

Abstract
Partitioning of membrane proteins into various types of microdomains is crucial for many cellular functions. Tetraspanin-enriched microdomains (TEMs) are a unique type of protein-based microdomain, clearly distinct from membrane rafts, and important for several cellular processes such as fusion, migration and signaling. Paradoxically, HIV-1 assembly/egress occurs at TEMs, yet the viral particles also incorporate raft lipids.
Using different quantitative microscopy approaches, we investigated the dynamic relationship between TEMs, membrane rafts and HIV-1 exit sites, focusing mainly on the tetraspanin CD9. Our results show that clustering of CD9 correlates with multimerization of the major viral structural component, Gag, at the plasma membrane. CD9 exhibited confined behavior and reduced lateral mobility at viral assembly sites, suggesting that Gag locally traps tetraspanins. In contrast, the raft lipid GM1 and the raft-associated protein CD55, while also recruited to assembly/budding sites, were only transiently trapped in these membrane areas. CD9 recruitment and confinement were found to be partially dependent on cholesterol, while those of CD55 were completely dependent on cholesterol. Importantly, our findings support the emerging concept that cellular and viral components, instead of clustering at preexisting microdomain platforms, direct the formation of distinct domains for the execution of specific functions.
Keywords: assembly, cholesterol, Gag, HIV-1, microdomain, raft, tetraspanin, virus
Compartmentalization of cellular membranes is important for the coordination of specialized processes such as signaling, adhesion, migration and fusion. Membrane rafts (formerly called lipid rafts), i.e. membrane protein-lipid clusters rich in sphingolipids and cholesterol, have been suggested to achieve such compartmentalization by recruiting specific proteins into plasma membrane (PM) microdomains. Rafts were originally thought to be relatively large [average diameter 100–200 nm, (1)] and stable entities. However, though some current definitions of rafts still give an upper limit of 200 nm (2), recent studies utilizing newly developed methods, such as super-resolution microscopy, have documented that basic raft entities are highly dynamic, transient and typically less than 50 nm in diameter (3,4). Further, it was proposed that aggregation of such small protein-lipid nanodomains leads to the formation of larger platforms for the organization of various cellular processes (5-7). Also, such lateral segregation is now thought to be driven in large part by protein–protein interactions (8,9).
One group of cellular proteins involved in the formation and organization of membrane molecule microclusters are the tetraspanins. By virtue of their multiple interactions with partner proteins and because of their strong propensity to multimerize, they are thought to create scaffolds in membranes, recruiting or excluding specific proteins needed for a particular cellular process (10-12). Studies from our laboratories and others provided a preliminary analysis of TEMs in fixed cells, and showed detectable clusters of tetraspanins present at the PM (13-15). Such platforms have been termed tetraspanin-enriched microdomains/areas (TEMs, TErMs or TEAs) (16-18). More recently, using single-molecule fluorescence microcopy and other analytical microscopy techniques in live cells, we and others confirmed the existence of small tetraspanin-based microdomains/platforms (18,19). It was shown that the tetraspanin CD9 could be confined in these platforms but also escaped and freely diffused in the PM. TEMs can therefore be viewed as stable platforms in position and shape but in permanent exchange with the rest of the membrane.
Tetraspanins are coregulators of many cellular functions including antigen presentation, cell–cell fusion, cell adhesion, cell migration and signaling (reviewed in 10,20-22). They also are co-opted by pathogenic microorganisms such as plasmodium (23), listeria (24) and various viruses. For example, both human T-lymphotrophic virus-1 (HTLV-1) Gag and HIV-1 Gag accumulate at, and bud through, PM TEMs containing CD9, CD63, CD81 and CD82 (14,25-29). Notably, recent work from our laboratory and others has documented functional roles of tetraspanins in regulating HIV-1-induced virus-cell and cell-to-cell fusion, budding and cell-to-cell transmission (30-33), although their putative role in viral release is still under debate (31,33-35). Importantly, how tetraspanin dynamics and their organization relate to specific membrane-based processes, such as viral assembly, remains largely unexplored.
Here, using multiple fluorescence microscopy approaches, we provide the first analysis, in live cells, of the distribution and dynamics of tetraspanins relative to HIV-1 assembly sites. Previous studies hypothesized that Gag assembly may be targeted to preexisting assembly platforms, either membrane rafts or TEMs (14,27,36-38). In contrast, in this study, we show that HIV-1 assembly leads to formation of new platforms and immobilization of CD9 at viral exit sites, while the mobility of the raft lipid GM1 and raft-associated protein CD55, which are also recruited to Gag assembly sites, is not locally restricted. Overall, these data are compatible with the concept that protein–protein interactions (here: multimerization of HIV-1 Gag) drive the formation of specific membrane microdomains.
Results
Previous studies clearly established that TEMs serve as exit sites for HIV-1. However, it remained unclear whether these were preexisting platforms to which Gag assembly was targeted or whether Gag assembly induced the formation of these platforms. To answer this question, we analyzed the native distribution and the dynamics of endogenous tetraspanins in live cells and using different imaging approaches.
Colocalization of CD9 and Gag in live cells
Multiple reports have shown that HIV-1 Gag and Env colocalize with tetraspanins at the PM. However, because all of these studies utilized bivalent antibodies to detect tetraspanins in fixed cells, we set out to determine whether colocalization of Gag and tetraspanins could still be detected in the absence of potential cross-linking or fixation artifacts. If no Gag was expressed, live cells labeled with monovalent Fab fragments exhibited a more diffuse distribution of CD9 compared to cells labeled with bivalent antibodies (Figure 1A,B), even if the bivalent labeling was carried out after fixation (data not shown). A distribution similar to monovalent labeling was obtained with CD9 fused to green fluorescent protein (GFP) (data not shown). To visualize Gag, HeLa cells were transfected with HIV-1 provirus (NL4-3) harboring matrix protein (MA) fused to GFP or monomeric red fluorescent protein (mRFP) (39), and live cells were labeled with anti-CD9 Fab and secondary Fabs. While much of the CD9 signal remained relatively diffuse in Gag-expressing cells, enrichment of CD9 at Gag assembly sites was readily observed (Figure 1C). Similar results were obtained in infected primary CD4+ T cells (Figure 1D). Overall, these results show that endogenous CD9 clusters at Gag assembly sites. They also document that bivalent labeling still represents a valid approach to study Gag–tetraspanin association, and this method, because of its convenience, was thus still used in some of the colocalization analyses in this study.
Figure 1. Clustering of PM CD9 by Gag in live cells.
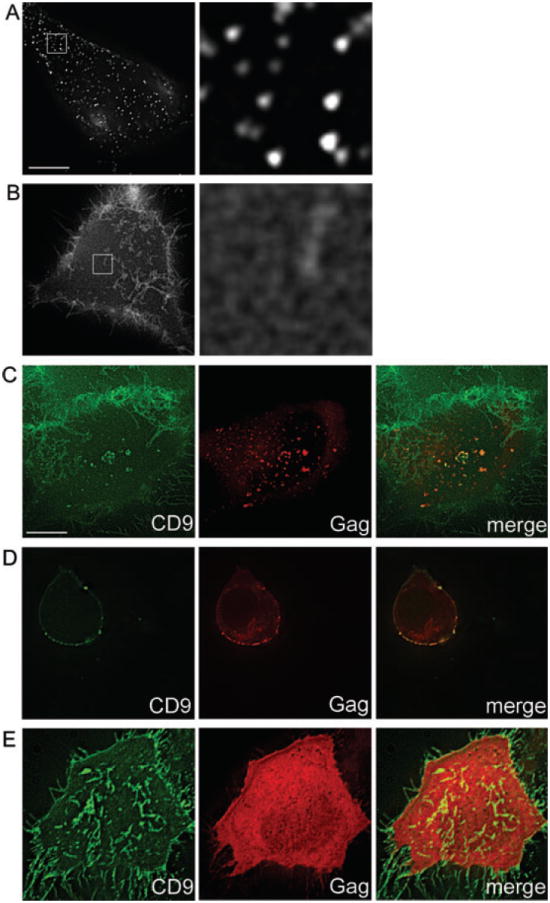
Live HeLa cells were labeled on ice using full-length anti-CD9 antibody and full-length secondary (A) or Fab anti-CD9 and Fab secondary (B) then imaged at 37°C. C) HeLa cells expressing NL4-3(MA-mRFP) were labeled with Fab anti-CD9 and Fab secondary on ice, then imaged at 37°C. D) Human CD4+ T cells infected with NL4-3(MA-GFP) were labeled as in (C). E) HeLa cells expressing Venus-fused monomeric Gag were labeled and imaged as in (C). Bottom optical Z-sections (i.e. cell–coverslip contact) are shown, except in (D), which shows middle sections. Images in (A and B) second column represent a 10× magnification of the boxed region. Scale bar represents 10 μm.
