time points. Bottom image shows a pixel-by-pixel calculation of the FRET signal of the donor TRPV1-CyPet with superimposed ROI used for analysis of %FRETN (0.2 ± 0.1%FRETN, n = 6). Scale bar, 10 μm. C, Top: Representative images showing pre-bleach and post-bleach fluorescence of YFP-tagged AKAP mutants in live COS7 cells. Scale bars: 10 μm. Bottom: Image of FRET N signal of the donor TRPV1-CyPet expressed as percentage increase in post-bleach signal relative to pre-bleach with representative ROI superimposed to show area where %FRET was analyzed. To enhance AKAP79-mediated trafficking of TRPV1 to the membrane and increase the FRET signal, cells were exposed to the PKC activator PMA (1 μm, applied for 10 min immediately before pre-bleach image). D, Average %FRETN signal of TRPV1 determined in outer region of cell including membrane (see Materials and Methods). AKAP mutants show a loss of interaction when amino acids 326–336 are missing. Dotted line shows control FRET signal between AKAP-CyPet and AKAP-YFP. *p < 0.05. E, TRPV1-CyPet intensity levels in the ROIs selected for %FRETN calculation are similar for all AKAP79 mutants.
Fluorescence of tagged proteins in live cells was detected using a Leica TCS-SP5 confocal microscope with a plan-apochromatic oil-immersion 1.4 NA 63× objective lens mounted on a DMI6000 inverted stand using the LAS-AF software, FRET acceptor photobleach module (Leica Microsystems). Acceptor photobleach was used to quantitatively measure FRET efficiency in the plasma membrane. HEPES-buffered extracellular solution, containing 1.8 mm CaCl2, 140 mm NaCl, 4 mm KCl, 1 mm MgCl2, 10 mm HEPES, and 5 mm d-glucose, pH 7.4, was used to replace the growth medium for imaging 24 h after transfection. Phorbol-12-myristate-13-acetate (PMA, 1 μm) was added for 10 min at 37°C to increase TRPV1 membrane expression. The donor, CyPet, was excited with a 405 nm diode laser and the emission was collected at 448–500 nm. The acceptor, YFP, was excited and bleached with an Argon laser at a wavelength of 514 nm; emission was collected at 518–580 nm. Fluorophores were imaged sequentially to avoid cross talk from spectral overlap. The acceptor was bleached using 70% Argon laser power for 20 s, which gave at least 80% bleach (Fig. 2A). COS7 cells present a very flat morphology when adhered (Fig. 2C; Hammond et al., 2009) and do not present a clearly defined membrane, unlike HEK cells (see Fig. 4), so a membrane region of interest was drawn by hand including the membrane and adjacent cytoplasm but excluding the central region of the cell (see Figs. 1, 2). The %FRET at the plasma membrane was calculated as described using FRETcalc for ImageJ (Stepensky, 2007). The donor image was thresholded to reduce noise in the ratio image by removing low-intensity pixels from the analysis (50% of pixels with lowest intensity removed). The mean acceptor background level was found to be 1.75 ± 0.25 (8-bit scale) so acceptor pixels with an intensity of <2 were excluded from the analysis of the donor image. The %FRET calculated for each cell was transformed to %FRETN by normalizing it to the product of the CyPet and YFP fluorescence intensities before bleaching (Sorkin et al., 2000; Willoughby et al., 2010).
Figure 4.
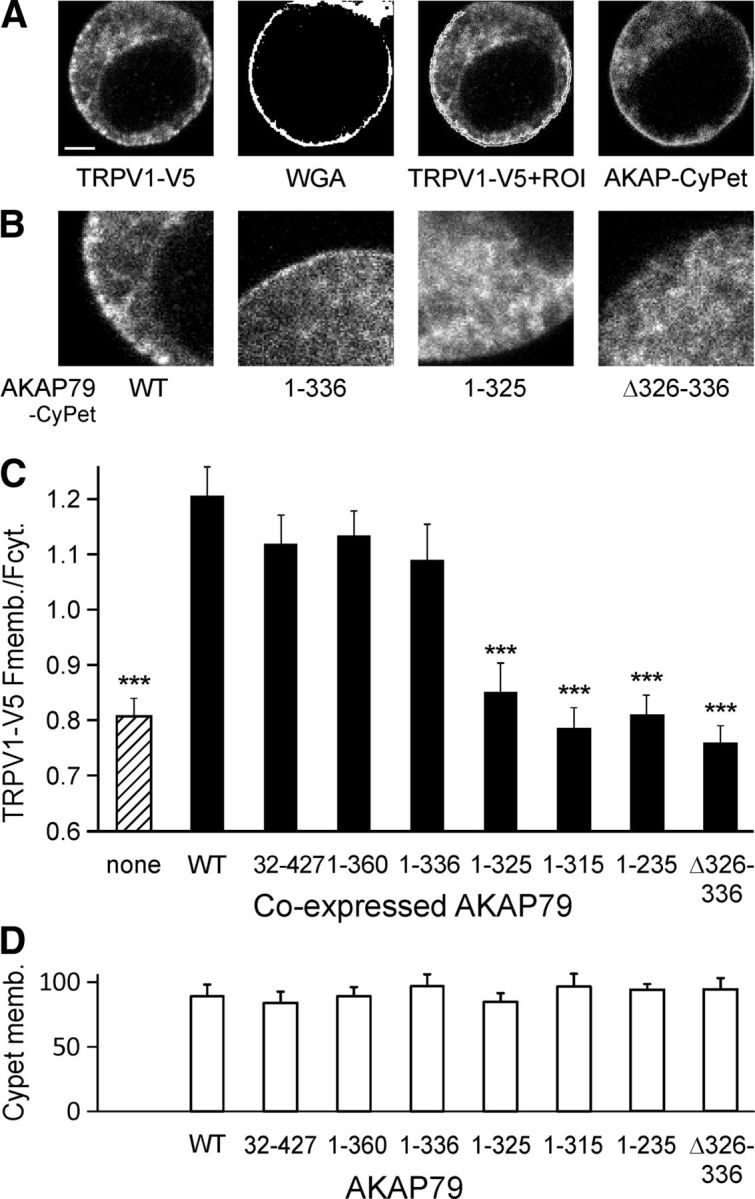
Trafficking of TRPV1 to surface membrane depends on binding to AKAP79. A, Example confocal images of single fixed HEK293 cell transfected with TRPV1–V5 and AKAP79-CyPet. TRPV1 is visualized with an anti-V5 antibody (left) and plasma membrane location determined with a brief exposure to WGA before fixation (WGA, middle left), from which a mask delineating the membrane is constructed (ROI, shown superimposed on TRPV1 image, middle right). AKAP79 coupled to CyPet is visualized in same cell (right). Scale bar, 5 μm. B, Representative fourfold enlargement of membrane segments showing TRPV1 distribution in the presence of WT AKAP79 and AKAP79 mutants as noted. C, TRPV1–V5 fluorescence signal at the plasma membrane relative to cytosol when coexpressed with AKAP79-CyPet mutants. Membrane expression of TRPV1 is reduced relative to WT AKAP79 when coexpressed with AKAP79 mutants lacking amino acids 326–336 (***p < 0.001) and is similar to cells without AKAP79 transfection. D, Intracellular distribution of AKAP79 is not affected in the mutations.
Figure 1.

