

Abstract
Hypertensive disorders are life-threatening diseases with high morbidity and mortality, affecting billions of individuals worldwide. A multitude of underlying conditions can contribute to hypertension, thus the need for a plethora of treatment options to identify the approach that best meets the needs of individual patients. As reviewed here a growing body of evidence indicates that i) autoantibodies that bind to and activate the major angiotensin receptor, AT1R exist in the circulation of patients with hypertensive disorders, ii) these autoantibodies contribute to disease pathophysiology, iii) antibody titers correlate to the severity of the disease, and iv) efforts to block or remove these pathogenic autoantibodies have therapeutic potential. These autoantibodies, termed AT1 agonistic autoantibodies (AT1-AA) have been extensively characterized in preeclampsia (PE), a life-threatening hypertensive condition of pregnancy. As reviewed here these autoantibodies cause symptoms of PE when injected into pregnant mice. Somewhat surprisingly, these antibodies also appear in three animal models of preeclampsia. However, the occurrence of AT1-AA is not restricted to pregnancy. These autoantibodies are prevalent among kidney transplant recipients who develop severe transplant rejection and malignant hypertension during the first week following transplantation. AT1-AA are also highly abundant among a group of patients with essential hypertension that are refractory to standard therapy. More recently these autoantibodies have been seen in patients with the autoimmune disease, systemic sclerosis. These three examples extend the clinical impact of AT1-AA beyond pregnancy. Research reviewed here raises the intriguing possibility that preeclampsia and other hypertensive conditions are autoimmune diseases characterized by the presence of pathogenic autoantibodies that activate the major angiotensin receptor, AT1R. These pathogenic autoantibodies could serve as pre-symptomatic biomarkers and therapeutic targets, thereby providing improved medical management for these conditions.
Keywords: hypertension, autoimmunity, AT1-AA, agonistic autoantibodies, preeclampsia
Introduction
Hypertensive disorders are life-threatening diseases with high morbidity and mortality, affecting billions of individuals worldwide. The pathogenesis of essential hypertension is multifactorial, with different underlying mechanisms contributing to the disease. Because of disease heterogeneity a variety of antihypertensive drugs are needed to tailor medical approaches to the specific needs of individual patients. Common areas of investigation for hypertension research include the vascular system, renal hemodynamics and renovascular hypertension, the endothelin system and the renin-angiotensin-aldosterone system. Here we review evidence suggesting that some forms of hypertension may have an underlying autoimmune component.
Autoimmune diseases are relatively common (5% of the US population) and include well-known diseases such as Type 1 diabetes, multiple sclerosis, rheumatoid arthritis and celiac disease. It is worth noting that in each case the autoimmune nature of the disease was not originally obvious and only became apparent after extensive investigation. Research reviewed here suggests that hypertensive disorders may result from the presence of agonistic autoantibodies that are directed to a specific epitope on the second extracellular of loop of the AT1R. The classic example of receptor activating autoantibodies and disease is Graves' hyperthyroidism, in which autoantibodies activate the thyroid-stimulating hormone receptor resulting in overproduction of thyroid hormones1, 2. Other compelling examples come from the cardiovascular literature and include: 1) agonistic autoantibodies targeting the cardiac β1-adrenergic receptor, which are associated with dilated cardiomyopathy3, 2) autoantibodies capable of activating α1-adrenergic receptors, associated with refractory hypertension4-6, and 3) autoantibodies that activate the major angiotensin II receptor, associated with preeclampsia7-9, malignant hypertension and renal allograft rejection10-12. Angiotensin receptor agonistic autoantibodies are the focus of this review.
Angiotensin receptor agonistic autoantibodies and preeclampsia
Preeclampsia (PE) is a life-threatening hypertensive condition of pregnancy and a leading cause of maternal and neonatal morbidity and mortality13, 14. The condition generally appears during the third trimester and is also characterized by proteinuria, inflammation and thrombosis. PE affects approximately 7% of pregnancies and accounts for 15% of premature births (180,000 in US). Current strategies for managing PE are inadequate and reflect a fundamental lack of understanding of the etiology and pathogenesis of the disorder. Numerous studies over the past 14 years have shown that women with PE possess autoantibodies with the ability to bind and activate the major angiotensin receptor, AT1R.
Early in vitro Studies
Agonistic autoantibodies to AT1R, were initially described by Wallukat et al. in 19997. In this seminal report the authors described the use of a rat neonatal cardiomyocyte contraction assay to detect the presence of AT1 agonsitic autoantibodies, termed AT1-AA. Receptor specificity was shown pharmacologically and by immunohistochemistry and western blotting. Peptide competition experiments were used to identify the precise epitope recognized by these autoantibodies, a seven amino acid peptide sequence located on the second extracellular loop of the receptor. Subsequent database analysis revealed that this amino acid sequence corresponded to a highly antigenic region present on the coat protein of parvovirus B19, a common and relatively benign human pathogen. This finding raised the possibility that AT1-AA arise in part as a result of molecular mimicry. However epidemiological studies rendered this explanation unlikely15, 16.
Although AT1-AA were initially detected by their ability to stimulate an increase in the beating rate of isolated neonatal rat cardiomyocytes numerous additional early studies showed that AT1-AA could activate AT1 receptors on a variety of cell types and provoke biological responses relevant to the pathophysiology of PE. Studies from multiple laboratories showed that AT1-AA may contribute to hypercoagulation by stimulating tissue factor production by vascular smooth muscle cells and monocytes17, as well as plasminogen activator inhibitor-1 (PAI-1) production from trophoblasts8 and mesangial cells18. Other studies showed that IgG from women with preeclampsia, in contrast to IgG from normotensive pregnant women, contributes to the production of reactive oxygen species by stimulating NADPH oxidase activity in vascular smooth muscle cells and human trophoblasts19. Finally, antibody-mediated AT1 receptor activation results in increased soluble Fms-like tyrosine kinase-1 (sFlt-1)20 and soluble endoglin (sEng)21 production from human trophoblasts and placental explants. In this way these antibodies may contribute to the anti-angiogenic state that is characteristic of PE.
Antibody transfer experiments in animals
Because these earlier studies were restricted to the use of cultured cells or tissue explants, they did not directly address the relevance of AT1-AAs to hypertension and proteinuria, the defining clinical features of PE. For this reason, an in vivo experimental approach was needed to determine if these autoantibodies could cause clinical features of PE. Using a classical antibody transfer approach Zhou et al.9 showed that the introduction of these autoantibodies into pregnant mice resulted in hypertension, proteinuria and a variety of other features of PE (Figure 1). Proteinuria was accompanied by a characteristic renal abnormality termed glomerular endotheliosis (endothelial cell swelling). These features appeared in pregnant mice following AT1-AA injection and were prevented by co-injection with losartan (an AT1R antagonist) or a 7-amino acid epitope peptide that corresponds to a highly antigenic site on the second extracellular loop of the AT1R. These results indicate that hypertension, proteinuria and renal pathology resulted from autoantibody-induced AT1 receptor activation in pregnant mice.
Figure 1.