Because Gag assembly appeared to be the driving factor behind CD9 redistribution and clustering, we tested whether multimerization of Gag was required for these phenomena. Cells expressing a multimerization-deficient Gag that can still be targeted to the PM (40) were labeled for CD9. In the absence of Gag multimerization, no clustering of CD9 by Gag was observed and the overall distribution of CD9 resembled that in untransfected cells (Figure 1E).
Kinetics of Gag colocalization with tetraspanins
To investigate the kinetics of Gag–tetraspanin association, HeLa cells expressing HIV-1 GagGFP [which colocalizes with tetraspanins in the absence of other viral components (14)] were fixed at various time-points post-transfection, stained with antibodies against CD9 and assessed for colocalization between these two antigens at the PM. Only cells displaying detectable PM Gag puncta [which are known to represent assembling and budding structures (41-47)] were included in the analysis. Colocalization of Gag puncta with CD9 increased over time (Figure 2A). However, because the level of GagGFP expression varied greatly from cell to cell, even at the same time-points, we refined our analysis by pooling data from different time-points and sorting cells by their total GagGFP intensity (Figure 2B), which, on a single cell level, is proportional to expression time (Figure 2C). We found that cells displaying more intense GagGFP signal (and thus those cells that have been expressing Gag for a longer time and/or at a faster rate) exhibited higher colocalization with CD9 (Figure 2B). Similar results were obtained for surface CD63 (data not shown). Representative images of cells displaying low and high colocalization of Gag with CD9 are shown (Figure 2D,E). Because colocalization was expressed as percentage of Gag+ pixels colocalizing with CD9 and not vice versa, it was not biased by the overall amount of Gag signal. Overall, these data strongly suggested that tetraspanin–Gag colocalization increases over time, i.e. as virus assembly proceeds, and that Gag assembly is not targeted to preexisting TEMs.
Figure 2. Gag-CD9 colocalization is increased at later time-points and at higher Gag expression levels.
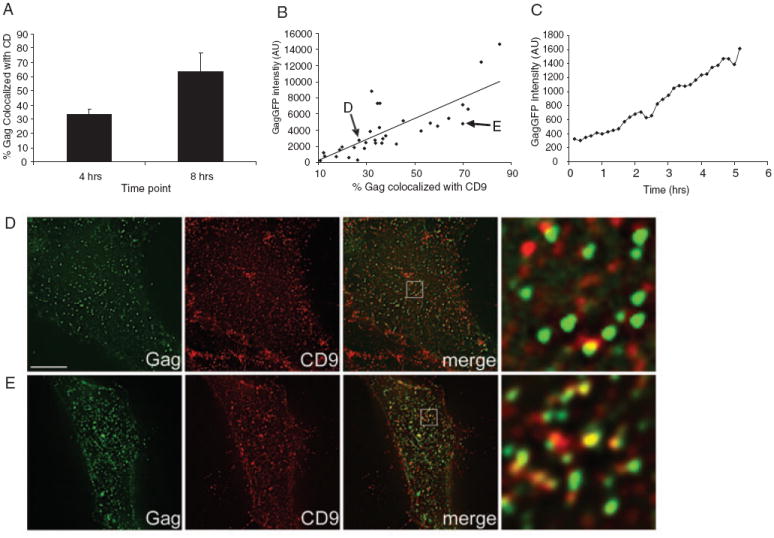
HeLa cells transfected with GagGFP were fixed at 4 or 8 h posttransfection, then surface labeled for CD9 and processed for colocalization analysis. A) Average colocalization of Gag and CD9 at 4 versus 8 h posttransfection (data represent the mean of 10 or more cells, error bars represent SEM) (see Materials and Methods). B) Colocalization data for individual cells from 4-, 8- and 16-h time-points were pooled and plotted as a function of mean Gag intensity. Each point represents a single cell – the cells displayed in (D) and (E) are indicated with arrows. A best fit line obtained using linear regression is displayed. C) HeLa cells expressing GagGFP were imaged continuously for 5 h at 37°C, one frame/10 min. The mean GFP intensity of a representative cell is displayed. Representative bottom sections of cells included for the analysis in (A), exhibiting low (26%, GFP intensity = 2755) and high (70%, GFP intensity = 4823) colocalization, are shown in (D) and (E), respectively. Gag intensity was scaled differently between (D) and (E), in order to enhance visualization of the weaker signals in (D). Images in right hand column represent 9.5× magnifications of the boxed area. Scale bar represents 10 μm. AU, arbitrary units.
HIV-1 Gag influences mobility of CD9 but not of the raft marker GM1
The observation that HIV-1 Gag clusters CD9 over time suggested that Gag induces CD9 trapping at assembly/budding sites. To test this hypothesis, we measured the mobility of CD9 at the population level by fluorescence recovery after photobleaching (FRAP) analysis. FRAP is commonly utilized to study the mobility of membrane proteins and lipids, and can yield important information about membrane compartmentalization (reviewed in 48). Here, FRAP measurements were used to calculate two parameters, the diffusion coefficient, D, and the mobile fraction, Mf, indicative of the fraction of molecules available for diffusion, for both CD9 and GM1. Vero cells were used in the FRAP experiments, because they express relatively high levels of CD9 (and GM1, see below) allowing for robust fluorescence bleaching and recovery. Further, colocalization between Gag and the former antigens was more apparent in these cells because of the low numbers of microvilli on the ventral side. Live cells were labeled with anti-CD9 Fab fragments on ice to visualize surface CD9, then FRAP experiments were carried out at 37°C. Representative FRAP analyses of single cells and average recovery curves (from multiple cells) for CD9 are shown in Figure 3. The mean Mf and D values from all FRAP experiments are summarized in Table S1.
Figure 3. Gag expression alters the mobility of CD9.
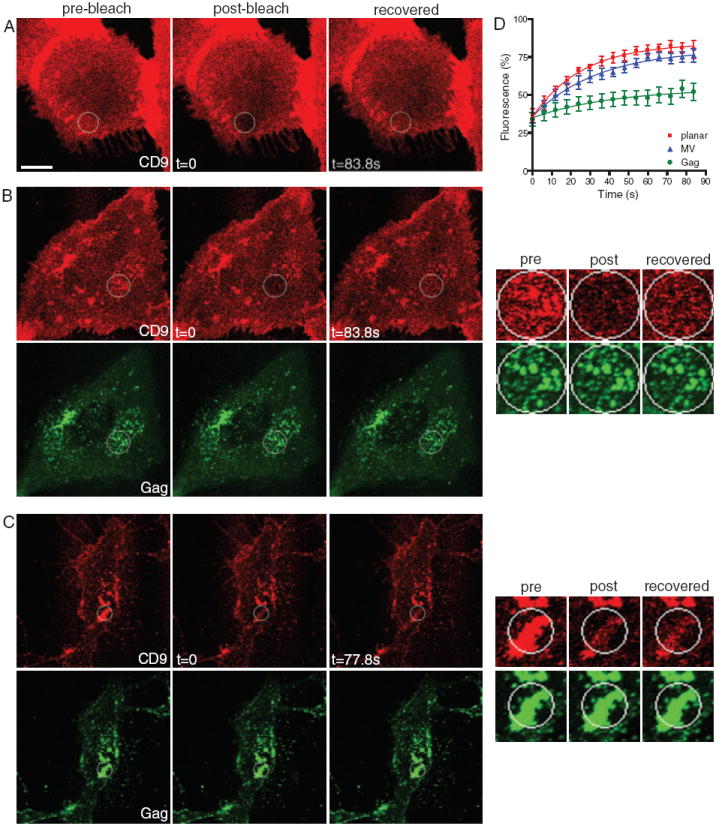
Representative FRAP analyses of untransfected Vero cells (A) or Vero cells expressing GagGFP (B) and (C) and labeled for surface CD9 with Fab primary and secondary. The cell shown in (C) expresses a higher amount of Gag than the cell in (B). The bleach ROIs are indicated with a circle. Images in right column in (B) and (C) represent a 3× magnification of a region surrounding the bleach ROI. Note that the bleach areas in (B) and (C) are different sizes. Scale bar represents 10 μm. D) Average recovery curves for CD9 in the absence of Gag [at planar membranes, red, and at microvilli (MV), blue] or in the presence of Gag (green). Values indicate mean fractional fluorescence ± SEM.