AKAP79 truncation and deletion constructs. A, Bars represent length of the constructs. The three membrane-targeting regions (31–52, 76–101, and 116–145) are contained in all constructs. B, Images of COS7 cells expressing AKAP79-YFP (green) and stained with membrane labeling WGA-Alexa 594 (red). Expression of AKAP79 WT and mutants fused to YFP colocalize almost entirely with plasma membrane WGA staining. The plasma membrane staining of COS7 cells extends to the cortical region of the cells due to their flat morphology. Scale bars: 10 μm.
Coimmunoprecipitation.
AKAP79-V5-His WT and truncations were expressed in HEK293 cells for 24–48 h. To harvest the proteins an ice-cold lysis solution was used, containing isotonic Tris-NaCl-Buffer (20 mm Tris and 150 mm NaCl), 1% NP-40, and 10% proteinase inhibitor mixture (Roche). After cell lysis, proteins were purified with nickel beads (Sigma) targeting the 6×His tag. HEK293 whole-cell extracts expressing TRPV1 were added. The lysis solution for TRPV1-expressing cells contained in addition 1 mm EDTA, which was excluded from the AKAP79-V5-His lysis solution due to interference with the Ni2+-resin binding. AKAP79-V5-His was precipitated with anti-V5 antibody (Invitrogen) and pulled down by Protein A agarose beads overnight at 4°C. Coprecipitation was detected by blotting with anti-TRPV1 antibody (Santa Cruz Biotechnology) and AKAP79 precipitation was verified using mouse anti-V5 (Invitrogen). Precipitation was normalized to the respective AKAP79-V5 intensity. For peptide inhibition experiments the test peptide was incubated with the cell extract of the protein being precipitated for 2 h at 4°C before adding the second protein extract, antibody, and beads. When TRPV1-V5 was precipitated, AKAP79 without a tag was used and was detected with an anti-AKAP79 antibody (Santa Cruz Biotechnology). Analysis was performed using ImageJ with the MBF plug-in collection (http://rsb.info.nih.gov/ij/).
Immunocytochemistry.
HEK293 cells were transfected with TRPV1-V5 and WT or truncation/deletion mutants of AKAP79-CyPet. TRPV1 transfection typically caused a more rounded morphology than is normal in HEK293 cells, particularly in highly expressing cells (Fig. 4), but TRPV1-expressing cells continued to be viable in culture. Two days after transfection the membrane was labeled with wheat germ agglutinin (WGA)-Alexa 594 (5 μg/ml; Invitrogen) for 10 min on ice, followed immediately by fixation with 4% paraformaldehyde and 0.2% glutaraldehyde for 20 min at room temperature, permeabilization with 0.2% Saponin, and blocking with 0.1% fish gelatin (Sigma-Aldrich) for 1 h. Anti-V5 mouse primary antibody (1:500) and goat anti-mouse Alexa 488 (1:1000; Molecular Probes) were used to detect TRPV1. Fluorescent antibodies and fused proteins, WGA and AKAP79-CyPet, were visualized using a confocal microscope as above. AKAP79-CyPet was excited with a 405 nm diode laser and emission detected between 448 and 500 nm; Alexa 488 (TRPV1) was excited using a 496 nm laser line and detected in the range of 516–580 nm; WGA-Alexa 594 was excited at 546 nm and detected in the range of 600–680 nm. Analysis was performed using ImageJ as above. To evaluate intracellular distribution, regions of interest (ROIs) of the plasma membrane and the cytoplasm were generated. The plasma membrane was determined by thresholding the WGA image. A dilation and subsequent erosion operation in ImageJ provides continuous ROIs corresponding to the plasma membrane (Fig. 4A). The cytoplasmic ROI was chosen excluding the nucleus, which had lower fluorescence intensity.
Whole-cell patch clamp.
C57BL/6 mouse DRG neurons were harvested and cultured overnight as above. Whole-cell voltage clamp was performed on small-medium diameter DRG neurons as described previously (Fischer et al., 2013). Briefly, HEPES-buffered saline solution was used as extracellular solution, but with 110 mm Na+ replaced with NMDG+ to reduce TRPV1 current amplitudes to improve the fidelity of patch-clamp recording. Membrane currents at −60 mV were low-pass filtered at 1 kHz and sampled at 2 kHz with an Axopatch 200B amplifier and pCLAMP 10 (Molecular Devices). Glass recording electrodes with a final resistance of 2.5–5.0 MΩ were filled with intracellular solution with or without AKAP79 (326–336) or AKAP79 Scramble (200 μm) peptides. After achieving the whole-cell configuration neurons were held at –60 mV for 13 min before PMA application to allow for diffusion of the peptide into the cell. Capsaicin (100 nm, applied every 45 s for a period of 5 s) and PMA (1 μm, applied for 30 s) were applied by a computer-controlled gravity-driven superfusion system.
Calcium imaging.
Calcium imaging was performed as described in Fischer et al. (2013). Briefly, DRG neurons or HEK293 cells transfected with TRPV1-V5 were seeded onto glass coverslips and after 24–48 h were loaded with fura-2 AM dye (3 μm, 30 min) in the presence of pluronic acid F-127 0.02% (Invitrogen) at 37°C and 5% CO2. Before imaging cells were exposed to HEPES-buffered extracellular solution containing 5 μm of the TAT-conjugated peptides (see below) for 20 min at room temperature. Coverslips were mounted on a Nikon Eclipse Ti-E inverted microscope with a 10× Plan Fluor objective and imaged with continuous superfusion of a gravity-driven extracellular solution or test solutions through a common outlet. Capsaicin responses (200 nm for HEK293 cells and 100 nm for DRGs) were quantified as the difference (Δ[Ca]i) between baseline, calculated over 10 s immediately before application of capsaicin, and peak calcium following capsaicin application. Capsaicin was applied six times for 1 s at intervals of 5 min. PMA (1 μm) or forskolin (10 μm) was applied for 180 s after the fourth capsaicin application. Ionomycin (5 μm) was applied to saturate calcium influx at the end of each protocol.
Behavioral experiments.
Male C57BL/6 mice 6–10 weeks of age were used. Mice were kept on a 12 h light/dark cycle with food and water ad libitum, in a temperature-controlled environment (21°C ± 1°C). Experimenters were blinded to the injection and only unblinded at the end of the experiment. All injections were performed using a 50 μl Hamilton syringe and a 31G needle. All licensed procedures were approved by the UK Home Office, adhering to the legislation of the Animals (Scientific Procedures) Act 1986.
Formalin and PMA models were performed as described in Fischer et al. (2013). In brief, mice were injected with peptide (10 μm in saline, 20 μl) in the left plantar hindpaw, and after 30 min a formaldehyde solution (4% formalin, 20 μl) was injected into the same site. The total time spent lifting, licking, and biting of the injected paw was recorded in 5 min intervals from 5 min before 60 min after injection. For PMA-induced nociception mice were habituated for 2 h in the test chambers on the day of the experiment. PMA (50 pmol/paw) and peptide (final concentration 10 μm) were mixed and injected into the left plantar hindpaw (20 μl). The total duration of licking and biting of the injected paw was measured in 10 min bins for 1 h after injection (Ferreira et al., 2005).
