

Abstract
STAT1 is a key component of Interferon (IFN)-γ and IFN-α signaling and mediates protection against mycobacteria, fungal, viral infections, and cancer. Dominant negative inhibitory as well as gain of function heterozygous STAT1 mutations demonstrate that IFN-γ driven cellular responses need to be tightly regulated to control infections. We describe an autosomal dominant mutation in the SH2 domain of STAT1 that disrupts protein phosphorylation, c.1961 T>A (M654K). The mutant allele does not permit STAT1 phosphorylation, and impairs STAT1 phosphorylation of the wild type allele. Protein dimerization is preserved but DNA binding activity, IFN-γ driven GAS-luciferase activity, and expression of IFN-γ target genes are reduced. IFN-α driven ISRE response, but not IFN-α driven GAS response, are preserved when cells are co-transfected with wild type and the mutant STAT1 constructs. M654K exerts a dominant negative effect on IFN-γ related immunity and is recessive for IFN-α induced immune function.
Keywords: STAT1, SH2 domain, mycobacterial disease, IFN-γ
INTRODUCTION
Signal transducer and activator of transcription (STAT) family members are latent transcription factors sharing a similar structure, containing N-terminal, central DNA binding, carboxy-terminal SH2-containing, and transactivation domains [1]. STAT1 is critical to both interferon (IFN)-γ and IFN-α signaling. Following IFN-γ binding to its receptor, STAT1 is phosphorylated resulting in homodimerization and nuclear translocation. Binding to specific DNA sequences known as gamma-activating sequences (GAS) in the promoters of IFN stimulated genes induces transcription [2]. The binding of IFN-α to its receptor results in heterodimerization of phosphorylated STAT1 and STAT2 molecules followed by association with another signaling molecule, interferon regulatory family-9 (IRF9) or p48. This heterotrimer then translocates to the nucleus, and binds to specific DNA sequences, the interferon stimulating response element (ISRE), and induces gene transcription [2]. Although IFN-γ and IFN-α signaling pathways seem quite distinct, there is considerable overlap [3].
Dominant negative mutations in STAT1 cause increased susceptibility to weakly virulent intracellular pathogens, such as Bacillus Calmette-Guérin (BCG) and nontuberculous mycobacteria (NTM) due to impaired IFN-γ activity [4]; patients with heterozygous mutations that are dominant negative for GAS activation and recessive for ISRE activation, mostly present with only mycobacterial disease and the clinical course of their infections is usually milder [5, 6]. On the other hand, autosomal recessive STAT1 mutations typically cause more profound defects in STAT1 and are therefore associated with impairment of both IFN-γ and IFN-α related immunity. The clinical picture of patients with recessive mutations is typically more severe and characterized by both viral and mycobacterial infections [7, 8]. We report a novel autosomal dominant negative mutation in the SH2 domain of STAT1 in a patient who presented with disseminated mycobacterial infection.
METHODS
Blood cell isolation and mutational analysis
All blood samples were collected under NIAID IRB-approved protocol. The parents of the patient provided written informed consent for study participation. Blood of healthy volunteers were obtained through the NIH Blood Bank (Dept. of Transfusion Medicine, National Institutes of Health, Bethesda, MD) in accordance with NIAID IRB-approved protocol of the National Institutes of Health.
For sequencing, genomic DNA and total RNA were extracted from EBV-transformed B cell lines or polymorphonuclear leukocytes. Primers spanning exons and flanking splice sites of human STAT1 and full-length cDNA were designed using Primer Select (DNAstar Lasergene). Genomic amplification was performed with Platinum PCR Supermix High Fidelity (Invitrogen). Sequencing was performed with Big Dye Terminators v3.1 (Applied Biosystems, Foster City, CA), run on an Applied Biosystems 3730XL sequencer and aligned to the consensus sequence NM_007315.3 using Sequencer software (Gene Codes).
