

Abstract
Hepatitis C virus (HCV) is a significant public health concern with approximately 160 million people infected worldwide 1. HCV infection often results in chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. No vaccine is available and current therapies are effective against certain, but not all, genotypes. HCV is an enveloped virus with two surface glycoproteins (E1 and E2). E2 binds to the host cell through interactions with scavenger receptor class B type I (SR-BI) and CD81, and serves as a target for neutralizing antibodies 2-4. Little is known about the molecular mechanism that mediates cell entry and membrane fusion, although E2 is predicted to be a class II viral fusion protein. Here we describe the structure of the E2 core domain in complex with an Fab at 2.4 Å resolution. The E2 core has a compact, globular domain structure, consisting mostly of beta strands and random coil with two small alpha helices. The strands are arranged in two, perpendicular sheets (A and B), which are held together by an extensive hydrophobic core and disulfide bonds. Sheet A has an IgG-like fold that is commonly found in viral and cellular proteins while sheet B represents a novel fold. Solution-based studies demonstrate that the full-length E2 ectodomain has a similar globular architecture and does not undergo significant conformational or oligomeric rearrangements upon exposure to low pH. Thus, the IgG-like fold is the only feature that E2 shares with class II membrane fusion proteins. These results provide unprecedented insights into HCV entry and will assist in developing an HCV vaccine and new inhibitors.
HCV envelope glycoprotein 2 (E2) is a type II transmembrane protein with an amino-terminal ectodomain connected to a carboxyl-terminal transmembrane helix through an amphipathic, alpha helical stem (Fig. 1a) 5,6. E2 is highly modified post translationlly with 9 to 11 N-linked glycosylation sites and 18 cysteine residues that are conserved across all genotypes. For ease of comparison with other genotypes, the cysteines and N-linked glycosylation sites will be referred to as C1 to C18 and N1 to N11, respectively, with residue numbers from the J6 (2a) genome given in parentheses. Full-length, E2 ectodomain (eE2) (384-656) was produced in HEK293T GnTI- cells by a lentiviral expression system and grown in an adherent cell bioreactor. The resulting eE2 protein is monomeric as determined by non-reducing SDS-PAGE and size exclusion chromatography (SEC) (Extended Data Fig. 1).
Figure 1. Overview of HCV E2.

a, Schematic representation of the HCV genome and E2 domain organization. Full-length eE2 and the crystallization construct are indicated by the black and grey bars, respectively. C, capsid protein; nonstructural protein (NS) 2-5B. Stars indicate the location of trypsin (blue), chymotrypsin (green) and GluC (magenta) cleavage sites. Ribbon diagram of the E2 core domain bound to Fab 2A12 (b) and alone (c and d). The view in d is a 90° rotation about a horizontal axis from c. The E2 polypeptide chain is colored from the N terminus (blue) to C terminus (red). e, Topology diagram of E2 core domain, detailing secondary structure elements, disulfide bonds (dashed lines), N-linked glycosylation sites and regions of disordered polypeptide (dotted lines).
Solution-based studies using limited proteolysis and hydrogen deuterium exchange (HDX) demonstrated that approximately 80 amino acids on the amino terminus (384-463) from hypervariable region (HVR) 1 through HVR2 are exposed and flexible. This region includes conserved sequences implicated in binding to the cellular receptors (SR-BI and CD81) as well as several epitopes for neutralizing antibodies (Fig. 1 and Extended Data Figs. 2, 3) 7-11. Various amino-terminal deletions were produced to minimize regions of disorder while preserving an even number of cysteines, potentially allowing them to form intramolecular disulfide bonds. All constructs were screened for aggregation by non-reducing SDS-PAGE and SEC. E2 core (456-656) is soluble, monomeric, and maintains similar secondary structure content when compared with eE2 as determined by reactivity towards HCV infected patient sera (Extended Data Fig. 4a-b) and circular dichroism (data not shown). However, in contrast to eE2, CD81 binding affinity and the efficiency of inhibition of HCVcc entry was diminished for the E2 core (Extended Data Fig. 4c-e). This suggests that the amino-terminus of eE2 is critical for CD81 interaction and likely undergoes a transition from disorder to order upon binding. Alternatively, the amino-terminal region may also be ordered through interactions with other factors, e.g. E1, apolipoproteins, lipids, cellular receptors, or antibodies.
