

Abstract
Cardiac myosin binding protein-C (cMyBP-C) is a regulatory protein of the contractile apparatus within the cardiac sarcomere. Ischemic injury to the heart during myocardial infarction (MI) results in the cleavage of cMyBP-C in a phosphorylation-dependent manner and release of an N-terminal fragment (C0C1f) into the circulation. C0C1f has been shown to be pathogenic within cardiac tissue, leading to the development of heart failure. Based on its high levels and early release into the circulation post-MI, C0C1f may serve as a novel biomarker for diagnosing MI more effectively than current clinically used biomarkers. Over time, circulating C0C1f could trigger an autoimmune response leading to myocarditis and progressive cardiac dysfunction. Given the importance of cMyBP-C phosphorylation state in the context of proteolytic cleavage and release into the circulation post-MI, understanding the posttranslational modifications (PTMs) of cMyBP-C would help in further elucidating the role of this protein in health and disease. Accordingly, recent studies have implemented the latest proteomics approaches to define the PTMs of cMyBP-C. The use of such proteomics assays may provide accurate quantitation of the levels of cMyBP-C in the circulation following MI, which could, in turn, demonstrate the efficacy of using plasma cMyBP-C as a cardiac-specific early biomarker of MI. In this review, we define the pathogenic and potential immunogenic effects of C0C1f on cardiac function in the post-MI heart. We also discuss the most advanced proteomics approaches now used to determine cMyBP-C PTMs with the aim of validating C0C1f as an early biomarker of MI.
Keywords: Autoantibodies, Biomarkers, cMyBP-C, Dilated cardiomyopathy, Myocardial infarction
1. Introduction
Acute myocardial infarction (MI) is clinically defined by myocardial necrosis in the setting of prolonged myocardial ischemia and it remains a leading cause of morbidity and mortality worldwide 1,2. Most MIs are caused by the occlusion of a coronary artery by thrombus formation following the rupture of an unstable atherosclerotic plaque (Fig. 1A) 3. Myocardium lying distal to the occlusion will not be supplied with oxygen and nutrients and reduced perfusion will limit washout of cellular waste products. An occlusion lasting more than 20 min generates irreversible myocardial necrosis that first occurs within the endocardium and expands throughout the myocardium and epicardium 4. Complete necrosis of the area at risk occurs within several hours and depends on such factors as the extent of collateral circulation and demand for oxygen and nutrients 4. Since the amount of tissue damage is directly related to the duration of the occlusion, the therapeutic strategy is to reopen the coronary artery as quickly as possible 3. Therefore, early detection of MI is imperative for successful treatment. Large transmural infarctions can typically be detected by electrocardiogram ST-segment elevation and are termed STEMI 5. However, a large proportion of infarctions do not result in clear ST-segment elevation 6. In these non-ST segment elevation (NSTEMI) patients, diagnosis is based on release of cardiac proteins (mostly cardiac troponins) into the circulation caused by necrotic cardiomyocytes 7. Unlike the ECG changes in STEMI patients, an increase in serum levels of the cardiac troponins is only detectable after several hours 2,8.
Figure 1.
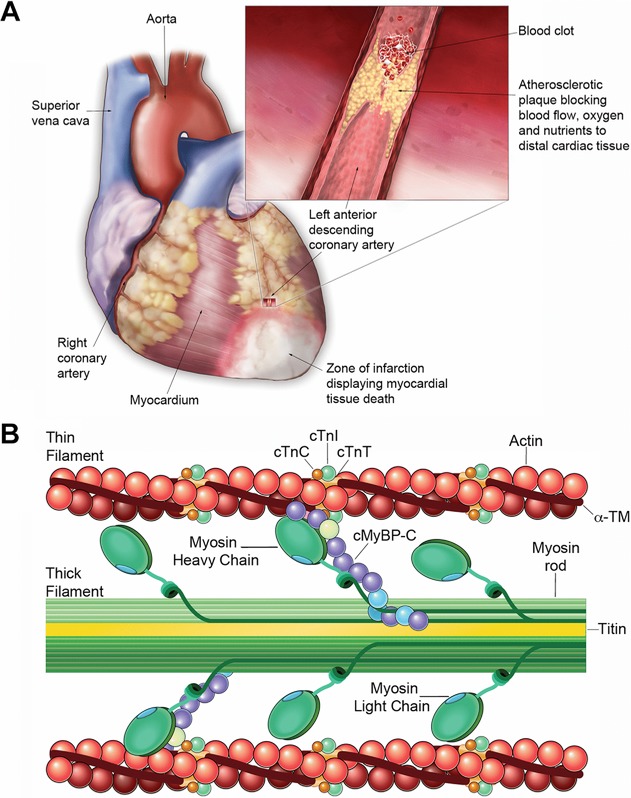
Cardiac morphology and sarcomere structure. (A) Anatomical diagram of the heart showing cardiac vasculature. Inset illustrating myocardial infarction (MI). A leading cause of MI is the rupture of an unstable cholesterol plaque promoting thrombus formation, inflammatory cell infiltration, and occlusion of a coronary artery. The loss of blood supply to distal myocardial tissue inhibits the supply of oxygen and nutrients within the infarct zone and leads to tissue necrosis following prolonged ischemia. The duration of occlusion determines the severity of tissue damage with irreversible necrosis occurring within several hours of the infarct. As such, identifying early markers of MI presence will be vital in the timely diagnosis and treatment of patients in the future. (B) Structure of cMyBP-C in the cardiac sarcomere. The cardiac sarcomere consists of the thick-filament proteins: myosin and titin, as well as the thin-filament proteins: actin, α-tropomyosin, and troponins I, C, and T. cMyBP-C is a sarcomeric protein that participates in stabilizing sarcomere structure and regulating the kinetics of crossbridge cycling by controlling the interaction between actin and myosin filaments.