The mutation in the STAT1 coding sequence was created using a STAT1 expression vector (OriGene technologies, Rockville, MD) as template (BioInnovatise Inc., Rockville, MD). STAT1-Myc tag or GFP-tagged constructs were created from the untagged STAT1 expression vector (BioInnovatise). STAT1-FLAG tag (Addgene plasmid 8691) was purchased from Addgene, Cambridge, MA (deposited by Dr. Jim Darnell) [9]. Plasmids encoding wild type (WT) STAT1 and the mutant constructs were isolated using the QIAprep maxiprep kit (QIAGEN) according to the manufacturer's recommendations; all mutations were verified by sequencing.
Cell lines
EBV-transformed B cell lines derived from patients and normal donors were maintained in RPMI 1640 with 20% fetal calf serum (FCS; Gibco BRL, Carlsbad, CA), 2mM L-glutamine, penicillin 100U/ml, 100μg/ml streptomycin (Gibco), at 37°C in a humidified 5% CO2 incubator. STAT1 deficient U3A cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% FCS, 2mM L-glutamine and antibiotics.
Transient transfection of U3A cells was done using the Amaxa nucleofector (Lonza, Walkersville, MD). Culture media were replaced 24 hours post-transfection and cells were either left untreated or stimulated with IFNs as indicated.
Flow cytometry
To assay STAT1 activation, EBV transformed B cells or transfected U3A cells (Amaxa nucleofector; Lonza, Walkersville, MD) were stimulated with IFN-γ (R&D System, Minneapolis, MN) 400 IU/ml or IFN-α (IFN-α2b, PBL Biomedical Laboratories, Piscataway, NJ) 1,000 IU/ml for 15 min, when cells were recovered, fixed and permeabilized in methanol. Cells were stained for total (Alexa647 conjugated anti-STAT1) and phosphorylated tyrosine Y701 STAT1 (Alexa488 conjugated anti-pSTAT1; BD Biosciences). For U3A cells, the levels of phosphorylation were assessed in the cells gated for the expression of total STAT1. Data were collected using FACS Caliber (BD Biosciences) and analyzed using FlowJo (Treestar).
Immunoprecipitation and Immunoblotting
For Western blot analysis (WB), EBV-B or transfected U3A cells, stimulated as described above, were lysed in buffer containing protease and phosphatase inhibitors (Calbiochem, Gibbstown, NJ). Samples were sonicated, equal amounts of proteins were run on a 10% SDS polyacrylamide gel and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (Invitrogen). After blocking, the membranes were incubated with the primary antibody, anti-pSTAT1 Tyr701 (Cell Signaling Technology, Danvers, MA) or anti-pSTAT2 Tyr690 (Abcam, Cambridge, MA), as indicated. Membranes were washed, incubated in the horseradish peroxidase-conjugated secondary antibody and signal detected using an enhanced chemiluminescence system (ECL; Amersham Biosciences, Piscataway, NJ). Blots were stripped and re-probed with anti-total STAT1 (Cell Signaling) or STAT2 (Millipore, Billerica, MA) antibodies, respectively, to assess protein loading. For detection of dimerization, STAT1 co-immunoprecipitation (IP) was evaluated using U3A cells co-transfected with FLAG- or myc-tagged WT or mutant STAT1 constructs and stimulated or not with IFN-γ (400 IU/ml, 30 min). Cell lysates were then precipitated with anti-myc or anti-FLAG antibodies (Sigma-Aldrich, St. Louis, MO) followed by protein G-Sepharose binding and immunobloting. Blots were probed with anti-FLAG, anti-Myc or anti-STAT1 antibodies. To evaluate STAT1 / STAT2 association, transfected U3Acells (WT and M654K STAT1) were stimulated with IFN-α (1,000 IU/ml, 30min) and IP:WB STAT1:STAT2.