In cells not expressing Gag, bleached regions of interest (ROIs) in planar membranes recovered relatively rapidly, though not completely, with an apparent D for CD9 of 0.213 μm/s2, and Mf of 76%. FRAP of microvilli-like structures enriched in CD9 yielded a lower (though not significantly, p > 0.05) apparent D (0.144 μm/s2) and a similar Mf (77%). However, in Gag-expressing cells, at planar membrane areas containing Gag puncta, CD9 exhibited a considerably lower D (0.137 μm/s2) and a drastically decreased Mf (29%), indicating that CD9 is trapped at Gag assembly sites. Consistent with this idea, CD9-enriched areas containing Gag puncta within the bleach ROI failed to recover to their initial intensity, whereas areas adjacent to Gag puncta within the same ROIs recovered most of their (diffuse) CD9 signal (see insets in Figure 3B,C). The amount of recovery in Gag-containing ROIs decreased with increasing amounts of Gag expression (cf. Figure 3, panels B and C). Typically, the ventral side of the cell was used for FRAP analysis, because it provided a large, flat and uniform membrane area. However, because previous studies have shown that HIV particles released from ventral side of cells can get trapped between the cell and the coverslip (41,43,47), we also performed FRAP experiments on the dorsal side of cells to rule out the possibility that the Gag puncta analyzed in our experiment represent detached, budded particles that cannot exchange proteins with the rest of the membrane. As on the ventral side, recovery of CD9 at Gag-containing structures on the dorsal side was also strongly reduced (data not shown).
Membrane rafts have been proposed to serve as platforms for many membrane-based processes, including budding of HIV-1 and other viruses (recently reviewed in 49). The glycosphingolipid GM1 was used as a marker of raft-like domains, because it is a prototypical component of membrane rafts (50), and has been reported to colocalize with HIV-1 Gag (37,38,51,52). Because Gag appeared to cluster and colocalize with GM1 (as previously reported) comparably to CD9 (also see Figure 4B), we also measured the mobility of this raft marker [labeled with fluorescently labeled cholera toxin subunit B (CTxB)] in the presence or absence of Gag. Overall, GM1 exhibited relatively slow diffusion and low recovery, likely because of the clustering and/or internalization induced by pentavalent CTxB, as previously reported (53,54). Interestingly, neither diffusion nor recovery of GM1 was significantly altered by the presence of Gag assembly sites (Figure 4 and Table S1). Further, GM1, like CD9, showed apparent accumulation at Gag-rich areas, but unlike CD9, this enrichment could be recovered after photobleaching (see insets in Figure 4B). Together, these results imply that, while both GM1 and CD9 are recruited to Gag assembly sites, CD9 becomes trapped but GM1 remains free to diffuse. Importantly, the fact that GM1 is still in exchange with the rest of the PM also shows that most of the Gag puncta analyzed in our experiments represent viral assembly sites that are still connected to the PM, as opposed to being released particles.
Figure 4. Mobility of GM1 is not altered by Gag.
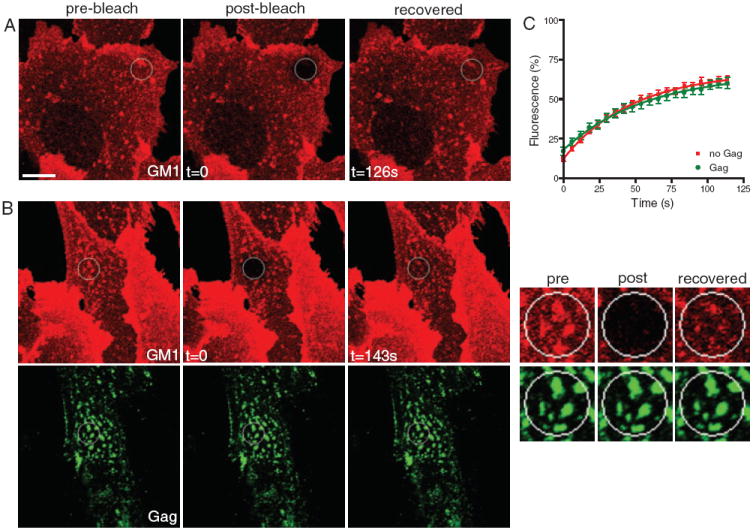
Representative FRAP analyses of untransfected Vero cells (A) or Vero cells expressing GagGFP (B) and labeled for surface GM1 with CTxB-Alexa 647. The bleach ROIs are indicated with a circle. Images in right column in (B) represent a 3× magnification of a region surrounding the bleach ROI. Scale bar represents 10 μm. C) Average recovery curves for GM1 in the absence (red) or in the presence of Gag (green). Values indicate mean fractional fluorescence ± SEM.
HIV-1 Gag differentially influences CD9 and CD55 dynamics and membrane partitioning at the single-molecule level
To corroborate and expand upon the population-level mobility measurements obtained by FRAP, single-molecule fluorescence experiments were performed to analyze the behavior of single CD9 molecules. Single molecules were tracked using Atto647-labeled Fab fragments with a high temporal (100 ms) and spatial (~50 nm) resolution, as in our previous work (18). As observed in PC3 cells, CD9 molecules exhibited three main types of diffusion in control GFP-expressing HeLa cells, namely Brownian (48%), confined (14%) and mixed (38%), a combination of Brownian and confined behavior (Figure 5; Table S2 for the distribution of diffusion coefficient and detailed values). In the absence of Gag expression, the mean value of CD9 apparent diffusion coefficient (ADC) was 0.24 ± 0.03 μm/s2, which is comparable to that previously obtained in PC3 cells and in concurrence with the D obtained in the FRAP measurements in this study. Upon expression of Gag, the percentage of CD9 molecules exhibiting confined motion increased significantly (from 14 to 34% of the total number of trajectories) at the expense of Brownian trajectories (from 48 to 27%) (Figure 5; Table S2). The amplitude of this increase was comparable to that of the mobile fraction measured in our FRAP experiments. Reflecting the increase in CD9 confinement, the mean ADC decreased to 0.16 ± 0.03 μm/s2. Comparable results were obtained with single molecule tracking (SMT) analysis of CD81, another tetraspanin recruited to Gag assembly sites, where expression of Gag induced trapping of CD81 (data not shown). Note that, while the confined fraction observed by single-molecule analysis was not quite as large as that observed by FRAP, the SMT analysis included the entire cell, which contained large areas of membrane not containing Gag, whereas the FRAP analyses focused on an ROI containing large amounts of Gag.
Figure 5. Single-molecule analysis of CD9, CD55 and CD46 behavior in the presence or absence of Gag.