The thermal threshold for paw withdrawal was assessed using a plantar aesthesiometer (Ugo Basile). Withdrawal latency was measured using a fixed infrared stimulus (50 a.u. of infrared intensity) with a cutoff latency of 15 s to prevent tissue damage. Mice were habituated in the testing chambers for 1 h on the day of the experiment. To assess inflammatory heat hyperalgesia, λ-carrageenan (2% in saline, 20 μl) or saline (20 μl) was injected into the left hindpaw of each animal 30 min after the test compound was injected at the same site (10 μm in saline, 20 μl). The paw withdrawal latency was assessed at 30, 60, 90, 120, 150, and 180 min after λ-carrageenan injection.
The mechanical threshold for paw withdrawal was assessed using a dynamic plantar aesthesiometer (Ugo Basile). A linear increase in force to 5 g was applied over 10 s, after which the force remained constant at 5 g for 30 s. Mice were habituated in the testing chambers for at least 2 h for each of 2 d before and on the day of the experiment. To assess inflammatory hyperalgesia, λ-carrageenan and test compounds were injected as above. Paw withdrawal force threshold (g) was assessed at 30, 60, 90, and 120 min after λ-carrageenan injection.
Chemicals.
Peptides of at least 80% purity were obtained from Thermo Fischer Scientific or Biomatik. Confirmation of purity and composition was obtained via HPLC and mass spectrometry. The AKAP79 (326–336) peptide sequence was KSKSEESKRME, which was scrambled to KEMRESKESKS. The TAT peptide (HIV transactivator of transcription) was added to the N terminus to achieve cell permeability, generating TAT-326–336 (YGRKKRRQRRRKSKSEESKRME). The scrambled version of the peptide was TAT-Scramble (YGRKKRRQRRRKEMRESKESKS). Capsaicin, formalin (37%, saturated formaldehyde solution), forskolin, PMA, and λ-carrageenan were obtained from Sigma-Aldrich.
Statistics and data analysis.
Two data groups containing at least 10 samples were compared with a paired or unpaired t test as appropriate. Three or more groups were compared by ANOVA, and a significant effect was further analyzed. Mutations were compared with the WT or controls with Dunnett's post hoc test; all other groups were compared with a Tukey post hoc test. Analysis was performed using Statistica 8 (Statsoft). Data are presented as mean ± SEM. Significance levels are as follows: n.s., nonsignificant; *,#p < 0.05, **,##p < 0.01, and ***,###p < 0.001.
Results
Localization of TRPV1 binding site on AKAP79
To locate the binding site of TRPV1 on AKAP79 we generated sequential truncations 1–384, 1–360, 1–336, 1–325, 1–315, and 1–235 of the C terminus of AKAP79, together with an N-terminal truncation 32–427 (Fig. 1A). The three membrane targeting domains (31–52, 76–101, and 116–145) were conserved to maintain the membrane localization of truncated proteins within the cell (Dell'Acqua et al., 1998), and the PKC binding site was preserved in all mutants (see Figs. 1, 8).
Figure 8.

Illustration of the binding of AKAP79 to TRPV1 and sequence homology of AKAP79 binding site. A, AKAP79 contains three membrane targeting domains: (31–52), (76–101), and (116–145). Approximately 85% of the AKAP79 protein is suspected to remain in a native unfolded and unordered “necklace with beads” conformation (Wang et al., 2006). PKC binds to residues 31–52 (Klauck et al., 1996). Adenylyl cyclase isoforms (purple diamond) bind via the membrane targeting subunit (residues 76–101) (Efendiev et al., 2010; Willoughby et al., 2010). PP2B is suggested to bind to residues 337–343 through a PxIxIT binding domain (Dell'Acqua et al., 2002; Gold et al., 2011). The RII subunit of PKA binds to residues 391–409, which form an amphipathic helix binding motif (Carr et al., 1992). Calmodulin and caldendrin bind within residues 61–101 (data not shown; Gorny et al., 2012). Cav1.2 binds through a leucine zipper within residues 408–427 (Hulme et al., 2003). MAGUKs PSD-95 and SAP-97 bind in the range of amino acids 153–315 (Colledge et al., 2000; Bhattacharyya et al., 2009). In the present study we show that TRPV1 interacts with AKAP79 via amino acids 326–336 (orange, shown interacting with TRPV1 C-terminal residues 736–745). The ankyrin repeats at the N terminus of TRPV1 are represented in brown and the reported PKC (T145, S502, T705, and S801) and PKA (S117, T145, T371, and S502) phosphorylation sites on hTRPV1 are noted (Bhave et al., 2002, 2003; Rathee et al., 2002; Mohapatra and Nau, 2005; Mohapatra et al., 2003; Zhang et al., 2008; Lee et al., 2012). B, Alignment of the human AKAP79 326–336 protein region (basic local alignment search tool, protein BLAST, NCBI) to murine homolog AKAP150 (644–654) and bovine homolog AKAP75 (327–337). No other human protein contains more than four sequential amino acids aligning to this region of AKAP79. Aside from TRPV1 no other binding partner is known to interact with this region on AKAP79.
We assessed the interaction between TRPV1 and the AKAP79 deletion mutants using three independent methods. First, CyPet and YFP were fused to the C termini of TRPV1 and AKAP79, respectively (see Materials and Methods), and the FRET between them was measured by acceptor (YFP) photobleach in live COS7 cells. Compared with WT no significant change in %FRETN at the plasma membrane was observed until amino acids 326–336 were removed, when FRET dropped abruptly to the control value (Fig. 2D, dotted line), which was observed for a negative control of FRET between AKAP79-CyPet and AKAP79-YFP (Oliveria et al., 2003). The difference between WT AKAP79 and each of the 1–325, 1–315, and 1–235 AKAP79 deletion mutants is significant (p < 0.036, n ≥ 12, Dunnett post hoc tests) and in each case is not different from the level of the negative control (p > 0.8, Dunnett post hoc test). The loss of FRET when amino acids 326–336 are deleted suggests that this domain alone may mediate interaction with TRPV1. In confirmation, FRET of an AKAP79 Δ326–336 deletion mutant with TRPV1 was also at the level of the negative control (Fig. 2D, last bar).
We next used coimmunoprecipitation to investigate the location of the TRPV1 binding site on AKAP79. His- and V5-tagged AKAP79 WT and mutants were expressed in HEK293 cells and purified by Ni2+ beads, and the ability of the purified protein to precipitate TRPV1 from a whole-cell lysate was explored in pull-down experiments using Protein A agarose beads coupled to a V5 antibody. The amount of bound protein was reduced for truncations 1–235, 1–315, and 1–325 compared with AKAP WT (p < 0.001, n ≥ 3, Dunnett post hoc tests; Fig. 3).
Figure 3.

Coimmunoprecipitation between AKAP79 mutants and TRPV1 identifies localization of interaction site. A, Representative blots showing pull down of TRPV1 by AKAP79 mutants with (1–336, 32–427, 1–384) and without (1–325, 1–315, 1–235) the binding site for TRPV1. Expression level of AKAP79 mutants was similar (lower blot). B, Intensity of TRPV1 band after immunoprecipitation, expressed relative to pull-down level of AKAP79 mutants and normalized to WT. Interaction dropped when amino acids 326–336 were not present. All bars represent the average of at least three repetitions. ***p < 0.001 compared with WT, ANOVA with post hoc Dunnett test.