Confocal microscopy
U3A cells were seeded onto coverslips in the 12-well plates (Costar), followed by transfection of plasmids encoding WT STAT1 and/or its mutant M654K with lipofectamine (Invitrogen). The following day, culture media was replaced and cells were either untreated or treated with IFN-γ (400 IU/ml, 15 min). Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% (w/v) Triton X-100 in PBS. Coverslips were incubated with the mouse anti-human STAT1 (BD biosciences), followed by a secondary staining with goat anti-mouse IgG conjugated to Alexa Flour-568. The nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Co-localization studies were done in a Leica SP5 confocal microscope (Leica Microsystems, Exton, PA) using a 63×-oil immersion objective NA 1.4. The images were collected sequentially and the data were analyzed using Leica software. For data presentation, the images were assembled in Adobe Photoshop CS3.
Nuclear extracts and nuclear complex binding
Nuclear extracts from transfected U3A cells stimulated or not with IFN-γ (400 IU/ml) or IFN-α (1,000 IU/ml), were prepared using the Panomics kit (Panomics, Fremont, CA). For determination of DNA binding activity, an ELISA-like colorimetric assay (TRANSAM, Active Motif, Carlsbad, CA) using a plate coated with a STAT1 binding oligonucleotide derived from the GAS sequence, was used according to the manufacturer's protocol. Absorbance was measured on a spectrophotometer at 450nm.
Reporter gene assay
U3A cells were transiently transfected with WT and/or mutant STAT1 expression constructs and a plasmid containing tandem IFN-response elements driving a luciferase reporter gene (1μg; HSV-thymidine kinase promoter; Panomics, Fremont, CA). A Renilla expression vector (0.03μg/ml) was co-transfected to serve as an internal control for transfection efficiency. Following overnight incubation, media were replaced and cells were stimulated or not with IFN-γ or IFN-α (1,000 IU/ml) for 6 h. Cells were resuspended in lysis buffer and luciferase activity was evaluated using a dual luciferase assay (Promega, Madison, WI). Relative luciferase units were normalized to Renilla activity. Data are expressed as fold increase in response to interferon over the non stimulated samples.
Real time PCR
Total RNA was extracted from cultured cells with the RNeasy mini kit (QIAGEN). For RT-PCR, 1 μg of total RNA was reverse transcribed and the resulting cDNA amplified by PCR using the ABI 7500 Sequencer using Taqman expression assays (Applied Biosystems). GAPDH was used as normalization control. The data were analyzed using the 2-ΔΔCT method.
Statistical analysis
Results are reported as mean ± standard error (SEM) unless otherwise stated. Differences between groups were assessed by the unpaired two-tailed Student's t-test using GraphPad Prism Software (San Diego, CA). The statistical significance level adopted was p < 0.05.
RESULTS
Case description and mutation
The patient was a 5-year-old boy, born to unrelated parents, who first presented at age 1 week with recurrent pneumonia. At 1.5 years, he presented with cervical lymphadenitis, fever and respiratory complaints. Computed tomography (CT) of the chest indicated massive mediastinal lymphadenopathy. Mycobacterium avium complex (MAC) was isolated from the cultures of lymph nodes, lung and bone marrow. Disseminated MAC infection was treated with ethambutol, azithromycin, rifampin, ciprofloxacin and amikacin. Amikacin was subsequently discontinued due to ototoxicity. IFN-γ was added by subcutaneous injection three times weekly. At 2.5 years the patient was referred to the National Institutes of Health (NIH) for evaluation. His CT scan showed right bronchial mucus plugging with atelectasis that improved on subsequent imaging; there was no significant lymphadenopathy. Repeated mycobacterial blood cultures were negative. In addition to IFN-γ injections, azithromycin, rifampin, and ciprofloxacin were continued. Ethambutol was discontinued due to concern for optic neuritis. He has remained well. He has been hospitalized once for an asthma exacerbation and received additional antibiotics for bacterial pharyngitis and otitis media. He has had typical childhood viral infections without complications. He had normal lymphocyte numbers and quantitative immunoglobulins.