Monoclonal antibodies were generated against recombinant eE2 and crystals of deglycosylated E2 core were produced in complex with an Fab (2A12) to 2.4 Å resolution (Fig. 1 and Extended Data Table 1). The complex structure was determined by molecular replacement using an Fab structure followed by iterative rounds of model building and refinement. The E2 core domain has a globular fold, consisting of mostly beta strands and random coil with two short alpha helices, which is consistent with previous spectroscopic studies of eE2 12,13. The protein contains two, four-stranded antiparallel beta sheets (termed sheets A and B), the planes of which are approximately perpendicular to each other. The four strands of the amino-terminal beta sheet (sheet A) are stabilized by two disulfide bonds, between strands 1 and 3 [C7 (510) and C8 (554)] and the amino terminal loop with strand 4 [C5 (496) and C9 (566)]. The loop between strands 2 and 3 contains sequences implicated in CD81 binding 14,15 and is flexible, similar to the amino-terminal CD81 binding sites, which were deleted. After strand 4, the polypeptide continues into a long, disordered loop before forming the first short helix (H1) followed by the second beta sheet (sheet B). A second short alpha helix (H2) is located between strands 6 and 7. A disulfide bond [C14 (611) and C16 (648)] between strand 6 and the carboxyl-terminal strand 8 further stabilizes the fold. The carboxyl-terminal strands (7 and 8) are the longest within the protein with approximately nine amino acids each and encompass the 2A12-binding site. 2A12 does not neutralize HCV infection, suggesting that the epitope is either buried within the particle or incapable of preventing entry (Extended Data Fig. 4f). The two beta sheets are held together by i) two disulfide bonds, connecting the loops before strand 1 and after H2 [C4 (488) with C15 (624)] as well as the loops after strand 4 and before H1 [C10 (571) and C13 (601)], and ii) an extensive hydrophobic core consisting of numerous aromatic residues (Extended Data Fig. 5).
HCV belongs to the genus Hepacivirus of the Flaviviridae family. Other members of the family include the flavivirus and pestivirus genera, which consist of arthropod-borne viruses and important livestock pathogens, respectively 16. The flavivirus envelope glycoprotein (E) is a class II fusion protein and HCV E2 was expected to have a similar fold 10,17,18. All class II fusion proteins have a common elongated structure, consisting of predominantly beta sheets, and exist as homo or heterodimers with the membrane-fusion, hydrophobic peptide buried at the dimer interface at neutral pH. Upon receptor binding and/or exposure to low pH, these proteins undergo self-rearrangement into stable trimers, exposing the fusion peptide and resulting in viral and host membrane fusion. Despite containing a similar extended organization, the recent structure of the pestivirus, bovine viral diarrhea virus (BVDV) E2 glycoprotein does not represent a typical class II fusion protein fold and lacks an apparent fusion peptide, suggesting it is unlikely to be a class II fusion protein 16,17,18.
Similar to the flavivirus and pestivirus glycoproteins, the HCV E2 core secondary structure consists of predominantly beta sheets and random coil. However, E2 core is a monomer with a compact globular shape, in contrast to the extended structures reported in other viruses. Solution-based small angle X-ray scattering (SAXS) was used to correlate the crystallographic core domain structure with fully glycosylated eE2 and various fragments. The ab initio SAXS envelopes of E2 core and eE2 are similar with approximately the same radius of gyration (Rg) (Fig. 2a-b and Extended Data Table 2). Glycosylation, which is missing in the E2 core crystal structure, represents roughly a third of the mass and accounts for the unmodeled areas of the envelopes. Interestingly, neither the Rg nor the elution profiles on SEC for fully glycosylated eE2 and E2 core changed significantly at pH 5.0 (Extended Data Fig. 6a-b). These results suggest that unlike class II membrane fusion proteins, E2 does not undergo significant structural rearrangements upon exposure to low pH.
Figure 2. Ab initio SAXS envelopes of E2 core, eE2ΔHVR1 and eE2.

SAXS envelopes of glycosylated E2 core (a), eE2 (b), eE2ΔHVR1 (c), and eE2ΔHVR1 in complex with CD81-LEL (d). The E2 core domain structure has been fitted into a and b. e, Superposition of the SAXS envelopes of eE2ΔHVR1 alone (c) and in complex with CD81-LEL (d), highlighting the approximate position of CD81-LEL.
SAXS was used to investigate the CD81 binding region on the E2 ectodomain. To simplify data interpretation, eE2ΔHVR1 was used since HCV lacking HVR1 remains infectious 16. The binding site of CD81 was identified by superimposing the SAXS envelopes of eE2ΔHVR1 alone and in complex with CD81-LEL (Fig. 2c-e). Although CD81-LEL is a dimer in solution (Extended Data Fig. 6c), the extra density in the SAXS envelope is more consistent with monomeric binding, however a dimer cannot be ruled out.
HCV E2 is modified by N-linked glycosylation, which is necessary for proper folding and immune invasion. E2 from the J6 genotype has 11 glycosylation sites. Four of the glycosylation sites are in the flexible amino terminal region, which were deleted, and seven are in the core domain (N5-N11). The location of N7, N8, N10 and N11 are modeled in the final E2 core structure. All of these glycans are present in loop areas, indicating that these sites are solvent exposed and flexible. Mutagenesis studies in HCVcc have shown that N6, N8, and N10 are integral for virus infectivity. Removal of the N6 site results in improved CD81 binding, while N8 and N10 mutations destabilize the protein and cause defective particle production 19. Both sheets have one critical glycosylation site, N8 in sheet A and N10 in sheet B. All four of the observed glycosylation sites are on the periphery of the core and are located on a highly basic surface (Fig. 3). The opposite surface is predominantly hydrophobic and highly conserved when compared to the basic surface. Furthermore, the epitope for antibodies (AR1, AR3A and AR3C) that inhibit E1E2 binding to CD81 is located at the interface of the hydrophobic and basic surfaces, including the N7 glycosylation site (Extended Data Fig. 7a). Interestingly, N7 is only 7 residues away from N6, which has a critical role in CD81 binding. Epitopes for antibodies (i.e., AR5) that block E1E2 heterodimerization are also found on the hydrophobic surface, making it highly plausible that this surface is interacting with E1 in the context of the viral particle 4.