In the healthy heart, contraction is regulated by a system that couples electrical excitation to elevated levels of intracellular calcium (Ca2+), triggering an interaction between actin and myosin filaments to generate crossbridge cycling and shortening of the sarcomere (Fig. 1B) 9. However, upon ischemic injury to the heart, the loss of myocardial tissue alters the contractile properties of myocytes leading to further myocyte death and exacerbating cardiac dysfunction 10. The heart attempts to compensate for the loss of contractile activity and diminished cardiac pump function by increasing myocyte Ca2+ transients 10. However, maintaining contractile function by increasing Ca2+ influx induces continuous pathological stress in the post-MI heart 10. This stress causes structural remodeling of the left ventricle and dysfunction leading to hypertrophy and eventually, dilation 10–12.
Our group recently found that the sarcomeric regulatory protein, cardiac myosin binding protein-C (cMyBP-C), is degraded into an N-terminal 40 kDa fragment (C0C1f) following MI and that this fragment is released into the circulation 13–15. Within cardiac tissue, C0C1f has been demonstrated to produce significant contractile dysfunction 13,16 and heart failure (HF) 15. Furthermore, previous studies have determined cMyBP-C to be immunogenic, triggering the production of autoantibodies (AAbs) that generated experimental autoimmune myocarditis (EAM) and dilated cardiomyopathy (DCM) in animal models 17–19. In support of this, the immunogenicity of circulating cardiac proteins has been studied in patients with DCM 20,21. As such, C0C1f may also be considered immunogenic within the circulation and could lead to post-MI myocarditis and DCM. Thus, the pathogenic and potentially immunogenic effects of C0C1f may augment cardiac dysfunction post-MI, but its immediate release following ischemic injury may also implicate cMyBP-C as a novel biomarker aiding in the early diagnosis and treatment of MI.
Overall, the status of cMyBP-C phosphorylation, proteolytic cleavage, and release into the circulation post-MI underscores the importance of understanding cMyBP-C regulation through PTM. Lately, several studies have used leading-edge proteomics approaches to define the PTMs of cMyBP-C and characterize their role in health and disease 22–25. Such proteomics assays may provide accurate quantitation of the levels of cMyBP-C in the circulation post-MI and could demonstrate the efficacy of using plasma cMyBP-C as an early cardiac-specific biomarker of MI 26.
2. Structure and function of cMyBP-C
The sarcomere is the functional unit of striated muscle consisting of thick and thin filaments that regulate contraction (Fig. 1B). cMyBP-C is a cardiac protein of 140.8 kDa that expresses specifically in the heart 27, stabilizing sarcomere structure and regulating actomyosin crossbridge formation 28–30. Structurally, cMyBP-C contains 12 domains, including a unique cardiac N-terminal domain (C0), an insertion loop of 28 residues within the C5 domain, and similar to other actin-binding proteins, a Pro-Ala-rich region between the C0 and C1 (Fig. 2A). A cardiac-specific M-domain with multiple phosphorylation sites lies between C1 and C2 that is phosphorylated by several kinases 31–35 (Fig. 2A). Several studies have determined the significance of cMyBP-C phosphorylation status in the context of its ability to regulate actomyosin crossbridge formation and hence, control force generation within the sarcomere 36–39. In addition, phosphorylation has been shown to protect cMyBP-C from proteolysis during ischemic injury to the heart 14, whereas dephosphorylation promotes its interaction with myosin and degradation. Moreover, mutations in the gene encoding cMyBP-C are associated with contractile dysfunction 40 and cause hypertrophic cardiomyopathy and DCM 41, suggesting the importance of increased studies that focus on cMyBP-C regulatory pathways.
Figure 2.
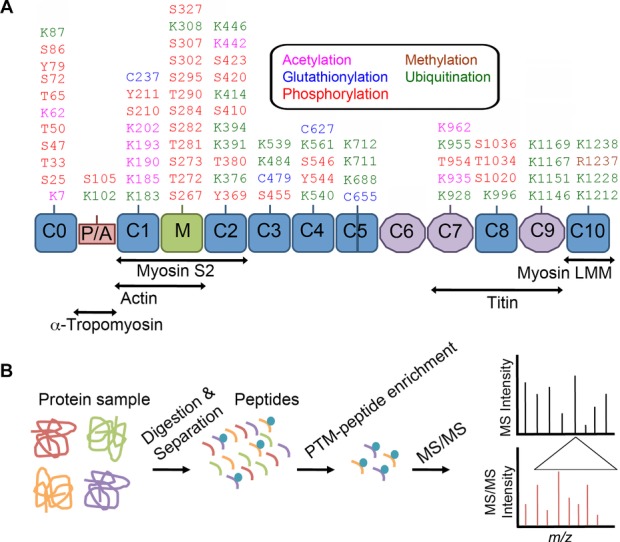
Mapping of PTMs in cMyBP-C by MS/MS. Elucidating the profile of PTMs that occur within cMyBP-C through proteomics approaches has been and will continue to be essential for determining how these modifications affect the role of cMyBP-C in health and disease. (A) Structurally, cMyBP-C consists of 12 domains of which 8 are immunoglobulin (IgC2) like domains (blue), and three are fibronectin (FN3) domains (purple) 65. Unique to cMyBP-C is a cardiac-specific C0 domain, a proline–alanine rich linker region (red), an insertion loop within the C5 domain, and a phosphorylation motif at the M-domain (green) 65. Sites of interaction between cMyBP-C domains and other sarcomeric proteins are shown below. Listed are the PTM sites within mouse cMyBP-C adapted from PhosphoSitePlus® analysis of cMyBP-C PTMs (http://www.phosphosite.org) 66 that have been determined using MS and other techniques. PTM residues are indicated in the text as acetylation (pink), glutathionylation (blue), methylation (brown), phosphorylation (red), and ubiquitination (green). (B) MS/MS represents a highly sensitive analytical tool for identifying the sites and types of PTMs within enriched peptides from a sample of tryptically digested and separated proteins and will be a critical proteomics approach in the future of cMyBP-C PTM discovery. The first MS analysis provides the mass of all peptides in a sample. Specific peptides can then be fragmented to generate ions that undergo another round of MS to determine fragment ion m/z values and peptide sequence by computational analysis 67. Based upon changes in the mass of modified amino acid residues, MS/MS allows for identification of the type and site of a PTM 67.
