

Abstract
Elucidating targets of physiological tumor metastasis suppressors can highlight key signaling pathways leading to invasion and metastasis. To identify downstream targets of the metastasis suppressor Raf Kinase Inhibitory Protein (RKIP/PEBP1), we utilized an integrated approach based upon statistical analysis of tumor gene expression data combined with experimental validation. Previous studies from our laboratory identified the architectural transcription factor and oncogene, HMGA2, as a target of inhibition by RKIP. Here we identify two signaling pathways that promote HMGA2-driven metastasis. Using both human breast tumor cells and an MMTV-Wnt mouse breast tumor model, we show that RKIP induces and HMGA2 inhibits expression of miR-200b; miR-200b directly inhibits expression of lysyl oxidase (LOX), leading to decreased invasion. RKIP also inhibits syndecan-2 (SDC2), which is aberrantly expressed in breast cancer, via down-regulation of HMGA2; but this mechanism is independent of miR-200. Depletion of SDC2 induces apoptosis and suppresses breast tumor growth and metastasis in mouse xenografts. RKIP, LOX, and SDC2 are coordinately regulated and collectively encompass a prognostic signature for metastasis-free survival in ER-negative breast cancer patients. Taken together, our findings reveal two novel signaling pathways targeted by the metastasis suppressor RKIP that regulate remodeling of the extracellular matrix and tumor survival.
Keywords: RKIP, HMGA2, LOX, SDC2, miR-200, breast, metastasis
Introduction
Metastasis is the key process that leads to death from solid tumors. The metastatic program comprises multiple steps including early events such as tumor cell migration, invasion and intravasation into vessels and later events leading to growth and colonization at distant organ sites1. A wealth of genomic and transcriptional data for tumors has recently become available, but the data are largely derived from primary tumors. A major challenge is how to utilize these resources to identify regulatory mechanisms that drive tumor invasion and metastasis.
One approach is to utilize natural suppressors of metastasis to identify their downstream targets. Metastasis suppressors are genes that do not significantly alter primary tumor growth but prevent cancer cells from undergoing metastasis2, 3. Raf-1 Kinase Inhibitory Protein (RKIP/PEBP1) functions as a metastasis suppressor in prostate4, 5, colorectal6, 7, and breast8-10 cancer, and is prognostic for survival in a number of tumors5, 6, 11. A member of the phosphatidylethanolamine binding protein (PEBP) family that is conserved throughout evolution, RKIP acts as an inhibitor of cellular kinase pathways in mammalian cells such as MAP kinase (MAPK). Raf-1 is inhibited upon RKIP binding, and phosphorylation of RKIP at S153 leads to RKIP release and Raf-1 activation12, 13. Previous studies from our laboratory have shown that RKIP suppresses breast tumor metastasis in mouse xenografts. RKIP inhibition of the MAPK pathway induces the microRNA let-7 that, in turn, inhibits synthesis of the architectural transcription factor high mobility group AT-hook 2 (HMGA2)10. HMGA2 promotes metastasis in part through a pathway involving induction of osteopontin and CXCR410.
To systematically identify downstream mediators of RKIP that regulate metastasis, we developed an integrated bioinformatics/experimental approach. We identified the microRNA miR-200b as a novel downstream target of RKIP and HMGA2, and further showed that miR-200b directly inhibits Lysyl Oxidase (LOX) expression. Previous studies have shown that miR-200 inhibits and LOX promotes breast tumor cell invasion and metastasis14-17. Expression of these genes is similarly correlated in patient tumors. Together these results identify an RKIP/HMGA2/miR-200b/LOX pathway that regulates breast cancer metastasis. Moreover, we demonstrated that syndecan-2 (SDC2) is another important effecter of RKIP and HMGA2 in regulating breast tumorigenesis. Depletion of SDC2 expression suppresses breast cancer cell survival and metastasis in mouse xenografts. Together, the RKIP/LOX/SDC2 genes form a prognostic signature for metastasis-free survival in ER-negative breast cancer patient populations.
Results
Identification of RKIP-mediated microRNAs and genes that regulate breast cancer metastasis
To systematically identify RKIP-mediated signaling pathways in suppression of breast cancer metastasis, we did gene and microRNA expression array analyses for 1833 cells with or without expression of S153E-RKIP (a more potent Raf-1 inhibitor). The 1833 cell-line is a highly metastatic, bone-tropic derivative of the human breast cancer cell-line MDA-MB-23118. Our previous studies have demonstrated that expression of S153E-RKIP in 1833 cells suppressed breast tumor invasion and metastasis both in culture and in mouse xenografts10. The present gene expression array analysis of 1833 cells expressing S153E-RKIP versus control identified 589 differentially expressed protein-coding genes based on the criteria: fold change > 1.25, Benjamini-Hochberg adjusted P < 0.05 and false discovery rate < 0.05 (Supplementary Figure S1A). Comparing this list with a previously published 101 bone metastasis signature (BMS) genes derived from a series of metastatic stages for a similar breast cancer cell type18, we found 20 overlapped genes between BMS and the 589 differentially expressed gene set. Interestingly, expression of these 20 genes is significantly reverted from a highly metastatic stage to relatively less aggressive status upon expression of RKIP in the breast cancer cells (Supplementary Figure S2). Gene Ontology analysis19 of the 589 RKIP-regulated genes showed that they are enriched in functional categories such as extracellular stimulus, cell migration, proliferation, apoptosis, blood vessel development, extracellular matrix and extracellular space (Supplementary Table S1). Pathway enrichment analysis19 of these differentially expressed genes showed that they are significantly enriched in inflammatory response, cell adhesion molecules, IL1R signaling, TGF-β signaling, and the NF-κB pathway (Supplementary Table S2). These results demonstrate that RKIP-mediated suppression of breast tumor metastasis involves many different signaling pathways.