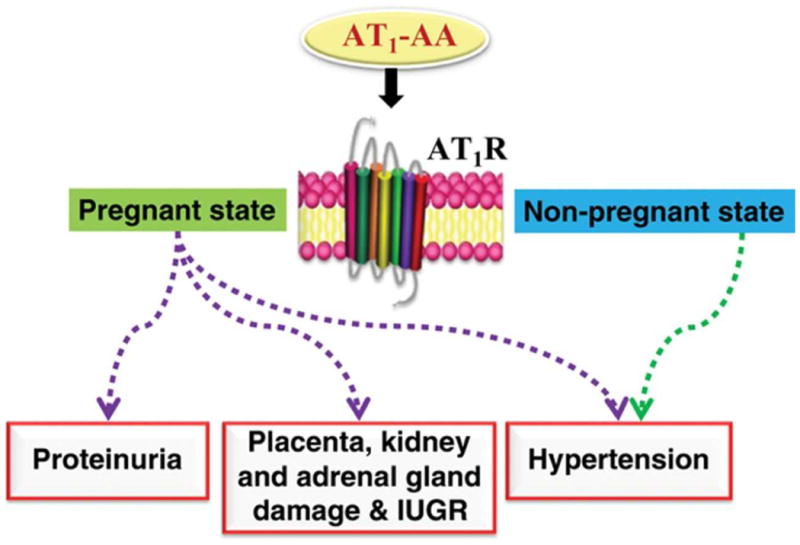
Pathophysiological role of AT1-AA in PE by antibody transfer experiments. IUGR: intrauterine growth restriction.
AT1-AA injection into non-pregnant mice also results in hypertension, but not the renal pathology observed in pregnant mice9. These results indicate that the anti-angiogenic action of excessive sFlt-1 production by the placenta is detrimental to renal endothelial function, resulting in glomerular endotheliosis and proteinuria (discussed in more detail below). The hypertensive effects of AT1-AA in the absence of pregnancy are consistent with the contribution of AT1-AA to essential and malignant hypertension reported by others (see later sections).
Because AT1-AA produce clinical features of preeclampsia when introduced into pregnant mice, it is likely that they contribute to symptoms of preeclampsia in the women from whom they were obtained. This view is supported by data showing that AT1-AAs are highly prevalent in preeclampsia7, 8, 15, 22 and that antibody titers correlate to the severity of the disease22. Thus, in vitro and in vivo findings with IgG from patients' sera suggest a pathophysiological role for these autoantibodies in preeclampsia and provide experimental support for the hypothesis that preeclampsia is an autoimmune condition characterized by the presence of disease-causing autoantibodies.
Antibody-induced pathogenic factors
Following the success of the initial antibody transfer experiments this mouse model of PE was used to identify numerous antibody-induced factors that contribute to disease pathophysiology. These include the inflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), the anti-angiogenic factors sFlt-1 and sEng, the vasoconstrictor endothelin-1 (ET-1), and complement component C3a. In each case it was possible to identify inhibitory strategies to block the action of these pathogenic mediators and achieve therapeutic benefit in the antibody-induced model of PE in pregnant mice. The results of these studies are summarized in Figure 2 and discussed below.
Figure 2.
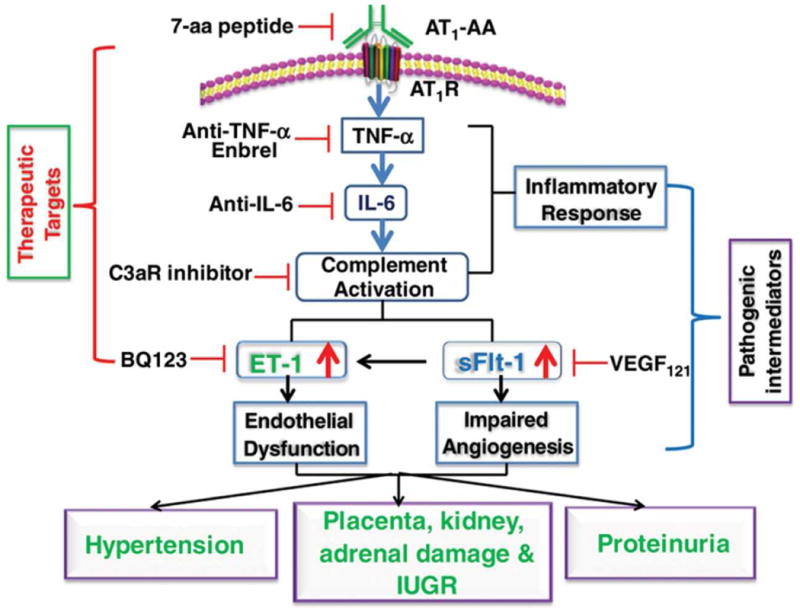
Pathogenic mediators of AT1-AA-induced PE in pregnant mice. Therapeutic strategies based on blocking the detrimental effects of AT1-AA-induced pathogenic mediators are illustrated.
Inflammatory cytokines (TNA-α and IL-6)
Circulating TNF-α levels are significantly elevated in women with PE and correlate with the level of AT1-AA bioactivity23. Injection of IgG from women with PE (in contrast to IgG from normotensive pregnant women) into pregnant mice results in elevated levels of TNF-α characteristic of women with PE21, 24. Co-injection of AT1-AA with a TNF-α neutralizing antibody, or a soluble form of the TNF-α receptor, significantly attenuates key features of preeclampsia, including hypertension and proteinuria. Renal damage and placental abnormalities were also decreased by TNF-α blockade. TNF-α blockade also resulted in a reduction in sFlt-1, sEng and IL-6, indicating that these biomolecules function downstream of TNF-α signaling21, 25. Thus, TNF-α functions downstream of autoantibody-mediated AT1R activation to promote clinical features of PE in pregnant mice.
IL-6 functions downstream of TNF-α signaling, and contributes to increased ET-1 production in pregnant mice25. IL-6 blockade inhibits the appearance of PE features in autoantibody-injected pregnant mice. Extending the data to human studies, Zhou et al.25 found that IL-6 was a key cytokine underlying autoantibody-mediated induction of ET-1 in human placental villous explants and that endothelial cells are a key source of ET-1. Overall, human and mouse studies show that AT1-AA contributes to elevated ET-1 production and that increased TNF-α/IL-6 signaling is a key mechanism underlying increased ET-1 production and subsequent maternal features of PE.
Direct evidence for the ability of inflammatory cytokines to contribute to clinical features of PE comes from experiments showing that the injection of TNA-α26, IL-627 or IL-1728 into pregnant rats or mice causes hypertension, proteinuria and other features of PE.
Complement component C3a
PE is associated with increased complement activation of undetermined causality. Use of the antibody transfer model of PE in pregnant mice has shown that autoantibody-mediated C3a receptor activation contributes to the pathogenesis of PE29. Hypertension and proteinuria in autoantibody-injected pregnant mice is significantly reduced by a complement C3a receptor-specific antagonist to interefere with C3A receptor signaling. Additional experiments showed that complement C3a receptor antagonism significantly attenuated autoantibody-induced sFlt-1 production, placental impairment and intrauterine growth restriction. Human studies demonstrated that C3 deposition is significantly elevated in the placentas of preeclamptic patients compared with normotensive controls, and that complement C3a receptor activation is a key mechanism underlying autoantibody-induced sFlt-1 secretion and decreased angiogenesis in cultured human villous explants. Thus, complement component C3a signaling through its receptor contributes to autoantibody-induced features of PE in pregnant mice. These studies are consistent with earlier studies showing that complement C3a functions downstream of Ang II to mediate hypertension and renal malfunction30.