Finally, we used the fact that overexpression of AKAP79 enhances trafficking of TRPV1 to the plasma membrane (Zhang et al., 2008) to identify the TRPV1 binding domain within AKAP79. The location of the plasma membrane was visualized using fluorescently labeled WGA-Alexa 594, which binds to surface membrane sialic acid residues and is not internalized with a brief exposure at cold temperatures (Fig. 4A, second image) and we calculated the ratio of mean TRPV1 fluorescence in the membrane to the mean value in the cytoplasm excluding the nucleus. Coexpression of WT AKAP79-CyPet or of truncations 32–427, 1–360, or 1–336 significantly increased trafficking of TRPV1 to the membrane compared with that without AKAP79 (Fig. 4B,C; ANOVA, F(8,240) = 19.2, p < 0.001, Dunnett post hoc tests). TRPV1 trafficking with truncations 1–235, 1–315, and 1–325 and the deletion mutant Δ326–336 was significantly lower than with WT AKAP79 (p < 0.001, Dunnett post hoc tests) and was indistinguishable from that seen without transfection of AKAP79 (Fig. 4C, first bar). The effect was not attributable to any difference in the ability of AKAP79 mutants to localize to the membrane (Fig. 4D). In summary, all three approaches identify the 11 aa domain between K326 and E336 as critical for the binding between AKAP79 and TRPV1.
A peptide mimicking the AKAP79 binding site disrupts AKAP79–TRPV1 interaction
We designed an 11-mer peptide with the same sequence as the 326–336 AKAP79 binding domain, localized in the experiments above, to determine whether the peptide can antagonize binding of AKAP79 to TRPV1. The AKAP79 (326–336) peptide was found to be effective in inhibiting binding in coimmunoprecipitation experiments, while a scrambled peptide of the same amino acid composition (Scramble) was ineffective (Fig. 5A). We next investigated the effectiveness of the blocking peptide in preventing sensitization of TRPV1 in DRG neurons under whole-cell patch clamp. Brief pulses of capsaicin elicited inward currents carried by TRPV1, and in control experiments a sixfold increase in current amplitude was seen when PKC was activated by application of PMA, a potent PKC activator (Fig. 5C, inset). When the AKAP79 (326–336) peptide was added to the pipette solution the PMA-induced sensitization of TRPV1 was significantly reduced compared with control (ANOVA, F(2,30) = 4.7, p = 0.015, Tukey post hoc test) while the scrambled peptide was ineffective (Fig. 5C).
Figure 5.
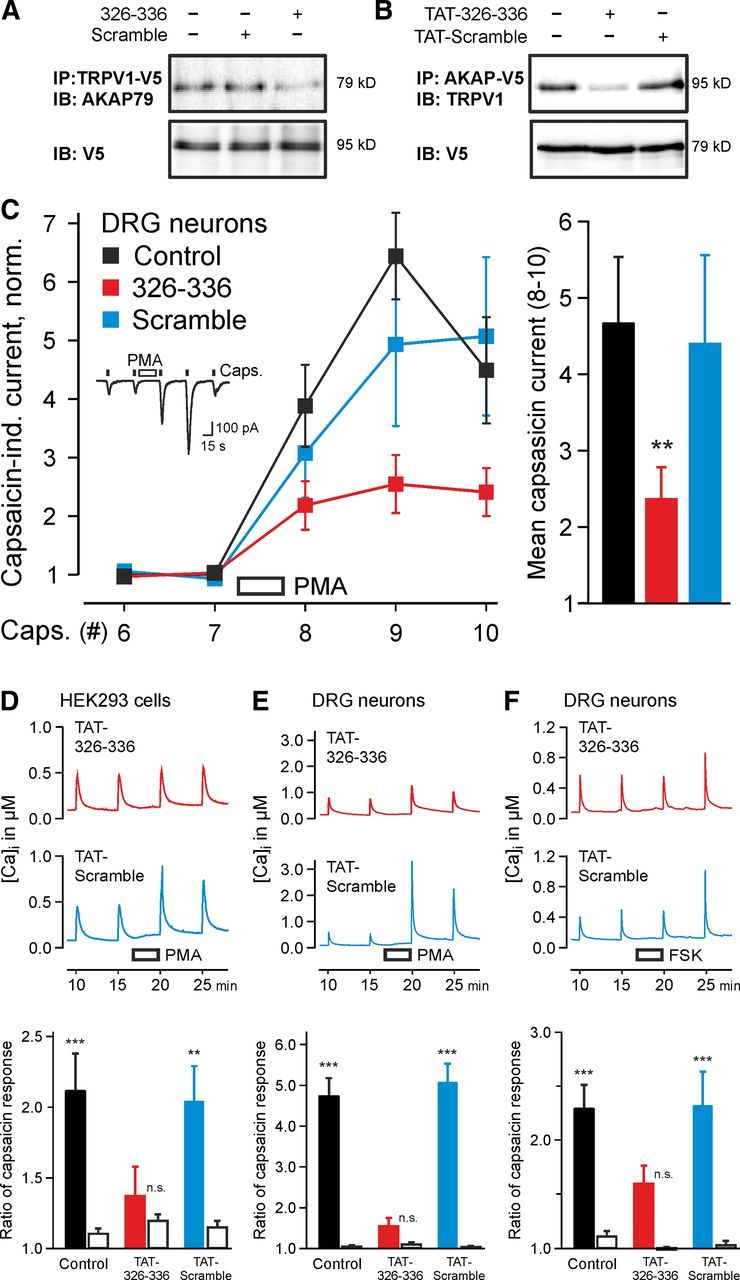
AKAP79 (326–336) peptide and cell-penetrating TAT-coupled peptide (TAT-326–336) inhibit binding of TRPV1 to AKAP79 and PKC- and PKA-mediated sensitization of TRPV1. A, Representative blot showing reduction of AKAP79 precipitation by TRPV1–V5 in presence of the active AKAP79 (326–336) peptide (KSKSEESKRME, 200 μm, last lane) compared with the scrambled version (KEMRESKESKS, 200 μm, middle lane) and the control without peptide (first lane). B, Representative blot showing the reduction of TRPV1 precipitation by AKAP79–V5 expressed in HEK293 cells with active TAT-326–336 peptide (100 μm) compared with TAT-Scramble (100 μm) and the positive control without peptide. C, Left, Inset shows inward currents in DRG neurons evoked by repetitive capsaicin application (100 nm, 5 s) without peptide in pipette. Application of PMA (1 μm, 30 s) before the eighth stimulation sensitized capsaicin-activated inward currents. Sensitization of TRPV1 is inhibited by the AKAP79 (326–336) peptide but not the scrambled analog in the pipette solution (200 μm, 13 min allowed for equilibration before PMA application) Right, Sensitization by PMA, calculated as mean of capsaicin-induced currents for applications 8, 9, 10, normalized to the mean of 6, 7 (n = 7–15, **p < 0.01). D, Calcium imaging of HEK293 cells loaded with fura-2 AM (3 μm, 30 min) and then exposed to extracellular solution containing a TAT-conjugated peptide (5 μm, 20 min). Six capsaicin pulses (100 nm, 1 s duration) applied at intervals of 5 min; average of responses 3–6 is displayed for n = 95–129 cells. PMA (1 μm) applied for 180 s before fifth capsaicin pulse. Bottom, Sensitization ratio (response 6/4, filled bars) compared with control ratio (responses 4/3, open bars). Sensitization is apparent in Control and TAT-Scramble exposed cells (p < 0.001 and p = 0.005), but not after exposure to TAT-326–336 (p = 0.35). E, Similar experiments with DRG neurons (n = 117–134). Sensitization is apparent in control and in cells exposed to TAT-Scramble (both p < 0.001), but not after exposure to TAT-326–336 (p = 0.91). F, Experimental design as in E but with FSK (10 μm) instead of PMA. As before, sensitization is apparent in Control and TAT-Scramble exposed cells (both p < 0.001), but not after exposure to TAT-326–336 (p = 0.08). **p < 0.01, ***p < 0.001, n.s. nonsignificant.
