

Background: Akt is a key regulator of mTORC1, functioning through phosphorylation of TSC2 and PRAS40.
Results: Downstream of Akt, IKKα directly phosphorylates mTOR to drive mTORC1 activation. Knock-out of IKKα suppresses mTORC1 activation in vivo.
Conclusion: IKKα is important in the activation of mTORC1 via direct phosphorylation.
Significance: Results provide insight into the ability of Akt to promote mTORC1 activity.
Keywords: Akt, Cell Proliferation, Mammalian Target of Rapamycin (mTOR), Phosphatase and Tensin Homolog (PTEN), Phosphorylation, IKK, Raptor
Abstract
The serine/threonine protein kinase Akt promotes cell survival, growth, and proliferation through phosphorylation of different downstream substrates. A key effector of Akt is the mammalian target of rapamycin (mTOR). Akt is known to stimulate mTORC1 activity through phosphorylation of tuberous sclerosis complex 2 (TSC2) and PRAS40, both negative regulators of mTOR activity. We previously reported that IκB kinase α (IKKα), a component of the kinase complex that leads to NF-κB activation, plays an important role in promoting mTORC1 activity downstream of activated Akt. Here, we demonstrate IKKα-dependent regulation of mTORC1 using multiple PTEN null cancer cell lines and an animal model with deletion of IKKα. Importantly, IKKα is shown to phosphorylate mTOR at serine 1415 in a manner dependent on Akt to promote mTORC1 activity. These results demonstrate that IKKα is an effector of Akt in promoting mTORC1 activity.
Introduction
The highly conserved serine/threonine kinase mTOR is a key regulator of metabolism and cell growth. Under dysregulated conditions, mTOR is involved in human diseases such as cancer and metabolic diseases and in aging (1–4). mTOR integrates signals from growth factors, hormones (such as insulin), and nutrients such as amino acids and glucose (1–4). mTOR exists in two structurally distinct complexes, mTORC1 and mTORC2. mTORC1 contains mTOR, Raptor, GβL, and PRAS40 (1). A primary function of mTORC1 is to control cell growth at least partly through its ability to phosphorylate S6K and 4EBP1 as well as IMP2, key regulators of mRNA translation (1, 2, 5, 6). Importantly, mTORC1 activity suppresses the induction of autophagy at least partly through the regulation of the ULK1 kinase (7). mTORC2 contains mTOR, mLST8, Rictor, and mSIN1 (1, 8). A primary function of mTORC2 is the phosphorylation and activation of Akt through phosphorylation at S473 (9) and the phosphorylation of SGK (10–12). Due to clinical importance and a further understanding of key metabolic and growth signaling events, dissection of the pathways regulating mTOR is essential.
Dysregulation of the serine/threonine protein kinase Akt (PKB) underlies the pathology of many human diseases. In cancers, Akt is constitutively activated through activating mutations in PI3K through up-regulated receptor-tyrosine kinase activation or after mutation or loss of PTEN (13, 14). Activated Akt phosphorylates key substrates to regulate different cell signaling pathways to promote cell survival, cell growth, and proliferation and energy metabolism (13, 15). A key effector downstream of Akt is the mTORC1 complex (1, 14–16). It has been shown that activated Akt activates mTORC1 through phosphorylation of tumor suppressor TSC2 to release inhibition of the GTPase Rheb leading to activation of mTORC1 (17–21). Additionally, Akt has been shown to phosphorylate PRAS40 to lead to TORC1 activation (22, 23). PRAS40 functions as a negative regulator of mTORC1 by inhibiting substrate interaction with the kinase complex (24). Additional mechanisms whereby Akt may promote mTORC1 have not been described.
The inducible transcription factor nuclear factor κB (NF-κB) is involved in immune and inflammatory responses and is often activated in human cancer (25–30). The IκB kinase (IKK)2 complex is composed of two catalytic subunits, IKKα and IKKβ, in association with a regulatory subunit IKKγ (NEMO) involved in transcriptional activation of NF-κB by phosphorylating the inhibitory molecule IκBα, leading to its degradation and the subsequent translocation of NF-κB to nucleus (28, 29). Recent studies demonstrated that several cytoplasmic and nuclear proteins distinct from NF-κB and IκBα are phosphorylated by IKKα or IKKβ to promote key growth regulatory responses (31–36). These findings considerably widen our knowledge of the biological roles of these kinases and indicate that a full understanding of the roles of IKKα and IKKβ will require the identification of key phosphorylation targets and the impact of these events on cell growth and metabolism.
We previously reported that IKKα associates with the mTORC1 complex to regulate mTORC1 kinase activity directed to S6K and 4E-BP1 in PTEN-deficient prostate cancer cells in an Akt-dependent manner (37). When IKKα is induced to interact with mTORC1, mTOR reciprocally activates IKK and NF-κB activity (38). Importantly, Hung and co-workers (31) showed that IKKβ phosphorylates TSC1 downstream of TNF to promote mTORC1 activity to drive angiogenesis. Additionally, we showed that IKKα is important for efficient induction of mTORC1 activity downstream of insulin and TNFα in an Akt-dependent manner (39). Taken together, these results indicate that IKKα is directed to control mTORC1 activity after exposure of cells to growth factors and cytokines and in response to Akt activation. Here we address a mechanism to explain the ability of IKKα to activate mTORC1 in the Akt pathway. Data are presented which show that mTOR is a direct substrate for the kinase activity of IKKα, targeting Ser-1415 in mTOR. Phosphorylation of mTOR controlled by IKKα is shown in multiple PTEN null cancer cell lines and in animal with deletion of IKKα. The phosphorylation of mTOR by IKKα promotes mTORC1 kinase activity and reduced affinity between Raptor and mTOR, previously shown to correlate with active mTORC1 (40, 41). The results provide further insight into the mechanism whereby Akt promotes mTORC1 activity and place IKKα as an effector of Akt activity.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies were obtained from the following sources. Antibodies against IKKα, IKKβ, mTOR, and GST were obtained from Upstate Biotechnology. Raptor, Rictor, and GβL antibodies were obtained from Bethyl Laboratories. Anti-HA and anti-FLAG antibodies were obtained from Roche Applied Science and Sigma, respectively. The phospho-IKKα-T23 antibody is from the Abcam (ab38515). Anti-actin was obtained from Calbiochem. The anti-myc (9E-10), anti-tubulin, anti-S6K, anti-PTEN, and control rabbit IgG as well as horseradish peroxidase-labeled anti-mouse and anti-rabbit secondary antibodies were from Santa Cruz Biotechnology. Phosphoserine antibody was from BD Transduction Laboratories. All other antibodies were from Cell Signaling. Purified IKKα protein was from Upstate Biotechnology. Phospho-mTOR-Ser-1415 antibody is generated by with 21st Century Biochemicals. Other reagents were obtained from the following sources. Protease and phosphatase inhibitor cocktails were from Roche Applied Science. CHAPS was from Pierce. Protein A and protein G-agarose beads were from Invitrogen. All radiochemicals used were obtained from PerkinElmer Life Sciences.
Plasmids
GST-IKKα WT and KM were gifts from J. Hutti. All other plasmids below were obtained via Addgene: the pRK5/Myc-mTOR, pRK5/Myc-mTOR-KD, and HA-Raptor vectors were from D. Sabatini; the pRK7/HA-S6K1, FLAG-4E-BP1, GST-S6K, and GST-4E-BP1 were from J. Bleni; the FLAG-mTOR WT and kinase-inactive vectors were from J. Chen.
cDNA Mutagenesis and Sequencing
Site-directed mutagenesis was performed using QuikChange II XL (Stratagene), and cDNA inserts in mutated plasmids were fully sequenced. We generated the following mutations in WT-myc-mTOR backbones: S1415A, S1418A, and S1415A/S1418A. Mutations of S1415A, S1418A, S1415E, and S145A/S1418A in FLAG-mTOR were generated by DNA Express Inc.
Cell Lines, Cell Culture, Transient Transfection
IKKα wild type and IKKα−/− MEFs were provided by I. Verma and M. Karin. Eker rat embryo fibroblast TSC2 wild type and TSC2−/− cells were from J. Cheng and originally from R. Yeung. HEK293T and HeLa as well as prostate cancer cell lines PC3 and LNCaP and other PTEN mutated cancer cell lines were from American Type Culture Collection (ATCC). All cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mmol/liter glutamine, and 100 units/ml penicillin and streptomycin (Invitrogen). Transfections were done using Polyfect Transfection Reagent (Qiagen) or Lipofectamine Plus (Invitrogen) following the manufacturer's instructions. 3–4 h after transfection, cells were recovered in full serum for 36 h or in full serum for 24 h and then serum-starved for 16–24 h as indicated.