We then used microRNA expression arrays to analyze small non-coding microRNAs that are regulated by RKIP. We identified 50 differentially expressed microRNAs in 1833 cells expressing S153E-RKIP versus control including 37 up-regulated and 13 down-regulated microRNAs (Supplementary Figures S1A and B). To identify target genes of a differentially expressed microRNAs, we first obtained a list of targets for a given microRNA from a combination of five major microRNA-target pair prediction programs/databases including PicTar20, TargetScanS21, miRanda22, miRBase Targets23, and PITA Top24 (Figure 1). If a gene in the list for a given microRNA negatively correlated with the microRNA expression and was differentially expressed upon RKIP expression in the breast cancer cells, it would be assigned to be one of the potential targets of the microRNA (Figure 1). To evaluate whether microRNA expression can be statistically correlated with RKIP expression in patient tumor samples, we analyzed correlation between RKIP and potential targets of a microRNA using Gene Set Analysis (GSA)25 (Figure 1). These analyses showed that miR-200b was the most significant microRNA (P < 4.8×10-5) that positively correlated with RKIP expression in a cohort of breast cancer patient samples (Br426).
Figure 1.

Bioinformatics scheme for selection of RKIP-mediated microRNA(s) and/or target gene(s) that may regulate breast tumor metastasis.
The miR-200 family is highly expressed in epithelial tissue26, and an aberrant decrease in expression of several family members has been implicated in breast cancer progression14. We analyzed the 79 predicted targets of miR-200b for an inverse correlation to RKIP expression in cell lines and tumors and a positive correlation to metastasis. These analyses revealed that LOX and SDC2 as potential targets of RKIP and miR-200b suppression are likely mediators of metastatic progression in breast cancer (Figure 1, Supplementary Figures S1B and C). LOX has previously been implicated as an initiator of elastin and collagen crosslinking that potentiates invasion and metastasis16, 17, 27; SDC2 is aberrantly expressed in breast cancer but its function is not known.
To evaluate the correlation between expression of miR-200b and its two predicted target genes LOX and SDC2 in cells, we analyzed their expression levels by qRT-PCR in three invasive human breast cancer cell lines including Hs578T, 1833 and MDA-MB-436 and one non-invasive line, MCF-7. MiR-200b expression was extremely low in 3 metastatic human breast cancer cell lines, but high in non-metastatic MCF-7 cells, while its two predicted targets LOX and SDC2 were inversely correlated with miR-200b expression (Supplementary Figure S1D). These results are consistent with LOX and SDC2 as pro-metastatic genes in breast cancer16, 17, 28.
RKIP induces miR-200b and inhibits LOX expression in breast cancer cells via HMGA2
To test the proposed RKIP/miR-200/LOX signaling pathway, we first investigated the regulation of miR-200b and LOX expression by RKIP. Consistent with the results from the gene and microRNA expression array analyses, qRT-PCR also showed a significant induction of miR-200b and inhibition of LOX expression upon expression of S153E-RKIP in 1833 cells (Figures 2a and b).
Figure 2.

Expression of RKIP or depletion of HMGA2 induces miR-200b and inhibits LOX expression in 1833 cells, a bone-tropic derivative of the ER-negative human breast cancer cell line MDA-MB-231. (a,b) Expression of RKIP induced miR-200b and inhibited LOX expression. 1833 cells were stably transduced with S153E-RKIP or vector control. Samples were analyzed by qRT-PCR and immunoblotting for: (a) miR-200b RNA; (b) LOX mRNA (upper panel), and RKIP and LOX protein (lower panel). (c) Significant negative correlation between RKIP and LOX expression in both ER-negative (n = 175) and ER-positive (n = 556) breast cancer patient data (Br731). (d,e) Depletion of HMGA2 induced miR-200b and inhibited LOX expression. 1833 cells were stably transduced with HMGA2 shRNA (shHMGA2) or scrambled shRNA (control). Samples were analyzed by qRT-PCR and immunoblotting for: (d) primary miR-200b transcript (pri-200b) and mature RNA (miR-200b); (e) HMGA2 and LOX mRNA (upper panel) and protein (lower panel). (f) Significant positive correlation between HMGA2 and LOX expression observed in ER-negative (n = 175, left) but not in ER-positive (n = 556, right) breast cancer patient data (Br731). (g,h,i) Loss of Hmga2 in MMTV-Wnt1 transgenic mouse breast tumors induced miR-200b and inhibited LOX expression. Wnt1 transgenic mice were crossed with Hmga2 specific knockout mice. Mouse primary breast tumors were obtained from Hmga2 wildtype (Hmga2+/+) or null (Hmga2-/-) mice: (g) Murine miR-200b RNA and (h) Murine Hmga2 and Lox mRNA analyzed by qRT-PCR; (i) Hematoxylin and eosin (H&E) (Left) and murine Lox protein (Right) analyzed by immunostaining. (a,b,d,e,g,h) human or murine GAPDH was used as the normalization control for mRNA and U6 snRNA was used as the normalization control for microRNA; Tubulin was a loading control for protein; Data are mean ± s.e., n = 3. *, P < 0.05; **, P < 0.01. (c,f) Correlations were determined by Pearson's correlation coefficient. P value was determined by Student t test.
To validate the relationship between RKIP and LOX clinically, we also analyzed an independent gene expression dataset of breast cancer patients that comprises 175 ER-negative and 556 ER-positive samples (Br731). As shown in Figure 2c, a significant negative correlation between RKIP and LOX expression was observed in both ER-negative and -positive patient datasets, and this negative relationship was even stronger in the ER-negative patient samples (Pearson correlation -0.31 vs. -0.21).