3. cMyBP-C is a substrate for calpain-mediated proteolytic cleavage
Throughout ischemia and in early reperfusion, increased levels of Ca2+ activate the Ca2+-dependent protease calpain, which degrades intracellular proteins, including contractile proteins, during myocardial necrosis 42. Postischemic dysfunction has been associated with Ca2+-dependent degradation of α-actin 43 and cardiac troponin-I (cTnI) 44. Models of ischemia–reperfusion (I–R) also show degradation of cardiac troponin-T and myosin light chain-1 45. Previous work has identified cMyBP-C as a substrate for calpains 46,47, and that dephosphorylation makes cMyBP-C susceptible to proteolysis, leading to the release of C0C1f fragments. MS/MS analyses revealed that the calpain cleavage site lies within the M-domain at 272-TSLAGAGRR-280 in mice and that cMyBP-C cleavage fragments could be readily detected in cardiac tissue 46. It was determined that C0C1f is generated and released into the circulation in an in vivo rat model of MI and in samples from post-MI patients 14. Furthermore, cMyBP-C dephosphorylation was found to correlate with its increased degradation and release in vitro and in ischemic areas of the in vivo MI heart 14. Given the role of cMyBP-C as a regulator of actomyosin interaction, its degradation may be involved in pathological dysfunction following ischemic injury to the heart 46.
4. Pathogenic consequences of C0C1f
Pathogenic consequences of cMyBP-C degradation in the heart post-MI could arise through disruption of normal full-length protein function, through direct pathogenicity of C0C1f within the sarcomere, or both. The pathogenic properties of C0C1f were determined upon adenoviral infection of neonatal rat ventricular cardiomyocytes and adult rabbit ventricular cardiomyocytes with full-length cMyBP-C, C0C1f, or the 110 kDa C-terminal fragment 13. This work revealed that C0C1f binds to the actin thin-filament within the cardiac sarcomere. Using LC-MS/MS analysis, it was determined that this fragment contains several acetylation sites that may be involved in regulating the interaction between actin and myosin filaments 13. Furthermore, C0C1f expression significantly elevated caspase-3 activity, as well as the release of lactate dehydrogenase, which is an indicator of increased cell injury, demonstrating the toxicity of this fragment to cardiomyocytes 13. C0C1f reduced cell viability, altered Ca2+ handling, and significantly decreased sarcomere length shortening and the velocities of contraction and relaxation by inhibiting actomyosin function 13. It was later established in vivo that the C0C1f fragment could cause cardiac dysfunction and HF 15. Most recently, Witayavanitkul et al. examined in vitro the association between C0C1f and altered contractility in human cardiac myofilaments. They found that C0C1f decreased myofilament Ca2+ sensitivity and maximum force generation, but increased cooperative activation, the kinetics of crossbridge cycling, and tension cost at short sarcomere lengths 16. Furthermore, it was shown that C0C1f interacted with actin and α-tropomyosin to exert its effects by reducing contractility, suggesting that reduced cardiac function following MI may result from C0C1f taking on the characteristics of a poison polypeptide by interfering with the ability of native cMyBP-C to interact normally with the thin filament 16. Overall, delineating the toxic effects of cMyBP-C degradation and subsequent release of C0C1f on sarcomere function has provided insight into the mechanisms underlying contractile dysfunction in MI patients, therefore preserving phosphorylation status is necessary to protect its degradation. While C0C1f is pathogenic within cardiac tissue, its release into the circulation following MI may also trigger an autoimmune response contributing to the development of debilitating myocarditis and progressive cardiac dysfunction.
5. Immunogenic response to released cMyBP-C post-MI
Autoimmune-mediated disease occurs when the body fails to distinguish its constituents as being self rather than harmful nonself, leading to the development of an immune response against its own healthy tissues 48,49. AAbs are antibodies that are produced by the immune system and target an individual's own proteins. The onset of myocarditis and DCM has been associated with the presence of circulating AAbs to cardiac contractile proteins 21. Indeed, the production of AAbs against released cTnI in patients with ischemic cardiomyopathy and DCM has been correlated with the appearance of myocarditis and the progression of DCM into HF 50. Similar to cTnI, released cMyBP-C may indirectly damage the myocardium and induce cardiac dysfunction by triggering an immunogenic response and the production of cMyBP-C-specific AAbs following MI. These AAbs may then play a role in the development of autoimmune myocarditis and the progression of DCM into HF.