RNA Interference
Small interfering RNA (siRNA) SMARTpool IKKα, Akt1, and Akt2 were from Dharmacon. Each of these represents four pooled SMART-selected siRNA duplexes that target the indicated gene. PC3 cells were transfected with the indicated SMARTpool siRNA or nonspecific control pool using DharmaFECT 1 reagent (Dharmacon) according to the manufacturer's instructions. In brief, 20 nmol/liter final concentration of siRNA was used to transfect cells at 60–70% confluency. Twenty-four hours after transfection, cells were recovered in full serum or were serum-starved 16 h before harvest. Cells were harvested 48–72 h after siRNA transfection.
Cell Lysis, Immunoblotting, and Coimmunoprecipitations
Cells growing in 100-mm dishes were rinsed twice with cold PBS and then lysed on ice for 20 min in 1 ml of lysis buffer (40 mmol/liter HEPES (pH 7.5), 120 mmol/liter NaCl, 1 mmol/liter EDTA, 10 mmol/liter pyrophosphate, 10 mmol/liter glycerophosphate, 50 mmol/liter NaF, 0.5 mmol/liter orthovanadate, and EDTA-free protease inhibitors; Roche Applied Science) containing 1% Triton X-100. After centrifugation at 13,000 × g for 10 min, samples containing 20–50 μg of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and proteins were transferred to Pure Nitrocellulose Membrane (Bio-Rad), blocked in 5% nonfat milk, and blotted with the indicated antibodies. For immunoprecipitation experiments, the lysis buffer contained 0.3% CHAPS instead of 1% Triton. Four micrograms of the indicated antibodies were added to the cleared cellular lysates and incubated with rotation for 6–16 h. Then 25 μl of protein G-agarose were added, and the incubation was continued for 1 h. Immunoprecipitates captured with protein G-agarose were washed 3 times with the CHAPS lysis buffer and twice by wash buffer A (50 mmol/liter HEPES, (pH 7.5), 150 mmol/liter NaCl) and boiled in 4× SDS sample buffer for Western blot.
In Vitro mTOR Kinase Assay
Transfected HEK293T cells were grown in 100-mm dishes for 48 h in DMEM containing 10% FBS and lysed in 1 ml of lysis buffer with 0.3% CHAPS. Half of total cell lysate was incubated with anti-mTOR or FLAG antibody for 3 h followed by another hour of incubation with 25 μl of protein G-agarose beads. Immunoprecipitates were washed twice by lysis buffer, twice by wash buffer B (20 mmol/liter Tris (pH 7.5), 500 mmol/liter NaCl, 1 mmol/liter EDTA, 20 mmol/liter β-glycerophosphate, 5 mmol/liter EGTA, 1 mmol/liter DTT, 1 mmol/liter orthovanadate, 40 mg/ml phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml leupeptin, 5 μg/ml pepstatin), once with wash buffer C (10 mmol/liter HEPES (pH 7.4), 50 mmol/liter glycerophosphate, 50 mmol/liter NaCl, 1 mmol/liter DTT, 1 mmol/liter orthovanadate, 40 mg/ml PMSF, 10 μg/ml leupeptin, 5 μg/ml pepstatin), and once with mTOR kinase assay buffer without ATP (10 mmol/liter HEPES (pH 7.4), 50 mmol/liter NaCl, 50 mmol/liter glycerophosphate, 1 mmol/liter DTT, 10 mmol/liter MgCl2, 4 mmol/liter MnCl2). Kinase assay toward recombinant GST-S6K1 (amino acids 308–400) in washed immunoprecipitates was done for 30 min at 30 °C in 30 μl of mTOR kinase buffer with 100 μmol/liter ATP unlabeled and 10 μCi [γ-32P]ATP (PerkinElmer Life Sciences). To stop the reaction, 6 μl of 4× SDS sample buffer was added to each reaction, which was boiled for 10 min. The reaction was then separated by 4–12% SDS-PAGE and transferred to polyvinylidene difluoride membranes. 32P incorporated into GST-S6K was assessed by autoradiography. In a cold in vitro kinase assay to GST-S6K, phosphorylation S6K was detected by phosphor-S6K-Thr-389 antibody.
In Vitro IKKα Kinase Assay
To map the phosphorylation sites in mTOR, the fragments of the mTOR coding sequence were cloned into pGEX vector (GE Healthcare). Purified GST-mTOR fusion proteins were immobilized to glutathione-agarose for kinase assay. Kinase assays were performed following a previously described protocol (Upstate Biotechnology). Kinase activity was determined by incubating purified IKKα with GST-mTOR fragments, GST-IκB, or immunoprecipitates of mTOR and Raptor from HEK 293T cells as indicated in the presence of 1 μCi ml−1 [γ-32P]ATP or cold ATP (100 μm) for 30 min at 30 °C. Reactions were resolved by SDS-PAGE (4–12%) and processed for autoradiography or protein immunoblotting.
Mass Spectrometric Analysis to Identify mTOR Phosphorylation Sites
Purified GST-mTOR-(1351–1650) protein was incubated with recombinant IKKα for cold kinase assay, resolved by SDS-PAGE, and stained with Coomassie Blue, excised from the gel, digested with trypsin, and analyzed by tandem mass spectrometry by Dr. John Asara of the Beth Israel Deaconess Medical Center.
Cell Proliferation Assays
The indicated transfected cells were plated in 6-well plates at a density of 2.0 × 104 cells/well. Cells were trypsinized and counted using a hematocytometer every day until confluency. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays were performed according to the manufacturer's recommendations (Promega, Madison, WI). The cells were plated in 96-well microtiter plates at a density of 1.0 × 103 cells/well in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. The number of cells at 1, 2, and 3 days was determined using a cell counter and the colorimetric CellTiter96 AQueous (MTS) assay (Promega). Results were depicted as absorbance at 490 nm as a function of time.
In Vivo Experimentation
The conditional IKKαloxp/loxp mice are described in Gareus et al. (42). Pb-cre mice were from Dr. Van Dyke's group. IKKαloxp/loxp/C+ males were bred to IKKαloxp/loxp females to obtain the IKKαloxp/loxp/C+ genotype. Male IKKαloxp/loxp/C+ mice between 6 and 15 weeks of age were used for experiments. All animal procedures were reviewed and approved by the University of North Carolina Institutional Animal Care and Use Committee.
Statistics
Data from the in vitro experiments are expressed as the mean ± S.E. from a minimum of three independent experiments. Comparison between groups were carried out by two-way analysis of variance or Student's t test, and a p value of <0.05 was considered significant.
RESULTS
IKKα Activates mTORC1 in a Kinase-dependent Manner
Our previous studies demonstrating the ability of IKKα to promote mTORC1 activity (37–39) prompted an examination of whether IKKα regulates mTORC1 through its kinase activity. To address this issue, S6K, a known mTOR substrate, was expressed as an HA-tagged version with FLAG-tagged IKKα wild type or kinase-inactive forms. Results from this experiment revealed that expression of IKKα wild type, but not a kinase mutant, enhances S6K phosphorylation at Thr-389 (Fig. 1A). Similarly, phosphorylation of 4E-BP1, another mTOR substrate, was enhanced with wild type IKKα expression but not with the kinase-dead variant (Fig. 1B). Next, we determined whether IKKα affects endogenous S6K phosphorylation and found that wild type IKKαsignificantly promoted, but kinase mutant IKKα inhibited, endogenous S6K phosphorylation in HEK 393T cells (Fig. 1C). These results demonstrate that IKKα activates mTOR in a kinase-dependent manner. To determine whether IKKα functions to promote mTOR activity through regulation of TSC2, it was determined whether IKKα affects the hyperactive mTORC1 signaling found in TSC2-null cells in which Rheb activates mTORC1 activity. It would be predicted that IKKα could not modulate mTORC1 activity in TSC2 null cells if IKKα regulates mTORC1 through TSC2. First, IKKα expression was silenced by siRNA-directed knockdown, and endogenous phosphorylation of S6K was analyzed. The data demonstrate that knockdown of IKKα significantly impaired mTORC1 activity in these cells (Fig. 1D). Moreover, expression of IKKα WT, but not kinase-inactive IKKα, promotes mTORC1 activity in both TSC2 wild type and TSC2-null cells (Fig. 1E). Collectively, the data indicate that IKKα activates mTORC1 in a kinase-dependent manner that is at least partly independent of TSC2. In addition, we tested the effects of wild type and kinase-inactive IKKα on insulin-induced mTORC1 activity. Endogenous IKKα was knocked down with siRNA (data not shown), and wild type and kinase-inactive FLAG-IKKα was transiently transfected into HeLa cells. As shown in Fig. 1F, insulin induces mTORC1 activity in IKKα wild type, but not the mutant, transfected cells (Fig. 1F). These data indicate that IKK also regulates insulin-induced mTORC1 activity in a kinase-dependent manner.