Anti-angiogenic factors (sFlt-1 and sEng)
A soluble form of the vascular endothelial growth factor receptor-1 (VEGFR1, also called sFlt-1) is elevated in the circulation of women with PE relative to normotensive pregnant women31, 32. As a decoy receptor, sFlt-1 blocks VEGF mediated signaling that is important for normal endothelial function and thereby contributes to hypertension and renal dysfunction. Siddiqui et al. used the autoantibody-injection model of PE in pregnant mice to assess the therapeutic potential of recombinant VEGF121, a relatively stable form of VEGF. Their results show that the infusion of recombinant murine VEGF121 attenuated autoantibody-induced hypertension and proteinuria and prevented renal histological impairment33. Impaired placental angiogenesis was also significantly improved as a result of rVEGR121 infusion. Siddiqui et al. also showed that infusion of rVEGF121 alleviates the detrimental effects of sFlt-1 on adrenal glands and the production of aldosterone34. Together, these studies show that infusion of rVEGF121 compensated for the excess sFlt-1 mediated interference with VEGF signaling. Multiple additional methods to block autoantibody-mediated induction of sFlt-1 showed therapeutic benefit. These include the use of C3a receptor antagonism29 or TNF-α blockade21. Thus, C3a and TNF-α receptor signaling appear to be downstream of AT1R signaling and upstream of sFlt-1 induction.
sEng, a soluble form of the transforming growth factor-β (TGF-β) receptor, is also elevated in the circulation of women with PE and interferes with TGF-β signaling resulting in reduced NO production and increased blood pressure35. sEng also functions in cooperation with sFlt-1 to induce the HELLP syndrome in animal models of PE. TNF-α blockade (neutralizing antibody or soluble TNF receptor) or the induction of heme oxygenase (HO) by hemin reduced autoantibody-mediated induction of sEng, and attenuated pathological features of PE21. Thus, HO functions to interfere with PE-IgG induction of sEng and sFlt-1.
Endothelin-1 (ET-1)
Elevated ET-1 is associated with PE and is believed to be a key terminal pathway of disease pathophysiology by activating the endothelin receptor, ETA36. The ETA receptor specific antagonist BQ123 significantly attenuated autoantibody-induced hypertension, proteinuria and renal damage in pregnant mice, demonstrating that autoantibody-induced ET-1 production contributes to pathophysiology in this pregnant mouse model25. Mechanistically, IL-6 functions downstream of TNF-α to induce ET-1 production in pregnant mice. Thus, either TNF-α blockade or IL-6 blockade reduced antibody-induced production of ET-1 and the associated hypertension (Figure 2).
AT1-AAs contribute to the placental abnormalities and fetal growth restriction associated with PE
PE is often associated with intrauterine growth restriction (IUGR). Growth-restricted fetuses have a higher incidence of mortality and morbidity than fetuses of normal growth and are at increased risk for future development of metabolic disorders such as hypertension, coronary heart disease, dyslipidemia, obesity, impaired glucose tolerance, Type 2 diabetes mellitus and many other diseases37-40. AT1-AA are present in the cord blood of women with preeclampsia and retain the ability to activate AT1Rs 24. Using the antibody transfer model of PE in pregnant mice Irani et al. showed that AT1-AAs cross the placenta and enter the fetal circulation where they are associated with small fetuses with renal and hepatic abnormalities24. AT1-AAs also induce apoptosis in the placentas of pregnant mice, human villous explants and human trophoblast cells in culture. Finally, autoantibody-induced IUGR and placental apoptosis are diminished by losartan or the autoantibody-neutralizing 7-amino acid epitope peptide. These studies highlight AT1-AA as a potential contributor to preeclampsia-associated IUGR and offer two possible underlying mechanisms: 1) a direct detrimental effect on fetal development by crossing the placenta and entering the fetal circulation, and 2) fetal growth restriction secondary to AT1-AA-induced placental damage.
Experimental induction of AT1-AA in animal models of preeclampsia
As reviewed above PE in humans is characterized by the presence of autoreactive antibodies, AT1-AA, that are able to induce numerous pathological factors that contribute to clinical features of PE when injected into pregnant mice (Figure 2)41, 42. Somewhat surprisingly, these antibodies also appear in three animal models of preeclampsia that are reviewed below (Figure 3).
Figure 3.
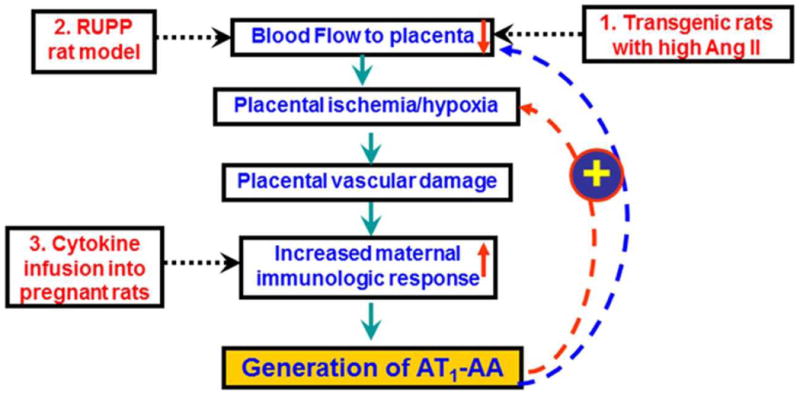
Summary of animal models of PE that produce AT1-AA.
Transgenic rodents engineered to produce excessive Ang II during the second half of pregnancy
When dams (mice or rats) harboring the human angiotensinogen gene are mated with males carrying the human renin gene, the dams develop severe hypertension, proteinuria and target organ damage that resembles PE43, 44. These features result from human renin produced by the placenta and released into the maternal circulation where it acts on human angiotensinogen produced and released from the maternal liver leading to increased circulating levels of Ang II during the second half of pregnancy. Working with the rat model, Dechend and colleagues demonstrated that agonistic antibodies directed at the AT1 receptor develop during the second half of pregnancy along with other features of PE43. Peptide competition experiments showed that the antibodies present in these pregnant rats are directed to the same epitope as the AT1-AA present in women with preeclampsia. The presence of these autoantibodies was initially shown by their chronotropic action in the neonatal cardiomyocyte assay and subsequently by antibody transfer experiments into pregnant rats45. The introduction of these autoantibodies into pregnant rats stimulated features of PE, including hypertension and proteinuria. In this regard the AT1-AA produced in pregnant rats resembles the AT1-AA produced in women with PE.
Surgically induced placental ischemia
Granger and colleagues developed a rat model of preeclampsia based on placental ischemia resulting from surgical reduction in uterine perfusion pressure (RUPP)46. Such experimentally manipulated gravid rats develop hypertension, proteinuria and other features of PE47, including elevated levels of sFlt148, sEng49, increased inflammatory cytokines (TNF-α50, and IL-651) and increased ET-152. Remarkably, RUPP rats also develop AT1-AA that contribute to pathophysiology in this animal model of PE53. RUPP-induced hypertension is inhibited by losartan but not by an angiotensin converting enzyme (ACE) inhibitor, consistent with a role for AT1-AA in this model of PE. Treatment of RUPP animals with rituximab, to prevent the mobilization of B lymphocytes inhibited the production of AT1-AA and reduced hypertension54. A role for CD4+ T helper cells in RUPP-induced hypertension and AT1-AA production has also been demonstrated by showing that AT1-AA play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from RUPP rats55, 56. The features of PE observed in RUPP rats are believed to be due to the production of AT1-AA and resulting activation of AT1R26, 57-60. Overall, features of PE seen in RUPP rats are remarkably similar to those seen in AT1-AA-injected pregnant mice as described above41, 42, 61.