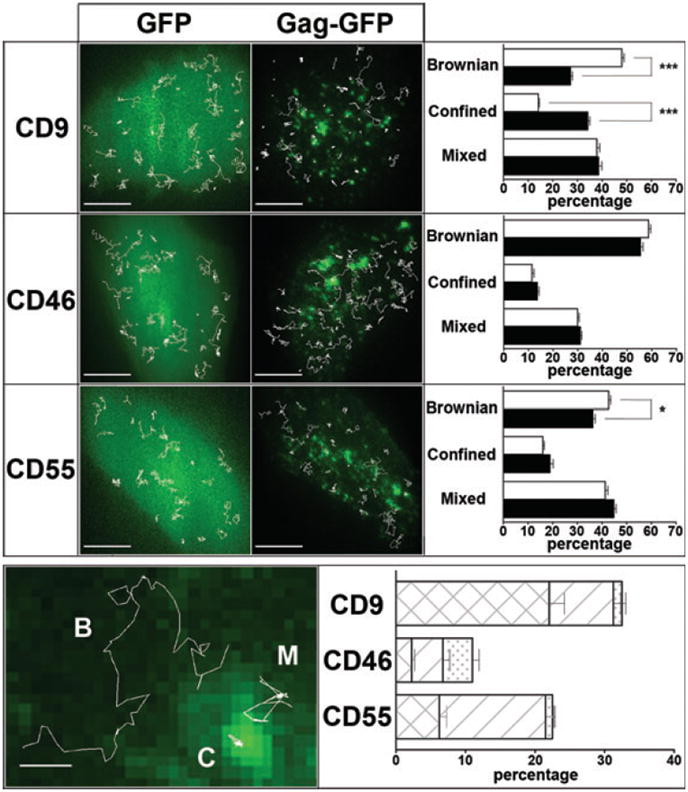
Upper panel: Micrographs on the left show superimposition of GFP and GagGFP fluorescence signal with several representative single molecule trajectories (white lines) obtained after tracking Atto647N-conjugated Fab fragments of anti-CD9, anti-CD46 or anti CD55 antibodies in HeLa cells. Histograms on the right represent the percentage of each diffusion mode (Brownian, mixed and confined) relative to the total number of trajectories for CD9, CD46 or CD55 for cells transfected with GFP (white bars) or with GagGFP (black bars). Error bars correspond to SEM. * and *** respectively indicate p values below 0.01 and 0.0001 for comparison of the diffusion modes percentage in GagGFP-expressing cells versus GFP-expressing control cells for CD9, CD46 and CD55, as determined by the Mann–Whitney U-test. Lower panel: The micrograph on the left shows CD9 representative trajectories with Brownian (B), mixed (M) or confined (C) behavior (white lines) superimposed with the GagGFP fluorescence ensemble labeling. Histograms on the right represent the percentage of trajectories that colocalized with Gag-enriched areas (see Materials and Methods) as well as the proportion of the different diffusion modes displayed at these Gag-enriched areas: confined (crosshatched bars), mixed (hatched bars) and Brownian (dotted bars).
To determine the generality of membrane protein trapping by Gag assembly, we also investigated the membrane behavior of CD46, a non-raft transmembrane protein, which is not confined by TEMs (18). The mean ADC of this protein (0.16 ± 0.02 μm/s2) as well as the percentage of the different diffusion modes were in the same range than those previously observed in PC3 cells (Figure 5; Table S1). However, compared to CD9, CD46 membrane behavior was mostly unaffected by Gag expression, with only a slight but non-significant increase in the number of confined trajectories (from 11 to 13% of the total number of trajectories) as well as slight decrease in the mean ADC value (from 0.16 to 0.15 μm2/s). These results show that trapping of CD9 at Gag assembly sites is a specific process.
Because Gag assembly also appeared to recruit raft-like domains and in order to parallel the FRAP experiments with GM1, we investigated the behavior of CD55, a putative raft-associated protein (18). CD55 ADC was slightly but significantly reduced in the presence of Gag (p < 0.01 using the Mann–Whitney test, Figure 5), as was the proportion of Brownian trajectories (from 43 to 36%, p < 0.01), while the percentage of confined and mixed trajectories was increased (respectively from 16 to 19% and 41 to 45%) but not significantly (Figure 5, upper panel). Taken together, these results show that CD9 and CD55 distribution of ADC were both modified upon Gag assembling but with a large difference in magnitude.
To confirm these findings, we analyzed the subpopulations of CD9, CD46 and CD55 trajectories that coincided with Gag-enriched areas (Figure 5, lower panel). Sixteen percent of the total CD9 trajectories colocalized with Gag (see Materials and Methods for details). As expected from the increase in confinement described above, most of these trajectories displayed a confined behavior (68%) (note that confinement areas not colocalized with Gag were often observed, presumably the same areas that were seen in the cells not expressing Gag). Interestingly, CD55 trajectories were less often associated with Gag (11%) as compared to CD9. More importantly, membrane behavior of CD55 in Gag areas was also different, with most of the trajectories (71%) exhibiting mixed diffusion, which corresponds to a transient confinement. As expected, CD46 trajectories that colocalized with Gag-enriched regions exhibited similar membrane behavior to that obtained with GFP-expressing control cells (upper panel of Figure 5). Altogether, these data show that, while CD9 and CD55 are specifically recruited into assembly sites, tetraspanins interact with Gag in a very different manner compared to raft components.
Differential dependence on cholesterol for enrichment and confinement of CD55 and CD9 at HIV-1 assembly sites
Because raft components and tetraspanins were clustered to HIV-1 Gag assembly sites, we tested whether cholesterol, a key structural component of membrane rafts and TEMs, was required for this clustering. Cells expressing HIV-1 GagGFP were briefly treated with cholesterol oxidase (COase) to decrease membrane cholesterol content, labeled with Fabs to visualize CD9, CD46 or CD55 (ensemble labeling conditions) and processed for colocalization analysis (Figure 6). In agreement with our results on trajectories that overlapped with Gag-enriched areas (Figure 5, lower panel), this colocalization analysis indicated that CD9 is associated with these areas (57%) as was CD55, although to a lesser degree (36%). Interestingly, when cells were treated with COase, the fraction of Gag that colocalized with CD9 partially decreased (from 57 to 38%). However, the overlap of Gag and CD55 was almost completely abolished (from 36 to 9%). In contrast, CD46 showed little colocalization with Gag-enriched areas (6%) and this association was not significantly affected by COase treatment (from 6 to 5%). These results suggest that the recruitment of CD9 to assembly sites only partially depends on cholesterol, while this lipid is fully required for CD55 association with Gag-enriched areas.
Figure 6. Influence of cholesterol on Gag-induced clustering of membrane components.
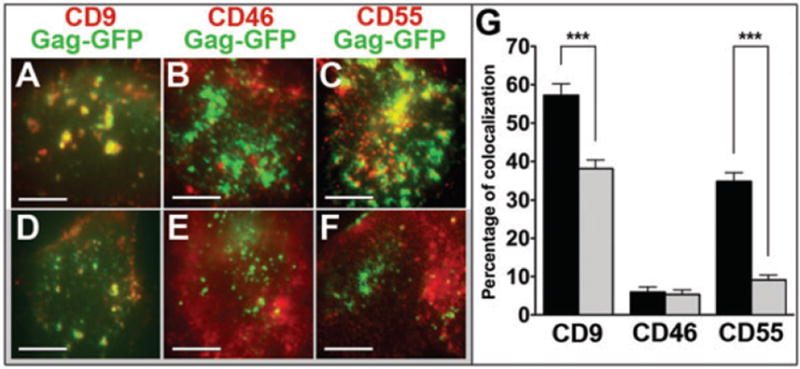
Colocalization fluorescence experiments between CD9, CD46 or CD55 (in red) with Gag-enriched areas (in green) in Gag-expressing HeLa cells treated (D, E, F) or not (A, B, C) with COase. Histograms (G) represent the percentage of colocalization between CD9, CD46 or CD55 with Gag-enriched areas (defined as the number of yellow pixels over the total number of yellow and green pixels) in HeLa cells treated (gray bars) or not (black bars) with COase. *** indicates p values below 0.0001 for comparison of the percentage of colocalization in GagGFP-expressing cells treated with COase versus GagGFP-expressing control cells for CD9, CD46 and CD55, as determined by the Mann–Whitney U-test.
The effect of COase treatment was then investigated at the single-molecule level (Figure 7). When membrane cholesterol content was decreased, the mobility of CD9, CD55 and CD46, as measured by their Brownian diffusion coefficient in the absence of Gag was slightly decreased, as previously observed with methyl-β-cyclodextrin treatment (18). This decrease was attributed to a general effect on membrane dynamics upon cholesterol depletion. While the effect of Gag expression, namely the increase in confined CD9 trajectories at the expense of Brownian trajectories, was reduced by cholesterol depletion, the expression of Gag still clearly increased the percentage of confined trajectories under these conditions (upper right panel of Figure 7 and Table S2). Meanwhile, the increase in CD55 confinement by Gag was almost completely abolished by cholesterol depletion (lower panel of Figure 7). The behavior of CD46 was not affected by cholesterol depletion because no effect of Gag expression was observed in the first place (middle panel of Figure 7). Taken together, these data suggest that CD9 confinement at Gag assembly sites is partially dependent on cholesterol, whereas the transient recruitment of CD55 in these areas is completely dependent on cholesterol.
Figure 7. Influence of cholesterol on Gag-induced trapping of CD9, CD46 and CD55 at the single-molecule level.