Previous work from our laboratory demonstrated that RKIP inhibits HMGA2 through induction of let-78, 10, and a potential interaction between let-7 and miR-200 via HMGA2 has been implied29, 30. Therefore, we determined whether RKIP induces miR-200b via down-regulation of HMGA2. HMGA2 depletion by shRNA in 1833 cells led to induction of both the miR-200b primary transcript and mature RNA (Figure 2d). HMGA2 depletion in 1833 cells also decreased LOX expression (Figure 2e). Similar regulation was observed in two other ER-negative breast cancer cell lines, MDA-MB-436 and MDA-MB-231 (Supplementary Figures S3A, B, D and E). Furthermore, transfection of the let-7g precursor into MDA-MB-436 cells significantly induced miR-200b and inhibited HMGA2 and LOX expression (Supplementary Figures S4A-D). Consistent with these results, we also noted a strong positive correlation between HMGA2 and LOX expression in the ER-negative, but not ER-positive, breast cancer patient dataset (Figure 2f).
To validate the regulation of miR-200b and LOX by HMGA2 in vivo, we used an MMTV-Wnt1 transgenic mouse model for breast cancer. Deletion of Hmga2 by crossing MMTV-Wnt1 mice with Hmga2-/- mice reduced tumor incidence and decreased tumor cell proliferation31. Analysis by qRT-PCR or immunohistochemistry showed a strong induction of miR-200b and inhibition of LOX expression in tumors from the MMTV-Wnt1/Hmga2-/- mice (Figures 2g-i). Together, these results reveal a novel signaling cascade whereby RKIP induces miR-200b and inhibits LOX expression through down-regulation of HMGA2.
MiR-200b directly inhibits LOX expression and breast cancer cell invasion
LOX is a predicted target of miR-200b through a potential binding site within the 3′UTR21. Consistent with this possibility, transfection of the miR-200b precursor into 1833, MDA-MB-231 or MDA-MB-436 cells significantly decreased LOX expression (Figure 3a; Supplementary Figures S5A and C-E). Conversely, inhibition of miR-200b in MCF-7 cells by anti-miR induced LOX expression (Figure 3b; Supplementary Figure S5B). To determine whether miR-200b directly binds to LOX, we performed luciferase reporter and mutagenesis assays. The 3′UTR fragment in the LOX mRNA containing the potential seed region for miR-200b was cloned into a luciferase reporter vector (LOX-wt-UTR). Co-transfection of miR-200b significantly inhibited luciferase expression from the LOX-wt-UTR reporter (Figures 3c and d) whereas mutation of the miR-200b binding seed region in LOX-wt-UTR (LOX-mut-UTR) restored luciferase activity (Figures 3c and d). Consistent with these results, analysis of gene expression in a cohort of 86 human breast tumors showed a significant negative correlation between miR-200b and LOX expression (Figure 3f). These results indicate that miR-200b directly binds to LOX mRNA and inhibits LOX expression.
Figure 3.

LOX is a direct target of miR-200b. (a) Transfection of 1833 cells with miR-200b precursor inhibited LOX expression: LOX mRNA was analyzed by qRT-PCR (upper panel) and protein by immunoblotting (lower panel). (b) Transfection of MCF-7 cells with anti-miR-200b induced LOX expression: LOX mRNA was analyzed by qRT-PCR (upper panel) and protein by immunoblotting (lower panel). (a,b) GAPDH as control. (c) Wildtype 3′UTR fragment in the LOX mRNA (LOX-wt-UTR) with the predicted binding site of miR-200b, and the mutant fragment (LOX-mut-UTR) showing the mutated sequence. (d) Luciferase and mutagenesis assay. LOX-wt-UTR and LOX-mut-UTR fragments were fused to a luciferase reporter (pMIR-REPORTER). Following co-transfection with miR-200b into cells, cell extracts were analyzed for luciferase activity as described in Methods. (e) Transfection of 1833 cells with miR-200b precursor inhibited cell invasion. Invasion assays were conducted as described in Methods. (a,b,d,e) Data are mean ± s.e., n = 3. (f) Significant negative correlation between miR-200b and LOX expression in breast cancer patients (Br86). P value is determined by Wilcoxon rank sum test.
To assess the functional role of miR-200b, we determined its effect on cell invasion. Transfection of the miR-200b precursor into 1833 or MDA-MB-436 cells significantly inhibited cell invasion (Figure 3e; Supplementary Figure S5F). However, we did not observe a significant change in cell invasion by inhibiting miR-200b with anti-miR in MCF-7 cells (data not shown), indicating that loss of miR-200b may not be sufficient to promote invasion in these cells. These results are consistent with previous studies showing that miR-200b can suppress early metastatic events during breast tumorigenesis14, 15, 32.
RKIP inhibits SDC2 expression via down-regulation of HMGA2
To confirm that SDC2 is also inhibited by RKIP, we examined SDC2 expression by qRT-PCR and immunoblotting in 1833, MDA-MB-436 and MDA-MB-231 cells stably transfected with S153E-RKIP. Similar to the regulation of LOX by RKIP (Figure 2b), SDC2 expression was decreased upon RKIP expression in these lines (Figure 4a; Supplementary Figures S3C and F). Conversely, RKIP depletion by shRNA in MDA-MB-435 cells significantly increased SDC2 expression (Figure 4b). Interestingly, depletion of RKIP in MDA-MB-435 cells also caused a significant induction of HMGA2 (Figure 4b). To test whether HMGA2 regulates SDC2, we stably depleted HMGA2 expression by shRNA in 1833, MDA-MB-436 and MDA-MB-231 cells and observed decreased SDC2 expression (Figure 4c; Supplementary Figures S3A, B, D and E). Transfection of the let-7g precursor into MDA-MB-436 cells also decreased SDC2 expression (Supplementary Figures S4A, C and D). Similar to Lox (Figure 2i), Sdc2 expression in MMTV-Wnt1 transgenic mouse breast tumors was inhibited upon loss of Hmga2 (Figure 4d). However, in contrast to LOX, transfection of breast cancer cells with the miR-200b precursor at a transfection efficiency of 70-80% (Supplementary Figures S5G-I) did not significantly alter SDC2 expression. Together, these data indicate that SDC2 is inhibited by RKIP and induced by HMGA2 through a miR200b-independent mechanism. Consistent with these results, we noted a strong positive correlation between HMGA2 and SDC2 expression in the ER-negative, but not in the ER-positive, breast cancer patient data (Figure 4e). Moreover, consistent with the observation that HMGA2 promotes both LOX and SDC2 expression, we observed a strong positive correlation between LOX and SDC2 expression in ER-negative breast cancer patient data (Supplementary Figure S4E).