FIGURE 1.

IKKα activates mTORC1 in a kinase-dependent manner independent of TSC2. A, effect of wild type and mutant IKKα on S6K phosphorylation. HEK 293T cells were transfected with HA-S6K, FLAG-IKKα wild type, or IKKα mutants, and HA immunoprecipitates (IP) and whole cell lysates (WCL) were analyzed with the indicated antibodies. The bands of phospho-S6K and S6K were quantified, and the ratio of pS6K/S6K was measured as indicated. The experiments were carried out on three separate occasions. B, effect of wild type and mutant IKKα on 4E-BP1 phosphorylation. HEK 293T cells were transfected with FLAG-4E-BP1, GST-IKKα wild type, or IKKα mutants, and FLAG immunoprecipitates and whole cell lysates were analyzed with the indicated antibody. The bands phospho-4E-BP1 and FLAG-4E-BP1 were quantified, and the ratio of p-4E-BP1/4E-BP1 was measured as described in A. Results are representative of three experimental repetitions. C, effect of wild type and mutant IKKα on endogenous S6K phosphorylation. HEK 293 cells were transfected with FLAG-IKKα wild type or IKKα mutants, and endogenous phospho-S6K, S6K, and expression of FLAG-IKKα were detected. The bands phospho-S6K and S6K were quantified, and the ratio of pS6K/S6K was measured as described in A. The results are representative of three experimental repetitions. D, Eker rat embryo fibroblast TSC2−/− cells were transfected with siRNA control or siIKKα, and endogenous phospho-S6K and expression of IKKα and Actin were detected. E, Eker rat embryo fibroblast TSC2+/+ and TSC2−/− cells were transfected with FLAG-IKKα wild type or IKKα mutants, and endogenous phospho-S6K, S6K, and expression of FLAG-IKKα was detected. The results are representative of three experimental experiments. F, HeLa cells were transfected with siRNA IKKα for 48 h and then expressed with FLAG-IKKα wild type or IKKα mutants, the cells were serum-deprived (16 h), incubated in the absence or presence of insulin (100 nm) for 15–30 min, and whole cell lysates were analyzed with indicated antibodies. The results are representative of three experimental repetitions.
IKKα Phosphorylates mTOR in Vitro and in Vivo
Previous studies demonstrated that IKKα interacts with mTORC1 downstream of active Akt (37), and the results above demonstrate that IKKα controls mTORC1 through its kinase activity. These results prompted us to investigate whether IKKα regulates mTOR through phosphorylation of a component of the mTORC1 complex. To this end it was determined whether IKKα can directly phosphorylate mTOR or Raptor. As expected, an antibody to mTOR immunoprecipitated mTOR, Raptor, and Rictor (Fig. 2A). An in vitro phosphorylation assay using the immunoprecipitate with purified IKKα led to phosphorylation of mTOR but not Raptor or Rictor. Similarly, an antibody to Raptor immunoprecipitated mTOR and Raptor but not Rictor. In vitro phosphorylation of this immunoprecipitate using IKKα yielded phosphorylation of mTOR but not Raptor or Rictor (Fig. 2A). To rule out the possibility that the phosphorylation of mTOR is due to mTOR autophosphorylation, FLAG-tagged wild type and kinase-inactive mTOR were expressed in 293T cells and immunoprecipitated with the FLAG antibody. As before, an in vitro IKKα kinase assay was performed which demonstrated that both wild type mTOR and kinase-inactive mTOR were phosphorylated by IKKα to a similar level indicating that the induced phosphorylation is not due to mTOR autophosphorylation (Fig. 2B). It was determined whether IKKα could directly phosphorylate mTOR in vitro in a nonradioactive kinase assay. As shown in Fig. 2C, recombinant IKKα phosphorylated immunoprecipitated mTOR as detected through recognition with an anti-phosphoserine antibody. Similarly, both immunoprecipitated WT and kinase-inactive mTOR were phosphorylated by IKKα (Fig. 2D). These data demonstrate that IKKα can phosphorylate mTOR in vitro. To determine whether IKKα phosphorylates mTOR in vivo, IKKα−/− MEFs were transfected with IKKα, and endogenous mTOR was subsequently immunoprecipitated. As with the in vitro studies, the anti-phosphoserine antibody showed significantly enhanced reactivity for mTOR after IKKα transfection (Fig. 2E). Previously we demonstrated that the PTEN null prostate cancer cell line PC3 exhibited strong mTOR activity that is controlled partly by IKKα (37). Consistent with this, endogenous mTOR exhibits phosphorylation as recognized by the anti-phosphoserine antibody, and this phosphorylation is reduced by siRNA knockdown of IKKα (Fig. 2F). These results indicate that mTOR is a direct substrate of the kinase activity of IKKα.
FIGURE 2.

IKKα phosphorylates mTOR in vitro and in vivo. A, IKKα phosphorylates mTOR in vitro. Endogenous mTOR or Raptor was immunoprecipitated (IP) with anti-mTOR or anti-Raptor from 293T cells and incubated with recombinant active IKKα and [γ-32P]ATP. Autoradiography was performed followed by immunoblotting with anti-mTOR, Raptor, and Rictor. B, FLAG-mTOR wild type and kinase dead (KD) were immunoprecipitated by anti-FLAG from 293T cells transfected and incubated with recombinant active IKKα and [γ-32P]ATP. Autoradiography was performed followed by immunoblotting with indicated antibodies. C, nonradioactive in vitro kinase assay. Endogenous mTOR was immunoprecipitated by anti-mTOR from 293T cells and probed with anti-phosphoserine antibody followed by anti-mTOR. D, nonradioactive in vitro kinase assay. FLAG-mTOR wild type and kinase dead were immunoprecipitated by anti-FLAG from 293T cells and blotted with anti-phospho-serine antibody followed by anti-FLAG. E, IKKα induces mTOR phosphorylation. IKKα−/− MEFs were transfected with HA-IKKα and lysed, and the endogenous mTOR was immunoprecipitated by anti-mTOR and blotted with anti-phosphoserine antibody followed by anti-mTOR. The whole cell lysates (WCL) also were tested by indicated antibodies. F, knockdown of IKKα decreases mTOR phosphorylation. PC3 cells were transfected with siRNA against IKKα, lysed, and immunoprecipitated with anti-mTOR. mTOR immunoprecipitate and whole cell lysates were blotted with the indicated antibodies.
IKKα Phosphorylates mTOR at Serine 1415
To identify a potential phosphorylation site(s) for IKKα on mTOR, nine fragments of mTOR that encompass the full-length protein were produced as GST fusions and were used as substrates in an IKKα-driven in vitro kinase assay. Results from this approach showed that the 1351–1650 fragment of mTOR was phosphorylated by IKKα, whereas other fragments were not phosphorylated significantly (Fig. 3A). When normalized to input, IκBα exhibits slightly higher phosphorylation by IKKα than mTOR (Fig. 3B). Moreover, further experimentation demonstrated that the purified wild type GST-IKKα, but not kinase-inactive IKKα, phosphorylates mTOR-(1351–1650) (Fig. 3C). These data ruled out the possibility that a contaminating kinase could phosphorylate mTOR. Using an in vitro IKKα kinase approach, mTOR-(1351–1650) was phosphorylated (nonradioactively) and was gel-purified for mass spectrometry analysis. The results demonstrated that both serine 1415 and serine 1418 of mTOR were phosphorylated by IKKα in vitro (Fig. 3D). Serines 1415 and 1418 are located in a conserved region of mTOR (Fig. 3E), and the sequence surrounding Ser-1415 exhibits homology with the human Foxo3a IKK site (33, 43). Mutation of Ser-1415 and Ser-1418 to alanines blocked in vitro phosphorylation of mTOR by IKKα (Fig. 3F), suggesting that this region is the predominant site of IKKα phosphorylation on mTOR. To investigate mTOR phosphorylation by IKKα in vivo, a rabbit polyclonal antibody against a peptide of mTOR including phospho-Ser-1415 was generated (p-mTOR-Ser-1415). Immunoblotting of exogenous immunoprecipitated WT and mutant mTOR revealed that the phospho-Ser-1415 antibody recognized the wild type protein but not mTOR mutated in serines 1415 and 1418 (Fig. 3G). PC3 prostate cancer cells have a high level of basal mTORC1 activity due to PTEN deletion and subsequent activation of Akt. Previously we showed that PC3 cells utilize IKKα to promote mTORC1 activity downstream of Akt (37). To determine if IKKα is involved in endogenous mTOR Ser-1415 phosphorylation, IKKα expression was silenced in PC3 cells, and endogenous Ser-1415 phosphorylation was detected. The results showed that Ser-1415 mTOR phosphorylation in PC3 cells is reduced when IKKα expression is knocked down with siRNA. Moreover, the decrease of phospho-mTOR-Ser-1415 is consistent with the reduction of phospho-S6K (Fig. 3H). Virtually identical results were found in other PTEN-null cancer cell lines (LNCaP, U87, Jurkat, K562, and U937) when IKKα expression was knocked down (Fig. 3H). Taken together, the results demonstrate that mTOR serine 1415 is a primary site of IKKα phosphorylation, although we cannot eliminate the possibility that there are other residues on mTOR that can be phosphorylated by IKKα.