Infusion of inflammatory cytokines
The Lamarca group has also shown that infusion of inflammatory cytokines (TNF-α, IL-6 or IL-17) into pregnant rats, but not virgin female rats, results in features of PE, including the production of AT1-AA26-28, 50.
Thus, AT1-AA have appeared in animal models of PE associated with genetically induced hypertension, surgically induced placental ischemia and cytokine infusion. The generation of these autoantibodies may be secondary to placental ischemia, vascular damage and increased maternal inflammatory response that is associated with preeclampsia. Experimental induction of uteroplacental ischemia62 and the infusion of inflammatory cytokines27, 53 appear to be promising experimental models to study the relationship between preeclampsia and autoimmunity. The animal models of experimentally induced autoantibody production also represent a well-defind and experimentally pliable system for understanding the molecular mechanisms responsible for autoimmunity (Figure 3).
The clinical impact of AT1-AA outside of pregnancy
The occurrence of AT1-AA is not restricted to pregnancy. These autoantibodies are prevalent among kidney transplant recipients that develop severe transplant rejection and malignant hypertension during the first week following transplantation. AT1-AA are also highly abundant among a group of patients with essential hypertension that are refractory to standard therapy. These two examples extend the clinical impact of AT1-AA beyond pregnancy and are reviewed below.
Acute vascular rejection associated with malignant hypertension
Dragun et al. investigated a group of 16 kidney-transplant recipients characterized by acute vascular rejection and malignant hypertension occurring during the first week after kidney transplantation10. Four of these individuals also had seizures. The combination of vascular pathology, hypertension and seizures prompted these investigators to consider the presence of AT1-AA, a pathogenic autoantibody previously observed in women with preeclampsia7, a condition also associated with vascular lesions, hypertension and seizures. The cardiomyocyte contraction assay was used to show that AT1-AAs were present in all 16 patients with malignant hypertension and not in a group of patients characterized with acute vascular rejection in the absence of malignant hypertension. Retrospective analysis of historic sera taken from patients prior to transplantation revealed the presence of AT1-AA in all 16 patients presenting with acute vascular rejection and malignant hypertension during the week following transplantation. Characterization of the AT1-AA present in these 16 individual revealed IgG1 and IgG3 autoantibodies directed at two distinct sites on the second extracellular loop of the AT1 receptor. One site was identical to the 7aa epitope observed in women with preeclampsia. Treatment of seven antibody-positive patients with plasmaphoresis, intravenous immune globulin and 100 mg losartan per day resulted in significantly improved allograft survival compared with that of patients receiving standard antirejection treatment. Subsequent analysis of 278 kidney transplant recipients performed at their center revealed a prevalence of AT1-AA associated vascular rejection episodes of 3.6% (i.e., ten patients). These findings are consistent with an earlier report indicating the presence of AT1-AA in some patients with malignant hypertension11 and more recent reports of others showing AT1-AA association with essential hypertension (see following section). Based on these studies it was suggested that individuals being considered for a kidney transplant should be tested for the presence of AT1-AA, to allow for patient-specific post-transplant medical care for those testing positive63-65. For this purpose a high-throughput cell based ELISA has recently been developed64.
Essential hypertension
In general it is possible to divide the underlying causality for hypertension into two broad categories; renal and vascular. The well-known renin, angiotensin, aldosterone system spans both categories and is the most common system affected in hypertension. Because alterations in the RAAS are common, angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are common therapeutic approaches. For some time is has been apparent that pathogenic alterations in the RAAS include the presence of autoantibodies capable of activating the major angiotensin receptor, AT1R. Liao and colleagues observed AT1-AA (which they term anti-AT1 receptor autoantibodies) in approximately 43% of patients with refractory hypertension66. The prevalence of AT1-AA among refractory hypertension was revised upward in more recent studies when autoantibody testing relied on using higher serum concentrations67. In the latter case an overall percentage of 59% of refractory hypertensive patients harbored AT1-AA, and among these patients the prevalence of AT1-AA increased with increasing blood pressure. The relationship of AT1-AA to systolic blood pressure ranged from 52% to 69%, with increasing blood pressure. Other studies from this group showed that these autoantibodies were able to activate AT1 receptors and initiate a chain of signaling events resulting in the proliferation of vascular smooth muscle cells and vascular remodeling.
Wei et al. conducted a clinical trial to test the possibility that AT1-AA contribute to hypertension12. The authors hypothesized that if AT1-AAs contribute to hypertension, then AT1-AA positive hypertensive patients would show a better response to angiotensin receptor blockers (ARBs) than ACE inhibitors compared to AT1-AA negative hypertensive patients. In a study involving 512 patients with roughly half on candesartan (an ARB) and half on imidapril (an ACE inhibitor), the results clearly showed that the ARB-based regimen is more effective in lowering blood pressure than an ACE inhibitor-based regimen in AT1-AA positive patients. The results of this clinical trial highlight the contribution of AT1-AAs to high blood pressure in these patients with refractory hypertension and that treatment with ARBs alleviates the hypertensive effects of these pathogenic autoantibodies more effectively than ACE inhibitors.
Systemic sclerosis
Systemic sclerosis (SSc) is a chronic systemic autoimmune disease characterized by fibrosis affecting the hands, arms and face. Progressive forms of the disease affect large areas of the skin and one or more internal organs (kidneys, esophagus, heart or lungs). Death occurs most often from pulmonary, cardiac or renal complications, secondary to hypertension. ACE inhibitors, ET-1 receptor blockers and ARBs reduce hypertension and alleviate some of the cardiac, renal and pulmonary manifestations of SSc. Based on these clinical features Dragun and colleagues68 were prompted to consider the possibility that receptor-activating autoantibodies were involved. To test this possibility they developed a cell-based ELISA to detect antibodies to the AT1R and the ET-1 receptor, ETA. Their results indicate that most SSc patients possess antibodies to each of these receptors. Antibodies to each receptor are biologically active and induce receptor-directed ERK1/2 phosphorylation and increased TGF-β gene expression in human microvascular endothelial cells. Ang II and ET-1 induce collagen synthesis via target receptor stimulation in fibroblasts, features that could be attributed to the AT1 and ETA receptor-activating autoantibodies in SSc. The biological effects of both autoantibodies were blocked by the respective receptor antagonists, providing additional evidence for antibody-mediated receptor activation. Higher antibody titers were associated with late complications such as pulmonary hypertension, lung fibrosis and digital ulcers. Thus, according to these authors, AT1R and ETAR agonistic autoantibodies may contribute to disease pathogenesis in SSc and link disease features including autoimmunity, vascular pathology and hypertension.