Figure 4.

Expression of RKIP or depletion of HMGA2 inhibits SDC2 expression. (a) RKIP inhibited SDC2 expression. 1833 cells were stably transduced with S153E-RKIP or vector control. Samples were analyzed by qRT-PCR for SDC2 mRNA (upper panel) or by immunoblotting for RKIP and SDC2 protein (lower panel). (b) Depletion of RKIP by RKIP shRNA induced HMGA2 and SDC2 expression. MDA-MB-435 cells were stably transduced with RKIP shRNA (shRKIP) or scrambled shRNA (Control). Samples were analyzed by qRT-PCR (upper panel) and immunoblotting (lower panel) for RKIP, HMGA2 and SDC2 expression. (c) Depletion of HMGA2 by HMGA2 shRNA inhibited SDC2 expression. 1833 cells were stably transduced with HMGA2 shRNA (shHMGA2) or scrambled shRNA (Control). Samples were analyzed by qRT-PCR (upper panel) and immunoblotting (lower panel) for HMGA2 and SDC2 expression. (d) Loss of Hmga2 in MMTV-Wnt1 transgenic mouse breast tumors inhibited SDC2 expression. Mouse primary breast tumors were obtained from MMTV-Wnt1/Hmga2+/+ or MMTV-Wnt1/Hmga2-/- mice. Murine Sdc2 protein was analyzed by immunostaining. (e) Significant positive correlation between HMGA2 and SDC2 expression observed in ER-negative (n = 175) (Left) but not in ER-positive (n = 556) (Right) breast cancer patient data (Br731). Correlations were determined by Pearson's correlation coefficient. P, Student t test. (a-c) GAPDH was used as the normalization control for mRNA; Tubulin was used as a loading control for protein. Data are mean ± s.e., n = 3. *, P < 0.05; **, P < 0.01.
SDC2 depletion inhibits breast cancer cell growth and invasion in vitro
Altered SDC2 expression has been detected in breast tumors28; however, its role in breast cancer progression has not been studied. We initially determined the effect of SDC2 expression on growth and invasion of breast tumor cells in vitro. Reducing SDC2 expression by shRNA in 1833 and MDA-MB-436 cells dramatically decreased both cell growth and cell invasion (Figures 5a-c; Supplementary Figures S6A, B and D). Moreover, depletion of SDC2 expression in MDA-MB-436 cells also promoted apoptosis (Supplementary Figure S6C).
Figure 5.

Depletion of SDC2 suppresses breast cancer cell growth, invasion, and metastasis. (a-c) Depletion of SDC2 inhibited cell growth and invasion in vitro. 1833 cells stably transduced with SDC2 shRNA (shSDC2) or scrambled shRNA (Control) were analyzed for: (a) SDC2 mRNA by qRT-PCR (upper panel) and protein by immunoblotting (lower panel); (b) Relative cell growth; or (c) Relative cell invasion. (d-i) Depletion of SDC2 suppressed breast tumor growth in vivo. 1833 cells stably transduced with shSDC2 or Control were orthotopically injected into the mammary fat pad of nude mice. Tumors were dissected at 6 weeks after implantation and analyzed: (d) by qRT-PCR for SDC2 mRNA; (e) by immunostaining for Hematoxylin and eosin (H&E) (Left) and SDC2 protein (Right); (f) by immunostaining for cleaved caspase-3. Representative images (Left) and quantification (Right) of apoptotic cells are shown; (g) Representative bioluminescence images of mice bearing 1833 cells treated as indicated; (h) Photograph of representative xenograft breast tumors of 1833 cells treated as indicated; (i) Xenograft breast tumors of 1833 cells treated as indicated and analyzed for tumor weight. Data are mean ± s.e., n = 7-10 per group. (j-l) Depletion of SDC2 suppressed breast tumor metastasis and enhanced overall survival. 1833 cells stably transduced with shSDC2 or Control were injected into the left ventricle of mice. Mice were imaged for luciferase activity after 3 weeks. (j) Representative bioluminescence images of mice with bone metastasis; (k) Quantification of bone colonization by 1833 cells treated as indicated. Data are mean ± s.e., n = 7-10 per group; (l) Kaplan-Meier survival analysis of mice over 8 weeks after injection of the tumor cells. (a-d) Data are mean ± s.e., n = 3.
SDC2 depletion suppresses breast tumor growth and metastasis in mouse xenografts
To elucidate the role of SDC2 in regulating breast tumorigenesis in vivo, we injected 1833 cells stably expressing control or SDC2 shRNA into the mammary fat pad of mice to test their effect on xenograft tumor growth. Consistent with our in vitro study (Figures 5a and b), SDC2 depletion (Figures 5d and e) significantly induced tumor cell apoptosis (Figure 5f) and suppressed xenograft tumor growth (Figures 5g-i).
To test the effect of SDC2 expression on breast tumor metastasis, we injected luciferase-labeled 1833 cells expressing control or SDC2 shRNA into the left ventricle of mice. After 3 weeks, mice were imaged for luciferase activity. The results indicated that SDC2 depletion dramatically decreased bone metastasis (Figures 5j and k), and significantly enhanced overall survival rate (Figure 5l).
