

Abstract
Interleukin 18 (IL-18) is a proinflammatory cytokine in the IL-1 family that has been implicated in a number of disease states. In animal models of acute myocardial infarction (AMI), pressure overload, and LPS-induced dysfunction, IL-18 regulates cardiomyocyte hypertrophy and induces cardiac contractile dysfunction and extracellular matrix remodeling. In patients, high IL-18 levels correlate with increased risk of developing cardiovascular disease (CVD) and with a worse prognosis in patients with established CVD. Two strategies have been used to counter the effects of IL-18:IL-18 binding protein (IL-18BP), a naturally occurring protein, and a neutralizing IL-18 antibody. Recombinant human IL-18BP (r-hIL-18BP) has been investigated in animal studies and in phase I/II clinical trials for psoriasis and rheumatoid arthritis. A phase II clinical trial using a humanized monoclonal IL-18 antibody for type 2 diabetes is ongoing. Here we review the literature regarding the role of IL-18 in AMI and heart failure and the evidence and challenges of using IL-18BP and blocking IL-18 antibodies as a therapeutic strategy in patients with heart disease.
INTRODUCTION
Heart failure (HF) is a clinical syndrome of impaired left ventricular function characterized by shortness of breath, fatigue and poor exercise tolerance (1). In 2010, hospital discharges for HF in the United States were estimated to be one million, and one in nine deaths had HF mentioned on the death certificate (2). Patient survival has improved in recent years, but the death rate remains unacceptably high, with approximately 50% of people diagnosed with HF dying within five years (2). Inflammation is a central component of the response to tissue stress and injury in the heart, coordinating remodeling and healing by promoting extracellular matrix remodeling, cell proliferation, cardiomyocyte hypertrophy and affecting cardiomyocyte contractility (3). While some inflammation is necessary for proper healing, increased inflammation appears to play a role in the predisposition to develop heart disease and may contribute to the disease severity and response to treatment (4–6). Increased inflammatory biomarkers correlate with HF severity and predict adverse prognosis (1,4,7). In experimental settings, the administration of proinflammatory cytokines promotes left ventricular dysfunction (1,8). However, antiinflammatory strategies in the treatment of HF are currently lacking, indicating that the inflammatory mechanisms involved in the development of HF are incompletely characterized.
INTERLEUKIN-18, AN IL-1 FAMILY MEMBER
Interleukin-18 (IL-18) is a proinflammatory cytokine that was first described in 1989 for its ability to induce interferon γ (IFN-γ) production (9). The cytokine was later cloned and found to have a synergistic effect with IL-12 in the production of IFN-γ from T cells, natural killer (NK) cells and macrophages (10,11). This synergism is thought to be the result of IL-12 inducing the expression of the IL-18 receptor on T cells (12). IL-18 stimulates the proliferation of T cells, making it functionally related to IL-12, but it is most similar structurally to the IL-1 family of cytokines, specifically IL-1β (11,13) (Table 1). Because of this structural similarity and other common characteristics shared with IL-1β, IL-18 is part of the IL-1 family. Like IL-1β, IL-18 has a secondary structure primarily consisting of β-sheets (11).
Table 1.
IL-18 and other members of the IL-1 family.
| Name | Receptor | Coreceptor | Decoy receptor | Soluble receptor/binding protein | Property |
|---|---|---|---|---|---|
| IL-1α | IL-1RI | IL-1RAcP | IL-1RII | sIL-1R2a | Proinflammatory |
| IL-1β | IL-1RI | IL-1RAcP | IL-1RII | sIL-1R2 | Proinflammatory |
| IL-1Ra | IL-1RI | N/A | IL-1RII | N/A | Antagonist for IL-1α and IL-1β |
| IL-18 | IL-18Rα | IL-18Rβ | N/A | IL-18BP | Proinflammatory |
Significance of intracellular binding of sIL-1R2 to IL-1α is not known.