Full length sequencing of STAT1 genomic and cDNA identified a heterozygous mutation c.1961 T>A in the SH2 domain, resulting in methionine to lysine at position 654 (M654K) (Fig. 1). His parents were mutation free.
Fig. 1.

STAT1 dominant negative and loss of function mutations. The N-terminal domain, coiled-coil domain, DNA binding domain, linker domain, SH2 domain, tail segment domain (TS), and transactivation domain (TAD) are represented; Y701, site of tyrosine phosphorylation. The dominant negative mutants are indicated above the protein and all recessive or loss of function mutations are below the protein. M654K is shown in red.
STAT1 phosphorylation and DNA binding
To investigate the ability of the M654K mutant to dimerize with WT STAT1 protein, U3A cells were co-transfected with Myc- and FLAG-tagged constructs followed by coimmunoprecipitation and immunoblotting. Cells stimulated or not with IFN-γ showed normal dimerization of the mutant protein to WT STAT1 (Fig. 2a).
Fig. 2.



Dimerization and activation of STAT1 protein. a For evaluation of STAT1 dimerization, U3A cells were transfected with WT and patient M654K STAT1 constructs tagged with either Myc or Flag. Proteins obtained from non stimulated (NS) or IFN-γ stimulated (400 IU/ml) cells were co-precipitated with antibodies against Myc or Flag and blotted for the detection of Myc or Flag. Blots were stripped and reprobed with anti-total STAT1 antibody; b For Western blot analysis, whole cell lysates obtained from transfected U3A cells stimulated or not (NS) with IFN-γ were blotted with anti-phosphorylated STAT1 (pSTAT1). Blots were stripped and reprobed with anti-STAT1 antibody; c Evaluation of U3A cells by flow cytometry confirmed the absent phosphorylation in the M654K transfected cells in response to IFN-γ. Lower phosphorylation is sustained in the co-transfected cells (M654K+WT). Histograms from one representative experiment are presented (right panel). The bars represent mean fold change (±SEM) in mean fluorescence value (MFV) observed in the stimulated cells relative to the unstimulated controls (uns) (n=3). **p<0.01; ***p<0.001 when compared to WT; d EBV transformed B cells obtained from healthy donors and from the STAT1 deficient patient (M654K) were stimulated with IFN-γ or IFN-α. Lysates were blotted with anti-phospho STAT1. Blots were treated as described above and are representative of three individual experiments. NS = non stimulated; e Flow cytometry assayed in EBV-B cells from patients (M654K STAT1 deficient and one patient with complete IFNGR1 deficiency) and two healthy donors. Histograms are included for each experimental condition (non stimulated, NS; IFN-γ 400 IU/ml; IFN-α 1,000 IU/ml). One representative experiment out of three is presented.
Activation of STAT1 was assayed in STAT1 deficient U3A cells. When transfected alone into the cells, M654K STAT1 was not phosphorylated (Tyr701) following stimulation with IFN-γ whereas WT STAT1 was (Fig. 2b). The previously described STAT1 mutation L706S STAT1, which disrupts tyrosine phosphorylation and predisposes to mycobacterial disease [6], also inhibits STAT1 phosphorylation. Co-transfection of M654K and WT STAT1 constructs partially restored IFN-γ responsiveness, consistent with this dominant mutant's inhibition of WT STAT1 activity. These results were confirmed by flow cytometry in the transfected cells (Fig. 2c). Experiments using EBV-B cells, immunoblotting (Fig. 2d) and flow cytometry (Fig. 2e), showed STAT1 phosphorylation (Y701) after stimulation with IFN-γ in the heterozygous patient cells (M654K; 27.2% positive cells) vs. healthy control cells (34.6%) as opposed to one patient with homozygous complete IFNGR1 deficiency (1.06%) (Fig. 2e), who had disseminated mycobacterial disease [10]. Activation of STAT1 in response to IFN-α occurred to similar extents in each group (82.9%, 81.2% and 70.2%, respectively). STAT2 tyrosine (Tyr690) phosphorylation was preserved in patient cells (not shown). Interestingly, IFN-γ induced nuclear translocation in the transfected cells was reduced when using the M654K construct alone, and it was detectable in co-transfection with WT STAT1 (Fig. 3).