Unlike cTnI, the immunogenic potential of cMyBP-C following MI has not been well documented; therefore, mechanistic data explaining how circulating cMyBP-C may induce AAb production and potential cardiac dysfunction in this post-MI period is limited. However, the strong reactivity of AAbs targeted to fragments of murine and human cMyBP-C that could produce animal models of EAM and DCM has been demonstrated 17–19. Kasahara et al. were the first to report the presence of cMyBP-C-reactive AAbs in the sera of patients with DCM 17. They further established the immunogenicity of cMyBP-C upon repeated injection of murine cMyBP-C fragments into various strains of mice, leading to the development of autoimmune myocarditis in a subset of animals 17. cMyBP-C-AAbs were found to react strongly with residues 205–916, the myocarditis-inducing region, but weakly with residues 945–1270, of murine cMyBP-C 17. Matsumoto et al. provoked severe EAM in rats upon immunization with recombinant human cMyBP-C fragments corresponding to the N-terminal residues 1–323 and 317–647, and mild EAM with the C-terminal residues 641–970 and 964–1274 18. Residues 615–647 were later found to be the most immunogenic with a peptide from this region causing moderate EAM. However, in order to produce DCM, they hypothesized the further requirement of a pathogenic mechanism sufficient to produce more severe inflammation and fibrosis 19. A major drawback of this study is the failure of Matsumoto et al. to further explore the antigenicity and immunogenicity of EAM-inducing residues 1–323 consisting of cMyBP-C domain C0 through an early part of the M-domain, largely constituted by the C0C1f fragment. Given that circulating cMyBP-C is able to induce EAM and DCM, it is possible that released C0C1f generates an immune response following MI, with the production of AAbs to this fragment promoting cardiac dysfunction. Particularly, important is the fact that cMyBP-C contains domains at its N-terminus that are present only in the cardiac isoform, indicating a potential cardiac specificity for AAbs developed to this region. Therefore, to further classify the immunogenic properties of cMyBP-C following MI, future studies should focus on the potential, production, and pathogenic roles of cMyBP-C-AAbs in the ischemic myocardium. While persistence of circulating cMyBP-C fragments may be immunogenic, C0C1f may also be beneficial in the early diagnosis and treatment of MI, as previously noted.
6. Clinical applications of cMyBP-C as a novel biomarker in the diagnosis of MI
To this point, we have largely defined the detrimental effects of released cMyBP-C on cardiac function in the post-MI heart. However, recent data implicate circulating C0C1f as having clinical utility and potentially serving as a novel biomarker for the early diagnosis of MI upon arrival of patients to the hospital 14,51. Following MI, released myofilament proteins are used as clinically relevant biomarkers for the detection of myocardial damage 52. While novel biomarkers that predict outcome following MI are being increasingly identified, many of them lack “ideal” cardiac specificity and diagnostic sensitivity. Unfortunately, the levels of current gold standard biomarkers, such as the cardiac troponins that are used for diagnosing NSTEMI, are undetectable in the early stages of MI when intervention is most beneficial 2,8. Detectable cTnI levels are present only at 6–12 h following infarction (Fig. 3) 51,53, resulting in patients having to wait longer for diagnosis and treatment. Furthermore, the cardiac troponins have a very long lifespan in the blood relative to other cardiac biomarkers, making them undesirable for diagnosing reinfarction, which is an acute MI that occurs within 28 days of the incident or recurrent MI 4. As early diagnosis of MI is vital for improved outcome, an immediate indicator of MI presence and severity between 0 and 6 h is urgently needed.
Figure 3.
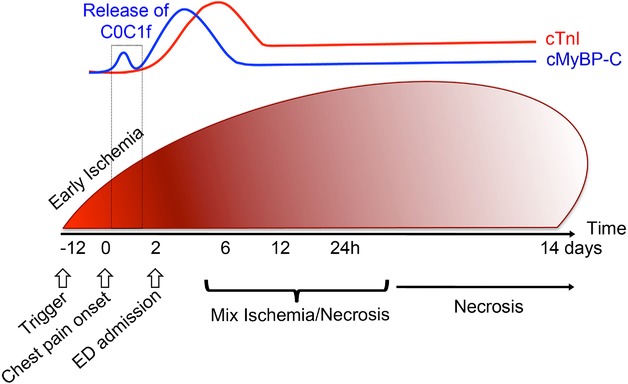
Schematic illustration of cMyBP-C release kinetics in the blood following MI. Cardiac ischemia occurs when the supply of blood to the heart does not appropriately respond to the demand required due to atherosclerosis in the anterior region of the heart and either partial or complete stenosis of a coronary artery by plaque rupture. Symptomatically, this could result in chest pain, which would send patients to the Emergency Department (ED). Prolonged ischemia leads to cellular necrosis, followed by acute MI. Currently, NSTEMI patients are diagnosed on the basis of increased blood levels of cTnI, but this marker of MI only becomes significantly elevated in the blood at 6–12 h after the onset of ischemia, causing critical diagnostic delays before treatment can be initiated. Recently, we detected the presence of cMyBP-C in the plasma of pigs within 30 min of MI 53, suggesting that cMyBP-C could be a sign of early ischemia in patients with NSTEMI. Interestingly, the C0C1f proteolytic fragment appears to be the first indication of an early ischemic event. Upper panel shows the release kinetics of circulating cMyBP-C and cTnI up to 14 days following ligation of the LAD coronary artery in swine as measured by immunoassay.
cMyBP-C has been identified as a potential biomarker for the timely diagnosis of MI based on its early release into the circulation in response to ischemic injury in rats, pigs, and humans 14,53,54. It was recently shown that plasma cMyBP-C levels significantly increased from baseline at 3 h and peaked at 6 h following ligation of a branch of the left anterior descending coronary artery in a porcine model of MI (Fig. 3) 53. In particular, cMyBP-C levels were detectable 30 min following left anterior descending ligation 53. Moreover, plasma cMyBP-C levels in humans with hypertrophic obstructive cardiomyopathy undergoing transcoronary ablation of septal hypertrophy were significantly increased from baseline after 4 h 53. This indicates the potential for cMyBP-C as an early biomarker of MI. We attempted to classify cMyBP-C as a bona fide biomarker of MI in comparison to the diagnostic capabilities of currently used gold standard biomarkers, i.e. cTnI or cardiac troponin-T. In the 65 MI patients studied, cMyBP-C levels were equal to, or greater than, those of other existing cardiac biomarkers, with cMyBP-C levels declining prior to those of the gold standard biomarker cTnI 51. Additionally, using proteomics analyses (Fig. 2B), such as 1D gel-LC-MS/MS, 2DE, and LTQ Orbitrap XL, Jacquet et al. identified cMyBP-C, among other candidate biomarkers of acute MI, in both its full-length form and as the C0C1f degradation product in coronary effluent collected after ischemia in isolated mouse hearts initially perfused with oxygenated protein-free buffer 55. cMyBP-C was detected in the plasma post-MI in vivo, signifying a profile in line with a biomarker of MI. Given that cMyBP-C has unique cardiac-specific domains, these results suggest that cMyBP-C could serve as both a sensitive and cardiac-specific biomarker for the accurate diagnosis of MI and that the application of proteomics approaches may help elucidate and validate this potential in future research 26.