Figure 3. Surface features of E2.

The surface of the E2 core domain colored for (a and d) electrostatic-1 potential: blue (basic), white (neutral), red (acidic) at ± 5 kTe-1 and (b and e) sequence identity (green) from the alignment in Extended Data Fig. 2. Ribbon diagram highlighting the location of the N-linked glycosylation sites are shown (c and f). The orientation of d, e, and f as well as a, b, and c are identical. The orientation in d, e, and f is rotated 180° about a horizontal axis from the view in a, b and c.
The precise roles played by E1 and E2 in membrane fusion are not fully understood. It has been predicted that amino acids 262-290 in E1 as well as 416-430, 504-522, and 604-624 in E2 are important for fusion 13,20,21. In the structure, the potential fusion regions in E2 (504-522 and 604-624) are located in secondary structure elements and therefore unlikely to serve as the fusion peptide. Furthermore, SEC and SAXS analyses at low pH indicate that E2 does not undergo oligomeric or structural rearrangement. Thus, it seems unlikely that E2 has a direct role in membrane fusion. However, it is possible that E1 alone or the E1E2 heterodimer plays a major role in the fusion process.
Structural comparison of the HCV E2 core domain with all known folds in the Protein Data Bank using the Dali server22 identified proteins with IgG-like folds similar to the amino-terminal sheet A, none of which are class II fusion proteins although IgG-like folds are common in these proteins. The server failed to identify any statistically significant structures to sheet B, suggesting a novel fold. During the review process of this manuscript, a structure for HCV E2 from genotype 1a was published 23. The core domain of both structures is highly similar with an RMSD of 0.8 Å for similar carbon alpha atoms. In summary, our biochemical and structural data provide valuable information towards defining the role of E2 and establishes a foundation for further studies in understanding HCV entry and infection.
Full Methods
Materials and Methods
J6 eE2 expression
eE2, eE2ΔHVR1 and E2 core domain encompasses residues 384-656, 413-656, and 456-656 from the HCV J6 genome, respectively. Due to incomplete deglycosylation at N7 (542) with EndoH, the crystallization construct contained an asparagine to glutamine mutation at this position. The expression constructs consisted of a CMV promoter, a prolactin signal sequence, E2 fragment, PreScission Protease cleavage site and a carboxyl-terminal protein-A (ProtA) tag. The entire prolactin-E2-ProtA sequence was PCR amplified and cloned into the pJG lentiviral vector (Dr. John Shires, Emory University).
Wild type and GnTI-HEK293T cells 24 (kindly provided by Dr. Davide Comoletti, Rutgers University Robert Wood Johnson Medical School) were maintained in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. One day prior to the planned transfection, a single T-225 monolayer flask was seeded with 6.0×106 HEK293T cells. 90 μg pJG-E2, 60 μg psPAX2 (HIV Gag-Pol packaging vector), 30 μg pMD2.G (VSV glycoprotein vector), and 450 μL of 2 M CaCl2 were mixed and brought to a final volume of 4.5 mL with ddH20. 4.5 mL of 2× HEPES buffered saline was added at room temperature. After two minute incubation, the mixture was added directly to HEK293T cells. After 6-8 hours, the media was replaced with DMEM with 10% FBS and 1% Antibiotic/antimycotic (A/A) media and incubated for another 70 hours.
Two days post transfection, 10,000 GnTI- HEK293T cells were seeded into a single well of a 96-well plate. The supernatant from the transfection, containing the recombinant lentiviruses, was harvested and centrifuged for 30 min at 4000 ×g at 4°C to pellet large cellular debris. Clarified supernatant was transferred to a Beckman Ultracentrifuge tube and virus was pelleted for 1.5 hours at 25,000 rpm (80,000 ×g) at 4°C in an SW28 rotor. Supernatant was discarded and the pellet resuspended in 120 μL of DMEM containing 20% FBS, 1% A/A, and 8 μg/mL of polybrene. 60 μL of virus suspension was added to the prepared GnTI-HEK293T cells and incubated overnight. Infected cells were expanded and ultimately seeded into an adherent cell bioreactor (Cesco Bioengineering) for long term growth and protein production.
eE2, eE2ΔHVR1, and E2 core purification
Cell supernatant containing E2-ProtA was centrifuged for 10 min at 7,000 ×g, filtered through a 0.22 μM membrane, and loaded onto an IgG FF column (GE Healthcare). The column was extensively washed with 20 mM sodium phosphate pH 7.0 and then equilibrated with 20 mM HEPES pH 7.5, 250 mM NaCl, and 5% glycerol. PreScission Protease was added into the column at approximately 400 μg per L of supernatant and incubated overnight at 4°C. For deglycosylation, the pH of the protein solution was adjusted using 1 M sodium citrate pH 5.5 to a final concentration of 100 mM. EndoH was added at a ratio of 1 mg per 2 mg of E2 and the reaction was incubated at room temperature for 3-4 hours. The deglycosylated proteins were desalted into 20 mM HEPES pH 7.5, 50 mM NaCl, and 5% glycerol and purified by heparin affinity followed by size exclusion chromatography over Superdex200 column. Final yields for all E2 proteins averaged 30 mg per liter of supernatant.