RKIP/LOX/SDC2 pathway is prognostic for metastasis-free survival in ER-negative breast cancer patients
Our results define two novel signaling cascades regulated by RKIP via its downstream target HMGA2. Upon expression of RKIP, HMGA2 protein is decreased8, 10, leading to up-regulation of miR-200b which binds and inhibits LOX expression. Moreover, down-regulated HMGA2 also decreases SDC2 expression. The resulting network represents the mechanism by which RKIP mediates suppression of breast tumor metastasis (Figure 6a).
Figure 6.
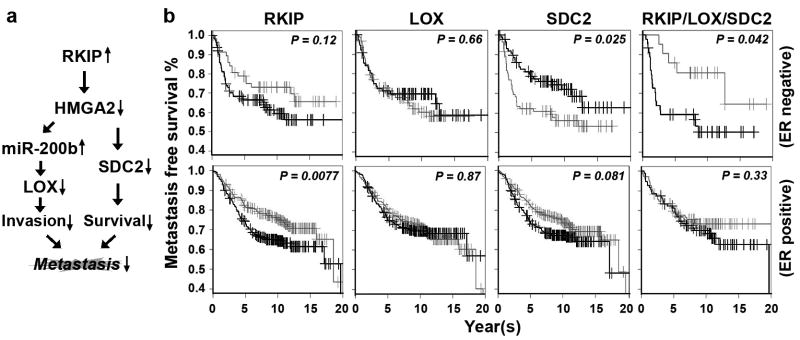
The RKIP/LOX/SDC2 signaling pathways regulate breast tumorigenesis and stratify ER-negative subtype breast cancer patients for metastasis-free survival. (a) Scheme illustrating RKIP/HMGA2/miR-200b/LOX and RKIP/HMGA2/SDC2 signaling pathways in breast tumorigenesis. (b) Kaplan-Meier analysis of gene expression data from Br731 including 175 ER-negative and 556 ER-positive samples. Patients were stratified for metastasis-free survival in ER-negative or ER-positive patients using gene expression for RKIP, LOX, or SDC2 individually or the combined pathways as indicated. Right panel (RKIP/LOX/SDC2): Light gray line, high RKIP and low LOX/SDC2; Dark gray line, low RKIP and high LOX/SDC2. Top right panel: light gray line, n=22; dark gray line, n=46. P, chi-square p value.
To evaluate the significance of the RKIP/LOX/SDC2 pathway in human breast cancer survival, we performed Kaplan-Meier analyses using the cohort of breast cancer patients Br731. When these genes are considered individually, neither expression of RKIP, LOX nor SDC2 can effectively stratify patients for metastasis-free survival in either ER-negative or ER-positive subtypes (Figure 6b, left panel). However, using the complete RKIP/LOX/SDC2 pathway (i.e. RKIP high, LOX/SDC2 low versus RKIP low, LOX/SDC2 high) we are able to stratify patients for metastasis-free survival in the ER-negative but not ER-positive cohort (Figure 6b, top-right panel). These results are consistent with the correlation analyses we conducted for RKIP, LOX and SDC2 gene expression using the same patient samples (Figures 2c, 2f and 4e).
Discussion
In the current study we used the metastasis suppressor RKIP as a tool to identify microRNAs and proteins that regulate tumor survival, invasion and metastasis in breast cancer. We showed that miR-200 can be induced by RKIP via down-regulation of HMGA2, and elevated expression of miR-200 directly inhibits LOX expression, leading to inhibition of breast tumor invasion. HMGA2 also induces SDC2 through a miR-200-independent mechanism, promoting breast tumor growth, survival and metastasis in mouse xenografts. Although both genes are regulated by HMGA2, the specific functions of LOX and SDC2 are likely to be different as each of them appears to play a key role in breast cancer progression. Moreover, the integrated RKIP/LOX/SDC2 gene signature can stratify patients and is prognostic for metastasis-free survival of ER-negative breast cancer patients.
The HMGA2/miR-200b/LOX axis plays an important role in the initial stages of breast tumor cell invasion and metastasis. The miR-200 family has five members and is organized into two clusters: mir-200a, miR-200b and miR-429 on chromosome 1, and miR-200c and miR-141 on chromosome 12. MiR-200c has been reported to inhibit the epithelial-to-mesenchymal tansition (EMT)14, 15, 29, an early stage of metastasis, by targeting E-cadherin transcriptional repressors ZEB1 and ZEB2. LOX is an extracellular copper enzyme that catalyzes formation of aldehydes from lysine residues in collagen and elastin precursors and initiates cross-linking of collagen and elastin27. LOX has been well studied in breast tumor progression16, 17, and its expression is often elevated in hypoxic breast tumors. Over-expression of LOX promotes breast tumor migration and invasion, while inhibition of LOX by shRNA abolishes metastasis but does not significantly affect cell growth17. LOX has also been reported to promote pre-metastatic niche formation and is associated with reduced overall survival16, 17. Interestingly, expression of LOX positively correlates with ZEB1 and ZEB2 expression in both ER-negative and -positive breast cancer patient data, consistent with the fact that both are direct targets of miR-200 (Supplementary Table S3). However, understanding the detailed relationship between ZEB1/2 and LOX would require further investigation. Together, miR-200 and LOX facilitate invasion through remodeling of the extracellular matrix.
A member of the syndecan family, SDC2 is a transmembrane (type I) heparan sulfate proteoglycan that functions as an integral membrane protein and receptor for extracellular matrix proteins 33. The syndecan family has four members: SDC1, 2, 3, and 4. SDC1 is the most studied member of the family and has been reported to play an important role in many types of cancer including breast; however there are a number of contradictory reports regarding the prognostic significance of epithelial SDC1 expression in breast carcinomas34, 35, which might be due to the different functional forms36. By contrast, SDC2 seems to be limited to very few cancer types such as pancreas37, colon38 and prostate39, and no functional studies related to breast cancer has been reported. As we observed for ER-negative breast cancer, SDC2 over-expression in prostate cancer strongly correlates with poor prognosis39. However, a recent report showed that SDC2 also functions as a suppressor of osteosarcomas through a mechanism involving endothelin-1/NFkB signaling and calpain-6 inhibition40. Therefore, the role of SDC2 in cancer can vary dependent upon the tissue or cell type. Our results show not only that SDC2 promotes breast tumor survival and metastasis but also identify two upstream regulators of SDC2, RKIP and HMGA2.