Mechanism of autoantibody-mediated AT1R activation
A large body of biochemical evidence published in recent years indicates that G protein coupled receptors (GPCRs) form homodimers, heterodimers, and possibly higher order oligomeric structures69-71. Agonist-induced dimerization has been shown for a number of GPCRs and in some cases is required for efficient signaling. Agonist induced activation of the AT1 receptor is accompanied by dimerization72, 73. Furthermore, the formation of stable covalently cross linked AT1 receptors is associated with enhanced signaling and prolonged activation74. Because receptor-activating antibodies are bivalent, it is possible that they exert their agonistic effect by cross-linking and thereby stabilizing receptor homodimers. Thus, we hypothesize that autoantibody-induced AT1 receptor activation is accompanied by receptor dimerization. Evidence in support of this view comes from studies of other receptor activating autoantibodies showing that bivalency is required for antibody induced activation of the β2-adrenergic receptor75, 76 and the M2 muscarinic receptor77. In each of these cases the original bivalent IgG autoantibody activated the receptor, the monovalent Fab fragments did not activate, whereas Fab fragments cross linked by anti-Fab fragment antibody had restored receptor activating ability. Another interesting feature of the agonistic autoantibodies to the β1-adrenergic and M2 muscarinic receptors is their ability to promote a sustained receptor activation for many hours without the customary desensitization observed with natural ligands75, 77-80. These features have not yet been examined for AT1-AA. A thorough knowledge of the mechanism of antibody-induced receptor activation may provide further useful insights for the development of therapeutic strategies to block the action of the antibodies, thereby reducing the detrimental effects of excessive receptor activation.
Factors contributing to autoantibody production
The immunological basis for the loss of self-tolerance that allows antibodies to develop against a particular seven amino acid epitope on the second extracellular loop is not understood. Molecular mimicry with a homologous sequence on human parvovirus has been considered for the origin of AT1-AA, but this view has not withstood the results of epidemiological data that failed to show a correlation between PE and prior parvovirus infection15, 16. Thus, if molecular mimicry does not account for the production of AT1-AA then it is necessary to have loss of self-tolerance by the humoral (antibody-mediated) and cellular (T-cell mediated) arms of the immune system. A common mechanism for loss of self-tolerance is posttranslational modification resulting in the creation of a neo-antigen that is not recognized as self by the immune system. It is estimated that 50-90% of the proteins in the human body are posttranslationally modified81-84. In the proper context, these modifications are necessary for the biological functions of a vast array of proteins and the effector function of the cells in which they reside. However, it is now clear that some post-translational modifications can create new self-antigens (neo-antigens) and therefore trigger specific antibody production under autoimmune conditions. It is possible that placental ischemia and the resulting tissue damage, inflammation and syncytial trophoblast shedding may create a favorable setting for autoimmunity. The fact that the placenta has the highest tissue level of AT1R may also contribute to enhanced immunogenicity in a setting of placental ischemia and oxidative stress. It is well known that a chronic inflammatory response activates the adaptive arm of the immune system and may create an environment that is permissive for autoimmunity. A role for helper T cells in this process should not be overlooked. Harrison and colleagues have proposed a proinflammatory immune response based on the production of neo-antigens as a result of protein oxidation, nitrosylation, fragmentation or posttranslational modification85. These neo-antigens are no longer recognized as self and are process by antigen presenting cells, such as dendritic cells, where peptide fragments are presented to T helper cells via MHC class II molecules. T cells activated in this way are equipped to provide immunological assistance to B cells producing AT1-AA. As discussed above, rodent models of PE that are based on placental ischemia (i.e. the RUPP model)53, 54, 56, 86 and a heightened inflammatory response (i.e. infusion of pro-inflammatory cytokines such as TNF, IL-6 and IL-17)26-28 are characterized by the presence of AT1-AA. These same conditions are associated with PE in humans (i.e. placental ischemia and a heightened inflammatory state) and are likely to contribute to AT1-AA production. Pregnant animal models in which surgical manipulation (i.e., reduction in uterine perfusion pressure) or cytokine infusion (TNF, IL-6, IL-17) result in production of AT1-AA represent ideal experimental systems to identify the underlying cause of autoantibody production.
A proinflammatory state is believed to contribute to the production of neo-antigens that are recognized as foreign and presented to T cells by
Concluding Remarks
As reviewed here a growing body of evidence indicates that i) autoantibodies capable of activating AT1Rs exist in the circulation of patients with hypertensive disorders, ii) these autoantibodies contribute to disease pathophysiology, iii) antibody titers correlate to the severity of the disease, and iv) efforts to block or remove these pathogenic autoantibodies has therapeutic benefit. The production of these pathogenic autoantibodies most likely precedes the onset of clinical symptoms, a possibility that highlights the autoantibodies as potentially valuable pre-symptomatic biomarkers. This view was validated by retrospective analysis of serum samples obtained from renal dialysis patients prior to kidney transplant10. These studies indicated that knowledge of AT1-AA prior to kidney transplantation would influence patient-specific medical care at the time of transplantation. In view of the considerable evidence indicating that AT1-AAs contribute to disease, they are also likely to be important therapeutic targets in the management of hypertensive disease. In the coming years we can expect to see continued development of improved immunological and functional tests to detect and quantify these pathogenic autoantibodies. Preliminary evidence shows that the removal of pathogenic antibodies by plasmaphoresis or immunoabsorption along with receptor antagonism provides therapeutic benefit10. It is encouraging to see initial promising results from these approaches for patients with antibody-mediated malignant hypertension and graft rejection. The results of clinical trials with patients with refractory hypertension show that AT1-AA-positive patients respond better to angiotensin receptor blockers than ACE inhibitors12, again illustrating the benefits of patient specific medical treatment based on identifying the specific underlying cause of the hypertension.
Research reviewed here raises the intriguing possibility that preeclampsia and other hypertensive conditions are autoimmune diseases characterized by the presence of pathogenic autoantibodies that activate the major angiotensin receptor, AT1R. These pathogenic autoantibodies could serve as pre-symptomatic biomarkers and therapeutic targets, thereby providing improved medical management for these conditions. For most autoimmune diseases it has been possible to identify distinct polymorphic alleles of the major histocompatibility complex (MHC) genes that are highly associated with the autoimmune condition. The tight linkage of specific MHC polymorphic genes and a particular autoimmune condition is presumed to be due to the preferential ability of certain MHC class II complexes to present the specific antigenic peptide to the T-cell receptor complex. Conversely, for related reasons, certain MHC class II genes are highly excluded in certain autoimmune diseases. Thus, if preeclampsia and other hypertensive conditions have a significant autoimmune component it may be possible to identify distinct MHC polymorphisms that serve as genetic markers to identify individuals at increased risk for hypertension.
Table I. Brief summary of diseases associated with AT1-AA and related.
| Diseases associated with AT1-AA | Animal studies |
|---|---|
| Preeclampsia | Transfer of purified AT1-AA or total lgG from women with PE to pregnant mice generates features of PE |
| Acute Vascular Rejection Associated with Malignant Hypertension | Infusion of AT1-AA purified from organ transplanted patients with malignant hypertension to rats causes kidney damage and hypertension |
| Essential Hypertension | None yet |
| Systemic Sclerosis | None yet |
Acknowledgments
Sources of Funding: This work was supported by National Institute of Health Grants HL076558 (to YX) and RC4HD067977 and HD34130 (to YX and REK). American Heart Association Grant 10GRNT3760081 (to YX) and China National Science Foundation 81228004 (to YX).
Footnotes
Nonstandard Abbreviations and Acronyms: None.