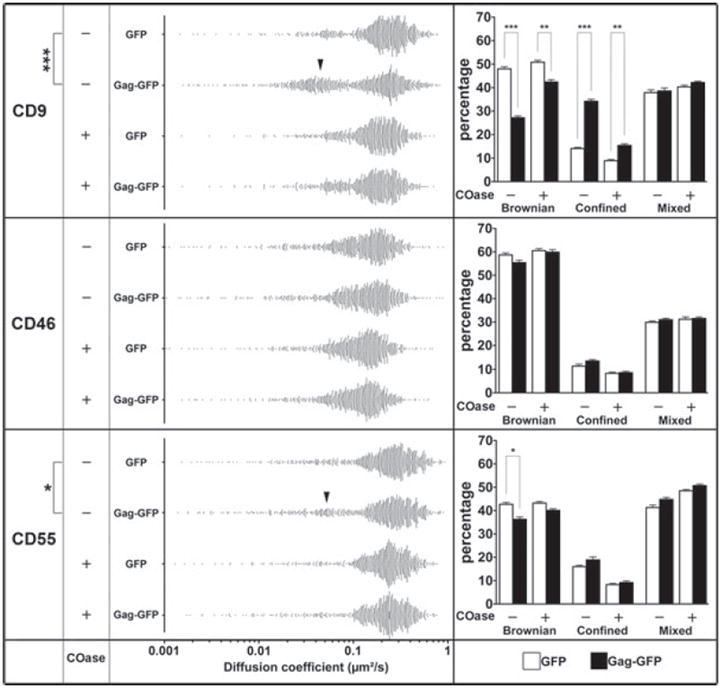
The left panels represent the distribution of all the apparent diffusion coefficients calculated from the MSD-τ plot for CD9, CD46 and CD55 in GFP-or Gag-GFP-expressing cells, treated or not with COase. Each point represents one trajectory. The black arrowheads highlight the increase of the confined trajectories. The histograms on the right represent the percentage of each diffusion mode (Brownian, mixed and confined) relative to the total number of trajectories for CD9, CD46 or CD55 for cells transfected with GFP (white bars) or with Gag-GFP (black bars), treated or not with COase. *, ** and *** respectively indicate a p value below 0.01, 0.001 and 0.0001 for comparison of the apparent diffusion coefficient (left panel) or percentage of diffusion modes (right panel) in GagGFP-expressing cells versus GFP-expressing control cells for CD9, CD46 and CD55, as determined by the Mann–Whitney U-test.
Discussion
While several studies have documented the presence of raft-like domains and TEMs at HIV-1 budding sites, the dynamics and the underlying mechanisms of their relationships have not been explored. In this study, we have employed different microscopy techniques to analyze the interactions between raft lipids and proteins (tetraspanins), and HIV-1 assembly sites. Our results indicate that HIV-1 assembly creates specialized microdomains, recruiting components of both rafts and TEMs. They also support the idea that protein composition (here Gag) can determine the specificity of microdomains (8,55,56).
Using monovalent antibody labeling of CD9, we were able to visualize the native distribution of endogenous CD9 in the PM of live cells. While the distribution of CD9 appeared more diffuse than when detected with bivalent antibodies, some punctate structures were detectable, although their number and size varied considerably depending on the cell type, presence or absence and of extracellular matrix, or imaging conditions [epifluorescence versus total internal reflectance fluorescence (TIRF)]. Importantly though, assembly of HIV-1 Gag at the PM clearly clustered CD9, whether detected by bivalent or monovalent methods, and this clustering depended on multimerization of Gag. Further, colocalization between Gag and tetraspanins gradually increased during Gag expression and assembly, suggesting that Gag assembly is not targeted to large preexisting tetraspanin platforms.
Because CD9 was locally enriched at Gag assembly sites, we hypothesized that Gag restricts the mobility of this protein. FRAP experiments indicated that CD9 is indeed locally trapped at Gag assembly sites, while raft lipid GM1 is still in exchange with the rest of the membrane, despite the fact that it is enriched at sites of assembly. It is likely that Gag assembly creates a local membrane microenvironment that can accommodate a higher concentration of raft lipids and proteins, without trapping them. The FRAP results were corroborated by SMT analyses, which also showed confinement of single CD9 molecules at Gag-rich areas.
Cholesterol depletion by COase treatment only partially reduced Gag-CD9 colocalization, but almost completely abolished colocalization between Gag and raft-associated protein CD55. Interestingly, the mobility of CD55 was also restricted by Gag, though to a lesser degree, and this confinement was completely reversed by cholesterol depletion, while confinement of CD9 was only partially dependent on cholesterol. Mobility of CD46, a non-raft membrane protein, which sometimes can be found associated with CD9 and other tetraspanins (57), was not significantly influenced by the presence of Gag. While CD46, like tetraspanins, has been shown to be incorporated into HIV-1 particles (58,59), the fact that this protein shows little trapping at Gag assembly sites suggests that it is incorporated into retroviral particles non-specifically, comparable to many other PM components (60).
Trapping of CD9 but not of raft components raises questions about the mechanisms that are involved in the lateral segregation of these proteins. Although CD81 and CD82 have been shown to interact (directly or indirectly) with HIV-1 Gag (31) and with HTLV-1 Gag (61), it was also shown that this latter interaction does not take place in the absence of Gag multimerization (25). If this is the case, it is not likely that direct and specific interactions between individual Gag and tetraspanin molecules, followed by multimerization of Gag, drive the clustering of tetraspanins at retroviral assembly sites. In an alternative scenario, the recruitment and coalescence of cholesterol-rich domains by Gag (as proposed in 49,51,62), indirectly recruits tetraspanins because of their interactions with cholesterol (63,64), thus providing the nucleation trigger, which is followed by tetraspanin retention due to tetraspanin homo- and heteromultimerization. This hypothesis can explain why CD9 molecules may be drastically confined, whereas CD55 molecules are only transiently trapped by Gag assemblies, probably reflecting their weaker interaction with these clusters. The coalescence of cholesterol-enriched domains is also in agreement with the observation that cholesterol is partially required for CD9-Gag colocalization and is necessary for CD55-Gag overlap. Note that CD9 has four membrane spanning domains, while CD55 is a glycosyl-phosphatidylinositol (GPI)-anchored protein, and thus CD9 has more potential to directly interact with the assembling Gag lattice at the inner leaflet of the PM, while the interaction with CD55 (and GM1) maybe more indirect, via their association with rafts-like domains.
Altogether, our results suggest that components of both raft-like domains and TEMs are actively enriched or stabilized at sites of Gag assembly. Such regulation is inconsistent with the notion that lipid- or protein-based membrane microdomains constitute stable, preformed platforms to which viral or cellular components are targeted (36,65,66), but instead supports the idea that specific (smaller) rafts and TEMs are clustered during HIV-1 budding, as proposed already by an early HIV-raft study (62), and as discussed earlier (49,51,67). Further, the gradual increase in Gag-TEM association over time (as shown in this report, see Figure 2) also argues against targeting of Gag to preassembled TEMs, a scenario that we previously favored (14). Instead, our current results suggest that large membrane domains enriched in TEM and raft components form only upon Gag expression, probably through the clustering of many smaller preformed nanodomains of different composition and behavior. This is analogous to how attachment of leukocytes to endothelial cells causes the coalescence of preformed tetraspanin-based nanoscale adhesion platforms into larger stable docking structures, where tetraspanins and their partners exhibit reduced mobility, but GPI-anchored GFP, a commonly used raft marker, diffuses freely (19).
The exquisite membrane compartmentalization by Gag described in this report suggests that HIV-1 (and likely many other enveloped viruses) have evolved to regulate both the lipid and protein composition of their assembly sites and subsequently released virions. Proper composition of egress sites probably helps viruses to achieve maximal fitness, e.g. by appropriately targeting viral budding, by maintaining virion stability and by enhancing virion infectivity. Consistent with this idea, our previous results suggest that the optimal levels of tetraspanins (in virus-producing cells or in virions) likely ensure efficient virus spread (32,33). Overall, our study presents the first dynamic view of the relationship between TEMs, rafts and HIV-1 assembly sites, providing new information about how this human pathogen reorganizes the PM at the microdomain level.