We designed a cell-permeable version of the AKAP79 (326–336) peptide by addition of a TAT peptide sequence (HIV transactivator of transcription 47–57, YGRKKRRQRRR) at the N terminus, to give AKAP79 (TAT-326–336). Coimmunoprecipitation experiments showed that the TAT-326–336 peptide interrupted the interaction between AKAP79 and TRPV1, while the TAT-Scramble peptide was ineffective (Fig. 5B). In calcium imaging experiments responses to capsaicin in TRPV1-expressing HEK cells were sensitized by PKC activation (Fig. 5D, black bar; p < 0.001). The addition of the AKAP79 (TAT-326–336) peptide disrupted sensitization of TRPV1 by PKC activation (Fig. 5D, red trace and red bar; p = 0.35). The scrambled analog (TAT-Scramble), on the other hand, did not reduce sensitization (Fig. 5D, blue trace and blue bar; p = 0.005). Notably, the concentration of the TAT-326–336 peptide in the external solution required to completely ablate sensitization was much lower than when added to the intracellular solution, showing that the TAT-coupled peptide accumulated within the cell during the 20 min loading period.
The cell-permeable peptide was also tested in similar experiments for its effect in blocking sensitization of TRPV1 in cultured DRG neurons. In these experiments strong sensitization of TRPV1 was observed following activation of PKC both with no peptide and after pre-exposure to TAT-Scramble (Fig. 5E; both p < 0.001), but with TAT-326–336 sensitization was abolished (Fig. 5E; p = 0.91). Sensitization of TRPV1 by PKA also depends on the TRPV1–AKAP79 interaction (Zhang et al., 2008). We found that addition of the adenylyl cyclase activator forskolin (FSK) caused a significant sensitization of TRPV1 responses, which was unaffected by the scrambled peptide (Fig. 5F; both p < 0.001) but was abolished by the cell-permeable AKAP79 (TAT-326–336) peptide (p = 0.08). In summary, the cell-permeable peptide mimicking the TRPV1 binding site on AKAP79 can effectively block sensitization of TRPV1 in DRG neurons caused by activation of either PKC or PKA.
Behavioral experiments
The AKAP79 (TAT-326–336) cell-penetrating peptide was tested in models of inflammatory pain to assess the effects of disrupting the sensitization of TRPV1 at the level of the whole animal. Injection of formalin causes two distinct phases of pain-related behavior: an initial phase, caused by a direct action of formalin on nociceptive nerve terminals, and a later phase caused by the release of inflammatory mediators, many of which activate PKC or PKA downstream of binding to their receptors (Dubuisson and Dennis, 1977; Macpherson et al., 2007; McNamara et al., 2007). We injected active or scrambled TAT-coupled peptides into the mouse hindpaw 30 min before a 4% formalin injection to test whether interrupting sensitization of TRPV1 reduced inflammatory pain. Neither peptide had any effect on the initial phase of pain behavior when compared with control injections without peptide, but the TAT-326–336 peptide significantly reduced the second phase of the formalin response compared both to control and TAT-Scramble (Fig. 6A; ANOVA, F(2,15) = 6.6, p = 0.023 and p = 0.013, Tukey post hoc tests). We also used a more specific test for involvement of PKC, in which the selective PKC activator PMA is injected into the paw, producing a single delayed phase of pain behavior with a similar time course to the second phase of the formalin test (Ferreira et al., 2005). In the PMA test the AKAP79 (TAT-326–336) peptide significantly reduced pain-related behavior compared with PMA alone or to the TAT-Scramble peptide (Fig. 6B; ANOVA, F(2,15) = 5.4, p = 0.024 and p = 0.040, Tukey post hoc tests).
Figure 6.
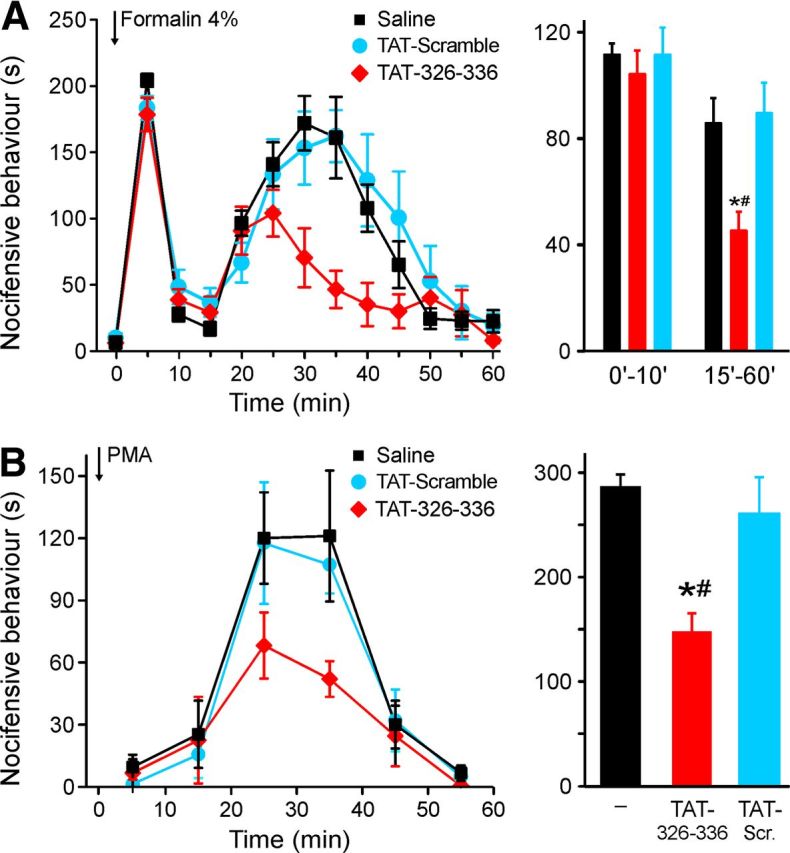
AKAP79 (TAT-326–336) blocks pain-related behavior evoked by formalin or PMA in mice. A, TAT-326–336 (10 μm, 20 μl) injected into the hindpaw 30 min before formalin injection into same site (4%, 20 μl) had no effect on phase I (0–10 min) but suppressed phase II (15–60 min) of pain-related behavior compared with pre-injection of saline or TAT-Scramble (10 μm, 20 μl). B, TAT-conjugated peptides as in A were coinjected with PMA (50 pmol/hindpaw). Integral of pain-related behavior (15–45 min) was attenuated after coinjection with AKAP79 (TAT-326–336), n = 6 for each test group, *p < 0.05 vs control, #p < 0.05 versus TAT-Scramble.