Disclosures: None
References
- 1.Smith BR, Hall R. Thyroid-stimulating immunoglobulins in graves' disease. Lancet. 1974;2:427–431. doi: 10.1016/s0140-6736(74)91815-7. [DOI] [PubMed] [Google Scholar]
- 2.Smith BR, Sanders J, Furmaniak J. Tsh receptor antibodies. Thyroid. 2007;17:923–938. doi: 10.1089/thy.2007.0239. [DOI] [PubMed] [Google Scholar]
- 3.Jahns R, Boivin V, Lohse MJ. Beta(1)-adrenergic receptor function, autoimmunity, and pathogenesis of dilated cardiomyopathy. Trends in cardiovascular medicine. 2006;16:20–24. doi: 10.1016/j.tcm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Fu ML, Herlitz H, Wallukat G, Hilme E, Hedner T, Hoebeke J, Hjalmarson A. Functional autoimmune epitope on alpha 1-adrenergic receptors in patients with malignant hypertension. Lancet. 1994;344:1660–1663. doi: 10.1016/s0140-6736(94)90456-1. [DOI] [PubMed] [Google Scholar]
- 5.Luther HP, Homuth V, Wallukat G. Alpha 1-adrenergic receptor antibodies in patients with primary hypertension. Hypertension. 1997;29:678–682. doi: 10.1161/01.hyp.29.2.678. [DOI] [PubMed] [Google Scholar]
- 6.Wenzel K, Haase H, Wallukat G, Derer W, Bartel S, Homuth V, Herse F, Hubner N, Schulz H, Janczikowski M, Lindschau C, Schroeder C, Verlohren S, Morano I, Muller DN, Luft FC, Dietz R, Dechend R, Karczewski P. Potential relevance of alpha(1)-adrenergic receptor autoantibodies in refractory hypertension. PloS one. 2008;3:e3742. doi: 10.1371/journal.pone.0003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin at1 receptor. The Journal of clinical investigation. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y, Wen H, Bobst S, Day MC, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. Journal of the Society for Gynecologic Investigation. 2003;10:82–93. doi: 10.1016/s1071-5576(02)00259-9. [DOI] [PubMed] [Google Scholar]
- 9.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nature medicine. 2008;14:855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, Kintscher U, Rudolph B, Hoebeke J, Eckert D, Mazak I, Plehm R, Schonemann C, Unger T, Budde K, Neumayer HH, Luft FC, Wallukat G. Angiotensin ii type 1-receptor activating antibodies in renal-allograft rejection. The New England journal of medicine. 2005;352:558–569. doi: 10.1056/NEJMoa035717. [DOI] [PubMed] [Google Scholar]
- 11.Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, Sjogren KG, Hjalmarson A, Muller-Esterl W, Hoebeke J. Autoantibodies against the angiotensin receptor (at1) in patients with hypertension. Journal of hypertension. 2000;18:945–953. doi: 10.1097/00004872-200018070-00017. [DOI] [PubMed] [Google Scholar]
- 12.Wei F, Jia XJ, Yu SQ, Gu Y, Wang L, Guo XM, Wang M, Zhu F, Cheng X, Wei YM, Zhou ZH, Fu M, Liao YH, Group S-AS Candesartan versus imidapril in hypertension: A randomised study to assess effects of anti-at1 receptor autoantibodies. Heart. 2011;97:479–484. doi: 10.1136/hrt.2009.192104. [DOI] [PubMed] [Google Scholar]
- 13.Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Annual review of pathology. 2010;5:173–192. doi: 10.1146/annurev-pathol-121808-102149. [DOI] [PubMed] [Google Scholar]
- 14.Roberts JM, Cooper DW. Pathogenesis and genetics of pre-eclampsia. Lancet. 2001;357:53–56. doi: 10.1016/s0140-6736(00)03577-7. [DOI] [PubMed] [Google Scholar]
- 15.Herse F, Verlohren S, Wenzel K, Pape J, Muller DN, Modrow S, Wallukat G, Luft FC, Redman CW, Dechend R. Prevalence of agonistic autoantibodies against the angiotensin ii type 1 receptor and soluble fms-like tyrosine kinase 1 in a gestational age-matched case study. Hypertension. 2009;53:393–398. doi: 10.1161/HYPERTENSIONAHA.108.124115. [DOI] [PubMed] [Google Scholar]
- 16.Stepan H, Wallukat G, Schultheiss HP, Faber R, Walther T. Is parvovirus b19 the cause for autoimmunity against the angiotensin ii type receptor? Journal of reproductive immunology. 2007;73:130–134. doi: 10.1016/j.jri.2006.08.084. [DOI] [PubMed] [Google Scholar]
- 17.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. At(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382–2387. doi: 10.1161/01.cir.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 18.Bobst SM, Day MC, Gilstrap LC, 3rd, Xia Y, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin-6 and plasminogen activator inhibitor-1 secretion. American journal of hypertension. 2005;18:330–336. doi: 10.1016/j.amjhyper.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Dechend R, Viedt C, Muller DN, Ugele B, Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, Fiebeler A, Homuth V, Dietz R, Haller H, Kreuzer J, Luft FC. At1 receptor agonistic antibodies from preeclamptic patients stimulate nadph oxidase. Circulation. 2003;107:1632–1639. doi: 10.1161/01.CIR.0000058200.90059.B1. [DOI] [PubMed] [Google Scholar]
- 20.Zhou CC, Ahmad S, Mi T, Abbasi S, Xia L, Day MC, Ramin SM, Ahmed A, Kellems RE, Xia Y. Autoantibody from women with preeclampsia induces soluble fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated t-cells signaling. Hypertension. 2008;51:1010–1019. doi: 10.1161/HYPERTENSIONAHA.107.097790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou CC, Irani RA, Zhang Y, Blackwell SC, Mi T, Wen J, Shelat H, Geng YJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody-mediated tumor necrosis factor-alpha induction contributes to increased soluble endoglin production in preeclampsia. Circulation. 2010;121:436–444. doi: 10.1161/CIRCULATIONAHA.109.902890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: Correlation with disease severity. Hypertension. 2010;55:386–393. doi: 10.1161/HYPERTENSIONAHA.109.140061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irani RA, Zhang Y, Zhou CC, Blackwell SC, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-alpha signaling. Hypertension. 2010;55:1246–1253. doi: 10.1161/HYPERTENSIONAHA.110.150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irani RA, Zhang Y, Blackwell SC, Zhou CC, Ramin SM, Kellems RE, Xia Y. The detrimental role of angiotensin receptor agonistic autoantibodies in intrauterine growth restriction seen in preeclampsia. The Journal of experimental medicine. 2009;206:2809–2822. doi: 10.1084/jem.20090872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou CC, Irani RA, Dai Y, Blackwell SC, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Autoantibody-mediated il-6-dependent endothelin-1 elevation underlies pathogenesis in a mouse model of preeclampsia. Journal of immunology. 2011;186:6024–6034. doi: 10.4049/jimmunol.1004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parrish MR, Murphy SR, Rutland S, Wallace K, Wenzel K, Wallukat G, Keiser S, Ray LF, Dechend R, Martin JN, Granger JP, LaMarca B. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin ii type i receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. American journal of hypertension. 2010;23:911–916. doi: 10.1038/ajh.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamarca B, Speed J, Ray LF, Cockrell K, Wallukat G, Dechend R, Granger J. Hypertension in response to il-6 during pregnancy: Role of at1-receptor activation. International journal of interferon, cytokine and mediator research : IJIM. 2011;2011:65–70. doi: 10.2147/IJICMR.S22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhillion P, Wallace K, Herse F, Scott J, Wallukat G, Heath J, Mosely J, Martin JN, Jr, Dechend R, LaMarca B. Il-17-mediated oxidative stress is an important stimulator of at1-aa and hypertension during pregnancy. American journal of physiology. Regulatory, integrative and comparative physiology. 2012;303:R353–358. doi: 10.1152/ajpregu.00051.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Irani RA, Zhang Y, Ramin SM, Blackwell SC, Tao L, Kellems RE, Xia Y. Autoantibody-mediated complement c3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension. 2012;60:712–721. doi: 10.1161/HYPERTENSIONAHA.112.191817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shagdarsuren E, Wellner M, Braesen JH, Park JK, Fiebeler A, Henke N, Dechend R, Gratze P, Luft FC, Muller DN. Complement activation in angiotensin ii-induced organ damage. Circulation research. 2005;97:716–724. doi: 10.1161/01.RES.0000182677.89816.38. [DOI] [PubMed] [Google Scholar]