Crystallization
A 1.1:1 molar ratio of E2 core domain to Fab was incubated for 1-2 hours at 4°C. The complex was purified over a Superdex200 column equilibrated with 20 mM HEPES pH 7.5 and 100 mM NaCl. The complex was concentrated to 5-7 mg/mL and crystals were grown by the hanging drop vapor diffusion method. Briefly, 2.5 μL of complex was mixed with an equal volume of reservoir solution, comprising of 22% (w/v) PEG 3350, 0.5 M MgCl2, 0.1 M HEPES pH 7.5, and 15% (v/v) dioxane. Initially, clusters of plate-like crystals grew in 3-4 days. Single, plate-like crystals were obtained via microseeding using a similar reservoir solution supplemented with 2% (v/v) formamide. Crystals were cryoprotected using reservoir solution with 24% (v/v) ethylene glycol and flash cooled in liquid nitrogen. Data was collected at a wavelength of 0.979 Å using beam line X25 of the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory.
Structure determination and refinement
The crystals belong to space group P21212 with cell parameters a= 85.96 Å, b= 194.57 Å, c= 37.92 Å. Phases were determined by the molecular replacement method using PHENIX 27 and the coordinates from chains A and B from PDB entry 2GSI. Unambiguous placement of the Fab heavy and light chains provided the necessary phases to extend the map to cover E2 core domain using iterative rounds of model building and density modification by COOT 28, PHENIX, REFMAC 29, and PARROT 30. The final model was built to a resolution of 2.40 Å, comprising residues 492-522, 538-571, and 596-649 of E2 from the J6 genome, 1-217 of 2A12 light chain, and 1-133, and 136-218 of 2A12 heavy chain with two N-linked, N-acetylglucosamine, six molecules of formamide and 141 solvents molecules. The model coordinates were refined to Rwork 0.217 and Rfree 0.269. Model validation demonstrated 95.0% of the residues located in the most favorable region of the Ramachandran plot with 4.8% in the generously allowed regions 27. Statistics of the data processing and structure refinement are summarized in Supplementary Table 1.
Small angle X-ray scattering (SAXS)
Glycosylated E2 proteins were purified over IgG and anion exchange columns. The proteins were equilibrated with either pH 7.5 buffer (50 mM HEPES pH 7.5, 250 mM NaCl and 1% glycerol) or pH 5.0 buffer (50 mM sodium citrate pH 5.0, 250 mM NaCl and 1% glycerol) by Superdex200 gel filtration column. Glycosylated eE2ΔHVR1 alone or complex with CD81-LEL (1:2 molar ratios) was purified using pH 7.5 buffer by gel filtration chromatography. Three concentrations of each protein were prepared along with their respective buffers as background control. SAXS data was collected on the SIBYLS beam line at the Advanced Light Source, Lawrence Berkeley National Laboratory. Sample analysis and processing was performed using BioSAXS RAW 25 and GNOM 31. The ab initio models were calculated using the application DAMMIF 32. Consensus models and the normalized spatial discrepancy (NSD) values were calculated by averaging 10 ab initio models using the application DAMAVER 33. X-ray structures of CD81 (PDB ID 1G8Q), HCV E2 core and ab initio models were aligned using the application SUPCOMB 34.
Hydrogen deuterium exchange
HD exchange experiments were conducted as described by Sharma et al (2009) 35. Briefly, 5 μl of deglycosylated eE2 (1.5 mg/mL), in 200 mM NaCl, 20 mM HEPES pH 7.5, was incubated with 15 μl of the same buffer made with 99.96% 2H2O (Cambridge Isotope Laboratories) for 10, 100, or 1000 seconds and quenched in 30 μl of 2 M urea, 0.8% formic acid and 50 mM tris(2-carboxyethyl)phosphine (TCEP). The reaction mixture was immediately frozen on dry ice until injection. For the zero time point experiment, the protein was incubated in the buffer made with 1H2O and then quenched and frozen. To correct background exchange, a completely deuterated sample was produced by incubating the protein with 100 mM TCEP in 99.96% 2H2O overnight before being quenched and frozen. Dionex RSLC with a C18 column (2.1×50 mm, 3 μm, Q-C18, 150A, CMP Scientific) and LTQ Velos Orbitrap pro were used for LC-MS analysis. The mass was measured using Orbitrap with resolution of 60,000 and mass range from 300-2000 with top10 MSMS performed. The LC-MS data were analyzed using HDexaminer 1.2.0 (Serra Analytics) with manual checking of each peptide afterwards.
Limited proteolysis
8-10 μg of deglycosylated eE2 protein was mixed with trypsin, chymotrypsin or GluC at 1:120 (w/w) ratio (endopeptidase:E2) and incubated at room temperature. Samples were taken at noted time points and analyzed by reducing SDS-PAGE, mass spectrometry and N-terminal sequencing.
