

Abstract
Family studies are consistent with genetic effects making substantial contributions to risk of psychiatric disorders such as schizophrenia, yet robust identification of specific genetic variants that explain variation in population risk had been disappointing until the advent of technologies that assay the entire genome in large samples. We highlight recent progress that has led to a better understanding of the number of risk variants in the population and the interaction of allele frequency and effect size. The emerging genetic architecture implies a large number of contributing loci (that is, a high genome-wide mutational target) and suggests that genetic risk to psychiatric disorders involves the combined effects of many common variants of small effect, as well as rare and de novo variants of large effect. The capture of a substantial proportion of genetic risk facilitates new study designs to investigate the combined effects of genes and the environment.
A genetic contribution to the risk of many psychiatric disorders has been well established by family studies1–5, but the nature of their genetic architectures has not. Genetic architecture refers to the number of genomic loci contributing to risk, the distribution of their allelic frequencies and effect sizes, and the interactions of alleles in and between genes, all of which contribute to the relationship between genotype and phenotype. Understanding genetic architecture is the foundation on which progress in dissecting etiology is built because it dictates which study designs for identifying risk variants are likely to be most successful. Here we review how recent studies that employed whole-genome genotyping or exome sequencing have contributed to a better understanding of the joint spectrum of allele frequencies and penetrance of risk variants for psychiatric disorders. We focus particular attention on schizophrenia (SCZ), the genetic dissection of which is further advanced than other disorders considered in this review, including Alzheimer’s disease (AD), anorexia nervosa (AN), attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), bipolar disorder (BPD), major depressive disorder (MDD), obsessive compulsive disorder (OCD) and Tourette’s syndrome (TS).
Psychiatric disorders are best classified as ‘complex traits’. The variability of such traits is a result of a large number of factors and can be dissected into sources of variation resulting from genetic factors, non-genetic factors and their interplay. With the exception of rare strictly monogenic disorders, almost any trait—for example, most common diseases, brain regional volume and the amount of DNA methylation of a particular gene in a particular tissue—can be considered a complex trait, so long as it can be measured and varies between individuals. In Box 1, we provide a primer on the genetic analysis of complex traits.
Box 1. Primer on genetic analysis of complex traits.
Before the availability of direct measures of genetic variation at the level of DNA, epidemiological studies on psychiatric disorders endeavored to detect robust associations between measurable environmental factors and risk of disease, whereas genetic epidemiologists attempted to quantify the risk in the population into genetic and non-genetic sources of variation. Notably, one can quantify the proportion of variation in the population resulting from genetic factors (the heritability) without knowing anything about how many or which genetic variants underlie the trait using the observed risk to relatives and contrasting that with what would be expected given the proportion of the genome those relatives share by descent. Epidemiological studies have identified a number of environmental risk factors, such as maternal infection during pregnancy and risk of schizophrenia to their children93. Reported recurrence risks to relatives are consistent with heritabilities of 40–80% for autism, schizophrenia and major depression. Genetic differences between individuals in their risk to psychiatric disorders are a result of one or more mostly unknown variants in the genomes of people. Such variants either segregate in the population or are de novo mutations not expressed in the parents. Segregating variants cause the observed similarity in risk between relatives, whereas new mutations do not, apart from rare exceptions such as mutations in monozygotic twins that arose before the embryo was split, and mutations shared by siblings as a result of germlinemosaicism.
The contribution of a specific genetic variant to trait variation in the population is determined by the combination of its allele frequency and its effect size. Variants are typically classified as common, low frequency or rare if their population frequencies are >0.01, between 0.01 and 0.001, and <0.001, respectively. At one extreme, rare variants of large effect are by definition carried by few individuals. They may have a very large effect on those people—for example if the variant is sufficient to cause disease—but tend to not contribute much of the variation in the population as a result of their rarity. At the other extreme, common variants of small effect are carried by lots of people, either in the heterozygous or homozygous state. The increased risk of disease to an individual from such variants may be trivial but the effect on population variance can be the same as for the rare variant of large effect (Fig. 3 in Box 1). For example, recurrent 22q11.21 deletions increase risk to schizophrenia from 1% to about 20–40%, yet are rare in the population (fewer than 1 in a 1,000 individuals carry the deletion), whereas a common variant in ZNF804A increases risk from 1 to 1.1% per allele, but is common (a proportion of 0.8 in the population has one or two alleles): both contribute about the same amount of variation in liability to schizophrenia (that is, r2 ~0.1%)77. The majority of risk alleles for schizophrenia, common and rare, are likely to explain substantially less of the variation than these examples (Fig. 3 in Box 1).
A positive family history is often assumed to be indicative of inherited risk variants, whereas ‘sporadic’ cases of disease (that is, those with a negative family history) are sometimes assumed to harbor new (de novo) high penetrance mutations. Although there is some evidence for this classification in SCZ (for example, see refs. 44,60), and although focusing on sporadic cases makes sense when searching for de novo mutations, it is worth noting that a negative family history is expected for highly polygenic, low prevalence disorders (for example, SCZ)94. Thus, under polygenicity, the delineation of affected individuals as familial and sporadic may be misleading. More data is needed to determine which genetic models are most appropriate for psychiatric disorders, but even for non-psychiatric diseases, such as amyotrophic lateral sclerosis, where there are clear-cut examples of Mendelian fully penetrant mutations, there is emerging consensus that the distinction between familial and sporadic amyotrophic lateral sclerosis is artificial95.
Geneticists have used a number of ‘whole genome’ toolboxes to try to discover genes and genetic variants that are associated with risk to psychiatric disorders: genetic epidemiology studies to quantify the combined effect of the entire genome to risk of disease, genome-wide linkage studies, GWASs and WES studies12. In the near future we can add whole-genome sequencing, which essentially will supersede genome-wide linkage studies, GWASs and WES studies.