The bioinformatics approach reported here enabled us to find novel signaling cascades and important mediators that regulate breast tumor growth and metastasis. We utilized a systematic approach to select microRNAs relevant both to our experimental system and to human breast cancer. Recently, we used a similar strategy based upon GSA to identify BACH1 a novel target of let-78, 41. However, there were several differences between the two approaches including (1) the use of bioinformatics to select miR-200b as an RKIP target; and (2) incorporation of tumor data relating to 'progression to metastasis' to choose important downstream genes (e.g. LOX and SDC2) in regulatory pathways and identify novel mediators of metastasis (e.g. SDC2).
The genes that we identified are regulated by RKIP and HMGA2. However, this GSA-based approach also identified genes that are co-regulated with downstream targets of RKIP. For instance, SDC2 is predicted to be and, in some cell types, may be a direct target of miR-200b. In our cells, SDC2 is coordinately regulated with miR-200b but not directly regulated by miR-200b. By contrast, LOX is a direct target of miR-200b in a number of breast cancer cell lines and in a genetically engineered mouse model. We did not observe effects of RKIP depletion on LOX or SDC2 levels in MCF10A cells (data not shown), suggesting that the RKIP pathway may have more impact on cells that have lost other regulatory controls. Furthermore, the decrease in LOX and SDC2 protein levels upon RKIP expression or HMGA2 depletion in breast cancer cells was in many cases greater than that observed for the gene transcripts (e.g. Figure 2e). This discrepency may be due to factors such as posttranscriptonal modification as well as differential rates of protein synthesis or degradation. Together, these results highlight the fact that signal transduction relationships are cell and tissue-dependent, requiring experimental validation in order to be applied to a specific tumor setting.
The present study generated a novel gene signature for high risk ER-negative patients. Our results demonstrate that building a gene signature upon a mechanistic network of coordinately regulated genes can be more effective for predicting metastasis-free survival than basing the signatures upon individual genes. Because this signature reflects key signaling pathways required for tumor survival and invasion, this cascade has the potential to identify patient subpopulations that would be responsive to treatments that target these gene networks.
Materials and Methods
Affymetrix gene arrays and Exiqon microRNA arrays
A total of 6 samples including 3 biological replicates of each (1833 cells expressing vector control or S153E-RKIP) were analyzed by Affymetrix GeneChip hgu133plus2.0 Array for gene expression and by Exiqon miRCURY™ LNA array v.11.0 for microRNA expression. The gene array analysis was conducted in the Functional Genomics Facility at Vanderbilt University. The quantified signals were normalized using Robust Multi-array Average42. R version 2.1143 and related packages from Bioconductor version 2.444 were used for the analysis of the normalized data. For microRNA array analysis, the samples were labeled using the miRCURY™ Hy3/Hy5 Power labeling kit and hybridized on a hybridization station. Each chip/slide contained two arrays, one for an individual sample labeled with Hy3 and another for an aliquot of the common reference pool (a mixture of all individual samples) labeled with Hy5. Background correction was conducted to remove non-biological contributions45. The quantified signals were normalized using the global lowess regression algorithm46. The expression values were log2(Hy3/Hy5) ratios, which were the average of replicated measurements of the normalized data on the same slide. Median-centering each miR across all arrays was performed for heatmap illustration. The data have been deposited in the Gene Expression Ominibus (GEO) repository with accession number GSE47379.
Generation of cell lines
Cell lines (1833, MDA-MB-231, MDA-MB-436, MDA-MB-435) stably expressing S153E-RKIP or depleting RKIP were generated and maintained as described in Ref.10. Stable knockdown of HMGA2 or SDC2 in cell lines were achieved by transducing the cells with HMGA2 or SDC2 shRNA or scrambled control in a pLKO.1 lentiviral vector (Open Biosystems). After transduction, cells were selected in 0.5 μg/ml puromycin.
RNA isolation and qRT-PCR analysis
Total RNA was isolated from cells using miRNeasy Mini Kit (Qiagen) following the manufacturer's instructions. QRT-PCR was performed as described10. The qPCR primers for human or murine RKIP, HMGA2, LOX, SDC2, GAPDH, primary miR-200b transcript, miR-200b and U6 snRNA, were Taqman and were purchased from Applied Biosystems.
Transient transfection with miR precursor or miR inhibitor
Cell lines were transiently transfected using HiPerFect transfection reagent (Qiagen) following the manufacturer's protocols. The transfection efficiency was approximately 70-80% (see Supplementary Figures S5H and I). MiR-200b and let-7g precursor were from Ambion. MiR-200b inhibitor (anti-miR-200b) was from Exiqon.
Immunoblotting
Cells were lysed and proteins were measured, separated and probed as described10. Antibodies specific for each protein are HMGA2 (Active Motif), LOX, SDC2 (Sigma), GAPDH (Abcam), and Tubulin (Santa Cruz Biotechnology). Anti-RKIP antisera used was generated by immunizing rabbits with purified GST-RKIP (a-RKIP).