IL-1β and IL-18 both are activated by caspase-1 following formation of the inflammasome. The inflammasome is a macromolecular structure consisting of (a) an intracellular NOD-like receptor (NLR) such as NLRP3, (b) an adaptor protein, apoptosis speck-like protein containing a caspase recruitment domain (ASC), and (c) procaspase-1 (14,15) (Figure 1). Accumulation of adenosine triphosphate (ATP) and cell debris after tissue injury, among other stimuli, acts as a danger signal and induces formation of the inflammasome resulting in the cleavage and activation of caspase-1, which subsequently cleaves the inactive precursor proteins pro-IL-1β and pro-IL-18 (16,17) (see Figure 1). Unlike pro-IL-1β, pro-IL-18 is constitutively expressed in unstimulated cells (18). Activation of the inflammasome occurs in different cell types in response to injury (19). After acute myocardial infarction (AMI), the inflammasome is formed in leukocytes, endothelial cells, fibroblasts and cardiomyocytes (15,20,21). Although following AMI, active IL-1β and IL-18 are increased in the ischemic myocardial tissue, in vitro cell studies have evidenced a different function of the inflammasome in the different cell types (22). Leukocytes produce much IL-1β and IL-18 and inflammasome positive leukocytes are found in the infarct area (15). In fibroblasts, the activation of the inflammasome represents a stimulus for myofibroblast differentiation and collagen synthesis by increasing the local production of IL-1β and IL-18 (21,23). In cardiomyocytes, the activation of the inflammasome leads to caspase-1 activation and pyroptosis, but not to IL-1β release.
Figure 1.

Overview of IL-18 signaling. Left: Stimulation with pathogen-associated molecular pattern molecules (PAMPs) and/or damage-associated molecular pattern molecules (DAMPs) (priming) followed by ATP’s binding the P2×7 receptor (trigger) results in formation of the inflammasome. Caspase-1 then cleaves pro-IL-1β and pro-IL-18 into their active forms. Right: The active IL-18 binds the IL-18Rα-IL-18Rβ receptor dimer or is sequestered by IL-18BP. Alternately, IL-37 binds IL-18Rα and SIGIRR, resulting in an antiinflammatory signal. After the active receptor complex is formed it recruits MyD88, IRAK, and TRAF6 which activates NFκB signaling causing an induction of secondary inflammatory mediators and increased inducible nitric oxide synthase (iNOS) production resulting in contractile dysfunction. The receptor also activates the PI3K-Akt-GATA4 pathway resulting in hypertrophy, and increases OPN resulting in fibrosis. The question mark in panel B highlights that whether the IL-18-mediated activation of IFN is direct or indirect is unknown.
Also, pro-IL-1β and pro-IL-18 can be activated by extracellular neutrophil proteinases such as neutrophil proteinase 3 (PR3) during tissue injury (24,25). Furthermore, pro-IL-18 can be cleaved by human chymase from mast cells at two sites, creating two unique species, only one of which is biologically active, p16 (26). The p16 fragment-induced IFN-γ production in vitro but with only approximately 20% of the activity of mature IL-18 (26). Adding supernatant from human CD8+ T cells containing granzyme B (GrB) resulted in cleavage at the same site as caspase-1 and resulted in active IL-18 (27). The proteases elastase and cathepsin G cleave and activate IL-1β and have been suggested to cleave IL-18 as well in human neutrophils, however this cleavage does not result in biologically active species (28,29). Although most IL-18 is soluble, IL-18 can exist as a membrane-bound protein in a subset of macrophages. Upon stimulation with lipopolysaccharide (LPS), these cells secrete soluble IL-18, suggesting an additional mechanism of IL-18 release and activation by a protease induced by LPS, possibly PR3 (30,31). PR3 could also be the mechanism by which pro-IL-1β and pro-IL-18 released from a dying cell are activated. Conversely, IL-18 is inactivated through cleavage by caspase-3 (32).