Segregating variants in the population that predispose to psychiatric disorders are the results of our evolutionary past. The evolutionary forces that shaped our modern genomes include mutation, natural selection and genetic drift. Natural selection acts on ‘net’ fitness, which is much more than fitness with respect to any individual disease. Although there are multiple theories of what our genomes should look like with respect to variants that predispose to psychiatric disorders and other complex traits6–8, testing their predictions awaits a better understanding of the genetic architecture of complex traits. Broadly speaking, for any given complex trait, the distribution of genetic variance explained as a function of minor allele frequency is expected to be uniform under a neutral evolutionary model, but should be shifted toward lower minor allele frequencies to the extent that alleles affecting the trait have been under selection9. Yet, irrespective of evolutionary arguments, we can study human genomes here and now to understand why some individuals are at higher risk of disease than others. The genomics revolution, particularly the ability to interrogate the entire genome through developments in high-throughput technologies such as genome-wide genotyping and sequencing, has provided the tools to dissect the genetic variation that was inferred from genetic epidemiological studies into contributions from individual variants.
Mutation, polymorphism, gene and pathway discovery
Important advances in psychiatric genetics have been made in recent years, with many replicated discoveries of common, rare and de novo risk factors that are converging on specific pathways and biological mechanisms. These successes have predominantly come about as a result of the experimental design of genome-wide association studies (GWASs)10,11 and, in particular, the international community combining resources and results across multiple GWASs and copy number variation (CNV) studies to maximize sample size and statistical power. Whole-exome sequencing (WES) has also begun to shed light on the role of rare and de novo coding sequence variation, particularly in autism. The flow of discoveries from these unbiased genome-wide methods stands in contrast to the weak and largely inconsistent findings from candidate gene studies12.
GWAS
At the time of writing this review, GWASs have identified >100 independent loci for SCZ13, 20 loci for AD14, eight loci for BIP15, one locus for ASD16, and none for other common psychiatric disorders, including ADHD17, AN18, MDD19, OCD20 and TS20 (Fig. 1). In addition, cross-disorder GWAS meta-analyses had identified three loci for a combined SCZ-BPD phenotype21 and four loci for a broader psychiatric phenotype spanning ASD, ADHD, BPD, MDD and SCZ22. These genetic findings surpass accepted standards for genome-wide significance (P < 5 × 10–8) and replication in human genetics; the stringent type 1 error threshold of 5 × 10–8 corresponds to a Bonferroni correction of 0.05 divided by 1,000,000 tests, which is the estimated number of independent common variants across the human genome23. The overt success for SCZ, the ‘flagship’ disorder of the Psychiatric Genomics Consortium (PGC)24, is largely explained by greater sample size (that is, >35,000 cases compared with a maximum of ~17,000 cases for any other disorder), which was achieved by the PGC combining data across >50 studies13. Indeed, no single locus had been robustly associated with SCZ when sample sizes were similar to those currently available for many other disorders (for example, ADHD: ~900 cases17). MDD is an exception, given that no discoveries were made in the most recent PGC mega-analysis19 (9,240 cases, 9,519 controls), a study similar in scale to the PGC’s initial SCZ GWAS (9,394 cases, 12,462 controls), which identified seven loci21. However, sample sizes approximately five-fold greater than SCZ are predicted to be necessary for GWAS discovery in MDD, mostly as a result of its high population prevalence25 (Fig. 2). Phenotypic heterogeneity may also be a factor contributing to differential success of GWAS in psychiatric disorders (discussed below). These considerations aside, the results for SCZ, the largest GWAS to date for any psychiatric disorder, show what might reasonably be expected for other disorders from larger studies that are currently in the pipeline.
Figure 1.

The trajectory of GWAS discoveries for schizophrenia and other psychiatric disorders in comparison to Crohn’s disease and inflammatory bowel disease. The number of independent genome-wide significant (P < 5 × 10–8) loci are plotted as a function of the numbers of cases in the largest meta/mega-analysis. The inset shows detail for studies with fewer than 6,000 cases. CD-IBD, Crohn’s disease and inflammatory bowel disease (including CD and ulcerative colitis); Cross-disorder: broad psychiatric phenotype spanning ASD, ADHD, BPD, MDD, SCZ; SCZ-BPD, SCZ and BPD.
Figure 2.

Not all GWASs are created equal under a polygenic architecture. For the same sample size, less common diseases have more power. Statistical power for detection of gene variants associated with disease for different disorders given the same sample size (type-1 error of 10–8, sample size of 10,000 cases and 10,000 controls). Different combinations of allele frequency and effect size can generate the same variance in liability. Examples of disorders with the prevalence shown are motor neuron disease (MND), SCZ and MDD.
The trajectory of GWAS discovery for SCZ, which increased from 1 locus26,27 to 7 (ref. 21) to 22 (ref. 28) to 62 (ref. 29) to >100 (ref. 13) as the number of cases increased from ~3,000 to >35,000, is not dissimilar to that of other (non-psychiatric) diseases (Fig. 1). Admittedly, late-onset diseases, including AD30 and age-related macular degeneration31, and some autoimmune diseases (for example, Crohn’s disease)32 achieved success earlier, often with first generation technologies such as linkage mapping and GWASs of small samples, but these discoveries involved risk alleles that explain an unusually large proportion of the total genetic risk (for example, APOE in AD). GWAS loci identified for SCZ, as with the majority of GWAS discoveries across the breadth of medicine, are very common (that is, the risk allele is often the major allele), have small effect size (for example, 1.1% absolute risk when the population prevalence is 1%) and individually explain only a small fraction of the total genetic risk (Fig. 3 in Box 1). The underlying genetic architecture is highly polygenic21,26,28, as we discuss below, a fact that has driven, and continues to drive, international efforts coordinated by the PGC to assemble ever-larger samples for meta-analyses33.
Figure 3.

Genetic discoveries for SCZ, irrespective of risk allele frequency, variant type (SNP or CNV) or discovery method (GWAS or CNV analysis), explain approximately the same proportion of the genetic variance. The red line defines a constant r2 of 0.04%, assuming a 1% prevalence of schizophrenia. Note that the population prevalence of some CNVs is set to 10–4 for convenience; some have not been observed in healthy controls and so the true allele frequency (and variance explained) will be lower.