7. cMyBP-C proteomics in health and disease
cMyBP-C has been demonstrated to contain multiple sites that are subject to PTM (Fig. 2A). Characterization of protein PTMs with lack of a priori knowledge remains a significant challenge. Recently, advanced proteomics approaches, such as MS/MS, have been utilized to assess cMyBP-C PTMs in health and disease (Fig. 2B) 22–25,56. Given that cMyBP-C contains 81 serine and 73 threonine residues, identifying specific phosphorylation sites has been a significant challenge 24. As such, Ge et al. made use of top-down electron capture dissociation MS as a powerful proteomics technique to identify and quantify all of the cMyBP-C phosphorylation sites using intact full-length (C0–C10) and truncated (C0–C1, C0–C4, and ΔC0–C1) forms of mouse cMyBP-C expressed in baculovirus 24. Sequential phosphorylations within the full-length sequence of cMyBP-C were determined using top-down and middle-down MS 24. From this work, they concluded that differences exist in posttranslational states between truncated and full-length cMyBP-C recombinant proteins and that these differences may alter both protein structure and function. cMyBP-C has been shown to be phosphorylated by PKA within its M-domain at Ser-275, Ser-284, Ser-304, and Ser-307 in humans 31,57. To further determine phosphorylation sites within cMyBP-C, Kuster et al. implemented MS/MS analysis of semipurified cMyBP-C from human heart tissue and identified a novel GSK3β phosphorylation site at Ser-133 within the cMyBP-C Pro-Ala-rich linker domain 35. This site was not phosphorylated by PKA, demonstrating the involvement of other kinases in the PTM of cMyBP-C.
While PTMs of myofilament proteins are vital for the regulation of cardiac function under normal physiological conditions 58, it is important to understand how PTMs become altered in disease. Yuan et al. suggested the involvement of PTM changes in the onset of myocardial stunning, which was defined by Braunwald and Kloner 59 as the “delayed recovery of regional myocardial contractile function after reperfusion despite the absence of irreversible damage and despite restoration of normal flow.” In a model of regional myocardial stunning in the canine heart, proteomic changes to cMyBP-C were determined using 2D-DIGE that detected altered phosphorylation of cMyBP-C in regional myocardial stunned tissue compared to nonischemic tissue 22. IMAC and MS/MS indicated the presence of three novel phosphorylation sites of the five sites identified in the C1–C2 linker of canine cMyBP-C upon phosphorylation mapping 22. In a rat model of global stunning, they determined an association between stunning and elevated cMyBP-C phosphorylation at a calcium/calmodulin-dependent kinase II site, which was prevented by ischemic preconditioning or low-calcium reperfusion, indicating a vital role for cMyBP-C phosphorylation in myocardial stunning 22. Subsequently, to determine how sarcomeric protein phosphorylation changes with development, Yuan et al. compared the sarcomeric phosphoproteomes of neonatal and adult rat hearts using a proteomics approach 23. Relative protein quantification and phosphorylation changes were determined by 2D-DIGE and phosphoprotein staining, which demonstrated elevated phosphorylation of neonatal cMyBP-C 23. The differential phosphorylation between neonatal and adult cMyBP-C was further characterized with high-resolution linear ion trap-FT (LTQ-FT) MS analysis of titanium dioxide (TiO2) enriched phosphopeptides labeled with 16O/18O during in-gel digestion, as well as by Western blot analysis. Their results indicated increased phosphorylation of cMyBP-C at Ser-295 and Ser-315, but decreased phosphorylation at Ser-320, in neonatal hearts compared to these sites in adult hearts 23. Thus, they concluded that differential phosphorylation changes within the same protein are regulated by development and the prevalent physiological state. Intriguingly, another potential phosphorylation site in cMyBP-C at Ser-297 was not basally phosphorylated in the rat heart. This finding seemed to contradict their earlier study in which Ser-297 phosphorylation was observed in canine hearts following I–R injury 22, indicating that phosphorylation of this site was canine specific or induced by the I–R injury.
cMyBP-C phosphorylation decreases during HF and pathologic hypertrophy 36, while the preservation of cMyBP-C phosphorylation by mutating the phosphorylation sites to aspartic acid sites, mimicking constitute active phosphorylation, appears to be cardioprotective following I–R injury 37. Although previous studies identified novel phosphorylation sites within cMyBP-C, a complete characterization of the cMyBP-C phosphoproteome was missing. To address this, Kooij et al. used two proteomics-based techniques in healthy and end-stage failing hearts to characterize the cMyBP-C phosphoproteome 25. Using LTQ-Orbitrap MS of TiO2-enriched cMyBP-C phosphopeptides following strong cation exchange chromatography, they were able to identify 17 phosphorylation sites on cMyBP-C in vivo, of which 9 were novel 25. Most phosphorylation sites were localized to domains C0–C2 of cMyBP-C. However, the physiological and pathological significance of many of these sites requires further characterization, as they are of uncertain significance and/or low occupancy. Based on the identified phosphorylation sites, SDS-PAGE revealed the M-domain residues Ser-284, Ser-286, and Thr-290 to be highly phosphorylated. Phosphorylation of Ser-284, the most highly phosphorylated among these residues, was significantly decreased in HF 25. These results indicate multiple new avenues by which cMyBP-C PTMs may regulate cardiac contractility and imply novel areas of research for understanding the role of cMyBP-C in the failing heart. Furthermore, the use of proteomics approaches to quantify additional cMyBP-C phosphosite-specific alterations within MI hearts may help to address the current challenges that exist in understanding the relationship between protein phosphorylation and cardiac function.