Production of monoclonal antibody 2A12
BALB/c mice were immunized intraperitoneally with 50 μg eE2 in either Complete Freund's Adjuvant (first immunization only), or incomplete Freund's Adujvant bi-weekly for eight weeks. A final immunization with 50 μg of eE2 was given intravenously four days prior to collection of splenocytes. Hybridomas were generated using a cloned HAT-sensitive mouse myeloma cell line as a fusion partner. Proliferating hybridomas were screened for their ability to bind eE2 via ELISA, at which point 2A12 was positively identified. Monoclonal antibodies were generated in the laboratory of Dr. Arash Grakoui (IACUC protocol number YER-2002369-070816GN, Emory University School of Medicine, principal investigator Dr. Arash Grakoui).
Generation and purification of 2A12 Fab
Hybridoma cells were expanded to a final volume of 2 L in spinner flasks at 100 rpm using Iscove's Modified Dulbecco's Medium, 10% ultra-low IgG FBS, 1% A/A, and 10 mM HEPES (Life Technologies). Cells were harvested at 2-3×106 cells/mL, centrifuged for 10 min at 7,000 ×g, filtered through a 0.22 μM membrane, and loaded onto a Protein G column (GE Healthcare Life Sciences). After completion, the column was washed with 20 mM sodium phosphate (pH 7.0) followed by phosphate buffered saline (PBS). The antibody was eluted with 0.05% TFA in 2 mL fractions into tubes containing 100 μL of 1 M Tris pH 7.5 for immediate pH neutralization. The eluted antibody was dialyzed into 20 mM sodium phosphate pH 7.0 and 10 mM EDTA. Insoluble papain was added to the antibody at 0.15 mg per 1 mg of antibody. Freshly prepared L-cysteine was added to the reaction to a final concentration of 20 mM and mixed at 37°C for 2 hours. The papain was removed by centrifugation at 3,500 ×g for 2 minutes and filtration through a 0.22 μM membrane. Fab was purified by subtractive chromatography over Protein A FF column and desalted into 20 mM Tris pH 8.0.
Sequencing Ig H and L chain gene segments of 2A12 antibody
Total RNA isolated from 2A12 hybridoma cells was reverse transcribed into cDNA using random hexamers. Expressed heavy (H) and light (L) chains were amplified using standard primers that are complimentary to all murine H and L chain gene segments 36. The PCR products were sequenced either directly or following cloning into pCR 2.1-TOPO vector (Life Technologies).
CD81 purification and binding assays
Human CD81 LEL (residues 112-202) was produced as a fusion with carboxyl-terminal protA tag in HEK293T cells using the same lentiviral expression system described for eE2. Cell culture supernatants were loaded onto an IgG FF column, washed with 20 mM sodium phosphate pH 7.0, eluted with 100 mM sodium citrate pH 3.0 containing 20 mM KCl and immediately neutralized with 1 M Tris pH 9.0. The protA tag was cleaved by PreScission Protease (GE Healthcare Life Sciences) in a ratio of 1:50 (w/w) followed by overnight dialysis in 20 mM HEPES pH 7.5, 250 mM NaCl, and 5% glycerol. High purity CD81 protein was obtained by anion exchange and size exclusion chromatography.
For binding studies, a 96-well plate (Nalgene Nunc, Thermo Fisher Scientific) was coated with 50 μg of CD81-LEL overnight at 4°C. All experiments were duplicated against BSA as a negative control. Plates were washed 3× with PBS containing 0.05% Tween 20 (PBS-T) and blocked with 3% (w/v) BSA in PBS-T for 1 hour at room temperature. 50 μL of eE2 or E2 core at different concentrations was added to appropriate wells and incubated overnight at 4°C. On day 3, the wells were washed 3× with PBS-T and incubated with mAb 2A12 cell supernatant for 1 hour at room temperature. Plates were washed 3× with PBS-T and incubated with anti-mouse-HRP conjugated antibody for 1 hour at room temperature. Finally, the plate was washed 5× with PBS-T. 50 μL of TMB substrate (ThermoFisher Scientific) was added to each well and incubated for 5 min, followed by the addition of 50 μL of 2 M sulfuric acid to stop the reaction. Absorbance readings were acquired at 450 nm using Softmax Pro software on a Spectra Max 250 (Molecular Devices).
Neutralization assay
Huh-7.5 cells were maintained in DMEM containing 10% FBS (Hyclone,) and 100 μg/mL of penicillin and streptomycin (Cellgro) at 37°C in 5% CO2. Naive Huh-7.5 cells were seeded at 6,000 cells per well in a 96-well plate. The following day, 100 μl of 2C1, 2A12, or H113 serially diluted in complete media were added per well at various concentrations. In parallel, 100 μL of eE2, E2 core, gp140, or CD81-LEL serially diluted in complete DMEM were added at varying concentrations beginning at 100 μg/mL. Cells were then infected with 100 μL of genotype 2a virus Cp7 encoding the Renilla luciferase gene 37. 72 hours post-infection, relative light units were measured on a Clarity 4.0 luminometer (Biotek) using the Renilla Luciferase Assay System (Promega).
Assessment of cellular cytotoxicity
Huh-7.5 cells were incubated with varying concentrations of protein as described above, beginning at 100 μg/mL. After 72 hours, cells were washed once with PBS, treated with trypsin, and harvested in 100 μL PBS. Cells were stained with 7-AAD according to the manufacturer's instructions (BD Biosciences) and analyzed using a BD LSR II and FlowJo software (Tree Star).