Materials and Methods
Cells, plasmids and transfections
HeLa and Vero cells were maintained in DMEM with 10% FBS. Primary human CD4+ T cells were obtained from Ficoll-separated whole blood (from healthy donors) by negative selection (Miltenyi Biotec, according to manufacturer’s instructions). Purity was typically 90–95%. CD4+ T cells were activated with 5 μg/mL phytohemagglutinin (PHA) for 24 h, then infected with NL4-3(MA-GFP) virus (see below). CD4+ T cells were maintained in RPMI with 20% FBS and 20 units/mL of interleukin-2 (IL-2).
The CD9-enhanced green fluorescent protein (EGFP) (in pEGFP-N1 vector, containing a mutation preventing the dimerization of EGFP) plasmid kindly provided by M. Hemler (DFCI) (68). The NL4-3(MA-EGFP) and NL4-3(MA-mRFP) proviruses were kindly provided by B. Müller (University of Heidelberg) (39). The codon-optimized untagged Gag and GagGFP-fusion constructs has been previously described (69). Venus-fused monomeric Gag construct was kindly provided by P. Spearman. This construct is myristoylated and can bind cellular membranes, albeit weakly, and contains mutations in the D (C-Terminal Domain) and N-terminal domain that completely abolish multimerization (40).
Transfections were performed using Lipofectamine 2000 (Invitrogen), according to manufacturer’s instructions.
Epifluorescence microscopy
HeLa cells were grown on glass-bottom dishes (Mattek). Cells were either labeled on ice or after fixation, as indicated in figure legends. Fixation was done in 4% paraformaldehyde (PFA) in PBS for 10 min, unless otherwise indicated. The following antibodies were used: mouse monoclonal anti-CD9 K41 (BACHEM), mouse monoclonal anti-CD63, H5C6 (Developmental Studies Hybridoma Bank); anti-Gag p6 rabbit serum, kind gift of D. Ott (NCI). Secondary antibodies were AlexaFluor 488, 594, or 647-conjugated and directed against the appropriate species (Invitrogen). Note that, while full-length anti-CD9 antibody K41 induces clustering of CD9 at physiological temperature (70,71), its Fab fragment completely lacks this activity (71), and was therefore suitable for live imaging, and the anti-CD9 SYB-1 antibody used in SMT experiments also lacks this activity.
Anti-CD9 Fab fragments were prepared using the ImmunoPure Fab Preparation Kit (Pierce), and their purity was tested by SDS–PAGE. When monovalent labeling was performed, fluorescein isothiocyanate (FITC)- or DyLight (488, 594 or 649)-conjugated anti-mouse Fab secondary antibodies (Jackson Immunoresearch) were used.
For GM1 labeling, cells were incubated on ice using AlexaFluor 488-conjugated CTxB (Invitrogen) for 30–60 min, fixed and incubated with anti-CTxB rabbit antibody (Invitrogen) to enhance clustering. For TfnR labeling, cells were incubated with transferrin conjugated to AlexaFluor 594 (Invitrogen) for 30 min on ice, then fixed.
Image acquisition and deconvolution were performed using either a DeltaVision deconvolution microscopy station (Applied Precision, Inc), as previously described (14), or a Nikon Eclipse Ti-E equipped with a Nikon 60x PlanApo 1.4 numerical aperture (NA) objective, Qimaging EXi Blue camera, image software NIS Elements 3.10 and AUTOQUANT 2.1.0 (deconvolution). For live imaging, cells were labeled on ice, then imaged at 37°C and 5% CO2 in a custom-built WeatherStation climate chamber (Applied Precision, Inc).
Colocalization analysis and image processing
Colocalization analysis was carried out using Volocity software essentially as described (14) or CoLocalizer Pro software. Briefly, deconvolved images representing a single bottom optical Z-section of a single cell were converted to tiffs and imported into Volocity software v3.7 (Improvision). The classifier module set to 2 or 3 SD above the mean pixel intensity was used to detect and define the bright puncta for each channel, while excluding the dimmer fluorescent signals. The total number of pixels overlapping between two different channels was divided by the total number of pixels for a chosen channel (as indicated in the figures), yielding percent colocalization. Colocalization values from at least 10 cells were averaged to give mean percent colocalization. Average pixel intensity measurements were obtained from corresponding images, also using the classifier module.
Final image assembly, cropping and resolution adjustments for figures, including magnification of boxed regions, was done using Adobe Photoshop.
FRAP analysis
Vero cells were plated on Mattek dishes coated with fibronectin (to minimize detachment of cells during photobleaching) and transfected with the GagGFP and untagged Gag expression vectors (1:1 ratio); 16–24 h posttransfection cells were labeled on ice with anti-CD9 Fab and DyLight 649-labeled secondary Fab (Jackson Immunoresearch), or with CTxB conjugated to Alexa 647 (Invitrogen). All FRAP experiments were performed on a Zeiss LSM 510 Meta confocal microscope at the Microscopy Imaging Facility at the University of Vermont. Cells were maintained at 37°C with a stage heater. Each dish was imaged for 20–40 min. A 100× PlanApochromat 1.4 NA oil immersion objective was used. GFP was excited with the Argon 488 laser line (3–15% transmission), while Alexa 647 and Dylight 649 were excited with the 633-nm He–Ne line (5–20% transmission), and emission was split and collected simultaneously for both channels. Single color experiments were performed to ensure no cross-talk between the two channels. Photobleaching was performed using the 633-nm laser line at full laser power for 200 iterations on a circular ROI approximately 3.5 μm in radius. Subsequent recovery images were collected using low laser power at 5-second intervals for approximately 2–3 min, until ROI recovery had reached a plateau. Only a single Z-section was bleached and imaged, typically the ventral side of the cell contacting the cover slip.
For FRAP calculations, the mean intensity over time of the bleach ROI, as well as a background ROI (containing no cells), and an unbleached ROI in the cell of interest were determined using the Zeiss LSM analysis software and exported into Microsoft Excel. The bleach ROI data were converted to fractional recovery (72), normalized by the overall fluorescence loss in the cell, as measured by the unbleached ROI. This data was imported into GraphPad Prism and fitted using non-linear regression (an exponential decay algorithm, Y = span × exp(−K × X) + plateau, with the half-time to recovery, t1/2 = 0.69/K). The span and plateau were used to calculate Mf. D was calculated using a simple two-dimensional diffusion model for a circular bleach ROI: D = 0.224 × (r2/t1/2) (73,74). This analysis was performed for each individual cell and the D and Mf values were averaged to get mean D and Mf. To yield average recovery curves, mean fractional recovery over time was averaged between multiple cells.
SMT experiments and analysis
SMT experiments were carried out as previously described (18). Briefly, cells expressing GFP or GagGFP [cotransfected with untagged Gag (1:1 ratio)] were incubated in DMEM at 37°C for 10 min with Atto647N-labeled Fab fragments of mAbs raised against CD9 (SYB-1), CD81 (TS81), CD46 (11C5) and CD55 (12A12) (18,75). For COase treatment, cells were incubated for 30 min with 1 UI/mL of the enzyme before labeling. A home-made objective-type TIRF setup allowing multicolor single-molecule imaging and equipped with a Plan Fluor 100x/1.45 NA objective (Zeiss, Le Peck, France Brattleboro, VT) was used. All the experiments were performed with a 100 ms integration time. For some experiments, to achieve a better specificity in the detection of the two fluorescent signals, alternating-laser excitation (ALEX) was performed using an acousto-optical tunable filter and controller (AOTF; AA Optoelectronics) (76).
All the movies were analyzed using a home-made software (named ‘PaTrack’) implemented in visual C++. Trajectories were constructed using the individual diffraction limited signal of each molecule. The center of each fluorescence peak was determined with subpixel resolution by fitting a two-dimensional elliptical Gaussian function. The two-dimensional trajectories of single molecules were constructed frame per frame. Only trajectories containing at least 40 points were retained. Diffusion coefficient values were determined from a linear fit to the MSD (mean square displacement)-τ plots between the first and the fourth points (D1–4) according to the equation MSD(t) = 4Dt.
The determination of the motional modes (Brownian, confined or directed) and parameters was performed as described by Kusumi et al. (77). For each trajectory, we first linearly fitted the MSD on the 10% first points in order to use sufficiently populated curves. If the MSD-τ plot shows positive or negative deviation from a straight line with a slope of 4D (Brownian diffusion), the MSD is respectively adjusted with a quadratic curve (4Dt + v2t2) (directed diffusion) or with an exponential curve (confined diffusion where L is the side of a square domain, the confinement diameter being related to L by dconf = (2/√π)L). For the mixed trajectory exhibiting a combination of Brownian and apparent confined motion mode, the trajectory was split and the MSD of each segment was adjusted with a linear or an exponential curve as described above.