- 31.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sflt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. The Journal of clinical investigation. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cerdeira AS, Karumanchi SA. Angiogenic factors in preeclampsia and related disorders. Cold Spring Harbor perspectives in medicine. 2012;2 doi: 10.1101/cshperspect.a006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siddiqui AH, Irani RA, Zhang Y, Dai Y, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Recombinant vascular endothelial growth factor 121 attenuates autoantibody-induced features of pre-eclampsia in pregnant mice. American journal of hypertension. 2011;24:606–612. doi: 10.1038/ajh.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siddiqui AH, Irani RA, Zhang W, Wang W, Blackwell SC, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody-mediated soluble fms-like tyrosine kinase-1 induction contributes to impaired adrenal vasculature and decreased aldosterone production in preeclampsia. Hypertension. 2013 doi: 10.1161/HYPERTENSIONAHA.111.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D'Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nature medicine. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 36.George EM, Palei AC, Granger JP. Endothelin as a final common pathway in the pathophysiology of preeclampsia: Therapeutic implications. Current opinion in nephrology and hypertension. 2012;21:157–162. doi: 10.1097/MNH.0b013e328350094b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barker DJ. In utero programming of chronic disease. Clin Sci (Lond) 1998;95:115–128. [PubMed] [Google Scholar]
- 38.Baum M, Ortiz L, Quan A. Fetal origins of cardiovascular disease. Current opinion in pediatrics. 2003;15:166–170. doi: 10.1097/00008480-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Godfrey KM, Barker DJ. Fetal nutrition and adult disease. The American journal of clinical nutrition. 2000;71:1344S–1352S. doi: 10.1093/ajcn/71.5.1344s. [DOI] [PubMed] [Google Scholar]
- 40.Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biology of reproduction. 2003;69:1–7. doi: 10.1095/biolreprod.102.014977. [DOI] [PubMed] [Google Scholar]
- 41.Xia Y, Kellems RE. Is preeclampsia an autoimmune disease? Clinical immunology (Orlando, Fla. 2009;133:1–12. doi: 10.1016/j.clim.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xia Y, Kellems RE. Receptor-activating autoantibodies and disease: Preeclampsia and beyond. Expert review of clinical immunology. 2011;7:659–674. doi: 10.1586/eci.11.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dechend R, Gratze P, Wallukat G, Shagdarsuren E, Plehm R, Brasen JH, Fiebeler A, Schneider W, Caluwaerts S, Vercruysse L, Pijnenborg R, Luft FC, Muller DN. Agonistic autoantibodies to the at1 receptor in a transgenic rat model of preeclampsia. Hypertension. 2005;45:742–746. doi: 10.1161/01.HYP.0000154785.50570.63. [DOI] [PubMed] [Google Scholar]
- 44.Takimoto E, Ishida J, Sugiyama F, Horiguchi H, Murakami K, Fukamizu A. Hypertension induced in pregnant mice by placental renin and maternal angiotensinogen. Science. 1996;274:995–998. doi: 10.1126/science.274.5289.995. [DOI] [PubMed] [Google Scholar]
- 45.LaMarca B, Parrish M, Ray LF, Murphy SR, Roberts L, Glover P, Wallukat G, Wenzel K, Cockrell K, Martin JN, Jr, Ryan MJ, Dechend R. Hypertension in response to autoantibodies to the angiotensin ii type i receptor (at1-aa) in pregnant rats: Role of endothelin-1. Hypertension. 2009;54:905–909. doi: 10.1161/HYPERTENSIONAHA.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D, Bennett W. Reduced uterine perfusion pressure (rupp) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods in molecular medicine. 2006;122:383–392. doi: 10.1385/1-59259-989-3:381. [DOI] [PubMed] [Google Scholar]
- 47.Alexander BT, Kassab SE, Miller MT, Abram SR, Reckelhoff JF, Bennett WA, Granger JP. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension. 2001;37:1191–1195. doi: 10.1161/01.hyp.37.4.1191. [DOI] [PubMed] [Google Scholar]
- 48.Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: Linking placental ischemia with endothelial dysfunction. American journal of physiology. Heart and circulatory physiology. 2008;294:H541–550. doi: 10.1152/ajpheart.01113.2007. [DOI] [PubMed] [Google Scholar]
- 49.Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension. 2009;53:399–403. doi: 10.1161/HYPERTENSIONAHA.108.123513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LaMarca B, Speed J, Fournier L, Babcock SA, Berry H, Cockrell K, Granger JP. Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: Effect of tumor necrosis factor-alpha blockade. Hypertension. 2008;52:1161–1167. doi: 10.1161/HYPERTENSIONAHA.108.120881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: Role of interleukin 6. Hypertension. 2006;48:711–716. doi: 10.1161/01.HYP.0000238442.33463.94. [DOI] [PubMed] [Google Scholar]
- 52.LaMarca BD, Alexander BT, Gilbert JS, Ryan MJ, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension in response to placental ischemia during pregnancy: A central role for endothelin? Gender medicine. 2008;5(Suppl A):S133–138. doi: 10.1016/j.genm.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type i receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension. 2008;52:1168–1172. doi: 10.1161/HYPERTENSIONAHA.108.120576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LaMarca B, Wallace K, Herse F, Wallukat G, Martin JN, Jr, Weimer A, Dechend R. Hypertension in response to placental ischemia during pregnancy: Role of b lymphocytes. Hypertension. 2011;57:865–871. doi: 10.1161/HYPERTENSIONAHA.110.167569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, Wilson M, Martin JN, Jr, LaMarca B. Cd4+ t-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension. 57:949–955. doi: 10.1161/HYPERTENSIONAHA.110.168344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Novotny SR, Wallace K, Heath J, Moseley J, Dhillon P, Weimer A, Wallukat G, Herse F, Wenzel K, Martin JN, Jr, Dechend R, Lamarca B. Activating autoantibodies to the angiotensin ii type i receptor play an important role in mediating hypertension in response to adoptive transfer of cd4+ t lymphocytes from placental ischemic rats. American journal of physiology. Regulatory, integrative and comparative physiology. 2012;302:R1197–1201. doi: 10.1152/ajpregu.00623.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.LaMarca B, Parrish MR, Wallace K. Agonistic autoantibodies to the angiotensin ii type i receptor cause pathophysiologic characteristics of preeclampsia. Gender medicine. 2012;9:139–146. doi: 10.1016/j.genm.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parrish MR, Ryan MJ, Glover P, Brewer J, Ray L, Dechend R, Martin JN, Jr, Lamarca BB. Angiotensin ii type 1 autoantibody induced hypertension during pregnancy is associated with renal endothelial dysfunction. Gender medicine. 8:184–188. doi: 10.1016/j.genm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LaMarca B, Wallace K, Granger J. Role of angiotensin ii type i receptor agonistic autoantibodies (at1-aa) in preeclampsia. Current opinion in pharmacology. 11:175–179. doi: 10.1016/j.coph.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lamarca B. The role of immune activation in contributing to vascular dysfunction and the pathophysiology of hypertension during preeclampsia. Minerva ginecologica. 2010;62:105–120. [PMC free article] [PubMed] [Google Scholar]