Fig. 3.

Evaluation of nuclear translocation. Nuclear translocation of the mutant STAT1 proteins was assessed in U3A cells transfected with WT STAT1 or M654K mutant, stimulated or not with IFN-γ (400 IU/ml, 15 min). The distribution of STAT1 in the nucleus was identified by primary staining against total STAT1 followed by a secondary staining with Alexa Fluor 568, together with the nuclear DAPI. Following stimulation, WT and WT+M654K STAT1 but not the M654K alone were detected at the nucleus as shown in the overlay image.
DNA binding activity of M654K STAT1 to the GAS oligonucleotide was determined in lysates of U3A cells transfected with the M654K construct and stimulated with IFN-γ or IFN-α. M654K showed markedly reduced GAS binding compared to WT (Fig. 4a). The dominant negative effect of M654K was confirmed following co-transfection with WT STAT1, which sustained the reduced DNA binding on IFN-γ and IFN-α response.
Fig. 4.


Effect of STAT1 mutation on DNA binding and transcriptional activity. a STAT1 DNA-binding activity was assayed in nuclear extracts isolated from U3A cells transfected with STAT1 WT, STAT1 mutant (M654K), or STAT1 mutant co-transfected with WT (M654K+WT) and stimulated or not with IFN-γ or IFN-α 1,000 IU/ml (30 min). Data are mean fold increase (± SD) detected in the stimulated cultures relative to the WT non stimulated (NS) from a total of five experiments; b STAT1 expression vectors (STAT1 WT, mutant M654K, M654K+WT) were transiently co-transfected with luciferase GAS reporter plasmids or c with luciferase ISRE reporter into U3A cells. Following stimulation with IFN-γ or IFN-α, cells were harvested and assayed using the dual luciferase reporter assay. Experiments were done in triplicate and results are mean fold increase (± SD) in the stimulated cells relative to the WT non stimulated (n=4).**p<0.01; ***p<0.001 when compared to WT.
Luciferase activity and gene induction
The transactivation activity of M654K STAT1 was evaluated in U3A cells co-transfected with WT and mutant constructs using GAS and ISRE response elements. In agreement with its diminished ability to bind DNA, the M654K construct alone showed no transactivation activity on GAS reporter constructs after stimulation with IFN-γ or IFN-α (Fig. 4b). The ISRE response was induced about 3-fold, indicating more preserved IFN-α than IFN-γ responsiveness (Fig. 4c). When WT and M654K constructs were co-transfected, normal GAS-driven luciferase activity was diminished, whereas ISRE driven luciferase activity to IFN-α and IFN-γ was essentially normal. Accordingly, experiments searching at STAT1 / STAT2 association performed in transfected U3A cells showed preserved association when compared WT and M654K STAT1 constructs (Fig. 5).
Fig. 5.
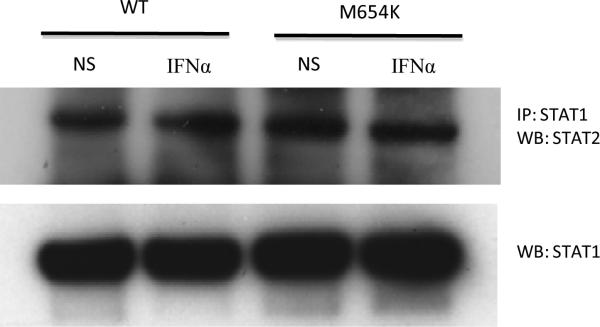
For evaluation of STAT1 / STAT2 association, U3A cells were transfected with WT STAT1 or M654K mutant constructs, left non stimulated (NS) or stimulated with IFN-α (1,000 IU/ml) for 30 min when cell lysates were prepared and co-precipitated with anti-STAT1 antibody and blotted for STAT2. Blots were stripped and reprobed with anti-total STAT1. One representative experiment out of two is presented.