Why should neuroscientists’ care about GWAS findings in psychiatric disorders given that identified loci have such small effects? First, although verified GWAS effects are usually small individually, their cumulative effect is not. Genetic profile scores derived from very large discovery samples can be applied to small target samples, such as those used in neuroscience studies, to facilitate more powerful experiments, for instance, by enabling stratification of samples according to upper and lower deciles of genetic risk. Second, there are now many examples of diseases for which GWAS has highlighted relevant biology (for example, see refs. 31,34), including known drug targets35,36. For instance, numerous genes identified in a GWAS meta-analysis for rheumatoid arthritis are the targets of known drugs that are effective therapies for this disease37, and the same is true for genes underlying natural variation in LDL levels that are the targets of statins38. Results from the latest GWAS for SCZ suggest that similar insights may be forthcoming in psychiatric disorders, given that identified loci include known targets of existing antipsychotics13. These examples indicate that, although GWAS loci have small effect size, they nonetheless may help identify targets for novel therapeutics38 or may identify existing drugs that can be repurposed for treatment of diseases that they were not initially developed to treat36. More broadly, and consistent with other complex diseases (for example, inflammatory bowel disease39) and traits (for example, height40) for which large numbers of genetic associations have been identified, the 100+ GWAS loci identified for SCZ converge on biological pathways relevant to disease etiology, including neuronal calcium ion channel signaling, altered immune function and MIR137-mediated post-transcriptional regulation of gene expression13. Functional investigation of the biological basis of genetic associations in these pathways represents a formidable challenge and will be far more difficult than that for rare variants with larger effects. We are in the very early days of addressing this challenge, but there is nonetheless potential for such efforts to make profound contributions to our understanding of disease pathogenesis.
Structural variation studies
At the opposite end of the risk allele frequency spectrum, rare and de novo submicroscopic chromosomal deletions and duplications termed CNVs have been implicated in a range of psychiatric disorders. The unbiased genome-wide analysis of large CNVs (>100 kb) has been facilitated by data from high-density genomic arrays used in GWAS. Over the past decade, such studies have established that large rare (<1% frequency in the population) CNVs, particularly those arising de novo in the parental germline, occur at higher frequencies in ASD41,42, SCZ43,44 and ADHD45 than in healthy controls, implying that some events contribute to risk. Rare CNVs have also been implicated in BPD, particularly early onset disease46, but the evidence is inconsistent47 and the contribution may be less than for ASD and SCZ. In addition to evidence for increased CNV burden, about a dozen recurrent CNVs have been identified, including deletions on chromosomes 2p16.3 (in the NRXN1 gene), 3q29 and 17q12, duplications on 7q11.23, 7q36.3 (in the VIPR2 gene), 15q11.2 and 16p13.11, and both deletions and duplications at 1q21.1, 15q13.3, 16p11.2 and 22q11.21 (refs. 45,48–51). At several loci there is evidence that ASD and SCZ are associated with reciprocal events; that is, deletions predispose to one disorder and duplications the other52. Most of the identified loci span multiple genes and confer risk for a range of psychiatric and neurodevelopmental disorders48,49, consistent with other evidence for shared genetic etiology across diagnostic boundaries. Odds ratios are high compared with common loci, ranging from ~2 to >20 (refs. 48,49), but their frequency is very low in the general population (that is, the majority have a frequency < 0.001) and so large samples are nonetheless required for discovery and replication (similar to that for GWAS). For some loci, penetrance for ASD appears to be higher in males than females (for example, 16p11.2)53, and de novo events in affected girls are more functionally disruptive than those in affected boys41,53. This suggests that girls have greater resistance to ASD from genetic factors, consistent with the ~4:1 male bias in incidence, although the mechanisms responsible for this pattern remain to be deciphered41,53.
WES
WES involves the targeted enrichment and high-throughput sequencing of all coding exons and non-coding RNAs in the genome. In recent years, the first WES studies investigating rare sequence variation in common psychiatric disorders have been published, with most attention to date on de novo mutations in ASD (for example, see refs. 54–57) and SCZ (for example, see refs. 58–60). WES studies of other psychiatric disorders are currently underway. Sequence-based discovery of genes involved in neurodevelopmental disorders is reviewed by Hoischen and Eichler61, so we focus here on implications of these studies for genetic architecture, with a brief overview of key findings.
Sample sizes for WES are still modest (for example, hundreds of families, up to ~5,000 cases and controls) compared with GWAS and CNV analyses, but results suggest an important, albeit limited (see below), role for de novo coding point mutations, particularly in early-onset neurodevelopmental disorders (for example, see refs. 54–57,62). Several key findings have emerged. First, about three quarters of de novo point mutations have a paternal origin and the mutation rate is correlated with paternal age at conception (for example, see refs. 63,64; Fig. 4). Advanced paternal age is a risk factor for a range of psychiatric disorders, including ASD and SCZ65, but it remains unclear whether this risk is explained by the increased mutation rate or whether other mechanisms (for example, delayed fatherhood as a result of high polygenic risk66; age-related epigenetic changes67) have a role. Second, ASD probands harbor an excess of gene-disrupting mutations (for example, stop gain/loss, splice altering, frameshift; Table 1), particularly in brain-expressed genes54,57. Notably, this enrichment is restricted to probands with low IQ68. Given that similar enrichment has been reported in intellectual disability (for example, see ref. 62), these findings raise the question of whether gene-disrupting de novo mutations are related to ASD per se or to intellectual impairment given ASD, or, conversely, to severe forms of ASD characterized by low IQ. Notably, in SCZ, the burden of gene-disrupting mutations is higher in SCZ probands with premorbid cognitive impairment compared with those without such impairment58, which is an interesting parallel to the enrichment of such mutations in ASD probands with low IQ. The available evidence does not support an overall excess of gene-disrupting de novo mutations in SCZ58, although one exception is a study (N = 146 case trios) involving SCZ probands that were carefully screened for a negative family history of psychosis60, and it is possible that disruptive mutations are enriched in such cases. These points aside, there is some evidence for an excess of disruptive mutations in SCZ in several postsynaptic gene sets (although see refs. 41,60 for exceptions). These include members of the activity-regulated cytoskeleton-associated protein (ARC) and NMDA receptor (NMDAR) gene complexes and in genes associated with the fragile-X mental retardation protein (FMRP)58. These gene sets were also enriched in a parallel SCZ case-control WES study59 and in ASD (NMDAR, FMRP)54 and intellectual disability (ARC, NMDAR, FMRP), pointing to substantial overlap of de novo signal between disorders58,59. An excess of disruptive mutations in SCZ has also been identified in genes involved in voltage-gated calcium ion channel signaling59, a notable overlap with GWAS findings13.