FIGURE 3.

IKKα phosphorylates mTOR at serine 1415 in vitro and in vivo. A, IKKα phosphorylates mTOR in vitro. Recombinant IKKα was incubated with GST-mTOR fragments for in vitro, radioactive IKKα kinase assays. Proteins from this assay were blotted with the indicated antibodies. WB, Western blot. B, IKKα phosphorylates GST-mTOR fragment and GST-IκBα in vitro. Recombinant IKKα was incubated with the indicated GST-mTOR fragments or GST-IκBα for in vitro IKKα kinase assays. C, purified IKKα used in A and B, and GST-IKKα wild type and kinase mutant were analyzed relative to their ability to phosphorylate GST-mTOR-(1351–1650). D, Ser-1415 and Ser-1418 are the primary direct IKKα phosphorylation sites in vitro. Purified GST-mTOR-1351–1650 was incubated with active IKKα for phosphorylation, and phosphorylation site mapping was determined by mass spectrometry. E, consensus IKKα phosphorylation sites on human mTOR and alignment with conserved sites on mouse and rat mTOR compared with human FOXO3a and IKK phosphorylation motif. F, IKKα phosphorylates mTOR in serine 1415 in vitro. Myc-tagged mTOR wild type and S1415A/S1418A mutants were immunoprecipitated (IP) by anti-Myc from 293T cells and used as substrates for in vitro kinase assay using recombinant active IKKα. Autoradiography was performed followed by immunoblotting with anti-Myc. G, HEK293T cells were cotransfected with Myc-mTOR (WT or S1415A) and FLAG-IKKα (WT) as indicated. The immunoprecipitates of Myc were analyzed with the phospho-mTOR-Ser-1415 antibody followed by FLAG antibody. WCL, whole cell lysates. H, PC3 and other PTEN-mutated cancer cell lines were transfected with siRNA against IKKα, and mTOR immunoprecipitates and whole cell lysates were analyzed with the indicated antibodies. I, wild type and IKKαLoxP/LoxP PB-Cre+ mice of 15 weeks of age were injected with insulin (intraperitoneally, 1 unit/kg) for 30 min, and protein lysates obtained from prostates were analyzed with the indicated antibodies.
Previously we found that IKKα also contributes to mTORC1 activity downstream of insulin and TNF in a manner dependent on Akt (39). To determine whether IKKα is involved in insulin-induced mTOR activation in vivo, wild type and prostate IKKα-deleted mice were treated with insulin for 30 min. The results showed that insulin treatment led to significant phosphorylation of mTOR (Ser-1415) and S6K (Thr-389) in wild type mice but not in IKKα-deleted mice. The data indicate that insulin induces IKKα to phosphorylate mTOR to promote mTORC1 activity in mice (Fig. 3I).
Phosphorylation of mTOR at Ser-1415 by IKKα Promotes mTORC1 Kinase Activity
Given the involvement of IKKα in the activation of mTORC1 and its induction of phosphorylation of mTOR, we next examined whether the promotion of mTORC1 activity by IKKα is through phosphorylation of mTOR. To compare WT and S1415A mTOR, a transient expression assay was performed using exogenously expressed HA-S6K as a marker of mTOR activity. Expression of wild type mTOR enhanced S6K phosphorylation, as expected; however, expression of mTOR S1415A or S1415A/S1418A led to the inability of mTOR to promote S6K phosphorylation (Fig. 4A, left panel). Mutation of mTOR at Ser-1418 did not block mTOR activity, consistent with a key role for Ser-1415 and not Ser-1418 phosphorylation. To address whether it is the mutation of Ser-1415 that affects mTOR activity, we generated a phosphomimetic mutant (mTOR S1415E). The ability of mTOR wild type, mTOR-S1415A (A-form), and a phosphomimetic mutant of mTOR-S1415E (E-form) to phosphorylate S6K was then compared. The results demonstrated that the E-form of mTOR has stronger ability to promote S6K phosphorylation as compared with wild type mTOR (Fig. 4A). As before, mTOR S1415A did not promote S6K phosphorylation. In addition, WT mTOR was activated by insulin to induce phosphorylation of exogenous S6K, whereas mTOR S1415A was not (Fig. 4B). Moreover, expression of wild type mTOR increased, whereas the A-form of mTOR was functionally impaired relative to the ability to induce endogenous S6K phosphorylation (Fig. 4C). We then compared wild type mTOR with the E-form relative to induction of endogenous S6K phosphorylation (Fig. 4D). The results demonstrate that the E-form phosphomimetic is more active than wild type mTOR in promoting endogenous S6K phosphorylation. Because PC3 cells exhibit potent mTOR activity dependent on elevated Akt activity, we chose to knock down endogenous mTOR in these cells (Fig. 4E, left panel) and then re-express mTOR wild type, S1415A, and S1415A/S1418A. Expression of WT mTOR promoted S6K phosphorylation in PC3 cells, whereas S1415A expression was ineffective at promoting this response (Fig. 4E, right panel). Interestingly, and consistent with previous results, mutation of Ser-1418 did not significantly affect the ability of mTOR to phosphorylate S6K (Fig. 4E). These results indicate that IKKα enhances the ability of mTOR to phosphorylate S6K through phosphorylation of mTOR at serine 1415 but not serine 1418. In further experiments, wild type mTOR enhanced, but S1415A mTOR abolished, IKKα-induced exogenous S6K phosphorylation (data not shown). These data indicate that IKKα promotes mTORC1 activity through direct phosphorylation of mTOR Ser-1415.
FIGURE 4.

IKKα regulates mTORC1 activity through phosphorylation of mTOR at serine 1415. A, mutation of IKKα phosphorylation site (S1415A) decreases mTOR activity. HA-S6K was cotransfected with wild type or various mTOR mutants in HEK293T cells as indicated. Phosphorylation of S6K-Thr-389 was determined in conjunction with expression levels of S6K, mTOR, and IKKα. IP, immunoprecipitated; WCL, whole cell lysates. B, IKKα phosphorylation of mTOR is involved in insulin-induced mTOR activation. HeLa cells were cotransfected with HA-S6K and FLAG-tagged wild type or various mTOR mutants as indicated, serum-starved overnight, stimulated with insulin, lysed, and analyzed with phospho-S6K-Thr-389 and other antibodies. C, mutation of IKKα phosphorylation sites with alanine substitution blocks endogenous mTOR activity. HEK 293T cells were transfected with wild type or various mTOR mutants as indicated. Endogenous phosphorylation of S6K-Thr-389 and Akt-Ser-473 was determined in conjunction with expression levels of S6K and FLAG-mTOR. D, HEK 293T cells were transfected with wild type or various mTOR mutants as indicated. Endogenous phosphorylation of S6K-Thr-389 was determined in conjunction with expression levels of S6K and FLAG-mTOR. E, PC3 cells were transfected with siRNA against mTOR and then wild type or various mTOR mutants as indicated 48 h after siRNA transfection, lysed, and analyzed with the indicated antibodies. F, expression of IKKα enhances in vitro mTOR kinase activity. HEK293T cells were cotransfected with HA-IKKα and FLAG-mTOR. FLAG-mTOR was IP with antibody, and mTOR kinase activity toward GST-S6K was determined in the immunoprecipitates. G, mutation of IKKα Ser-1415 by alanine substitution decreases mTOR kinase activity. FLAG-mTOR (WT and S1415A) was transfected in HEK293T cells and lysed. Kinase activity of FLAG immunoprecipitates toward GST-S6K were measured by phospho-S6K-Thr-389 antibody.