In agreement with these data, mRNA expression of typical IFN-γ target genes (CXCL9 and CXCL10), evaluated by qRT-PCR, was reduced in U3A cells transfected with the mutant construct alone or when co-transfected with the WT STAT1 and stimulated with IFN-γ (400 IU/ml) or IFN-α (1,000 IU/ml) for 3 h (Fig. 6a). On the other hand, the expression of the typical IFN-α induced genes (MX1 and OAS2) was sustained in the heterozygous cells (Fig. 6b).
Fig. 6.


Analysis of gene expression. U3A cells co-transfected with WT and/or patient construct (M654K) were stimulated or not with IFN-γ or IFN-α for 3 h. Expression of IFN target genes, a CXCL9, CXCL10, b MX1, OAS2 was evaluated in the transfected cells by real time PCR. Results are expressed as mean fold induction (± SD) of four individual experiments. *p <0.05; **p < 0.01; ***p < 0.001 when compared to WT.
DISCUSSION
We report a patient with a novel heterozygous mutation in the SH2 domain (M654K) of STAT1 associated with disseminated MAC infection. Transfection of U3A cells with the mutant construct alone showed no STAT1 phosphorylation in response to IFN-γ but preserved response to IFN-α and preserved STAT1: STAT2 association. In co-transfected U3A cells (WT + mutant construct M654K) or patient EBV-B cells (heterozygous), IFN-γ induced STAT1 phosphorylation and activation was partially affected, despite sustained capacity for protein dimerization and normal response to IFN-α. This phenotype is easily explained since STAT1 homodimers are formed in response to both IFN-γ and IFN-α [2]. Around 25% of STAT1 dimers formed in response to IFN-γ stimulation are dimers between WT molecules, therefore it is not so surprising that some nuclear translocation and also other functional effects can be observed following stimulation. In addition, 50% of the dimers formed between STAT1 and STAT2 in response to IFN-α are dimers containing WT-STAT1. These data reinforce the dominant negative effect of the mutation on IFN-γ / GAS related immunity. Similarly the ISRE activity driven following IFN-α was compromised in U3A cells transfected with the mutant construct alone. Co-transfection of mutant and WT constructs resulted in recovery of the IFN-α response.
The SH2 domain of STAT1 is required for the recruitment of latent STAT1 molecules to the activated interferon receptors [11]. In addition, the domain is thought to be crucial for dimerization of activated STAT1 molecules via reciprocal interactions between SH2 domains and phosphorylated Tyr701 residues [1]. Following cell activation and nuclear translocation, STAT1 dimers trigger high affinity DNA binding and gene transcription [1]. Accordingly, mutations in the SH2 domain of STAT1 have been associated with compromised STAT dimerization and nuclear translocation in vitro [11]. Moreover, mutations affecting the SH2 domain of STAT3 have been reported to diminish tyrosine phosphorylation [12], confirming that this domain plays a crucial role in the activation of STATs.
STAT1 mutations can lead to a wide range of clinical manifestations [4, 13]. Recently, dominant gain of function mutations in the coiled coil domain of STAT1 have been described as causing susceptibility to chronic mucocutaneous candidiasis [14, 15]. On the other hand, the previously identified dominant negative or loss of function comprise 10 mutations in STAT1 reported in 13 patients, involving the tail segment [6], DNA binding domain [5], the coiled coil domain [16], and the N-terminal region [17]. Another mutation is located between the SH2 domain and tail segment [18]. The three previously reported recessive mutations in the SH2 domain, 1758_1759delAG, L600P, 1928insA [7, 8], and a recently described STAT1 splicing mutation [19] were all associated with severe impairment of GAS and ISRE responses. In line with these observations, the clinical picture of those patients was characterized by mycobacterial as well as viral infections. Interestingly, one of the patients was able to clear the attenuated polio type II virus vaccine that was given at 2 months of age as well as a parainfluenza II and rhinovirus infections [7].