While cMyBP-C phosphorylation is a critical modification, the identification of other cMyBP-C PTMs through the use of proteomics assays will define novel roles for this protein in the regulation of cardiac function. For example, cMyBP-C has been shown to undergo S-glutathionylation, an oxidative modification, in animal models of diastolic dysfunction that correlates with alterations in crossbridge kinetics and may possibly influence contractile dynamics 60. Upon tetrahydrobiopterin treatment of animals with diastolic dysfunction, a reduction in cMyBP-C glutathionylation, and improved kinetics of crossbridge turnover were observed, suggesting that PTM of myofilament proteins can regulate cardiac relaxation 61. Patel et al. determined the residues within cMyBP-C that were glutathionylated by mimicking S-glutathionylation of myofilament proteins in vitro and using Western blot and MS/MS to analyze these proteins 56. Western blot analysis revealed elevated glutathionylation of cMyBP-C with MS/MS narrowing the glutathionylated residues to cysteines 479, 655, and 627 56. Functionally, it was indicated that S-glutathionylation of cMyBP-C has direct effects on elevating Ca2+-sensitivity of the myofilament 56. In addition to glutathionylation, it has recently been demonstrated that oxidative modification of cMyBP-C may be in the form of carbonylation 62. Using doxorubicin-induced oxidative stress, Aryal et al. demonstrated through MS analysis that cMyBP-C is carbonylated under cardiotoxic conditions in a rat model of spontaneous hypertension 62. During doxorubicin-induced oxidative stress, they confirmed that cMyBP-C is carbonylated and degraded within the proteasome, suggesting a role for cMyBP-C carbonylation in the identification of oxidative stress induced cardiotoxicity 62. Together, these studies demonstrate the utility of proteomics approaches in the identification and characterization of new cMyBP-C PTMs and implicate novel roles for cMyBP-C domains that were never before thought to have functional significance.
8. Conclusion and future directions
The data supporting release of cMyBP-C following MI and the damaging effects of the C0C1f peptide on cardiac function have illuminated the pathogenic roles of this sarcomeric protein following ischemic injury. Therefore, development of a therapeutic strategy that targets C0C1f may provide protection from this cardiac dysfunction. Furthermore, the release of C0C1f following MI has the potential to induce an immune response and the production of cMyBP-C-AAbs that could lead to the progression of autoimmune myocarditis to DCM and HF. However, the production of such AAbs and their pathogenic effects in these disease states remain the subjects of future research.
The use of proteomics approaches to define the PTMs of cMyBP-C and hence, the function of this cardiac protein in health and disease may provide a more complete understanding of the roles of cMyBP-C following MI and in the failing heart. The early release of cMyBP-C into the circulation after MI highlights its role as a potentially new and beneficial biomarker for the early clinical diagnosis of MI when patients arrive at the hospital. While it is evident that the early accurate detection of MI will likely require the use of several biomarkers in a “multimarker” regimen 63,64, the addition of cMyBP-C as a cardiac-specific biomarker of MI has the potential to shorten the time required for clinical diagnosis, allowing for much improved survival outcomes.
Acknowledgments
The work was supported by National Institutes of Health grants R01HL105826 and K02HL114749 to SS.
The authors have declared no conflict of interest.
Glossary
- AAb
autoantibody
- cMyBP-C
cardiac myosin binding protein-C
- cTnI
cardiac troponin-I
- DCM
dilated cardiomyopathy
- EAM
experimental autoimmune myocarditis
- HF
heart failure
- I–R
ischemia-reperfusion
- MI
myocardial infarction
9 References
- 1.Ahmad M. Biomarkers in acute myocardial infarction. J. Clin. Exp. 2012;3:222. [Google Scholar]
- 2.Reichlin T, Hochholzer W, Bassetti S, Steuer S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N. Engl. J. Med. 2009;361:858–867. doi: 10.1056/NEJMoa0900428. [DOI] [PubMed] [Google Scholar]
- 3.Sotolongo RP, Smith ML, Margolis WS. Coronary angioplasty in emergency treatment of myocardial infarction in a community-hospital setting. Tex. Heart Inst. J. 1990;17:31–36. [PMC free article] [PubMed] [Google Scholar]
- 4.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, et al. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012;60:1581–1598. doi: 10.1016/j.jacc.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Kim SE, Lee JH, Park DG, Han KR, et al. Acute myocardial infarction by right coronary artery occlusion presenting as precordial ST elevation on electrocardiography. Korean Circ. J. 2010;40:536–538. doi: 10.4070/kcj.2010.40.10.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Winter RJ, Tijssen JG. Non-ST-segment elevation myocardial infarction: revascularization for everyone. JACC Cardiovasc. Interv. 2012;5:903–905. doi: 10.1016/j.jcin.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Hall TS, Hallen J, Agewall S, Atar D, et al. Changes in diagnosing non-ST-segment elevation myocardial infarction after the introduction of a new high-sensitivity cardiac troponin T assay: a single-centre experience. Clin. Lab. 2012;58:1029–1036. [PubMed] [Google Scholar]
- 8.Ferguson JL. Beckett GJ, Stoddart M, Walker SW, et al. Myocardial infarction redefined: the new ACC/ESC definition, based on cardiac troponin, increases the apparent incidence of infarction. Heart. 2002;88:343–347. doi: 10.1136/heart.88.4.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Chen X, Gao E, MacDonnell SM, et al. Increasing cardiac contractility after myocardial infarction exacerbates cardiac injury and pump dysfunction. Circ. Res. 2010;107:800–809. doi: 10.1161/CIRCRESAHA.110.219220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta S, Prahash AJ, Anand IS. Myocyte contractile function is intact in the post-infarct remodeled rat heart despite molecular alterations. Cardiovasc. Res. 2000;48:77–88. doi: 10.1016/s0008-6363(00)00160-7. [DOI] [PubMed] [Google Scholar]