- 61.Irani RA, Xia Y. Renin angiotensin signaling in normal pregnancy and preeclampsia. Seminars in nephrology. 2011;31:47–58. doi: 10.1016/j.semnephrol.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, LaMarca B, Reckelhoff JF. A model of preeclampsia in rats: The reduced uterine perfusion pressure (rupp) model. American journal of physiology. Heart and circulatory physiology. 2012;303:H1–8. doi: 10.1152/ajpheart.00117.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dragun D, Catar R, Kusch A, Heidecke H, Philippe A. Non-hla-antibodies targeting angiotensin type 1 receptor and antibody mediated rejection. Human immunology. 2012;73:1282–1286. doi: 10.1016/j.humimm.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 64.Dragun D, Philippe A, Catar R. Role of non-hla antibodies in organ transplantation. Current opinion in organ transplantation. 2012;17:440–445. doi: 10.1097/MOT.0b013e328355f12b. [DOI] [PubMed] [Google Scholar]
- 65.Hiemann NE, Meyer R, Wellnhofer E, Schoenemann C, Heidecke H, Lachmann N, Hetzer R, Dragun D. Non-hla antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplantation. 2012;94:919–924. doi: 10.1097/TP.0b013e3182692ad2. [DOI] [PubMed] [Google Scholar]
- 66.Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against at1-receptor and alpha1-adrenergic receptor in patients with hypertension. Hypertension research : official journal of the Japanese Society of Hypertension. 2002;25:641–646. doi: 10.1291/hypres.25.641. [DOI] [PubMed] [Google Scholar]
- 67.Zhu F, Sun Y, Wang M, Ma S, Chen X, Cao A, Chen F, Qiu Y, Liao Y. Correlation between hla-drb1, hla-dqb1 polymorphism and autoantibodies against angiotensin at(1) receptors in chinese patients with essential hypertension. Clinical cardiology. 2011;34:302–308. doi: 10.1002/clc.20852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riemekasten G, Philippe A, Nather M, Slowinski T, Muller DN, Heidecke H, Matucci-Cerinic M, Czirjak L, Lukitsch I, Becker M, Kill A, van Laar JM, Catar R, Luft FC, Burmester GR, Hegner B, Dragun D. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Annals of the rheumatic diseases. 2011;70:530–536. doi: 10.1136/ard.2010.135772. [DOI] [PubMed] [Google Scholar]
- 69.Bai M. Dimerization of g-protein-coupled receptors: Roles in signal transduction. Cellular signalling. 2004;16:175–186. doi: 10.1016/s0898-6568(03)00128-1. [DOI] [PubMed] [Google Scholar]
- 70.Breitwieser GE. G protein-coupled receptor oligomerization: Implications for g protein activation and cell signaling. Circulation research. 2004;94:17–27. doi: 10.1161/01.RES.0000110420.68526.19. [DOI] [PubMed] [Google Scholar]
- 71.Terrillon S, Bouvier M. Roles of g-protein-coupled receptor dimerization. EMBO reports. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased at(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin ii responsiveness. Nature medicine. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- 73.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin ii at2 receptor is an at1 receptor antagonist. The Journal of biological chemistry. 2001;276:39721–39726. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 74.AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U. Factor xiiia transglutaminase crosslinks at1 receptor dimers of monocytes at the onset of atherosclerosis. Cell. 2004;119:343–354. doi: 10.1016/j.cell.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 75.Wallukat G, Muller J, Podlowski S, Nissen E, Morwinski R, Hetzer R. Agonist-like beta-adrenoceptor antibodies in heart failure. The American journal of cardiology. 1999;83:75H–79H. doi: 10.1016/s0002-9149(99)00265-9. [DOI] [PubMed] [Google Scholar]
- 76.Mijares A, Lebesgue D, Wallukat G, Hoebeke J. From agonist to antagonist: Fab fragments of an agonist-like monoclonal anti-beta(2)-adrenoceptor antibody behave as antagonists. Molecular pharmacology. 2000;58:373–379. doi: 10.1124/mol.58.2.373. [DOI] [PubMed] [Google Scholar]
- 77.Wallukat G, Fu HM, Matsui S, Hjalmarson A, Fu ML. Autoantibodies against m2 muscarinic receptors in patients with cardiomyopathy display non-desensitized agonist-like effects. Life sciences. 1999;64:465–469. doi: 10.1016/s0024-3205(98)00589-x. [DOI] [PubMed] [Google Scholar]
- 78.Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J. Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the beta 1-adrenoceptor with positive chronotropic effect. Circulation. 1994;89:2760–2767. doi: 10.1161/01.cir.89.6.2760. [DOI] [PubMed] [Google Scholar]
- 79.Wallukat G, Fu ML, Magnusson Y, Hjalmarson A, Hoebeke J, Wollenberger A. Agonistic effects of anti-peptide antibodies and autoantibodies directed against adrenergic and cholinergic receptors: Absence of desensitization. Blood pressure. Supplement. 1996;3:31–36. [PubMed] [Google Scholar]
- 80.Wallukat G, Podlowski S, Nissen E, Morwinski R, Csonka C, Tosaki A, Blasig IE. Functional and structural characterization of anti-beta1-adrenoceptor autoantibodies of spontaneously hypertensive rats. Molecular and cellular biochemistry. 2003;251:67–75. [PubMed] [Google Scholar]
- 81.Doyle HA, Mamula MJ. Autoantigenesis: The evolution of protein modifications in autoimmune disease. Current opinion in immunology. 2012;24:112–118. doi: 10.1016/j.coi.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Doyle HA, Mamula MJ. Post-translational protein modifications in antigen recognition and autoimmunity. Trends in immunology. 2001;22:443–449. doi: 10.1016/s1471-4906(01)01976-7. [DOI] [PubMed] [Google Scholar]
- 83.Doyle HA, Mamula MJ. Posttranslational protein modifications: New flavors in the menu of autoantigens. Current opinion in rheumatology. 2002;14:244–249. doi: 10.1097/00002281-200205000-00009. [DOI] [PubMed] [Google Scholar]
- 84.Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Annals of the New York Academy of Sciences. 2005;1050:1–9. doi: 10.1196/annals.1313.001. [DOI] [PubMed] [Google Scholar]
- 85.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, Wilson M, Martin JN, Jr, LaMarca B. Cd4+ t-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension. 2011;57:949–955. doi: 10.1161/HYPERTENSIONAHA.110.168344. [DOI] [PMC free article] [PubMed] [Google Scholar]