Supplementary Material
Summary of mobility measurements by FRAP.
Apparent diffusion coefficients and diffusion modes of CD9, CD46 and CD55.
Acknowledgments
We are grateful to Martin Hemler, Paul Spearman, Barbara Müller and Hans-Georg Kraeusslich for providing plasmids, Eric Rubinstein for providing SYB-1,11C5 and 12A12 monoclonal antibodies and to Linnea Olofson for her technical help. Members of the Thali lab and Alan Howe are acknowledged for critical reading of the manuscript. Chris Huston is acknowledged for the use of The Nikon Eclipse Ti-E instrument, which was purchased with funds from NIAID ARRA supplement R01 A1072021-0251.
This work was supported by NIH grants R56 AI047727 and R01 AI 080302 to M. T., ANR # 06-BLAN-0378 to P. E. M., as well as a Vermont Genetics Network Graduate Fellowship and T32 AI055402 Training Grant to D.K., P. R. is a recipient from the French Ministry for Research and Technology.
Footnotes
Supporting Information
Additional Supporting Information may be found in the online version of this article:
References
- 1.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Pike LJ. Rafts defined: a report on the Keystone Symposium on lipid rafts and cell function. J Lipid Res. 2006;47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Goswami D, Gowrishankar K, Bilgrami S, Ghosh S, Raghupathy R, Chadda R, Vishwakarma R, Rao M, Mayor S. Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell. 2008;135:1085–1097. doi: 10.1016/j.cell.2008.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, von Middendorff C, Schonle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson K, Mouritsen OG, Anderson RG. Lipid rafts: at a crossroad between cell biology and physics. Nat Cell Biol. 2007;9:7–14. doi: 10.1038/ncb0107-7. [DOI] [PubMed] [Google Scholar]
- 6.Kusumi A, Koyama-Honda I, Suzuki K. Molecular dynamics and interactions for creation of stimulation-induced stabilized rafts from small unstable steady-state rafts. Traffic (Copenhagen, Denmark) 2004;5:213–230. doi: 10.1111/j.1600-0854.2004.0178.x. [DOI] [PubMed] [Google Scholar]
- 7.Harding AS, Hancock JF. Using plasma membrane nanoclusters to build better signaling circuits. Trends Cell Biol. 2008;18:364–371. doi: 10.1016/j.tcb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Douglass AD, Vale RD. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 2005;121:937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieber JJ, Willig KI, Kutzner C, Gerding-Reimers C, Harke B, Donnert G, Rammner B, Eggeling C, Hell SW, Grubmuller H, Lang T. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- 10.Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 11.Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J. 2009;420:133–154. doi: 10.1042/BJ20082422. [DOI] [PubMed] [Google Scholar]
- 12.Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 2009;19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Berditchevski F, Odintsova E. Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J Cell Biol. 1999;146:477–492. doi: 10.1083/jcb.146.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nydegger S, Khurana S, Krementsov DN, Foti M, Thali M. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J Cell Biol. 2006;173:795–807. doi: 10.1083/jcb.200508165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Unternaehrer JJ, Chow A, Pypaert M, Inaba K, Mellman I. The tetraspanin CD9 mediates lateral association of MHC class II molecules on the dendritic cell surface. Proc Natl Acad Sci U S A. 2007;104:234–239. doi: 10.1073/pnas.0609665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol. 2003;19:397–422. doi: 10.1146/annurev.cellbio.19.111301.153609. [DOI] [PubMed] [Google Scholar]
- 17.Berditchevski F, Odintsova E. Tetraspanins as regulators of protein trafficking. Traffic. 2007;8:89–96. doi: 10.1111/j.1600-0854.2006.00515.x. [DOI] [PubMed] [Google Scholar]
- 18.Espenel C, Margeat E, Dosset P, Arduise C, Le Grimellec C, Royer CA, Boucheix C, Rubinstein E, Milhiet PE. Single-molecule analysis of CD9 dynamics and partitioning reveals multiple modes of interaction in the tetraspanin web. J Cell Biol. 2008;182:765–776. doi: 10.1083/jcb.200803010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barreiro O, Zamai M, Yanez-Mo M, Tejera E, Lopez-Romero P, Monk PN, Gratton E, Caiolfa VR, Sanchez-Madrid F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol. 2008;183:527–542. doi: 10.1083/jcb.200805076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemler ME. Specific tetraspanin functions. J Cell Biol. 2001;155:1103–1107. doi: 10.1083/jcb.200108061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol. 2005;5:136–148. doi: 10.1038/nri1548. [DOI] [PubMed] [Google Scholar]
- 22.Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005;20:218–224. doi: 10.1152/physiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- 23.Silvie O, Rubinstein E, Franetich JF, Prenant M, Belnoue E, Renia L, Hannoun L, Eling W, Levy S, Boucheix C, Mazier D. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat Med. 2003;9:93–96. doi: 10.1038/nm808. [DOI] [PubMed] [Google Scholar]
- 24.Tham TN, Gouin E, Rubinstein E, Boucheix C, Cossart P, Pizarro-Cerda J. The tetraspanin Cd81 is required for Listeria monocytogenes invasion. Infect Immun. 2009;9:9. doi: 10.1128/IAI.00661-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazurov D, Heidecker G, Derse D. HTLV-1 Gag protein associates with CD82 tetraspanin microdomains at the plasma membrane. Virology. 2006;346:194–204. doi: 10.1016/j.virol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 26.Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. 2006;172:923–935. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jolly C, Sattentau QJ. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J Virol. 2007;81:7873–7884. doi: 10.1128/JVI.01845-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Krausslich HG. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007;3:e36. doi: 10.1371/journal.ppat.0030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turville SG, Aravantinou M, Stossel H, Romani N, Robbiani M. Resolution of de novo HIV production and trafficking in immature dendritic cells. Nat Methods. 2008;5:75–85. doi: 10.1038/nmeth1137. [DOI] [PubMed] [Google Scholar]
- 30.Sato K, Aoki J, Misawa N, Daikoku E, Sano K, Tanaka Y, Koyanagi Y. Modulation of human immunodeficiency virus type 1 infectivity through incorporation of tetraspanin proteins. J Virol. 2008;82:1021–1033. doi: 10.1128/JVI.01044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grigorov B, Attuil-Audenis V, Perugi F, Nedelec M, Watson S, Pique C, Darlix JL, Conjeaud H, Muriaux D. A role for CD81 on the late steps of HIV-1 replication in a chronically infected T cell line. Retrovirology. 2009;6:28. doi: 10.1186/1742-4690-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weng J, Krementsov DN, Khurana S, Roy NH, Thali M. Formation of syncytia is repressed by tetraspanins in HIV-1 producing cells. J Virol. 2009;83:7467–7474. doi: 10.1128/JVI.00163-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krementsov DN, Weng J, Lambele M, Roy NH, Thali M. Tetraspanins regulate cell-to-cell transmission of HIV-1. Retrovirology. 2009;6:64. doi: 10.1186/1742-4690-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz-Mateos E, Pelchen-Matthews A, Deneka M, Marsh M. CD63 is not required for production of infectious human immunodeficiency virus type 1 in human macrophages. J Virol. 2008;82:4751–4761. doi: 10.1128/JVI.02320-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Dziuba N, Friedrich B, von Lindern J, Murray JL, Rojo DR, Hodge TW, O’Brien WA, Ferguson MR. A critical role for CD63 in HIV replication and infection of macrophages and cell lines. Virology. 2008;379:191–196. doi: 10.1016/j.virol.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung K, Kim J-O, Ganesh L, Kabat J, Schwartz O, Nabel GJ. HIV-1 assembly: viral glycoproteins segregate quantally to lipid rafts that associate individually with HIV-1 capsids and virions. Cell Host Microbe. 2008;3:285–292. doi: 10.1016/j.chom.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jolly C, Sattentau QJ. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J Virol. 2005;79:12088–12094. doi: 10.1128/JVI.79.18.12088-12094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen DH, Hildreth JE. Evidence for budding of human immunod-eficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74:3264–3272. doi: 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich HG. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol. 2004;78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dou J, Wang JJ, Chen X, Li H, Ding L, Spearman P. Characterization of a myristoylated, monomeric HIV Gag protein. Virology. 2009;387:341–352. doi: 10.1016/j.virol.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larson DR, Johnson MC, Webb WW, Vogt VM. Visualization of retrovirus budding with correlated light and electron microscopy. Proc Natl Acad Sci U S A. 2005;102:15453–15458. doi: 10.1073/pnas.0504812102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol. 2007;81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jouvenet N, Bieniasz PD, Simon SM. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature. 2008;454:236–240. doi: 10.1038/nature06998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez CY, Hope TJ. Mobility of human immunodeficiency virus type 1 Pr55Gag in living cells. J Virol. 2006;80:8796–8806. doi: 10.1128/JVI.02159-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 47.Ivanchenko S, Godinez WJ, Lampe M, Krausslich HG, Eils R, Rohr K, Brauchle C, Muller B, Lamb DC. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009;5:e1000652. doi: 10.1371/journal.ppat.1000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Lagerholm BC, Yang B, Jacobson K. Methods to measure the lateral diffusion of membrane lipids and proteins. Methods. 2006;39:147–153. doi: 10.1016/j.ymeth.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 49.Waheed AA, Freed EO. Lipids and membrane microdomains in HIV-1 replication. Virus Res. 2009;143(2):162–176. doi: 10.1016/j.virusres.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dietrich C, Volovyk ZN, Levi M, Thompson NL, Jacobson K. Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc Natl Acad Sci U S A. 2001;98:10642–10647. doi: 10.1073/pnas.191168698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ono A, Freed EO. Role of lipid rafts in virus replication. Adv Virus Res. 2005;64:311–358. doi: 10.1016/S0065-3527(05)64010-9. [DOI] [PubMed] [Google Scholar]
- 52.Holm K, Weclewicz K, Hewson R, Suomalainen M. Human immunod-eficiency virus type 1 assembly and lipid rafts: Pr55(gag) associates with membrane domains that are largely resistant to Brij98 but sensitive to Triton X-100. J Virol. 2003;77:4805–4817. doi: 10.1128/JVI.77.8.4805-4817.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pinaud F, Michalet X, Iyer G, Margeat E, Moore H-P, Weiss S. Dynamic partitioning of a glycosyl-phosphatidylinositol-anchored protein in glycosphingolipid-rich microdomains imaged by single-quantum dot tracking. Traffic (Copenhagen, Denmark) 2009;10:691–712. doi: 10.1111/j.1600-0854.2009.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. 2000;2:582–592. doi: 10.1038/35023524. [DOI] [PubMed] [Google Scholar]
- 56.Gomez-Mouton C, Abad JL, Mira E, Lacalle RA, Gallardo E, Jimenez-Baranda S, Illa I, Bernad A, Manes S, Martinez AC. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc Natl Acad Sci U S A. 2001;98:9642–9647. doi: 10.1073/pnas.171160298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lozahic S, Christiansen D, Manie S, Gerlier D, Billard M, Boucheix C, Rubinstein E. CD46 (membrane cofactor protein) associates with multiple beta1 integrins and tetraspans. Eur J Immunol. 2000;30:900–907. doi: 10.1002/1521-4141(200003)30:3<900::AID-IMMU900>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 58.Montefiori DC, Cornell RJ, Zhou JY, Zhou JT, Hirsch VM, Johnson PR. Complement control proteins, CD46, CD55, and CD59, as common surface constituents of human and simian immunodeficiency viruses and possible targets for vaccine protection. Virology. 1994;205:82–92. doi: 10.1006/viro.1994.1622. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan BL, Knopoff EJ, Saifuddin M, Takefman DM, Saarloos MN, Sha BE, Spear GT. Susceptibility of HIV-1 plasma virus to complement-mediated lysis. Evidence for a role in clearance of virus in vivo. J Immunol. 1996;157:1791–1798. [PubMed] [Google Scholar]
- 60.Hammarstedt M, Wallengren K, Pedersen KW, Roos N, Garoff H. Minimal exclusion of plasma membrane proteins during retroviral envelope formation. Proc Natl Acad Sci U S A. 2000;97:7527–7532. doi: 10.1073/pnas.120051597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mazurov D, Heidecker G, Derse D. The inner loop of tetraspanins CD82 and CD81 mediates interactions with human T cell lymphotrophic virus type 1 Gag protein. J Biol Chem. 2007;282:3896–3903. doi: 10.1074/jbc.M607322200. [DOI] [PubMed] [Google Scholar]
- 62.Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A. 2001;98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Charrin S, Manie S, Thiele C, Billard M, Gerlier D, Boucheix C, Rubinstein E. A physical and functional link between cholesterol and tetraspanins. Eur J Immunol. 2003;33:2479–2489. doi: 10.1002/eji.200323884. [DOI] [PubMed] [Google Scholar]
- 64.Delaguillaumie A, Harriague J, Kohanna S, Bismuth G, Rubinstein E, Seigneuret M, Conjeaud H. Tetraspanin CD82 controls the association of cholesterol-dependent microdomains with the actin cytoskeleton in T lymphocytes: relevance to co-stimulation. J Cell Sci. 2004;117:5269–5282. doi: 10.1242/jcs.01380. [DOI] [PubMed] [Google Scholar]
- 65.Pickl WF, Pimentel-Muinos FX, Seed B. Lipid rafts and pseudotyping. J Virol. 2001;75:7175–7183. doi: 10.1128/JVI.75.15.7175-7183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Briggs JA, Wilk T, Fuller SD. Do lipid rafts mediate virus assembly and pseudotyping? J Gen Virol. 2003;84:757–768. doi: 10.1099/vir.0.18779-0. [DOI] [PubMed] [Google Scholar]
- 67.Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A. 2006;103(8):2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kovalenko OV, Yang X, Kolesnikova TV, Hemler ME. Evidence for specific tetraspanin homodimers: inhibition of palmitoylation makes cysteine residues available for cross-linking. Biochem J. 2004;377:407–417. doi: 10.1042/BJ20031037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. HIV-1 egress is gated through late endosomal membranes. Traffic. 2003;4:902–910. doi: 10.1046/j.1600-0854.2003.00145.x. [DOI] [PubMed] [Google Scholar]
- 70.Khurana S, Krementsov DN, de Parseval A, Elder JH, Foti M, Thali M. Human immunodeficiency virus type 1 and influenza virus exit via different membrane microdomains. J Virol. 2007;81:12630–12640. doi: 10.1128/JVI.01255-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singethan K, Muller N, Schubert S, Luttge D, Krementsov DN, Khurana SR, Krohne G, Schneider-Schaulies S, Thali M, Schneider-Schaulies J. CD9 clustering and formation of microvilli zippers between contacting cells regulates virus-induced cell fusion. Traffic. 2008;9:924–935. doi: 10.1111/j.1600-0854.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Snapp EL, Altan N, Lippincott-Schwartz J. Measuring protein mobility by photobleaching GFP chimeras in living cells. Current Protocols in Cell Biology [Electronic Resource] (John Wiley, USA) 2003;Chapter 21(Unit 21) doi: 10.1002/0471143030.cb2101s19. [DOI] [PubMed] [Google Scholar]
- 73.Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soumpasis DM. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 1983;41:95–97. doi: 10.1016/S0006-3495(83)84410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charrin S, Le Naour F, Labas V, Billard M, Le Caer J-P, Emile J-F, Petit M-A, Boucheix C, Rubinstein E. EWI-2 is a new component of the tetraspanin web in hepatocytes and lymphoid cells. Biochem J. 2003;373:409–421. doi: 10.1042/BJ20030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Margeat E, Kapanidis AN, Tinnefeld P, Wang Y, Mukhopadhyay J, Ebright RH, Weiss S. Direct observation of abortive initiation and promoter escape within single immobilized transcription complexes. Biophys J. 2006;90:1419–1431. doi: 10.1529/biophysj.105.069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kusumi A, Sako Y, Yamamoto M. Confined lateral diffusion of membrane receptors as studied by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys J. 1993;65:2021–2040. doi: 10.1016/S0006-3495(93)81253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of mobility measurements by FRAP.
Apparent diffusion coefficients and diffusion modes of CD9, CD46 and CD55.