We next investigated the role of phosphorylation of Ser-1415 mTOR by IKKα in regulating catalytic activity of mTORC1. To examine whether IKKα promotes mTOR kinase activity, HEK293T cells were cotransfected with FLAG-mTOR and HA-IKKα, and FLAG-mTOR was immunoprecipitated for an in vitro kinase assay using GST-S6K. Phosphorylation of S6K (Thr-389) was significantly increased with mTOR co-transfected with IKKα relative to mTOR without IKKα cotransfection (Fig. 4F). To compare wild type and S1415A mTOR in phosphorylating GST-S6K in vitro, kinase assays of the immunoprecipitated wild type mTOR and mTOR A1415A were performed using GST-S6K as the substrate, and the phosphorylation of Thr-389 were detected by the phospho-S6K antibody. A dramatic reduction of S6K Thr(P)-389 was observed with S1415A mTOR as compared with wild type mTOR (Fig. 4G). These data indicate that phosphorylation of mTOR Ser-1415 promotes mTOR catalytic activity.
IKKα-dependent Effects on mTOR and Raptor Interaction
Previous studies have observed that nutrients such as amino acids and glucose, which activate mTORC1, lead to a reduction in the affinity (but not stoichiometry) of mTOR-Raptor association, as measured by coimmunoprecipitation (40, 41). These observations suggest that a conformational change or modification within the mTORC1 complex is related to mTORC1 activity. To determine whether IKKα modulates mTOR-raptor interaction, IKKα−/− MEFs and HEK293T cells were transfected with wild type HA-IKKα, and mTOR was immunoprecipitated from the cell lysates. The results demonstrate that IKKα destabilizes the interaction of mTOR with Raptor but did not affect mTOR-GβL interaction (Fig. 5A). Additionally, IKKα expression did not change mTOR-Rictor interaction in the mTORC2 complex (Fig. 5A). Next, to determine if IKKα could alter mTOR-raptor interaction in vitro, recombinant IKKα was incubated with mTOR immunoprecipitated from HEK 393T cells in an in vitro IKKα kinase assay. The results demonstrated that the affinity of interaction of mTOR and Raptor decreased in the presence of IKKα (Fig. 5B) consistent with the observation found in vivo. Further experiments showed that expression of wild type IKKα, but not kinase-inactive IKKα, weakened the mTOR-raptor interaction (Fig. 5C). To further address the ability of IKKα to modulate mTOR-raptor interaction, HA-Raptor was transfected with WT or S1415A FLAG-mTOR. Results from this experiment showed that wild type mTOR exhibits weaker interaction with Raptor as compared S1415A mTOR (Fig. 5D). Moreover, the E-form mTOR shows reduced association with exogenous and endogenous Raptor as compared with wild type mTOR (Fig. 5, E and F). These results imply that IKKα regulates mTOR-raptor interaction through phosphorylation of mTOR. To address this hypothesis, IKKα was transfected with myc-mTOR and HA-Raptor in 293T cells, and mTOR was immunoprecipitated. These data revealed that IKKα-induced mTOR phosphorylation was accompanied by a decreased mTOR-Raptor association (Fig. 5G). Consistent with this result, knockdown of IKKα decreased mTOR Ser-1415 phosphorylation and enhanced mTOR-Raptor interaction (Fig. 5H). Conversely, overexpression of IKKα increased mTOR phosphorylation and weakened the mTOR-Raptor interaction (Fig. 5I). These data indicate that IKKα-induced Ser-1415 phosphorylation promotes a reduced affinity of Raptor within the mTORC1 complex.
FIGURE 5.

IKKα phosphorylation of mTOR at serine 1415 modulates association with Raptor downstream of Akt. A, IKKα activity modulates mTOR-Raptor interaction. MEFs IKKα−/− and 293 cells were transfected with different amounts of HA-IKKα in full serum. Lysates were immunoprecipitated (IP) with anti-mTOR and blotted with mTOR, raptor, Rictor, and GβL antibodies. B, IKKα weakens mTOR-Raptor interaction in vitro. mTOR immunoprecipitates from HEK293T cells were incubated with recombinant IKKα and unlabeled ATP in IKK kinase buffer for 30 min and washed with lysis buffer three times, blotted with mTOR, Raptor, Rictor, and GβL antibodies, respectively. C, HEK293 cells were transfected with FLAG-IKKα (WT or mutant), lysed, and immunoprecipitated with anti-mTOR and blotted with mTOR and Raptor antibodies. D, 293 cells and PC3 cells were cotransfected with FLAG-mTOR WT or the S1415A mutant with HA-Raptor, immunoprecipitated with anti-FLAG, and blotted with FLAG and HA antibodies, respectively. WCL, whole cell lysates. E, 293 cells were cotransfected with FLAG-mTOR WT or S1415E with HA-Raptor, immunoprecipitated with anti-FLAG, and blotted with FLAG and HA antibodies, respectively. F, 293 cells were cotransfected with FLAG-mTOR WT or S1415E with HA-Raptor, immunoprecipitated with anti-FLAG, and blotted with FLAG and HA antibodies, respectively. G, IKKα mediates mTOR phosphorylation and mTOR-Raptor interaction in PC3 cells. siRNA to IKKα was transfected. Lysates were immunoprecipitated with anti-mTOR and blotted with phospho-mTOR-Ser-1415 antibody and followed by other antibodies. Additionally, whole cell lysates were analyzed with the indicated antibodies. H, IKKα mediates mTOR phosphorylation and mTOR-Raptor interaction in PC3 cells. siRNA to IKKα was transfected. Lysates were immunoprecipitated with anti-mTOR and blotted with phospho-mTOR-Ser-1415 antibody and followed by other antibodies. Additionally, whole cell lysates were analyzed with indicated antibodies. I, IKKα mediates mTOR phosphorylation and mTOR-Raptor interaction in PC3 cells. HA-IKKα was transfected in PC3 cells. Lysates were immunoprecipitated with anti-mTOR and blotted with phospho-mTOR-Ser-1415 antibody and followed by other antibodies. Additionally, whole cell lysates were analyzed with the indicated antibodies.
IKKα Modulates mTOR-Raptor Interaction Downstream of Akt
Our previous studies demonstrated that Akt promotes IKKα association with mTORC1, which prompted us to investigate whether Akt controls IKKα phosphorylation and regulation of mTORC1. To address this point, starved HeLa cells were treated with insulin, and mTOR association with Raptor and IKKα was measured. Results from this experiment demonstrated that insulin induces reduced affinity of interaction between Raptor and mTOR while inducing the association between IKKα and mTOR, which are consistent with phosphorylation of both Akt and S6K (Fig. 6A). Inhibition of PI3K/Akt suppresses the reduced interaction between mTOR and Raptor and blocked the association between IKKα and mTOR (Fig. 6B) while blocking Akt phosphorylation as well as the Akt-dependent phosphorylation of TSC2. IKKα was expressed with tagged mTOR and Raptor in HeLa cells, and stimulation with insulin led to enhanced phosphorylation of mTOR at Ser-1415 along with enhanced interaction between IKKα and mTOR (Fig. 6C). Inhibition of PI3K/Akt blocked the insulin-induced phosphorylation of mTOR and promoted mTOR-Raptor interaction (Fig. 6D). These data indicate that insulin, functioning through Akt, controls IKKα association with and phosphorylation of mTOR to activate mTORC1. To further address the ability of Akt to regulate mTORC1 activity and phosphorylation, additional experiments were performed. Inhibition of PI3K/Akt in PC3 cells reduced mTOR phosphorylation at Ser-1415 and promoted Raptor interaction with mTOR (Fig. 6E). Expression of PTEN in the PTEN-negative PC3 cells reduces mTOR Ser-1415 phosphorylation and leads to enhanced mTOR-Raptor interaction (Fig. 6F). Knockdown of either Akt1 or Akt2 by siRNA decreased mTOR phosphorylation at Ser-1415 and enhanced mTOR-Raptor interaction (Fig. 6G). These data demonstrated that the IKKα-mediated mTOR-Raptor interaction is regulated by Akt.