Cloning of luciferase reporter plasmid, pMSCVPIG-miR-200b plasmid and luciferase reporter assay
Wildtype (LOX-wt-UTR) and mutated (LOX-mut-UTR) 3′UTR fragment (Figure 3c) from the human LOX gene were designed using TargetScanS program21 and synthesized by GenScript. Two fragments were fused into a luciferase reporter plasmid pMIR-REPORTER (Ambion) at SpeI/HindIII sites, respectively. The miR-200b precursor was synthesized by GenScript, and was then ligated into the pMSCVPIG vector at XhoI/EcoRI sites. All constructs were confirmed by sequencing. Luciferase reporter assays were conducted as described47. Briefly, MCF-7 cells were plated in triplicate; for each well, pMIR-REPORTER bearing LOX-wt-UTR or LOX-mut-UTR and pMSCVPIG-miR-200b or control were co-transfected together with pMIR-REPORT™ Beta-galactosidase Reporter Control Vector (Ambion) into cells using Effectene transfection reagent (Qiagen). Beta-galactosidase Reporter served as the internal control for normalization of transfection efficiency across the wells. Cells were lysed, and firefly luciferase and Beta-galactosidase activities were measured after 48 h transfection using Dual-Light® Combined Reporter Gene Assay System (Applied Biosystems).
In vitro cell proliferation, apoptosis and invasion assays
Cell proliferation assays were performed using CellTiter-Blues Assay Kit (Promega) as described10. Cell apoptosis was assessed using ApoLive-Glo Multiplex Assay Kit (Promega) following the manufacturer's manual as described48. Invasion assays were performed as described49.
Animal studies
Treatment of mice was done in accordance with a protocol approved by The University of Chicago Animal Care and Use Committee. Xenograft breast tumor growth and bone metastasis assay have been described in Ref.10. Briefly, 106 cells were orthotopically injected into the mammary fat pad of nude mice for tumor growth assay; or 105 cells were injected into the left ventricle of nude mice for bone metastasis assay. Mice were imaged for luciferase activity after 3 weeks. After 6 weeks, tumor tissues were dissected, fixed and embedded.
MMTV-Wnt1 Hmga2 knockout mice
Wnt1 transgenic mice in the Hmga2 wildtype (Hmga2+/+) or null (Hmga2-/-) genetic backgrounds have been described31. Wnt1 and Hmga2 loci have been confirmed by PCR-based genotyping.
Immunostaining
Immunostaining for paraffin-embedded tumor samples was performed by Human Tissue Resource Centre Core Facility at The University of Chicago. Sample sections were stained with haematoxylin and eosin (H&E, Abcam), anti-Lox (Abcam), anti-Sdc2 (R&D Systems), or anti-Cleaved Caspase-3 (Cell Signaling) antibody.
Statistical analysis of experimental results
Samples were analyzed using the two-sample Student t test assuming equal variances (two-tailed). P values were calculated for samples from three independent experiments unless otherwise indicated. Gene ontology or pathway enrichment analysis was performed using the DAVID software19.
Target gene analysis
Five microRNA-target prediction programs/databases including PicTar20, TargetScanS (http://www.targetscan.org)21, miRanda (http://www.microrna.org)22, miRBase Targets (http://microrna.sanger.ac.uk)23, and PITA TOP (http://genie.weizmann.ac.il/pubs/mir07/)24 were used for identification of potential targets of microRNAs.
Gene set analysis
To test whether predicted targets (gene sets) of microRNAs were enriched in response to RKIP expression in human breast cancer cell lines or breast cancer patient samples, we used Gene Set Analysis software as implemented in the “GSA” R package version 1.025. The “maxmean” test statistic was used to test enrichment with p values and false discovery rates based on 1000 permutations. For re-standardization, we used a method that combines gene randomization and sample permutation to correct permutation values of the test statistic and to take into account the overall distribution of individual test statistics; the entire data set was used rather than only the genes in the gene sets tested. For testing gene set enrichment in cell lines, “two class unpaired” type was used for the response variable (control versus RKIP-expressing cells). For testing gene set enrichment in breast cancer patient samples, “Quantitative” type was set for the response variable RKIP. Values for RKIP expression were generated by averaging all probe expression corresponding to the gene RKIP in the microarrays. The GSA score for each individual gene relative to the response variable (e.g. RKIP) is a t-statistic calculated based on the entire expression dataset.
Patient data and Kaplan-Meier analysis
Expression data50-55 and clinical information for breast cancer patient samples were downloaded either from the relative publication websites or from http://www.ncbi.nlm.nih.gov/geo. The data were organized into 3 sets based on the platform that the arrays were performed: Br86 includes 86 gene and microRNA arrays for the same patient samples, Br426 includes 426 microarrays, and Br731 includes 731 microarrays. Correlation and Kaplan-Meier analysis were performed using stat and survival package in R43 respectively.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by NIH grant GM087630 to MRR and DOD Predoctoral Traineeship Award W81XWH-10-1-0396 to MS. We also thank Eva Eves for technical support and helpful discussion.
Footnotes
Author contributions: M.S. and M.R. designed research; M.S., S.G., P.C., C.F., and D.S. performed research; C.C. and K.C. contributed new reagents or analytic tools; M.S. analyzed data; M.S. and M.R. wrote the paper, and all the authors helped to revise the manuscript.
Conflict Of Interest: The authors declare no conflict of interest.