The sensitivity to heat is enhanced by inflammation, an effect that is partly reversed by genetic deletion or pharmacological block of TRPV1 (Caterina et al., 2000; Davis et al., 2000; Pomonis et al., 2003). We examined the effect of the TAT-326–336 peptide in the carrageenan-induced model of inflammatory heat hyperalgesia (Fig. 7A). Carrageenan caused a robust heat hyperalgesia (black line, solid diamonds), which was unaffected by the injection of the TAT-Scramble peptide (blue line, solid circles). The active TAT-326–336 peptide, however, inhibited inflammatory heat hyperalgesia by ∼50% (red line, solid diamonds) when compared with either carrageenan alone or carrageenan with injection of TAT-Scramble peptide (p < 0.001 and p < 0.001, n = 12, Tukey post hoc tests). Importantly, the active peptide had no effect on the response to the heat stimulus in the absence of inflammation (red and black open diamonds, p = 0.99, n = 11, Tukey post hoc test).
Figure 7.
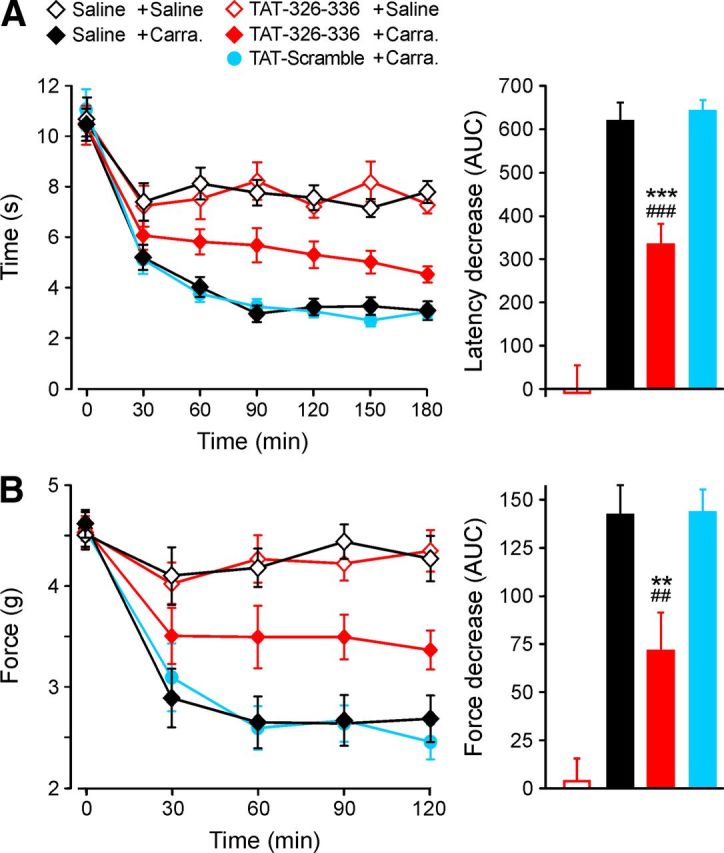
AKAP79 (TAT-326–336) blocks inflammatory thermal and mechanical hyperalgesia without affecting acute thresholds. TAT-326–336, TAT-Scramble (10 μm in saline, 20 μl), or saline (20 μl) injected 30 min before λ-carrageenan (2%, 20 μl) or saline. Baseline (point at 0 min) measured immediately before carrageenan injection. A, Latency for paw withdrawal to radiant heat stimulus after λ-carrageenan injection. Right, Shows the difference of area under the curve (AUC; 30–180 min) between saline control and (from left) Tat-326–336 + saline, saline + carrageenan, TAT-326–336 + carrageenan, and TAT-scramble + carrageenan. B, Threshold mechanical force for paw withdrawal after saline or λ-carrageenan injection. Right, Shows difference of AUC (30–120 min) between saline control and test samples as in A (n = 11–12 for each group, **p < 0.01 and ***p < 0.001 vs control, ##p < 0.01 and ###p < 0.001 vs TAT-Scramble).
We also tested the ability of the AKAP79 (TAT-326–336) peptide to inhibit inflammatory mechanical hyperalgesia, which is known to be inhibited by selective blockers of TRPV1 (Pomonis et al., 2003; Walker et al., 2003; Tang et al., 2007). The withdrawal threshold to a mechanical stimulus showed robust hyperalgesia following injection of carrageenan (Fig. 7B, black line, open diamonds), and the time course was unaffected by injection of the TAT-Scramble peptide (blue line, open circles). The active TAT-326–336 peptide, however, reduced hyperalgesia by ∼50% (red line, solid diamonds) when compared with either carrageenan alone or carrageenan with injection of TAT-Scramble peptide (p = 0.008 and p = 0.007, respectively, n = 12, Tukey post hoc tests), similar to the effect of a potent and specific blocker of TRPV1 (Pomonis et al., 2003; Tang et al., 2007). The active peptide had no effect on noninflamed responses to mechanical stimuli (red and black open diamonds, p = 0.99, n = 12, Tukey post hoc tests).
Discussion
The interaction between TRPV1 and the scaffolding protein AKAP79 is crucial for PKC- and PKA-dependent sensitization of TRPV1 (Jeske et al., 2008, 2009; Schnizler et al., 2008; Zhang et al., 2008). AKAP79 contains binding sites for several enzymes important in signaling cascades, including PKA, PKC, PP2B (calcineurin), and adenylate cyclase among others, and in turn assembles these enzymes into signaling complexes with downstream targets either by binding directly to the target (for example, in the case of the ion channel CaV1.2) or by binding via an intermediate protein such as PSD-95 or SAP-97 (Fig. 8A; further details in legend). Phosphorylation of several residues on TRPV1 has been implicated in sensitization (Fig. 8A) but to date only residues S502 and S801 (Zhang et al., 2008; Lee et al., 2012) have been shown to play a role in AKAP79/150-mediated phosphorylation.
Mapping the binding site of TRPV1 on AKAP79 is essential for an understanding of how this scaffolding protein interacts with its targets. A unique TRPV1 interaction site on AKAP79 may open up the possibility of selectively disrupting inflammatory hyperalgesia, without interfering with the interaction of AKAP79 with other downstream targets. By sequential C-terminal truncation of the AKAP79 protein a sequence of 11 aa, 326–336, was identified in experiments with FRET acceptor photobleach, coimmunoprecipitation, and TRPV1 trafficking (Figs. 2–4) as responsible for binding TRPV1. Deletion of this sequence confirmed the domain as the TRPV1 binding site in FRET and trafficking experiments (Figs. 2, 4), while a competitor peptide of the same sequence was found to inhibit sensitization of TRPV1 (Fig. 5). The TRPV1 interaction site identified (Fig. 8A) is adjacent to the reported PP2B binding site (Dell'Acqua et al., 2002; Gold et al., 2011), and does not overlap with any other reported binding site of AKAP79/150 interaction partners (Sanderson and Dell'Acqua, 2011). We have not completely excluded the possibility that an intermediary protein, such as a MAGUK, may be involved in the interaction between AKAP79 and TRPV1, though the similarity in the size of the binding site on TRPV1 (10 residues, Fischer et al., 2013) and AKAP79 (11 residues, this paper) may argue for a direct interaction.
In coimmunoprecipitation experiments with both truncations (Fig. 3) and competitor peptides (Fig. 5) we did not observe a complete abolition of TRPV1 binding, which might suggest the existence of a second binding site, but could equally be due either to nonspecific binding or to the AKAP79 truncations dimerizing with endogenous AKAP79 expressed in HEK293 cells (Gold et al., 2011). A possible presence of another TRPV1 binding site on AKAP79 is not supported by other results, in which deletion of the 326–336 domain reduced FRET and trafficking to the membrane to the level of the negative controls (Figs. 2, 4).