We describe here the first autosomal dominant mutation in the SH2 domain of STAT1 associated with a dominant negative effect on IFN-γ related immunity but clinically adequate IFN-α induced immune function. The M654K mutation led to relatively mild mycobacterial infection responsive to anti-mycobacterial therapy with IFN-γ supplementation. The patient cleared his usual childhood viral infections, which all ran benign clinical courses.
ACKNOWLEDGEMENTS
The research was supported by the Division of Intramural Research of National Institute of Allergy and Infectious Diseases, National Institutes of Health. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
REFERENCES
- 1.Wenta N, Strauss H, Meyer S, Vinkemeier U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci USA. 2008;105:9238–43. doi: 10.1073/pnas.0802130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 3.Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem. 2006;281:14111–8. doi: 10.1074/jbc.M511797200. [DOI] [PubMed] [Google Scholar]
- 4.Averbuch D, Chapgier A, Boisson-Dupuis S, Casanova JL, Engelhard D. The clinical spectrum of patients with deficiency of Signal Transducer and Activator of Transcription1. Pediatr Infect Dis J. 2011;30:352–5. doi: 10.1097/INF.0b013e3181fdff4a. [DOI] [PubMed] [Google Scholar]
- 5.Chapgier A, Boisson-Dupuis S, Jouanguy E, Vogt G, Feinberg J, Prochnicka-Chalufour A, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006;2:e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 7.Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, et al. Human complete STAT1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol. 2006;176:5078–83. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- 8.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, et al. Nat Genet. 2003;Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency.33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 9.Horvath CM, Wen Z, Darnell JE., Jr A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995;15:984–94. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 10.Holland SM, Dorman SE, Kwon A, Pitha-Rowe IF, Frucht DM, Gerstberger SM, et al. Abnormal regulation of interferon-gamma, interleukin-12, and tumor necrosis factor-alpha in human interferon-gamma receptor 1 deficiency. J Infect Dis. 1998;178:1095–104. doi: 10.1086/515670. [DOI] [PubMed] [Google Scholar]
- 11.Mowen K, David M. Role of the STAT1-SH2 domain and STAT2 in the activation and nuclear translocation of STAT1. J Biol Chem. 1998;273:30073–6. doi: 10.1074/jbc.273.46.30073. [DOI] [PubMed] [Google Scholar]
- 12.Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122:181–7. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu HQ, Fisher-Hoch SP, McCormick JB. Molecular immunity to mycobacteria: knowledge from the mutation and phenotype spectrum analysis of Mendelian susceptibility to mycobacterial diseases. Int J Infect Dis. 2011;15:e305–13. doi: 10.1016/j.ijid.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candiasis. N Engl J Med. 2011;365:1806–15. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208:1635–48. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong XF, Ciancanelli M, Al-Hajjar S, et al. A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood. 2010;116:5895–906. doi: 10.1182/blood-2010-04-280586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kristensen IA, Veirum JE, Moller BK, Christiansen M. Novel STAT1 alleles in a patient with impaired resistance to mycobacteria. J Clin Immunol. 2011;31:265–71. doi: 10.1007/s10875-010-9480-8. [DOI] [PubMed] [Google Scholar]
- 18.Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, Feinberg J, et al. A partial form of recessive STAT1 deficiency in humans. J Clin Invest. 2009;119:1502–14. doi: 10.1172/JCI37083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vairo D, Tassone L, Tabellini G, Tamassia N, Gasperini S, Bazzoni F, et al. Severe impairment of IFNγ and IFNα responses in cells of a patient with a novel STAT1 splicing mutation. Blood. 2011;118:1806–17. doi: 10.1182/blood-2011-01-330571. [DOI] [PubMed] [Google Scholar]