Figure 4.

Paternal age at child’s conception is associated with the burden of de novo mutations in the child’s genome (Poisson regression, P < 2 × 10–16, linear slope = 1.75 mutations per year). Data are from refs. 63 (N = 78) and 64 (N = 10).
Table 1.
De novo gene-disrupting mutations are enriched in ASD
A third noteworthy finding is that specific genes have been identified in ASD and SCZ with recurrent gene-disrupting de novo mutations that exceed the number expected by chance. In SCZ, a single recurrence has been reported in the TAF13 gene58, whereas, for ASD, a total of seven genes with recurrences have been identified in 1,043 families16,21,26. These are important findings given that specific mutations in single genes are expected to be more amenable to functional follow-up than GWAS loci or multigenic CNVs. Attempts to identify specific genes using gene-based testing of segregating rare (as opposed to de novo) variants in WES case-control studies of SCZ59 and ASD69 have so far been unsuccessful (although see ref. 70), and much larger sample sizes, equivalent to those in current GWASs, are likely to be necessary59. This also applies to whole-genome sequencing studies, which examine variants that exist between as well as within genes, which are expected to become increasingly prevalent in coming years.
Synthesis: genetic architecture and mutational target size
The results from recent GWAS, CNV and WES studies are enabling, for the first time, empirical assessment of fundamental questions about the genetic architecture of psychiatric disorders. For each disorder, how many loci are there? What are their frequencies and effect sizes? Taken together, how much of the heritability can be accounted for? How do the different disorders compare and how do they compare to non-psychiatric diseases? To what extent does the genetic contribution to one disorder overlap with others? And what is the evidence for gene-gene interaction? These are important questions because, without this knowledge, the field is operating in the dark when it comes to dissecting genetic etiology. Newly developed statistical methods that assess the joint contribution of common genetic variation across the genome are beginning to provide initial answers to these questions. The available evidence suggests that psychiatric disorders, including SCZ71, BPD71, ASD71, ADHD71, MDD71, OCD20, TS20 and AD72, are highly polygenic, with between one third and a half of the heritability being explained by common genetic variation distributed across the genome. In the case of SCZ, an estimated 8,300 independent common loci collectively account for up to 50% of the genetic risk28. A high degree of polygenicity (that is, high number of contributing genes or mutational target size) appears to be typical for most complex traits (for example, height73) and common diseases (for example, Crohn’s disease74, type I diabetes74) examined to date75. However, although a substantial proportion of the genetic risk for some autoimmune diseases is a result of alleles that individually explain a substantial proportion of the heritability (for example, 1%), currently available data suggest that the polygenic risk for schizophrenia is dominated by alleles with very small effects (that is, 0.05% or less; Fig. 3 in Box 1). This is probably true for most psychiatric disorders (with AD being an exception). Whole-genome analysis methods also support substantial overlap of common genetic variation between disorders, in most instances consistent with clinical overlap and evidence for comorbidity from epidemiological studies.
A role for rare variants in psychiatric disorders has been well established by CNV and WES studies, but the overall contribution of rare variation is less well understood compared with common variation because coverage and sample sizes are not yet optimal. This is also true for low-frequency variation. A substantial contribution is possible if the heritability not explained by common variation (for example, half to two thirds for most disorders) is predominantly a result of rare and low-frequency variants, a question that will only be fully resolved by whole-genome sequencing studies that completely ascertain all genetic variation in large samples of cases and controls. In the interim, the PGC has developed the PsychChip, a single nucleotide polymorphism (SNP) array that features ~250,000 functional protein-coding variants, most of which are low frequency, in addition to high-density coverage of known and suggestive psychiatric disorder loci and a genome-wide backbone of common variation (enabling integration with existing data sets). In excess of 100,000 cases across multiple psychiatric disorders will be genotyped by the PGC with this array in the coming year, which should provide initial insights into the role of low-frequency variation in psychiatric disorders.
The overall contribution of de novo CNVs and coding mutations is also an open question, but the majority of new mutations do not contribute to heritability estimates76, which are high for most psychiatric disorders (that is, ~0.6–0.8). Currently available WES data for ASD are consistent with ~5–10% of affected individuals harboring a damaging de novo point mutation with an odds ratio of ~5–20 (ref. 55). Importantly, in the context of using WES to assist clinical diagnosis, a genetic model for ASD involving fully penetrant mutations in many genes can be rejected55 (although a proportion of cases may harbor new highly penetrant mutations). Existing CNV and WES studies are consistent with GWAS evidence for a large mutational target in psychiatric disorders. The data on de novo coding mutations in ASD suggests that between 370 (ref. 54) and 1,000 (ref. 57) genes contribute to disease, and similar figures have been reported in schizophrenia (for example, N = 850 genes)60.
Although it is clear that genetic variation for psychiatric illness spans the entire spectrum of allele frequencies, there is evidence that the relative importance of rare and common variation may differ between disorders. For instance, SNPs with a minor allele frequency <0.05 explain 21% of the heritability for TS, compared with 0% for OCD (in analyses that compared the two case samples to a common control), despite substantial overlap of common variation between these disorders20. Studies that ascertain rare and low-frequency variation in large samples will help to clarify the distribution of genetic variation as a function of allele frequency in these and other disorders.