We considered a number of possible complicating factors in the experiments described here. First, the reduction of FRET between TRPV1 and AKAP79 truncations 1–235, 1–315, and 1–325 (Fig. 2) was not due to a change in trafficking of TRPV1 to the membrane, as observed in the intracellular localization experiments (Fig. 4), because TRPV1-CyPet levels are similar in the ROIs selected (Fig. 2E). Second, we note that AKAP79 mutants 1–360 and 1–336, which lack a PKA binding site, increased trafficking of TRPV1 to the membrane in a similar way to WT AKAP79 (Fig. 4C), while in biotinylation studies of trafficking of TRPV1 deletion of either the PKC or the PKA binding site was found to reduce TRPV1 in the surface membrane (Zhang et al., 2008). While we have not elucidated the reason for this discrepancy, it is possible that truncation of AKAP79, as used in the present study, has a different effect from selective deletion of the PKA binding site.
The AKAP79 326–336 domain is highly charged and is conserved, apart from two amino acids, among AKAP79 mammalian homologs (Fig. 8B). The two amino acid difference from human to mouse, S327P and K328R, does not appear to affect the action of the peptide in blocking the AKAP79–TRPV1 interaction, because a peptide based on the human sequence blocked sensitization of TRPV1 in mouse neurons (Fig. 5) and was an effective analgesic in behavioral experiments (Figs. 6, 7). Both the TRPV1 736–745 binding site for AKAP79 (Fischer et al., 2013), and the corresponding binding site on AKAP79 at residues 326–336, contain many charged residues, suggesting an electrostatic basis for the interaction. Our results suggest that the AKAP79 (326–336) peptide could be used as a basis for developing novel small molecule inhibitors of inflammatory hyperalgesia, without side effects such as hyperthermia and elevation of heat pain threshold, which have been observed in clinical trials of TRPV1 antagonists. The AKAP79 326–336 domain appears not to overlap with known binding sites for other downstream targets (Fig. 8A), which suggests that an antagonist may not affect other signaling cascades, but further studies of specificity would be required to confirm this. AKAP79 has been implicated in several other cellular processes, such as synaptic plasticity via its effects on long-term depression and long-term potentiation, and in the interaction with β2-adrenergic receptors (Fraser et al., 2000; Gomez et al., 2002; Tavalin et al., 2002), and a confirmation that the peptide directed against the TRPV1 interaction domain does not affect other critical processes is clearly essential.
Blocking the actions of inflammatory mediators on TRPV1, or inhibiting downstream signaling kinases, has been shown to reduce sensitization in vitro and thermal hyperalgesia in vivo (Cesare and McNaughton, 1996; Lopshire and Nicol, 1998; Vellani et al., 2001; Moriyama et al., 2005; Lin et al., 2006; Zhang et al., 2008). AKAP79/150 plays a crucial role in this process, because genetic deletion or knockdown of AKAP150 (the murine analog of AKAP79) reduced thermal hyperalgesia in inflammatory models in vivo (Jeske et al., 2009). By the use of a peptide mimicking the 326–336 site on AKAP79 we demonstrate here that disruption of the AKAP79–TRPV1 interaction reduces both sensitization of TRPV1 by PKC and PKA in vitro and inflammatory hyperalgesia in vivo. In the formalin test the first phase of pain behavior is attributed to a direct action of formalin on nociceptive terminals, while the second phase is caused by release of inflammatory mediators, many of which activate PKC or PKA downstream of binding to their receptors (Dubuisson and Dennis, 1977; Macpherson et al., 2007; McNamara et al., 2007). The AKAP79 (TAT-326–336) peptide reduced the second phase by ∼50% but did not affect the first phase, consistent with an action on sensitization without an effect on nociception itself. In the PMA test, where PKC is directly activated, pain behavior was also reduced by ∼50%. In more direct tests of inflammatory hyperalgesia we induced inflammation by injection of carrageenan and found that the peptide reduced both heat and mechanical hyperalgesia by ∼50%, a result that is comparable to the reduction observed with direct block of TRPV1 (Pomonis et al., 2003; Walker et al., 2003; Tang et al., 2007). A reduction of mechanical hyperalgesia may appear surprising, as TRPV1 is not believed to be directly sensitive to mechanical stimuli, but may be attributed to an indirect effect of discharge in TRPV1-expressing nociceptive fibers in establishing mechanical hyperalgesia via a process of central sensitization (Emery et al., 2011). It has been suggested that TRPV1-deficient mice may not exhibit a reduction in mechanical hyperalgesia (Caterina et al., 2000). However, TRPV1-deficient mice do exhibit a reduction in the sensitivity to stretch in colonic afferents (Jones et al., 2005) while selective TRPV1 antagonists have been shown to cause a reduction in both thermal and mechanical hyperalgesia (Pomonis et al., 2003; Walker et al., 2003; Tang et al., 2007). The results presented in our study suggest that TRPV1 plays a role in mechanical hyperalgesia and, moreover, that the effect is mediated by AKAP79.
An interesting area for future study will be to determine whether the AKAP79 (326–336) peptide has any effect on core temperature, a side effect that has largely halted development of TRPV1 blockers. We note that the peptide does not affect the sensitivity of TRPV1 to capsaicin in whole-cell patch-clamp and calcium imaging experiments in vitro, and does not affect acute thresholds to thermal and mechanical stimuli in vivo, in contrast to the effects on acute pain thresholds seen with several TRPV1 antagonists (Xia et al., 2011).
Footnotes
Work in the McNaughton lab was funded by grants from the Biotechnology and Biological Sciences Research Council and the Newton Trust to P.A.M., a Feodor-Lynen Fellowship to M.J.M.F., and a PhD studentship from the Cambridge Overseas Trust to J.B. We thank Xuming Zhang and Gerald Hammond for experimental advice, John Holdich for the CFP-YFP plasmid, Andrew Charlton for assistance with behavioural experiments, Chun-Hsiang Tan and Sergio Lainez for assistance with experiments on DRG neurons, and David Stepensky for a FRETcalc update.
The authors declare no competing financial interests.