FIGURE 6.

IKKα-mediated mTOR-Raptor interaction is regulated by Akt. A, HeLa cells were serum-deprived (16 h), incubated in the absence or presence of insulin (100 nm) for 15–30 min, and lysed. mTOR immunoprecipitates (IP) and whole cell lysates (WCL) were immunoblotted as indicated. B, HeLa cells were serum-deprived (16 h), pretreated with or without LY 294000 (LY), incubated in the absence or presence of insulin for 30 min, and lysed. mTOR immunoprecipitates and whole cell lysates were immunoblotted as indicated antibodies. C and D, HeLa cells were cotransfected with Myc-mTOR, HA-Raptor, and FLAG-IKKα and then serum-deprived (16 h), pretreated with or without LY 294000, incubated in the absence or presence of insulin for 30 min as indicated, and mTOR immunoprecipitates and whole cell lysates were immunoblotted as the indicated antibodies. E, PI3K inhibitor, LY 294000, blocks IKKα regulation of mTORC1. PC3 cells were treated with LY 294000 for the indicated times, lysed, immunoprecipitated with mTOR antibody, and immunoblotted with antibodies as indicated. F and G, PC3 cells were transfected with PTEN (F) and siRNA against Akt1 or Akt2 (G), and mTOR immunoprecipitates and whole cell lysates were analyzed with the indicated antibodies. H, PC3 cells were treated with PI3K inhibitor, LY 294000, at the indicated times, lysed, immunoprecipitated by IKKα antibodies, and immunoblotted with the indicated antibodies. I, 293T cells were co-transfected as indicated and immunoprecipitated with FLAG antibody and immunoblotted with the indicated antibodies. J, PC3 cells were transfected with FLAG-IKKα wild type or mutant, and whole cell lysates were analyzed with antibodies as indicated.
It has been reported that Akt phosphorylates IKKα at threonine 23 to activate NF-κB in response to TNFα treatment or DNA damage (46). Thus, we determine if Akt regulates IKKα-mTORC1 interaction and promotes mTORC1 activity via phosphorylation of IKKα at threonine 23. To examine if Akt phosphorylates IKKα Thr-23, we treated PC3 prostate cancer cells, which have high basal levels of Akt and IKKα, with a PI3K inhibitor, immunoprecipitated IKKα, and immunoblotted with a phospho-IKKα-Thr-23 antibody. Phosphorylation of IKKα at Thr-23 was reduced by the PI3K inhibitor (Fig. 6H). The results are consistent with the ability of Akt to phosphorylate IKKα at Thr-23. To determine if Akt promotes an interaction between IKKα and mTOR via phosphorylation at Thr-23, Akt was transfected with myc-mTOR and with either FLAG-tagged WT IKKα or T23A IKKα. The results show that Akt promotes an interaction between both WT and the T23A mutant IKKα (Fig. 6I). Consistent with these results, transfection of IKKα-T23A enhances mTORC1 activity identically to that generated by WT IKKα (Fig. 6J). These data indicate that Akt-mediated IKKα activation of mTORC1 is not through phosphorylation of IKKα at Thr-23.
IKKα Phosphorylates mTOR to Promote Cell Proliferation
Previously it was shown that mTOR phosphorylation is involved in cell growth and cell proliferation in certain cells (41). Given the functional interaction of IKKα and mTORC1, we hypothesized that IKKα will induce prostate cancer cell proliferation through mTOR phosphorylation. To determine whether IKKα and mTORC1 affect PC3 cell proliferation, siRNA was targeted against IKKα, mTOR, and Raptor, and cell proliferation (cell number) was measured at 48 h after transfection. Consistent with previous studies (38), knockdown of IKKα, mTOR, and Raptor impaired PC3 cell proliferation (Fig. 7A). To determine if Ser-1415 mTOR phosphorylation may contribute to growth of PC3 cells, WT mTOR or S1415A mTOR was stably expressed in PC3 cells (Fig. 7B). Consistent with our hypothesis that phosphorylation of mTOR by IKKα is important for mTOR function, expression of WT mTOR activated and mTOR S1415A reduced endogenous phosphorylation of S6K and 4E-BP1 (Fig. 6B). Expression of WT mTOR led to an increase in cell number, whereas expression of S1415A reduced cell number (Fig. 7C). Additionally, the phosphomimetic S1415E mTOR further promotes cell proliferation as compared with wild type mTOR (Fig. 7D). Next, mTOR wild type and the S1415A mutant were cotransfected with IKKα, and cell proliferation was measured at 3 days after transfection. Results demonstrate that transfection of IKKα with mTOR wild type, but not the S1415A mutant, enhances cell proliferation over IKKα expression alone (Fig. 7E). Collectively, the data indicate that IKKα promotes PTEN-deficient cancer cell proliferation through mTORC1 by mTOR phosphorylation.
FIGURE 7.

IKKα phosphorylation of mTOR promotes PTEN null and Akt active PC3 prostate cancer cell proliferation. A, knockdown of IKKα and mTOR decreases cell proliferation. PC3 cells were transfected with siRNA against IKKα, mTOR, and Raptor and plated in 6-well plates (2.0 × 104) 48 h posttransfection, and cell numbers were counted using a hemocytometer after 3 days. The numbers were calculated and presented as the mean ± S.D. from triplicates. B, stably transfected PC3 cells (mTOR-WT or mTOR-S1415A) were lysed and analyzed with antibodies as indicated. C, the stably transfected cells (pcDNA3, mTOR-WT, and mTOR-S1415A) were plated in 6 well plates (10 × 104), and cell numbers were counted using a hemocytometer every day for 3 days. The numbers were calculated and are presented as the mean ± S.D. from triplicates. The single asterisk indicates statistical significance compared with controls (t test, p < 0.05), and the double asterisks indicate p < 0.01. D, PC3 cells were transfected with mTOR wild type and its mutants, plated in 6-well plates (15 × 104) 48 h posttransfection, and cell numbers were counted using a hemocytometer in 3 days. Statistical analysis was performed as described in A and C. E, PC3 cells were transfected with IKKα and mTOR wild type and its mutants as indicated, and plated in 6-well plates (10 × 104) 48 h posttransfection, and cell numbers were counted using a hemocytometer after 3 days followed by statistical analysis. The single asterisk indicates statistical significance compared with controls (t test, p < 0.05), and the double asterisks indicate p < 0.01.
DISCUSSION
mTOR is a key effector of the cell growth- and metabolic-promoting functions of Akt (1, 2, 13). Previous studies have demonstrated that Akt activates mTORC1 through phosphorylation and subsequent inhibition of TSC2 (17, 18), promoting Rheb activation of mTORC1 (21). More recent reports have shown that PRAS40 is a negative regulator of mTORC1 and that Akt phosphorylates PRAS40 to relieve its inhibitory function on mTORC1 (22, 23). Although our earlier studies indicated that IKKα is important for the induction of mTORC1 activity downstream of Akt-induced signaling (37), there was no mechanism to explain how IKKα functions in this pathway. Although phosphorylation of mTOR has been shown previously to occur at Ser-2448, Ser-2481, Thr-2446, Ser-1261, Ser-2159, and Thr-2164 (41), the mechanisms and significance remains unclear. Here we demonstrate that IKKα directly phosphorylates mTOR in the mTORC1 complex at Ser-1415, downstream of activated Akt, to stimulate kinase activity. Taken together, our data and that of others demonstrate that Akt induces multiple steps in mTOR activation: (i) phosphorylation of TSC2, (ii) phosphorylation of PRAS40, and (iii) induction of association of IKKα with mTOR to drive mTOR phosphorylation at Ser-1415 to promote mTORC1 kinase activity (Fig. 8).
FIGURE 8.
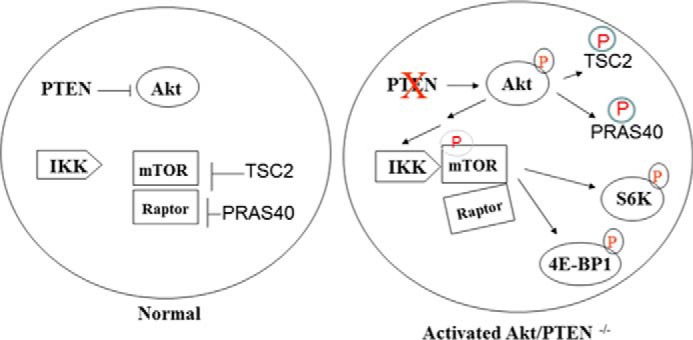
A proposed model of IKKα regulation of mTORC1 downstream of activated Akt.