References
- 1.Massague J. Sorting out breast-cancer gene signatures. N Engl J Med. 2007;356:294–297. doi: 10.1056/NEJMe068292. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 3.Smith SC, Theodorescu D. Learning therapeutic lessons from metastasis suppressor proteins. Nat Rev Cancer. 2009;9:253–264. doi: 10.1038/nrc2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, et al. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95:878–889. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 5.Fu Z, Kitagawa Y, Shen R, Shah R, Mehra R, Rhodes D, et al. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2006;66:248–256. doi: 10.1002/pros.20319. [DOI] [PubMed] [Google Scholar]
- 6.Al-Mulla F, Hagan S, Behbehani AI, Bitar MS, George SS, Going JJ, et al. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J Clin Oncol. 2006;24:5672–5679. doi: 10.1200/JCO.2006.07.5499. [DOI] [PubMed] [Google Scholar]
- 7.Minoo P, Zlobec I, Baker K, Tornillo L, Terracciano L, Jass JR, et al. Loss of raf-1 kinase inhibitor protein expression is associated with tumor progression and metastasis in colorectal cancer. Am J Clin Pathol. 2007;127:820–827. doi: 10.1309/5D7MM22DAVGDT1R8. [DOI] [PubMed] [Google Scholar]
- 8.Yun J, Frankenberger CA, Kuo WL, Boelens MC, Eves EM, Cheng N, et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J. 2011 doi: 10.1038/emboj.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagan S, Al-Mulla F, Mallon E, Oien K, Ferrier R, Gusterson B, et al. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin Cancer Res. 2005;11:7392–7397. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 10.Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM, et al. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. The EMBO journal. 2009;28:347–358. doi: 10.1038/emboj.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu YF, Yi Y, Qiu SJ, Gao Q, Li YW, Dai CX, et al. PEBP1 downregulation is associated to poor prognosis in HCC related to hepatitis B infection. J Hepatol. 2010;53:872–879. doi: 10.1016/j.jhep.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 12.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 13.Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–13068. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- 14.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 15.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 17.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 19.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 20.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nature genetics. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 21.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 22.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nature genetics. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 25.Efron B, Tibshirani R. On testing the significance of sets of genes. Ann Appl Stat. 2007;1:107–129. [Google Scholar]
- 26.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 27.Csiszar K. Lysyl oxidases: a novel multifunctional amine oxidase family. Prog Nucleic Acid Res Mol Biol. 2001;70:1–32. doi: 10.1016/s0079-6603(01)70012-8. [DOI] [PubMed] [Google Scholar]
- 28.Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, et al. Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer research. 2006;66:5278–5286. doi: 10.1158/0008-5472.CAN-05-4610. [DOI] [PubMed] [Google Scholar]
- 29.Peter ME. Let-7 and miR-200 microRNAs: Guardians against pluripotency and cancer progression. Cell Cycle. 2009;8 doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, VandenBoom TG, 2nd, Kong D, Wang Z, Ali S, Philip PA, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer research. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morishita A, Zaidi M, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, et al. HMGA2 is an in vivo driver of tumor metastasis. Cancer research. 2013 doi: 10.1158/0008-5472.CAN-12-3848. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 33.Lambaerts K, Wilcox-Adelman SA, Zimmermann P. The signaling mechanisms of syndecan heparan sulfate proteoglycans. Curr Opin Cell Biol. 2009;21:662–669. doi: 10.1016/j.ceb.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbareschi M, Maisonneuve P, Aldovini D, Cangi MG, Pecciarini L, Angelo Mauri F, et al. High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer. 2003;98:474–483. doi: 10.1002/cncr.11515. [DOI] [PubMed] [Google Scholar]
- 35.Loussouarn D, Campion L, Sagan C, Frenel JS, Dravet F, Classe JM, et al. Prognostic impact of syndecan-1 expression in invasive ductal breast carcinomas. British journal of cancer. 2008;98:1993–1998. doi: 10.1038/sj.bjc.6604400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolova V, Koo CY, Ibrahim SA, Wang Z, Spillmann D, Dreier R, et al. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis. 2009;30:397–407. doi: 10.1093/carcin/bgp001. [DOI] [PubMed] [Google Scholar]
- 37.De Oliveira T, Abiatari I, Raulefs S, Sauliunaite D, Erkan M, Kong B, et al. Syndecan-2 promotes perineural invasion and cooperates with K-ras to induce an invasive pancreatic cancer cell phenotype. Mol Cancer. 2012;11:19. doi: 10.1186/1476-4598-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park H, Kim Y, Lim Y, Han I, Oh ES. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J Biol Chem. 2002;277:29730–29736. doi: 10.1074/jbc.M202435200. [DOI] [PubMed] [Google Scholar]
- 39.Popovic A, Demirovic A, Spajic B, Stimac G, Kruslin B, Tomas D. Expression and prognostic role of syndecan-2 in prostate cancer. Prostate Cancer Prostatic Dis. 2010;13:78–82. doi: 10.1038/pcan.2009.43. [DOI] [PubMed] [Google Scholar]
- 40.Marion A, Dieudonne FX, Patino-Garcia A, Lecanda F, Marie PJ, Modrowski D. Calpain-6 is an endothelin-1 signaling dependent protective factor in chemoresistant osteosarcoma. Int J Cancer. 2012;130:2514–2525. doi: 10.1002/ijc.26246. [DOI] [PubMed] [Google Scholar]
- 41.Minn AJ, Bevilacqua E, Yun J, Rosner MR. Identification of novel metastasis suppressor signaling pathways for breast cancer. Cell Cycle. 2012;11:2452–2457. doi: 10.4161/cc.20624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Team RDC. R: A language and environment for statistical computing 2009 [Google Scholar]
- 44.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ritchie ME, Silver J, Oshlack A, Holmes M, Diyagama D, Holloway A, et al. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 46.Berger JA, Hautaniemi S, Jarvinen AK, Edgren H, Mitra SK, Astola J. Optimized LOWESS normalization parameter selection for DNA microarray data. BMC Bioinformatics. 2004;5:194. doi: 10.1186/1471-2105-5-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci U S A. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang X, Huang H, Li Z, Li Y, Wang X, Gurbuxani S, et al. Blockade of miR-150 Maturation by MLL-Fusion/MYC/LIN-28 Is Required for MLL-Associated Leukemia. Cancer Cell. 2012;22:524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1305172110. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Minn AJ, Gupta GP, Padua D, Bos P, Nguyen DX, Nuyten D, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A. 2007;104:6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enerly E, Steinfeld I, Kleivi K, Leivonen SK, Aure MR, Russnes HG, et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One. 2011;6:e16915. doi: 10.1371/journal.pone.0016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13:3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.