Human plasma ELISA
96 well enzyme immunoassay plates (ThermoFisher Scientific) were coated overnight at 4° with 50 μL of a 1 μg/mL solution of eE2 or E2 core diluted in 0.1 M Na2CO3. Plates were washed twice with PBS-T and then blocked for one hour at 37 °C in PBS-T containing 10% fetal calf serum (HyClone). Blood samples were collected in heparin tubes (Becton Dickinson) and plasma was isolated and frozen at -80 °C. Plasma was serially diluted in a binding solution composed of 0.1% (v/v) normal goat serum in PBS-T (Jackson ImmunoResearch Laboratories). 100 μL of sample were added per well and incubated at room temperature for 90 minutes. Following eight washes with PBS, 100 μL of mouse anti-human IgG biotin antibody (Mabtech) diluted 1:20,000 in binding solution were added per well and incubated 1 hour at room temperature. Following five additional washes with PBS, 100 μL streptavidin-horseradish peroxidase (HRP) conjugate was added to each well at a 1:2,000 dilution in binding buffer and incubated for 45 minutes at room temperature (Mabtech). Absorbance was measured and analyzed using a VersaMax microplate reader and SoftMax Pro software (Molecular Devices) following five washes and the addition of tetramethylbenzidine substrate solution (Ebioscience). Human sera were isolated from whole-blood samples informed consent was obtained for all subjects. (IRB no. 1358-2004, Emory University School of Medicine, principal investigator Dr. Arash Grakoui).
Alignment
Secondary structures were assigned using the program DSSP 38. Sequences were obtained from the National Center for Biotechnology Information (NCBI) using the following accession numbers: J6 ADV40003.1, H77 ACA53555.1, J8 P26661.3, S52 AEB71616.2, ED43 AEB71617.2, SA13 AEB71618.2, HK6a AEB71619.2, QC69 ACM69041.1. The E2 sequences were aligned with multiple alignment using fast fourier transform (MAFFT) 39 and edited for figure generation using JalView version 2 40.
Extended Data
Extended Data Figure 1. eE2, eE2ΔHVR1 and E2 core are highly soluble and monomeric in solution.
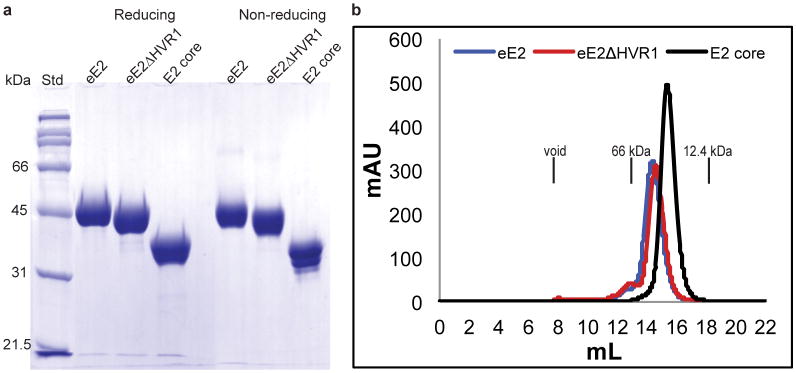
(a) A comparison of proteins under reducing and non-reducing conditions is shown by a 10% SDS-PAGE gel with protein standards (Std). (b) Size exclusion chromatography of eE2, eE2ΔHVR1 and E2 core proteins on a Superdex200 gel filtration column. The elution positions of the void volume (>200kDa), albumin (66kDa) and cytochrome C (12.4kDa) are indicated. Molecular weights of eE2, eE2ΔHVR1 and E2 core are ∼46kDa, ∼42kDa and ∼32kDa respectively.
Extended Data Figure 2. eE2 sequence alignment.

Red bent arrows indicate the amino- and carboxyl-terminal boundaries of the E2 crystallization construct. Cylinders and arrows represent α-helices and β-strands, respectively, and are colored according to cartoon representation in Fig.1. CD81 binding regions are bracketed in red; hypervariable regions are bracketed in black. SR-BI binds to HVR1. The stars indicate the location of trypsin (blue), chymotrypsin (green) and GluC (magenta) cleavage sites. The binding sites of neutralizing antibodies for which structural information is available are colored orange for HCV1 and AP33, blue for mAb #8, and purple for HC34-1 and HC34-17.
Extended Data Figure 3. Hydrogen deuterium exchange and limited proteolysis of eE2.

(a) The percentage hydrogen deuterium exchange shown at 10, 100 and 1000 seconds time points. The secondary structure of E2 core is placed above to emphasize flexible regions. A red arrow indicates the E2 core N-terminus. Extra residues (grey) on N- and C-terminus come from the vector. Potential cleavage sites for trypsin (blue), chymotrypsin (green) and GluC (magenta) are indicated by stars. The color pattern indicates the percentage of exchange. Grey areas are the regions of no coverage. (b) Digestion of deglycosylated eE2 with chymotrypsin (left) and GluC (right) reveals a shift from the ∼35kDa untreated protein (0 min) to ∼25kDa post-digestion. Samples were taken at the indicated time points and analyzed by reducing 12% SDS-PAGE gel. Molecular weight protein standards (Std) are indicated. The bands were analyzed by N-terminal sequencing and mass spectrometry.