The heritability explained by individual common and rare variants identified to date is approximately the same77 as a result of an inverse relationship between allele frequency and effect size (Fig. 3 in Box 1). This relationship is predicted by evolutionary theory due to the action of selection driving alleles with large effect size to lower frequency6, although it is important to note that many rare and low-frequency variants with small effect size are also likely to exist. The precise nature of the joint distribution of risk allele frequencies and effect sizes is yet to be elucidated.
Relationship between disorders: new insights from genomic data
In most other branches of medicine, diagnosis has moved on from classification based on self-reported symptoms and clinical observation to incorporate more objective tests of disease78. Although the classification system in psychiatry has been standardized and regularly revised since the 1980s, the biological validity of the current diagnosis system is unknown. Probing underlying biology is problematic when ‘case-ness’ is often the only consistently recorded phenotypic data available for large-scale analyses78. Both comorbidity between disorders and heterogeneity within disorders may be a reflection of the diagnostic label in the context of the current nosology. Dimensional scales have been proposed (for example, see ref. 79), but have not been widely adopted. A genetic contribution to the relationship between psychiatric disorders has been established from both clinical (for example, see ref. 80) and epidemiological (for example, see refs. 1,2) studies, showing increased risk of disorders in the relatives of those affected. However, accumulating large enough samples to achieve accurate results is a limiting factor.
The genomics era has provided powerful approaches to explore the genetic relationship between disorders in new ways. Specific variants associated with multiple disorders and cross-disorder phenotypes have been identified21,22, as discussed above. These studies are too underpowered to identify the majority of shared genetic factors, but currently available GWAS samples collected independently for different disorders can be used to interrogate the genetic relationship between disorders at a genome-wide level. In this paradigm, although all individuals are unrelated in the classical sense, they nonetheless share genomic segments through common ancestors. Thus, genome-wide genotypes can be used to determine whether cases are genetically more similar to each other (that is, across disorders) than to controls74 and to calculate the correlation between disorders on the basis of genome-wide SNPs (the SNP correlation)81. Given that the data sets have been collected independently, any relationship detected between disorders is directly attributable to shared DNA variants and contamination by shared environmental factors seems unlikely. The estimate of the genome-wide SNP correlation applied to SCZ and BPD data was 0.68 (s.e. of 0.04), implying important shared genetic etiology (Fig. 5)71. Theoretical modeling of potential misdiagnosis between disorders82,83 cannot explain the high correlation84. In contrast, a high genetic correlation between disorders is likely to generate individuals with mixed symptoms that may lead to changing diagnoses under the current nosology71. It is noteworthy that SNP correlations calculated between independently collected SCZ data sets were ~0.9 (Fig. 5), implying that the diagnostic classification system does indeed split out a genetically more homogeneous subset from the psychiatric spectrum71. Notably, the SNP correlations calculated between BPD sets were more variable (0.55–0.88), perhaps suggesting more genetic heterogeneity within BPD (Fig. 5). The same method has been applied to all pairs of disorders in the psychiatric genomics consortium (that is, SCZ, BPD, MDD, ADHD and ASD)71. These analyses demonstrated a moderate genetic relationship tagged by common polymorphisms between MDD and both BPD (0.47, s.e. 0.06) and SCZ (0.43, s.e. 0.06), implying an underlying genetic vulnerability to psychiatric disorders traditionally considered as adult onset disorders (Fig. 5).
Figure 5.
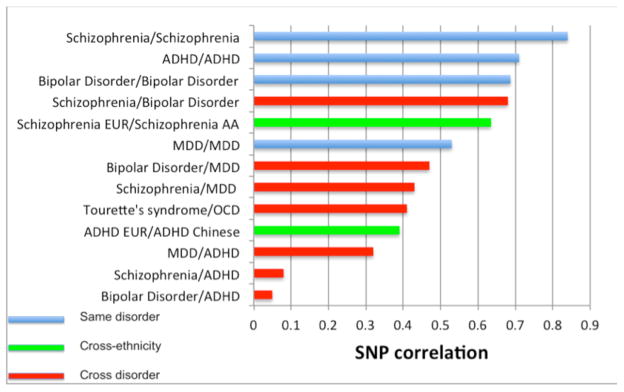
Quantifying the genetic relationship between independent data sets through the SNP correlation20,71,91,92. AA, African American; CHN, Chinese; EUR, European including European American.
Revisiting old questions with new data
The availability of genome-wide genetic data facilitates much more than gene and pathway discovery. It allows new experimental designs to address old scientific questions with new data. One example is the study of pleiotropy, whereby genes or genetic variants influence more than one trait. Pleiotropy is widespread, not just in humans, but across species85. However, pleiotropy has been difficult to study in humans with family designs because multiple diseases and disorders are unlikely to co-segregate in families with sufficient prevalence, even if there are shared genetic factors. The cross-disorder study, described above, is one specific example of a population-based study that links individuals by genome-wide similarity. Notably, in these designs, different individuals can be measured for different traits. These methods are being used to explore epidemiological puzzles such as different rates of immune disorders and cancers in people with and without schizophrenia (for example, see ref. 86).
The importance of genotype × environment interactions is another longstanding question that can be addressed with new genome-wide data. Such interactions have been suggested for psychiatric disorders, but we are not aware of any statistically significant and replicable interactions. The likely reason is that samples to date have been small and most studies have focused on only a small subset of candidate gene polymorphisms87. Large genetically and environmentally informative samples will be needed to achieve the statistical power required given small expected effects. The findings from whole-genome studies can overcome some of these limitations by using the aggregate of multiple genetic risk factors identified in large genotyped samples to obtain a better estimate of genotype and applying this in smaller genotyped samples that are informative for relevant environmental risk factors. This design could be used to test whether genotype and environment act multiplicatively on risk or whether there is a real interaction88.
Another hypothesis that was posed many years ago is the relationship between paternal age and risk of disease in offspring, but data on de novo mutation rates and the identification of specific new mutations has not been feasible until recently. The availability of direct estimates of the de novo mutation rate from sequencing studies in families offers new opportunities for evaluating the relationship between paternal age, new mutations and the increased risk of ASD, SCZ and other disorders.