Regulation of mTORC1 appears to occur partly through regulated interaction between Raptor and mTOR. After stimulation with nutrients, mTOR and Raptor exhibit a weakened interaction within the mTORC1 complex as compared with the inactive state of the kinase complex (40), indicating an alteration in the complex. A recent report (41) demonstrates that mTOR phosphorylation at Ser-2159 and Thr-2164, which is found basally in 293 cells, promotes reduced interaction found between mTOR and Raptor and between PRAS40 and mTOR in response to insulin stimulation. Consistent with results indicating that activated mTORC1 exhibits reduced affinity between mTOR and Raptor, our data indicate that Akt promotes reduced mTOR-Raptor interaction and that this involves IKKα and IKKα-directed Ser-1415 phosphorylation. These data support the conclusion that IKKα controls mTOR-Raptor interaction through mTOR phosphorylation downstream of Akt, although we cannot rule out the possibility that IKKα may be involved in other mechanisms to regulate mTORC1. Our data support a model whereby Akt controls mTORC1 activity through IKKα-directed phosphorylation of mTOR along with the known responses involving TSC2 and PRAS40 (Fig. 8).
Previously it has been shown that activated Akt leads to activation of NF-κB transactivation potential which involves IKK (44), and our data indicate that the Akt-induced association between IKKα and mTORC1 leads to activation of NF-κB which involves IKKα and IKKβ (38). Gustin et al. (45) showed that Akt promotes processing of the NF-κB2/p100 precursor to the p52 form, a process known to involve IKKα. This group also reported that Akt phosphorylates IKKα at Thr-23 to promote its activity (46). Thus, links between Akt and IKK/NF-κB pathways are known to occur. However, a mechanism to explain how Akt induces association between IKKα and the mTORC1 kinase complex are not presently understood and are a focus on ongoing research. In this regard we found Akt phosphorylates IKKα at Thr-23 (consistent with the work of Ozes et al. (46)) but that a T23A mutant of IKKα is promoted to interact with the mTORC1 complex downstream of Akt identically to that of wild type IKKα (Fig. 6, H–J), indicating that phosphorylation of IKKα at Thr-23 is not a determining factor in promoting mTORC1-IKKα interaction or mTORC1 activity. The data that Akt promotes the interaction between IKKα and mTORC1 are consistent with results that demonstrate that the ability of IKKα to drive mTORC1 is independent of the regulatory function TSC2 (Fig. 1, D and E). Thus, we propose that modulation of TSC2 activity is not a component of the ability of IKKα to regulate mTORC1, although it is possible that IKKα can modulate TSC2 as well as another key step in mTORC1 activation. It will be important to determine a mechanism whereby Akt promotes the association between IKKα and the mTORC1 complex to drive mTORC1 phosphorylation and activity.
We recently showed that amino acid deprivation of non-transformed cells leads to IKK-dependent phosphorylation of the p85 subunit of PI3K in the C-terminal SH2 domain (34). This phosphorylation blocks the ability of certain tyrosine-phosphorylated effector proteins to bind to p85, which suppresses PI3K activity as well as downstream Akt and mTOR activity. Under these conditions IKK functions to suppress Akt and mTOR, consistent with an early event in the promotion of autophagy induced by IKK (47, 48). In contrast, IKKα but not IKKβ functions downstream of activated Akt to promote mTOR activity via direct phosphorylation. It will be also important to determine if unique pools of IKKα are involved in these distinct responses or whether IKK broadly shifts from negative regulation of PI3K and subsequent downstream Akt and mTOR to a positive regulation of mTOR when Akt is active.
Both mTOR and Akt are important in tumorigenesis through their ability to promote cell growth and to suppress cell death (1, 13, 49–52). Majumder and Sellers (51) showed that the expansion of AKT-driven prostate epithelial cells requires mTOR-dependent survival signaling and that mTOR inhibition by rapamycin reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Hay and co-workers (53) reported that Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner in murine mammary tumor virus-v-H-Ras-induced tumors and in skin carcinogenesis, and the reduction in mTORC1 but not Akt activity impaired cell proliferation and susceptibility to oncogenic transformation. Guertin et al. (54) showed that mTORC2, with its role in promoting Akt activation, is critical for prostate cancer development following PTEN loss (and see below). Prostatic intraepithelial neoplasia was observed in Pten+/− mice; however, no progression to invasive cancer was observed (55, 56). Heterozygosity of Pten cooperates with a number of engineered secondary events to enhance the oncogenic phenotype. Interestingly, despite the convergence of PTEN and TSC2 on a common downstream signaling pathway (mTOR), reduction of Tsc2 cooperates to induce invasive prostate cancers in Pten+/− mice (51, 57). Additionally, Pten heterozygosity cooperates with Rheb overexpression to markedly promote prostate tumorigenesis through mTOR (58). These studies indicate that mTOR, potentially both as the mTORC1 and mTORC2 complexes, can promote oncogenesis in different settings. Our data indicated that IKKα promotes cell proliferation through phosphorylation of mTOR at serine 1415 downstream of Akt in PTEN null cancer cell lines. It will be critical to determine if IKKα controls key regulatory pathways including mTOR in the PTEN−/− prostate tumor to promote oncogenesis.
Acknowledgments
We thank Dr. Jin Cheng for constructs and cells and Dominic Moore for statistical analysis. A special thanks to Dr. Jessica Hutti for technical advice and to members of the Baldwin laboratory for valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants CA75080 (to A. S. B.), AI35098 (to A. S. B.), CA73756 (to A. S. B.), and K99CA149178 (to H. C. D.). Additional support was provided by Department of Defense Postdoctoral (DOD) Fellowship PC060420 (to H. C. D.) and by the Samuel Waxman Cancer Research Foundation (to A. S. B.).
- IKK
- IκB kinase
- TSC2
- tuberous sclerosis complex 2.