- 12.Dhalla NS, Kaura D, Liu X, Beamish RE. Mechanisms of subcellular remodelling in post-infarct heart failure. EXS. 1996;76:463–477. doi: 10.1007/978-3-0348-8988-9_28. [DOI] [PubMed] [Google Scholar]
- 13.Govindan S, Sarkey J, Ji X, Sundaresan NR, et al. Pathogenic properties of the N-terminal region of cardiac myosin binding protein-C in vitro. J. Muscle Res. Cell. Motil. 2012;33:17–30. doi: 10.1007/s10974-012-9292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Govindan S, McElligott A, Muthusamy S, Nair N, et al. Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction. J. Mol. Cell. Cardiol. 2012;52:154–164. doi: 10.1016/j.yjmcc.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Razzaque MA, Gupta M, Osinska H, Gulick J, et al. An endogenously produced fragment of cardiac myosin-binding protein C is pathogenic and can lead to heart failure. Circ. Res. 2013;113:553–561. doi: 10.1161/CIRCRESAHA.113.301225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witayavanitkul N, Ait Mou Y, Kuster DW, Khairallah RJ, et al. Myocardial infarction-induced N-terminal fragment of cMyBP-C impairs myofilament function in human myocardium. J. Biol. Chem. 2014;289:8818–8827. doi: 10.1074/jbc.M113.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasahara H, Itoh M, Sugiyama T, Kido N, et al. Autoimmune myocarditis induced in mice by cardiac C-protein. Cloning of complementary DNA encoding murine cardiac C-protein and partial characterization of the antigenic peptides. J. Clin. Invest. 1994;94:1026–1036. doi: 10.1172/JCI117416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumoto Y, Tsukada Y, Miyakoshi A, Sakuma H, et al. C protein-induced myocarditis and subsequent dilated cardiomyopathy: rescue from death and prevention of dilated cardiomyopathy by chemokine receptor DNA therapy. J. Immunol. 2004;173:3535–3541. doi: 10.4049/jimmunol.173.5.3535. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto Y, Park IK, Kohyama K. B-cell epitope spreading is a critical step for the switch from C-protein-induced myocarditis to dilated cardiomyopathy. Am. J. Pathol. 2007;170:43–51. doi: 10.2353/ajpath.2007.060544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leuschner F, Li J, Goser S, Reinhardt L, Ottl R, et al. Absence of auto-antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur. Heart J. 2008;29:1949–1955. doi: 10.1093/eurheartj/ehn268. [DOI] [PubMed] [Google Scholar]
- 21.Kaya Z, Leib C, Katus HA. Autoantibodies in heart failure and cardiac dysfunction. Circ. Res. 2012;110:145–158. doi: 10.1161/CIRCRESAHA.111.243360. [DOI] [PubMed] [Google Scholar]
- 22.Yuan C, Guo Y, Ravi R, Przyklenk K, et al. Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts—evidence for novel phosphorylation sites. Proteomics. 2006;6:4176–4186. doi: 10.1002/pmic.200500894. [DOI] [PubMed] [Google Scholar]
- 23.Yuan C, Sheng Q, Tang H, Li Y, et al. Quantitative comparison of sarcomeric phosphoproteomes of neonatal and adult rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H647–H656. doi: 10.1152/ajpheart.00357.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge Y, Rybakova IN, Xu Q, Moss RL. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc. Natl. Acad. Sci. USA. 2009;106:12658–12663. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kooij V, Holewinski RJ, Murphy AM, Van Eyk JE. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J. Mol. Cell. Cardiol. 2013;60:116–120. doi: 10.1016/j.yjmcc.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sadayappan S. Cardiac myosin binding protein-C: a potential early-stage, cardiac-specific biomarker of ischemia-reperfusion injury. Biomark. Med. 2012;6:69–72. doi: 10.2217/bmm.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin B, Govindan S, Lee K, Zhao P, et al. Cardiac myosin binding protein-C plays no regulatory role in skeletal muscle structure and function. PloS One. 2013;8:e69671. doi: 10.1371/journal.pone.0069671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadayappan S, de Tombe PP. Cardiac myosin binding protein-C: redefining its structure and function. Biophys. Rev. 2012;4:93–106. doi: 10.1007/s12551-012-0067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadayappan S, de Tombe PP. Cardiac myosin binding protein-C as a central target of cardiac sarcomere signaling: a special mini review series. Pflugers Archiv. 2014;466:195–200. doi: 10.1007/s00424-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch. Biochem. Biophys. 1998;358:313–319. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 33.Shaffer JF, Jia W, Lear JA, Harris SP. PKA phosphorylates serine 307 of murine cardiac myosin binding protein-C in vitro. Biophys. J. 2008;96:500a. [Google Scholar]
- 34.Sadayappan S, Gulick J, Osinska H, Barefield D, et al. A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 2011;109:141–150. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuster DW, Sequeira V, Najafi A, Boontje NM, et al. GSK3beta phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication. Circ. Res. 2013;112:633–639. doi: 10.1161/CIRCRESAHA.112.275602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sadayappan S, Gulick J, Osinska H, Martin LA, et al. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, et al. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong CW, Stelzer JE, Greaser ML, Powers PA, et al. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ. Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer BM, Sadayappan S, Wang Y, Weith AE, et al. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophys. J. 2011;101:1661–1669. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barefield D, Kumar M, de Tombe PP. Sadayappan S. Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2014;306:H807–H815. doi: 10.1152/ajpheart.00913.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuster DW, Sadayappan S. MYBPC3's alternate ending: consequences and therapeutic implications of a highly prevalent 25 bp deletion mutation. Pflugers Archiv. 2014;466:207–213. doi: 10.1007/s00424-013-1417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kusuoka H, Marban E. Cellular mechanisms of myocardial stunning. Annu. Rev. Physiol. 1992;54:243–256. doi: 10.1146/annurev.ph.54.030192.001331. [DOI] [PubMed] [Google Scholar]