Conclusions
The genomics era has demonstrated the value of large data sets and international consortia and has narrowed down the genetic architecture dramatically. In Box 2, we summarize the current state of knowledge of genetics in psychiatric disorders. Efforts to accumulate and process very large samples for genotyping and sequencing are underway for multiple disorders and we can be confident that many additional genetic associations, common and rare, will be identified. The rate-limiting step for the field is now the availability (or lack thereof) of standardized phenotype data in large cohorts. The development of innovative ‘next-generation phenotyping’ methods for the quick and cost-effective collection of phenotype data in large samples should be a priority. Such efforts will require international collaboration, and should be geared toward testable hypotheses in evolution and epidemiology. Possibilities include the investigation of genotype × environment interactions (provided the right environment risk factors can be identified), and the use of Mendelian randomization to infer causal effects of modifiable environmental risk factors. A third possibility relates to natural selection, for instance, comparing fecundity in those with large de novo CNVs versus those without. This type of approach is likely to be complemented by endophenotype studies, including brain imaging89, and by efforts to characterize the functional basis of genetic associations. Together, these approaches, by refining our understanding of etiology, hold the promise of better treatments and prevention for a group of disorders that account for about one third of the global burden of disease90.
Box 2. What we know and don’t know about the genetics of psychiatric disorders.
Heritability estimates for most psychiatric disorders are high, ranging from 0.4–0.8 (refs. 1–4). These estimates are consistent across studies and cannot be explained by perceived ‘flaws’ in twin analyses.
Psychiatric disorders are highly polygenic (for example, ref. 71), meaning that a large number of genomic loci contribute to risk (that is, the genome-wide mutational target is high). No single variant explains more than a small fraction (for example, ~0.1%) of the genetic variance (with the exception of AD, for which much of the variance is explained by a few common loci with large effects), which implies that large sample sizes are required for genetic discoveries77.
Genetic variation for psychiatric disorders spans the entire allelic spectrum from common to rare, and includes structural variation (CNVs) as well as SNPs. GWASs have identified >100 common loci for SCZ13 and smaller numbers for other disorders14–16. These findings have survived rigorous attempts at falsification and cannot be explained by population stratification. About a dozen rare CNVs have been identified, many in association with multiple disorders48.
De novo mutations, including CNVs41–44,46,49 and single base mutations54–58,60, are also important risk factors, and emerging evidence suggests that presence of these variants in the context of psychiatric disorders is associated with impaired cognition, consistent with evidence of de novo mutations in intellectual disability. The high heritability of most disorders limits the potential contribution of new mutations, the majority of which do not contribute to heritability estimates.
Common risk loci (MAF > 0.05) are associated with small effect size (odds ratio ~1.05–1.2), whereas identified rare variants, including de novo mutations, have larger effects (odds ratio ~2 to >20). Common variants with odds ratios >1.5 can be ruled out for most disorders (excepting AD). Conversely, rare variants with smaller effects (odds ratio < 2) are likely to exist, but current studies lack the power to detect them. Low-frequency variants are poorly surveyed, but initial studies in SCZ suggest few if any “Goldilocks” variants (low-frequency variants with odds ratios ~1.5–2; that is, not too big, not too small) exist59.
Genetic variation tagged by common SNPs accounts for between one-third and half of the heritability for most disorders (for example, see refs. 19,26,28,71). These estimates are not inflated by population stratification and largely resolve the controversy of missing heritability (that is, that GWAS findings explain only a small proportion of the heritability estimated by family studies), as they imply that much of the heritability is merely hidden (that is, many common loci exist that do not surpass the threshold for genome-wide significance in GWAS).
The contribution of rare variation is less well understood because coverage and sample size are not yet optimal, but it may be substantial if variation not explained by common variation (that is, half to one third) is a result of rare variants. Larger studies and improved ascertainment of rare variation (for example, by whole-exome and whole-genome sequencing) will be needed to clarify the overall distribution of genetic variance as a function of allele frequency.
There is strong evidence for sharing of common genetic variation (pleiotropy) between disorders, particularly SCZ, BPD and MDD71. Rare and de novo CNVs also exhibit substantial overlap between disorders96, and there is emerging evidence that the same may be true for rare and de novo point mutations58,59. The latter is in the form of shared enrichment across disorders of mutations in particular gene sets (for example, targets of FMRP), although whether this overlap extends to the level of specific mutations is currently unclear.
No fully penetrant mutations for SCZ have been identified, and the idea that SCZ is a set of monogenic disorders can be ruled out. Single mutations sufficient to cause illness are known for ASD, but, similar to SCZ, the idea that all cases can be explained by single mutations is inconsistent with the empirical data.
Although it’s clear that many common and rare variants exist for psychiatric disorders, we do not understand how these variants combine to influence disease pathogenesis and phenotypic heterogeneity. One possibility is that all risk variants, common and rare, contribute to an underlying liability for disease with individuals exceeding a threshold being affected. This could involve background common variation increasing or decreasing the effects of one or multiple rare high penetrance mutations, although other possible mechanisms exist and differentiating between them represents a major future challenge for the field.
There are few (if any) causal variants known and the precise joint distribution of effect size and risk allele frequency, although coming into focus, is currently unknown. The interpretation of empirically derived joint distributions will depend on the biological validity of the diagnostic constructs.