REFERENCES
- 1. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes, and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howell J. J., Manning B. D. (2011) mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 22, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inoki K., Kim J., Guan K.-L. (2012) AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 52, 381–400 [DOI] [PubMed] [Google Scholar]
- 4. Jewell J. L., Guan K. L. (2013) Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 38, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 6. Dai N., Rapley J., Angel M., Yanik M. F., Blower M. D., Avruch J. (2011) mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 25, 1159–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim J., Kundu M., Viollet B., Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cybulski N, Hall M. N. (2009) TOR complex 2: a signaling pathway of its own. Trends Biochem. Sci. 34, 620–627 [DOI] [PubMed] [Google Scholar]
- 9. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 10. García-Martínez J. M., Alessi D. R. (2008) mTORC2 controls hydrophobic motif phosphorylation and activation of SGK1. Biochem. J. 416, 375–385 [DOI] [PubMed] [Google Scholar]
- 11. Yan L., Mieulet V., Lamb R. (2008) mTORC2 is the hydrophobic motif kinase for SGK1. Biochem. J. 416, e19–e21 [DOI] [PubMed] [Google Scholar]
- 12. Alessi D. R., Pearce L. R., García-Martínez J. M. (2009) New insights into mTOR signaling: mTORC2 and beyond. Sci. Signal. 2, pe27. [DOI] [PubMed] [Google Scholar]
- 13. Shaw R. J., Cantley L. C. (2006) Ras, PI3K, and mTOR signaling controls tumor growth. Nature 441, 424–430 [DOI] [PubMed] [Google Scholar]
- 14. Rosen N., She Q. B. (2006) Akt and cancer: is it all mTOR? Cancer Cell 10, 254–256 [DOI] [PubMed] [Google Scholar]
- 15. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhaskar P. T., Hay N. (2007) The two TORCs and Akt. Dev. Cell 12, 487–502 [DOI] [PubMed] [Google Scholar]
- 17. Manning B. D., Tee A. R., Logsdon M. N., Blenis J., Cantley L. C. (2002) Identification of the TSC2 tumor suppressor gene product tuberin as a target of the PI3K/Akt pathway. Mol. Cell 10, 151–162 [DOI] [PubMed] [Google Scholar]
- 18. Inoki K., Li Y., Zhu T., Wu J., Guan K.-L. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nat. Cell Biol. 4, 648–657 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y., Gao X., Saucedo L. J., Ru B., Edgar B. A., Pan D. (2003) Rheb is a direct target of the tuberous sclerosis tumor suppressor proteins. Nat. Cell Biol. 5, 578–581 [DOI] [PubMed] [Google Scholar]
- 20. Long X., Lin Y., Ortiz-Vega S., Yonezawa K., Avruch J. (2005) Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713 [DOI] [PubMed] [Google Scholar]
- 21. Avruch J., Long X., Lin Y., Ortiz-Vega S., Rapley J., Papageorgiou A., Oshiro N., Kikkawa U. (2009) Activation of mTORC1 in two steps: Rheb-GTP activation of catalytic function and increased binding of substrates to Raptor. Biochem. Soc. Trans. 37, 223–226 [DOI] [PubMed] [Google Scholar]
- 22. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 23. Vander Haar E., Lee S. I., Bandhakavi S., Griffin T. J., Kim D.-H. (2007) Insulin signaling to mTOR mediated by Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323 [DOI] [PubMed] [Google Scholar]
- 24. Wang L., Harris T. E., Roth R. A., Lawrence J. C. (2007) PRAS40 regulates mTORC1 kinase activity by functioning as a director inhibitor of substrate binding. J. Biol. Chem. 282, 20036–20044 [DOI] [PubMed] [Google Scholar]
- 25. Karin M. (2006) Nuclear factor-κB in cancer development and progression. Nature 441, 431–436 [DOI] [PubMed] [Google Scholar]
- 26. Staudt L. M. (2010) Oncogenic activation of NF-κB. Cold Spring Harb. Perspect. Biol. 2, a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ben-Neriah Y., Karin M. (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 [DOI] [PubMed] [Google Scholar]
- 28. Hayden M. S., Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perkins N. D. (2012) The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer. 12, 121–132 [DOI] [PubMed] [Google Scholar]
- 30. Baldwin A. S. (2012) Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol. Rev. 246, 327–345 [DOI] [PubMed] [Google Scholar]
- 31. Lee D. F., Kuo H. P., Chen C. T., Hsu J. M., Chou C. K., Wei Y., Sun H. L., Li L. Y., Ping B., Huang W. C., He X., Hung J. Y., Lai C. C., Ding Q., Su J. L., Yang J. Y., Sahin A. A., Hortobagyi G. N., Tsai F. J., Tsai C. H., Hung M. C. (2007) IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455 [DOI] [PubMed] [Google Scholar]
- 32. Huang W. C., Ju T. K., Hung M. C., Chen C. C. (2007) Phosphorylation of CBP by IKKα promotes cell growth by switching the binding preference of CBP from p53 to NF-κB. Mol. Cell 26, 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hutti J. E., Turk B. E., Asara J. M., Ma A., Cantley L. C., Abbott D. W. (2007) IκB kinase β phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-κB pathway. Mol. Cell. Biol. 27, 7451–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Comb W. C., Hutti J. E., Cogswell P., Cantley L. C., Baldwin A. S. (2012) p85α SH2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and Akt in response to cellular starvation. Mol. Cell 45, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marinis J. M., Hutti J. E., Homer C. R., Cobb B. A., Cantley L. C., McDonald C., Abbott D. W. (2012) IκB kinase α phosphorylation of TRAF4 down-regulates innate immune signaling. Mol. Cell. Biol. 32, 2479–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu M., Lee D.-F., Chen C.-T., Yen C.-J., Li L.-Y., Lee H.-J., Chang C.-J., Chang W.-C., Hsu J.-M., Kuo H.-P., Xia W., Wei Y., Chiu P. C., Chou C. K., Du Y., Dhar D., Karin M., Chen C. H., Hung M. C. (2012) IKKα activation of Notch links tumorigenesis via FoxA2 suppression. Mol. Cell 45, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dan H. C., Adli M., Baldwin A. S. (2007) Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by IKKα. Cancer Res. 67, 6263–6269 [DOI] [PubMed] [Google Scholar]
- 38. Dan H. C., Cooper M. J., Cogswell P. C., Duncan J. A., Ting J. P., Baldwin A. S. (2008) Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22, 1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dan H. C., Baldwin A. S. (2008) Differential involvement of IKKα and -β in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J. Immunol. 180, 7582–7589 [DOI] [PubMed] [Google Scholar]
- 40. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 41. Ekim B., Magnuson B., Acosta-Jaquez H. A., Keller J. A., Feener E. P., Fingar D. C. (2011) mTOR kinase domain phosphorylation promotes mTORC1 signaling, cell growth, and cell cycle progression. Mol. Cell. Biol. 31, 2787–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gareus R., Huth M., Breiden B., Nenci A., Rösch N., Haase I., Bloch W., Sandhoff K., Pasparakis M. (2007) Normal epidermal differentiation but impaired skin-barrier formation upon keratinocyte-restricted IKK1 ablation. Nat. Cell Biol. 9, 461–469 [DOI] [PubMed] [Google Scholar]
- 43. Hu M. C., Lee D. F., Xia W., Golfman L. S., Ou-Yang F., Yang J. Y., Zou Y., Bao S., Hanada N., Saso H., Kobayashi R., Hung M. C. (2004) IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117, 225–237 [DOI] [PubMed] [Google Scholar]
- 44. Madrid L. V., Wang C. Y., Guttridge D. C., Schottelius A. J., Baldwin A. S., Jr., Mayo M. W. (2000) Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-κB. Mol. Cell. Biol. 20, 1626–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gustin J. A., Korgaonkar C. K., Pincheira R., Li Q., Donner D. B. (2006) Akt regulates basal and induced processing of NF-κB2 (p100) to p52. J. Biol. Chem. 281, 16473–16481 [DOI] [PubMed] [Google Scholar]
- 46. Ozes O. N., Mayo L. D., Gustin J. A., Pfeffer S. R., Pfeffer L. M., Donner D. B. (1999) NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401, 82–85 [DOI] [PubMed] [Google Scholar]
- 47. Criollo A., Senovilla L., Authier H., Maiuri M. C., Morselli E., Vitale I., Kepp O., Tasdemir E., Galluzzi L., Shen S., Tailler M., Delahaye N., Tesniere A., De Stefano D., Younes A. B., Harper F., Pierron G., Lavandero S., Zitvogel L., Israel A., Baud V., Kroemer G. (2010) The IKK complex contributes to the induction of autophagy. EMBO J. 29, 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Comb W. C., Cogswell P., Sitcheran R., Baldwin A. S. (2011) IKK-dependent, NF-κB-independent control of autophagic gene expression. Oncogene 30, 1727–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vivanco I., Sawyers C. L. (2002) The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 [DOI] [PubMed] [Google Scholar]
- 50. Hay N. (2005) The Akt-mTOR tango and its relevance to cancer. Cancer Cell 8, 179–183 [DOI] [PubMed] [Google Scholar]
- 51. Majumder P. K., Febbo P. G., Bikoff R., Berger R., Xue Q., McMahon L. M., Manola J., Brugarolas J., McDonnell T. J., Golub T. R., Loda M., Lane H. A., Sellers W. R. (2004) mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 10, 594–601 [DOI] [PubMed] [Google Scholar]
- 52. Plas D. R., Thompson C. B. (2005) Akt-dependent transformation: there is more to growth than just surviving. Oncogene 24, 7435–7442 [DOI] [PubMed] [Google Scholar]
- 53. Skeen J. E., Bhaskar P. T., Chen C.-C., Chen W. S., Peng X.-D., Nogueira V., Hahn-Windgassen A., Kiyokawa H., Hay N. (2006) Akt defiency impairs normal cell proliferation and suppressesoncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell 10, 269–280 [DOI] [PubMed] [Google Scholar]
- 54. Guertin D. A., Stevens D. M., Saitoh M., Kinkel S., Crosby K., Sheen J. H., Mullholland D. J., Magnuson M. A., Wu H., Sabatini D. M. (2009) (mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Di Cristofano A., De Acetis M., Koff A., Cordon-Cardo C., Pandolfi PP. (2001) Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet. 27, 222–224 [DOI] [PubMed] [Google Scholar]
- 56. You M. J., Castrillon D. H., Bastian B. C., O'Hagan R. C., Bosenberg M. W., Parsons R., Chin L., DePinho R. A. (2002) Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 99, 1455–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ma L., Teruya-Feldstein J., Behrendt N., Chen Z., Noda T., Hino O., Cordon-Cardo C., Pandolfi P. P. (2005) Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 19, 1779–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nardella C., Chen Z., Salmena L., Carracedo A., Alimonti A., Egia A., Carver B., Gerald W., Cordon-Cardo C., Pandolfi P. P. (2008) Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 22, 2172–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]