- 43.Matsumura Y, Kusuoka H, Inoue M, Hori M, et al. Protective effect of the protease inhibitor leupeptin against myocardial stunning. J. Cardiovasc. Pharmacol. 1993;22:135–142. doi: 10.1097/00005344-199307000-00021. [DOI] [PubMed] [Google Scholar]
- 44.Gao WD, Atar D, Liu Y, Perez NG, et al. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ. Res. 1997;80:393–399. [PubMed] [Google Scholar]
- 45.Van Eyk JE, Powers F, Law W, Larue C, et al. Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts: identification of degradation products and effects on the pCa-force relation. Circ. Res. 1998;82:261–271. doi: 10.1161/01.res.82.2.261. [DOI] [PubMed] [Google Scholar]
- 46.Sadayappan S, Greis KD, Robbins J. Phosphorylation-dependent proteolysis and pathogenesis of cardiac myosin binding protein-C. J. Mol. Cell. Cardiol. 2008;44:S44. [Google Scholar]
- 47.Previs MJ, Beck Previs S, Gulick J, Robbins J, et al. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delogu LG, Deidda S, Delitala G, Manetti R. Infectious diseases and autoimmunity. J. Infect. Dev. Ctries. 2011;5:679–687. doi: 10.3855/jidc.2061. [DOI] [PubMed] [Google Scholar]
- 49.Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584–589. doi: 10.1038/nature03723. [DOI] [PubMed] [Google Scholar]
- 50.Doesch AO, Mueller S, Nelles M, Konstandin M, et al. Impact of troponin I-autoantibodies in chronic dilated and ischemic cardiomyopathy. Basic. Res. Cardiol. 2011;106:25–35. doi: 10.1007/s00395-010-0126-z. [DOI] [PubMed] [Google Scholar]
- 51.Govindan S, Kuster DW, Lin B, Kahn DJ, et al. Increase in cardiac myosin binding protein-C plasma levels is a sensitive and cardiac-specific biomarker of myocardial infarction. Am. J. Cardiovasc. Dis. 2013;3:60–70. [PMC free article] [PubMed] [Google Scholar]
- 52.Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined—a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J. Am. Coll. Cardiol. 2000;36:959–969. doi: 10.1016/s0735-1097(00)00804-4. [DOI] [PubMed] [Google Scholar]
- 53.Kuster DW, Cardenas-Ospina A, Miller L, Liebetrau C, et al. Release kinetics of circulating cardiac myosin binding protein-C following cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 2014;306:H547–556. doi: 10.1152/ajpheart.00846.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuster DW, Barefield D, Govindan S, Sadayappan S. A sensitive and specific quantitation method for determination of serum cardiac myosin binding protein-C by electrochemiluminescence immunoassay. J. Vis. Exp. 2013;78:e50786. doi: 10.3791/50786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacquet S, Yin X, Sicard P, Clark J, et al. Identification of cardiac myosin-binding protein C as a candidate biomarker of myocardial infarction by proteomics analysis. Mol. Cell. Proteomics. 2009;8:2687–2699. doi: 10.1074/mcp.M900176-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel BG, Wilder T, Solaro RJ. Novel control of cardiac myofilament response to calcium by S-glutathionylation at specific sites of myosin binding protein C. Front. Physiol. 2013;4:336. doi: 10.3389/fphys.2013.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Copeland O, Sadayappan S, Messer AE, Steinen GJ, et al. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 2010;49:1003–1011. doi: 10.1016/j.yjmcc.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 58.Yuan C, Ravi R, Murphy AM. Discovery of disease-induced post-translational modifications in cardiac contractile proteins. Curr. Opin. Mol. Ther. 2005;7:234–239. [PubMed] [Google Scholar]
- 59.Braunwald E, Kloner RA. The stunned myocardium: prolonged, postischemic ventricular dysfunction. Circulation. 1982;66:1146–1149. doi: 10.1161/01.cir.66.6.1146. [DOI] [PubMed] [Google Scholar]
- 60.Lovelock JD, Monasky MM, Jeong EM, Lardin HA, et al. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ. Res. 2012;110:841–850. doi: 10.1161/CIRCRESAHA.111.258251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeong EM, Monasky MM, Gu L, Taglieri DM, et al. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J. Mol. Cell. Cardiol. 2013;56:44–54. doi: 10.1016/j.yjmcc.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aryal B, Jeong J, Rao VA. Doxorubicin-induced carbonylation and degradation of cardiac myosin binding protein C promote cardiotoxicity. Proc. Natl. Acad. Sci. USA. 2014;111:2011–2016. doi: 10.1073/pnas.1321783111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jurlander B, Clemmensen P, Wagner GS, Grande P. Very early diagnosis and risk stratification of patients admitted with suspected acute myocardial infarction by the combined evaluation of a single serum value of cardiac troponin-T, myoglobin, and creatine kinase MB(mass) Eur. Heart J. 2000;21:382–389. doi: 10.1053/euhj.1999.1760. [DOI] [PubMed] [Google Scholar]
- 64.McCord J, Nowak RM, McCullough PA, Foreback C, et al. Ninety-minute exclusion of acute myocardial infarction by use of quantitative point-of-care testing of myoglobin and troponin I. Circulation. 2001;104:1483–1488. doi: 10.1161/hc3801.096336. [DOI] [PubMed] [Google Scholar]
- 65.van Dijk SJ, Paalberends ER, Najafi A, Michels M, et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- 66.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larsen MR, Trelle MB, Thingholm TE, Jensen ON. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Biotechniques. 2006;40:790–798. doi: 10.2144/000112201. [DOI] [PubMed] [Google Scholar]