Acknowledgments
This work was supported by the US National Institutes of Health (GM099568 and GM075091 to P.M.V.), the National Institute of Mental Health (K01MH085812 and R01MH100141 to M.C.K.), the Australian Research Council (FT0991360 to N.R.W.) and the Australian National Health and Medical Research Council (APP1011506, APP1048853 and APP1067795 to P.M.V., APP1011506 and APP1047956 to N.R.W., APP1067795 to J.G.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 2010;167:1357–1363. doi: 10.1176/appi.ajp.2010.10020223. [DOI] [PubMed] [Google Scholar]
- 2.Lichtenstein P, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:255–274. doi: 10.1002/ajmg.b.31159. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 5.Wray NR, Gottesman Using summary data from the Danish national registers to estimate heritabilities for schizophrenia, bipolar disorder, and major depressive disorder. Front Genet. 2012;3:118. doi: 10.3389/fgene.2012.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eyre-Walker A. Evolution in health and medicine Sackler colloquium: Genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc Natl Acad Sci USA. 2010;107 (Suppl 1):1752–1756. doi: 10.1073/pnas.0906182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller MC, Miller G. Resolving the paradox of common, harmful, heritable mental disorders: which evolutionary genetic models work best? Behav Brain Sci. 2006;29:385–452. doi: 10.1017/S0140525X06009095. [DOI] [PubMed] [Google Scholar]
- 8.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 9.Hill WG, Goddard ME, Visscher PM. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 2008;4:e1000008. doi: 10.1371/journal.pgen.1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corvin A, Craddock N, Sullivan PF. Genome-wide association studies: a primer. Psychol Med. 2010;40:1063–1077. doi: 10.1017/S0033291709991723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarroll SA, Feng G, Hyman SE. Genome-scale neurogenetics: methodology and meaning. Nat Neurosci. 2014;17:756–763. doi: 10.1038/nn.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Psychiatric Genomics Consortium Schizophrenia Working Group. Psychiatric Genomics Consortium quadruples schizophrenia GWAS sample-size to 35,000 cases and 47,000 controls. inXXIst World Congress of Psychiatric Genetics: Redefining Mental Illness Through Genetics; Boston. 2013. This conference paper reported the identification of more than 100 loci for schizophrenia, the “flagship” disorder of the Psychiatric Genomics Consortium. The results show what might be achieved for other psychiatric disorders as sample sizes increase. [Google Scholar]
- 14.European Alzheimer’s Disease Initiative et al. Meta-analysis of 74, 046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charney AW, et al. Bipolar disorder GWAS of 13,741 cases and 19,762 controls identifies eight genome-wide significant hits and implicates genes targeted by FMRP protein. inXXIst World Congress of Psychiatric Genetics: Redefining Mental Illness Through Genetics; Boston. 2013. [Google Scholar]
- 16.Anney R, et al. Meta-analysis of European ancestry individuals with autism spectrum disorder reveals strong association 3′ of the astrotactin 2 (ASTN2) gene locus on chromosome 9. inXXIst World Congress of Psychiatric Genetics: Redefining Mental Illness Through Genetics; Boston. 2013. [Google Scholar]
- 17.Neale BM, et al. Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:906–920. doi: 10.1016/j.jaac.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, et al. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol Psychiatry. 2011;16:949–959. doi: 10.1038/mp.2010.107. [DOI] [PubMed] [Google Scholar]
- 19.Ripke S, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis LK, et al. Partitioning the heritability of tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet. 2013;9:e1003864. doi: 10.1371/journal.pgen.1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cross-Disorder Group of the Psychiatric Genomics Consortium et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genome-wide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan PF. The psychiatric GWAS consortium: big science comes to psychiatry. Neuron. 2010;68:182–186. doi: 10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wray NR, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis and lessons learned. Mol Psychiatry. 2012;17:36–48. doi: 10.1038/mp.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. This is a landmark study that demonstrated a substantial contribution to risk of schizophrenia from common variation distributed throughout the genome. It also revealed evidence for genetic overlap between schizophrenia and bipolar disorder involving many common loci of small effect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi J, et al. Common variants on chromosome 6p22. are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ripke S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson-Schmidt H, et al. Am J Med Genet B Neuropsychiatr Genet; Selected rapporteur summaries from the XX World Congress of Psychiatric Genetics; Hamburg, Germany. October 14–18, 2012; 2013. pp. 96–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 31.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellcome Trust Case Control Consortium. Genome-wide association study of 14, 000 cases of seven common diseases and 3, 000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan P. Don’t give up on GWAS. Mol Psychiatry. 2012;17:2–3. doi: 10.1038/mp.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12:581–594. doi: 10.1038/nrd4051. [DOI] [PubMed] [Google Scholar]
- 36.Sanseau P, et al. Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30:317–320. doi: 10.1038/nbt.2151. [DOI] [PubMed] [Google Scholar]
- 37.Okada Y, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lango Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilman SR, et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signaling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 45.Williams NM, et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. 2010;376:1401–1408. doi: 10.1016/S0140-6736(10)61109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malhotra D, et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergen SE, et al. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol Psychiatry. 2012;17:880–886. doi: 10.1038/mp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levinson DF, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. This is the largest meta-analysis of copy number variation in schizophrenia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.3 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vacic V, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams NM, et al. Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13. Am J Psychiatry. 2012;169:195–204. doi: 10.1176/appi.ajp.2011.11060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crespi B, Stead P, Elliot M. Evolution in health and medicine Sackler colloquium: comparative genomics of autism and schizophrenia. Proc Natl Acad Sci USA. 2010;107 (Suppl 1):1736–1741. doi: 10.1073/pnas.0906080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 54.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99. doi: 10.1016/j.neuron.2012.04.009. This is the largest of four recent studies reporting the results of WES in autism families, and the only study other than Sanders et al. (2012) to use a large sample of family controls. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. This is one of four studies reporting the results of WES in autism families. Collectively, these studies have identified six brain-expressed genes harboring recurrent de novo gene-disrupting mutations that are now strong candidate genes for autism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50. doi: 10.1038/nature10989. This is one of four studies reporting the results of WES in autism families. The authors report recurrent gene-disrupting mutations in the CHD8 gene in unrelated probands, an observation that is highly unlikely to have occurred by chance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. This study, one of four that report results from WES in autism families, used a discordant sibling family design to demonstrate an enrichment of de novo gene-disrupting mutations in autism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. This is the largest WES study of de novo mutations in schizophrenia to date. The authors report an enrichment of de novo mutations in genes encoding for post-synaptic proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purcell S, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. Together with the parallel family-based study by Fromer et al. (2014), this large case-control WES study demonstrated a burden of rare mutations in specific sets of post-synaptic genes in schizophrenia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu B, et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci. 2014;17:764–772. doi: 10.1038/nn.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.deLigt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 63.Kong A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. This paper demonstrated that the majority of de novo point mutations originate in the paternal germline, and that the de novo mutation rate is positively correlated with paternal age. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Michaelson JJ, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGrath JJ, et al. A comprehensive assessment of parental age and psychiatric disorders. JAMA Psychiatry. 2014;71:301–309. doi: 10.1001/jamapsychiatry.2013.4081. [DOI] [PubMed] [Google Scholar]
- 66.Petersen L, Mortensen PB, Pedersen CB. Paternal age at birth of first child and risk of schizophrenia. Am J Psychiatry. 2011;168:82–88. doi: 10.1176/appi.ajp.2010.10020252. [DOI] [PubMed] [Google Scholar]
- 67.Perrin MC, Brown AS, Malaspina D. Aberrant epigenetic regulation could explain the relationship of paternal age to schizophrenia. Schizophr Bull. 2007;33:1270–1273. doi: 10.1093/schbul/sbm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Samocha KE, Robinson E, Neale B, Daly M. Statistical evaluation of de novo variation implicates a distinct etiologic subtype of autism. inXXIst World Congress of Psychiatric Genetics: Redefining Mental Illness Through Genetics; Boston. 2013. [Google Scholar]
- 69.Liu L, et al. Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013;9:e1003443. doi: 10.1371/journal.pgen.1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ionita-Laza I, et al. Scan statistic-based analysis of exome sequencing data identifies FAN1 at 15q13.3 as a susceptibility gene for schizophrenia and autism. Proc Natl Acad Sci USA. 2014;111:343–348. doi: 10.1073/pnas.1309475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee SH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. This paper reported evidence for overlap of common genetic variation (that is, pleiotropy) between key psychiatric disorders, including SCZ and BIP (high), SCZ and MDD (moderate), MDD and BIP (moderate), MDD and ADHD (moderate) and SCZ and ASD (low) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee SH, et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22:832–841. doi: 10.1093/hmg/dds491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. This is a landmark paper that described methods for estimation of heritability for quantitative traits based on genetic relationships among individuals inferred from genome-wide data on common SNPs. Applied to height, the classic model trait for human quantitative genetics, the authors demonstrate that a substantial proportion of the heritability can be explained by many common loci throughout the genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, et al. Ubiquitous polygenicity of human complex traits: genome-wide analysis of 49 traits in Koreans. PLoS Genet. 2013;9:e1003355. doi: 10.1371/journal.pgen.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gratten J, Visscher PM, Mowry BJ, Wray NR. Interpreting the role of de novo protein-coding mutations in neuropsychiatric disease. Nat Genet. 2013;45:234–238. doi: 10.1038/ng.2555. [DOI] [PubMed] [Google Scholar]
- 77.Visscher PM, Goddard ME, Derks EM, Wray NR. Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol Psychiatry. 2012;17:474–485. doi: 10.1038/mp.2011.65. [DOI] [PubMed] [Google Scholar]
- 78.Kapur S, Phillips AG, Insel TR. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol Psychiatry. 2012;17:1174–1179. doi: 10.1038/mp.2012.105. [DOI] [PubMed] [Google Scholar]
- 79.Allardyce J, Suppes T, Van Os J. Dimensions and the psychosis phenotype. Int J Methods PsychiatrRes. 2007;16 (Suppl 1):S34–S40. doi: 10.1002/mpr.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kendler KS, Gardner CO. The risk for psychiatric disorders in relatives of schizophrenic and control probands: a comparison of three independent studies. Psychol Med. 1997;27:411–419. doi: 10.1017/s003329179600445x. [DOI] [PubMed] [Google Scholar]
- 81.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism–derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2542. doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bromet EJ, et al. Diagnostic shifts during the decade following first admission for psychosis. Am J Psychiatry. 2011;168:1186–1194. doi: 10.1176/appi.ajp.2011.11010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laursen TM, Agerbo E, Pedersen CB. Bipolar disorder, schizoaffective disorder, and schizophrenia overlap: a new comorbidity index. J Clin Psychiatry. 2009;70:1432–1438. doi: 10.4088/JCP.08m04807. [DOI] [PubMed] [Google Scholar]
- 84.Wray NR, Lee SH, Kendler KS. Impact of diagnostic misclassification on estimation of genetic correlations using genome-wide genotypes. Eur J Hum Genet. 2012;20:668–674. doi: 10.1038/ejhg.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sinauer Associates; 1998. [Google Scholar]
- 86.Ji J, et al. Incidence of cancer in patients with schizophrenia and their first-degree relatives: a population-based study in Sweden. Schizophr Bull. 2013;39:527–536. doi: 10.1093/schbul/sbs065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry. 2011;168:1041–1049. doi: 10.1176/appi.ajp.2011.11020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McGrath JJ, Mortensen PB, Visscher PM, Wray NR. Where GWAS and epidemiology meet: opportunities for the simultaneous study of genetic and environmental risk factors in schizophrenia. Schizophr Bull. 2013;39:955–959. doi: 10.1093/schbul/sbt108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Medland SE, Jahanshad N, Neale BM, Thompson PM. Whole genome analyses of whole-brain data: working within an expanded search space. Nat Neurosci. 2014;17:791–800. doi: 10.1038/nn.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.WHO. The Global Burden of Disease: 2004 Update. WHO Press; 2008. [Google Scholar]
- 91.de Candia TR, et al. Additive genetic variation in schizophrenia risk is shared by populations of African and European descent. Am J Hum Genet. 2013;93:463–470. doi: 10.1016/j.ajhg.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang L, et al. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: genome-wide association study of both common and rare variants. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:419–430. doi: 10.1002/ajmg.b.32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sullivan PF. The genetics of schizophrenia. PLoS Med. 2005;2:e212. doi: 10.1371/journal.pmed.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang J, Visscher PM, Wray NR. Sporadic cases are the norm for complex disease. Eur J Hum Genet. 2010;18:1039–1043. doi: 10.1038/ejhg.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Turner MR, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25:528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]