References
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/S0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992;267:16816–16823. [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci U S A. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/S0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Dodge KL, Tavalin SJ, Scott JD. Mapping the protein phosphatase-2B anchoring site on AKAP79. Binding and inhibition of phosphatase activity are mediated by residues 315–360. J Biol Chem. 2002;277:48796–48802. doi: 10.1074/jbc.M207833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson D, Dennis SG. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 1977;4:161–174. doi: 10.1016/0304-3959(77)90130-0. [DOI] [PubMed] [Google Scholar]
- Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem. 2010;285:14450–14458. doi: 10.1074/jbc.M110.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333:1462–1466. doi: 10.1126/science.1206243. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Triches KM, Medeiros R, Calixto JB. Mechanisms involved in the nociception produced by peripheral protein kinase c activation in mice. Pain. 2005;117:171–181. doi: 10.1016/j.pain.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Fischer MJ, Btesh J, McNaughton PA. Disrupting sensitization of TRPV1 inhibits inflammatory hyperalgesia. J Neurosci. 2013;33:7407–7414. doi: 10.1523/JNEUROSCI.3721-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD. Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol. 2000;10:409–412. doi: 10.1016/S0960-9822(00)00419-X. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, Alvarez F, Bak A, Darling M, Gore A, Jang GR, Kesslak JP, Ni L, Norman MH, Palluconi G, Rose MJ, Salfi M, Tan E, Romanovsky AA, Banfield C, et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain. 2008;136:202–210. doi: 10.1016/j.pain.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Bannon AW, Surapaneni S, Hovland DN, Jr, Lehto SG, Gore A, Juan T, Deng H, Han B, Klionsky L, Kuang R, Le A, Tamir R, Wang J, Youngblood B, Zhu D, Norman MH, Magal E, Treanor JJ, Louis JC. The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J Neurosci. 2007;27:3366–3374. doi: 10.1523/JNEUROSCI.4833-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Stengel F, Nygren PJ, Weisbrod CR, Bruce JE, Robinson CV, Barford D, Scott JD. Architecture and dynamics of an A-kinase anchoring protein 79 (AKAP79) signaling complex. Proc Natl Acad Sci U S A. 2011;108:6426–6431. doi: 10.1073/pnas.1014400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny X, Mikhaylova M, Seeger C, Reddy PP, Reissner C, Schott BH, Helena Danielson U, Kreutz MR, Seidenbecher C. AKAP79/150 interacts with the neuronal calcium-binding protein caldendrin. J Neurochem. 2012;122:714–726. doi: 10.1111/j.1471-4159.2012.07828.x. [DOI] [PubMed] [Google Scholar]
- Hammond GR, Schiavo G, Irvine RF. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2) Biochem J. 2009;422:23–35. doi: 10.1042/BJ20090428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA. Inflammatory pain: the cellular basis of heat hyperalgesia. Curr Neuropharmacol. 2006a;4:197–206. doi: 10.2174/157015906778019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA. Modulation of temperature-sensitive TRP channels. Semin Cell Dev Biol. 2006b;17:638–645. doi: 10.1016/j.semcdb.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske NA, Diogenes A, Ruparel NB, Fehrenbacher JC, Henry M, Akopian AN, Hargreaves KM. A-kinase anchoring protein mediates TRPV1 thermal hyperalgesia through PKA phosphorylation of TRPV1. Pain. 2008;138:604–616. doi: 10.1016/j.pain.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske NA, Patwardhan AM, Ruparel NB, Akopian AN, Shapiro MS, Henry MA. A-kinase anchoring protein 150 controls protein kinase C-mediated phosphorylation and sensitization of TRPV1. Pain. 2009;146:301–307. doi: 10.1016/j.pain.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RC, III, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25:10981–10989. doi: 10.1523/JNEUROSCI.0703-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- Lee J, Chung MK, Ro JY. Activation of NMDA receptors leads to phosphorylation of TRPV1 S800 by protein kinase C and A-Kinase anchoring protein 150 in rat trigeminal ganglia. Biochem Biophys Res Commun. 2012;424:358–363. doi: 10.1016/j.bbrc.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CR, Amaya F, Barrett L, Wang H, Takada J, Samad TA, Woolf CJ. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther. 2006;319:1096–1103. doi: 10.1124/jpet.106.105569. [DOI] [PubMed] [Google Scholar]
- Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. J Neurosci. 1998;18:6081–6092. doi: 10.1523/JNEUROSCI.18-16-06081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson LJ, Xiao B, Kwan KY, Petrus MJ, Dubin AE, Hwang S, Cravatt B, Corey DP, Patapoutian A. An ion channel essential for sensing chemical damage. J Neurosci. 2007;27:11412–11415. doi: 10.1523/JNEUROSCI.3600-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem. 2005;280:13424–13432. doi: 10.1074/jbc.M410917200. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Wang SY, Wang GK, Nau C. A tyrosine residue in TM6 of the Vanilloid Receptor TRPV1 involved in desensitization and calcium permeability of capsaicin-activated currents. Mol Cell Neurosci. 2003;23:314–324. doi: 10.1016/S1044-7431(03)00054-X. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, Tominaga T, Narumiya S, Tominaga M. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. 2005;1:3. doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML. Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomonis JD, Harrison JE, Mark L, Bristol DR, Valenzano KJ, Walker K. N-(4-Tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: II. In vivo characterization in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;306:387–393. doi: 10.1124/jpet.102.046268. [DOI] [PubMed] [Google Scholar]
- Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M. PKA/AKAP/VR-1 module: a common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci. 2002;22:4740–4745. doi: 10.1523/JNEUROSCI.22-11-04740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnizler K, Shutov LP, Van Kanegan MJ, Merrill MA, Nichols B, McKnight GS, Strack S, Hell JW, Usachev YM. Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J Neurosci. 2008;28:4904–4917. doi: 10.1523/JNEUROSCI.0233-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A, McClure M, Huang F, Carter R. Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr Biol. 2000;10:1395–1398. doi: 10.1016/S0960-9822(00)00785-5. [DOI] [PubMed] [Google Scholar]
- Steiner AA, Turek VF, Almeida MC, Burmeister JJ, Oliveira DL, Roberts JL, Bannon AW, Norman MH, Louis JC, Treanor JJ, Gavva NR, Romanovsky AA. Nonthermal activation of transient receptor potential vanilloid-1 channels in abdominal viscera tonically inhibits autonomic cold-defense effectors. J Neurosci. 2007;27:7459–7468. doi: 10.1523/JNEUROSCI.1483-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepensky D. FRETcalc plugin for calculation of FRET in noncontinuous intracellular compartments. Biochem Biophys Res Commun. 2007;359:752–758. doi: 10.1016/j.bbrc.2007.05.180. [DOI] [PubMed] [Google Scholar]
- Tang L, Chen Y, Chen Z, Blumberg PM, Kozikowski AP, Wang ZJ. Antinociceptive pharmacology of N-(4-chlorobenzyl)-N′-(4-hydroxy-3-iodo-5-methoxybenzyl) thiourea, a high-affinity competitive antagonist of the transient receptor potential vanilloid 1 receptor. J Pharmacol Exp Ther. 2007;321:791–798. doi: 10.1124/jpet.106.117572. [DOI] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vay L, Gu C, McNaughton PA. The thermo-TRP ion channel family: properties and therapeutic implications. Br J Pharmacol. 2012;165:787–801. doi: 10.1111/j.1476-5381.2011.01601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KM, Urban L, Medhurst SJ, Patel S, Panesar M, Fox AJ, McIntyre P. The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;304:56–62. doi: 10.1124/jpet.102.042010. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen Y, Chen M, Xu W. AKAPs competing peptide HT31 disrupts the inhibitory effect of PKA on RhoA activity. Oncol Rep. 2006;16:755–761. [PubMed] [Google Scholar]
- Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, Everett KL, Ciruela A, Cooper DM. AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J Biol Chem. 2010;285:20328–20342. doi: 10.1074/jbc.M110.120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R, Samad TA, Btesh J, Jiang LH, Kays I, Stjernborg L, Dekker N. TRPV1 signaling: mechanistic understanding and therapeutic potential. Curr Top Med Chem. 2011;11:2180–2191. doi: 10.2174/156802611796904843. [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, McNaughton PA. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron. 2008;59:450–461. doi: 10.1016/j.neuron.2008.05.015. [DOI] [PubMed] [Google Scholar]