Extended Data Figure 4. Functional analyses of eE2 and E2 core.

(b) Antibodies from patient sera infected with HCV genotype 2 show a concentration dependent binding to eE2 (red) whereas healthy donor sera exhibits only background binding (black). (b) Similar binding is observed for E2 core. The measurements were done in triplicate with the error bars representing the standard error of the mean (SEM). (c) E2 core (light grey) shows reduced binding to CD81 when compared to eE2 (dark grey) by an ELISA. Bars with stripes indicate E2 binding to a negative control, BSA. The solid black bar indicates CD81 binding to PBS, used to verify the absence of background. The measurements were done in triplicate with the error bars representing the SEM. (d) eE2 (blue) and CD81 LEL (positive control, grey) inhibit the infection. E2 core (red) shows reduced inhibition. HIV gp140 (black) expressed in the same system, was used as a negative control. The measurements were done in triplicate with the error bars representing the SEM. (e) To rule out the possibility of toxic effects from the recombinant proteins, the cell viability was measured as described in Material and Methods, using similar protein concentrations as in d. (f) In an ELISA, 2A12 (red), and an irrelevant antibody, H113 (grey), fail to neutralize HCVcc infection. 2C1 (positive control, black), a mouse monoclonal antibody that binds to the disordered amino-terminal region of eE2, blocks infection. The measurements were done in triplicate with the error bars representing the SEM.
Extended Data Figure 5. E2 core contains an extensive hydrophobic core.

Sheets A and B are held together by an extensive hydrophobic core composed of mostly aromatic amino acids (green) and five disulfide bonds (yellow).
Extended Data Figure 6. eE2 and E2 core do not undergo oligomeric changes at low pH.

An overlay of eE2 (a) and E2 core (b) elution profiles from Superdex200 gel filtration at pH 7.5 (blue) and pH 5.0 (red). The expected void volume and observed elution positions of individual proteins are indicated. (c) The SAXS envelope of CD81 LEL fit with a dimer crystal structure (PDB ID 1G8Q).
Extended Data Figure 7. Epitope mapping of conformational antibodies on E2 core surface.

(a) Surface epitopes of AR1 (orange) is shown. AR1 blocks the E1E2 heterodimer binding to CD81. AR5A (purple) inhibits E1E2 heterodimerization and is mapped on a well conserved hydrophobic surface of the core. (b) Surface of E2 core colored by electrostatic potential with conserved regions (blue). The view in a and b is identical.
Extended Data Table 1. Summary of the X-ray Crystallographic Analyses.
| Data collection | |
| Wavelength | 0.979 Å |
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 85.96, 194.57, 37.92 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 24.69-2.40 (2.49-2.40)* |
| Rmerge | 0.118 (1.042) |
| Rpim | 0.054 (0.490) |
| I/sI | 9.6 (1.9) |
| Completeness (%) | 100 (100) |
| Redundancy | 5.7 (5.3) |
| Refinement | |
| Resolution (Å) | 24.69-2.40 |
| No. unique reflections | 24349 (2517) |
| Rwork/Rfree | 0.217/0.269 |
| No. atoms | |
| Protein/glycans | 4162 |
| Solvent | 137 |
| Ions | 18 |
| B-factors (Å2) | |
| Protein/glycan | 54 |
| Solvent | 45 |
| Ions | 47 |
| R.M.S deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.14 |
| Ramachandran favored (%) | 95.0 |
| Ramachandran outliers (%) | 0.2 |
Highest resolution shell is shown in parenthesis.
Extended Data Table 2. Summary of SAXS Analyses.
| Protein | Rg (Å) | Dmax (Å) | NSD |
| E2 core | 27.8±0.03 | 95 | 0.705±0.026 |
| eE2ΔHVR1 | 28.9±0.05 | 101 | 0.696±0.075 |
| eE2 | 28.2±0.02 | 84 | 0.702±0.054 |
| eE2ΔHVR1 + CD81 | 36.8±0.22 | 127 | 0.714±0.026 |
| CD81 | 18.8±0.04 | 64 | 0.554±0.007 |
| E2 core pH 5.0 | 27.8±0.07 | 95 | - |
| eE2 pH 5.0 | 27.8±0.07 | 95 | - |
Supplementary Material
Acknowledgments
We acknowledge access to the X25 beamline at NSLS and thank the NSLS staff. NSLS is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. We thank J. Tainer and J. Perry for their support and access to SIBYLS beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory. We thank E. Arnold, J. Bonanno S.K. Burley, J. Chiu, D. Comoletti, E. Elrod, F. Jiang, S. Khare, A. Shatkin, A. Stock, J. Shires, and A. Thanou for providing helpful comments and assistance. Special thanks to C.M. Rice for providing J6 HCV clone and support. This work was supported by a Yerkes Research Center Base Grant RR-00165 (A.G) and NIH grants P50 GM103368 (J.M.), R01 AI080659 (J.M.) PHS AI070101 (A.G), and DK083356 (A.G). A.G.K. was supported by a grant from the New Jersey Commission On Cancer Research (DFHS13CRP001).
Footnotes
Authors' contributions. The project was initiated, designed and supervised by A.G. and J.M. A.G.K., J.W., A.V.Z., A.C., and S.A.Y. designed protein constructs and established purification protocols. A.G.K prepared all protein crystals. The mouse monoclonal antibody was produced by H.S. and A.G., and sequenced by C.D.B. and J.J. A.A.P. and A.G. performed the virus neutralization and patient sera ELISA. J.M., M.M., and A.G.K. collected, processed, and analyzed the X-ray crystallographic and SAXS data. A.G.K, J.W., and J.M. wrote the paper with contributions from all authors.
Author information. Reprints and permission information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of this article at www.nature.com/nature.
The coordinates and structure factors have been deposited to the Protein Data Bank under accession code (4NX3).
Supplementary information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Lavanchy D. Evolving epidemiology of hepatitis C virus. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2011;17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 2.Pileri P, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 3.Scarselli E, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. Embo J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sautto G, Tarr AW, Mancini N, Clementi M. Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clinical & developmental immunology. 2013;2013:450963. doi: 10.1155/2013/450963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michalak JP, et al. Characterization of truncated forms of hepatitis C virus glycoproteins. J Gen Virol. 1997;78(Pt 9):2299–2306. doi: 10.1099/0022-1317-78-9-2299. [DOI] [PubMed] [Google Scholar]
- 6.Drummer HE, Poumbourios P. Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J Biol Chem. 2004;279:30066–30072. doi: 10.1074/jbc.M405098200. [DOI] [PubMed] [Google Scholar]
- 7.Wahid A, Dubuisson J. Virus-neutralizing antibodies to hepatitis C virus. Journal of viral hepatitis. 2013;20:369–376. doi: 10.1111/jvh.12094. [DOI] [PubMed] [Google Scholar]
- 8.Keck ZY, et al. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 2012;8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong L, et al. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J Virol. 2012;86:13085–13088. doi: 10.1128/JVI.01939-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong L, et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A. 2012;109:9499–9504. doi: 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng L, et al. Structural evidence for a bifurcated mode of action in the antibody-mediated neutralization of hepatitis C virus. Proc Natl Acad Sci U S A. 2013;110:7418–7422. doi: 10.1073/pnas.1305306110. d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whidby J, et al. Blocking hepatitis C virus infection with recombinant form of envelope protein 2 ectodomain. J Virol. 2009;83:11078–11089. doi: 10.1128/JVI.00800-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krey T, et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010;6:e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keck ZY, et al. Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. J Virol. 2005;79:13199–13208. doi: 10.1128/JVI.79.21.13199-13208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rothwangl KB, Manicassamy B, Uprichard SL, Rong L. Dissecting the role of putative CD81 binding regions of E2 in mediating HCV entry: putative CD81 binding region 1 is not involved in CD81 binding. Virol J. 2008;5:46. doi: 10.1186/1743-422X-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindenbach BD, Thiel HJ, Rice CM. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott Williams & Wilkins; 2007. pp. 1101–1152. [Google Scholar]
- 17.White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Critical reviews in biochemistry and molecular biology. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaney MC, Rey FA. Class II enveloped viruses. Cellular microbiology. 2011;13:1451–1459. doi: 10.1111/j.1462-5822.2011.01653.x. [DOI] [PubMed] [Google Scholar]
- 19.Helle F, et al. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol. 2010;84:11905–11915. doi: 10.1128/JVI.01548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavillette D, et al. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol. 2007;81:8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li HF, Huang CH, Ai LS, Chuang CK, Chen SS. Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. Journal of biomedical science. 2009;16:89. doi: 10.1186/1423-0127-16-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong L, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science. 2013;342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen SS, Moller M, Gillilan RE. High-throughput biological small-angle X-ray scattering with a robotically loaded capillary cell. J Appl Cryst. 2012;45:213–223. doi: 10.1107/S0021889812000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petoukhov MV, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Cryst. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang KY, Cowtan K, Main P. Combining constraints for electron-density modification. Methods in enzymology. 1997;277:53–64. doi: 10.1016/s0076-6879(97)77006-x. [DOI] [PubMed] [Google Scholar]
- 31.Semenyuk AV, Svergun DI. GNOM - a program package for small-angle scattering data processing. Journal of Applied Crystallography. 1991;24:537–540. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozin MB, Svergun D. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 2001;34:33–41. [Google Scholar]
- 35.Sharma S, et al. Construct optimization for protein NMR structure analysis using amide hydrogen/deuterium exchange mass spectrometry. Proteins. 2009;76:882–894. doi: 10.1002/prot.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiller T, Busse CE, Wardemann H. Cloning and expression of murine Ig genes from single B cells. Journal of immunological methods. 2009;350:183–193. doi: 10.1016/j.jim.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Mateu G, Donis RO, Wakita T, Bukh J, Grakoui A. Intragenotypic JFH1 based recombinant hepatitis C virus produces high levels of infectious particles but causes increased cell death. Virology. 2008;376:397–407. doi: 10.1016/j.virol.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 39.Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.