INTERLEUKIN-18 SIGNALING
Similar to the receptors for IL-1 and other cytokines, the IL-18 receptor (IL-18R) is a dimer composed of IL-18Rα, a low affinity binding chain, and IL-18Rβ (also known as accessory protein-like [AcPL]) that binds the IL-18/IL-18Rα complex (33,34) (see Figure 1). Both receptor subunits are members of the IL-1 receptor family; IL-18Rα is also known as IL-1 receptor related protein (IL-1rp) (35). IL-37, on the other hand, binds IL-18Rα, but does not recruit IL-18Rβ, and instead recruits TIR8, also known as single immunoglobulin interleukin-1 receptor–related molecule (SIGIRR), containing a Toll/IL-1R (TIR) domain (see Figure 1) (36–38). Since an active receptor complex is not formed, there is no signaling through MyD88. Once the active receptor complex is assembled, IL-18R shares many downstream signaling pathways with the IL-1 receptor (39). Both the IL-1 and IL-18 receptors can recruit IL-1 receptor-associated kinase (IRAK) and MyD88, an intracellular adaptor molecule required for IRAK phosphorylation and for IRAK interaction with the receptor. Mice lacking MyD88 have decreased T cell proliferation and production of inflammatory cytokines in response to IL-1 and impaired IFN-γ, nuclear factor κB (NFκB), and c-Jun N-terminal kinase (JNK) signaling in response to IL-18 (40). Similar signaling disruptions were seen when IL-18 was given to IRAK-deficient helper T cell type 1 (Th1) cells and IRAK knockout mice, suggesting an important role for IRAK in IL-18 signaling (41). Like the IL-1 receptor, the IL-18R recruits tumor necrosis factor receptor–associated factor-6 (TRAF6) which allows for the activation of NF-κB and its translocation to the nucleus (42) (see Figure 1). IL-18 also activates p38 mitogen-activated protein kinase (MAPK) (43). A direct comparison of IL-1β and IL-18 signaling showed that IL-18 preferentially activates p38-MAPK and has minimal effects on the induction of cyclooxygenase-2 (COX-2) which mediates fever through synthesis of prostaglandin E2, whereas IL-1β induces a rapid and intense COX-2 expression (39).
In the search for a soluble IL-18 receptor, an IL-18 binding protein (IL-18BP) was found by running samples of human urine through an IL-18 agarose column (44). IL-18BP is able to block the activity of both human and murine IL-18 and prevent LPS-induced IFN-γ production in mouse splenocytes (44). IL-18BP is not a member of the IL-1 or IL-18 receptor families, and has six naturally occurring isoforms (44,45) (Figure 2). Human IL-18BPa (hIL-18BPa) has the greatest affinity for IL-18, and adding supernatants from cells expressing this protein to human and murine spleen cells resulted in a 90% decrease and 80% decrease in IL-18 activity, respectively (45). Human IL-18BPc (hIL-18BPc) and murine IL-18BPd (mIL-18BPd) show similar results, while murine IL-18BPc (mIL-18BPc) is specific for murine IL-18, and human IL-18BPb (hIL-18BPb) and IL-18BPd (hIL-18BPd) lack the complete Ig domain and therefore cannot bind IL-18 (45).
Figure 2.

Structures of the different IL-18BP isoforms. Immunoglobulin (IG) domains are in green, potential binding motifs in red and signal peptide in black (44,45).
INTERLEUKIN-18 LEVELS IN PATIENTS WITH HEART DISEASE
There is growing evidence for a role of IL-18 in human myocardial infarction, HF and other forms of heart disease. IL-18 expression is increased in human atherosclerotic plaques collected during carotid endarterectomy and its accumulation is associated with plaque destabilization (46). A recent study conducted in the community-based cohort of Framingham, however, reported no significant correlations between the levels of IL-18 and subclinical aortic atherosclerosis (47). High serum levels of IL-18 were associated with an increased risk of developing cardiovascular disease (CVD) in the general population, increased mortality in HF patients and development of congestive HF and AMI in patients with acute coronary syndromes (48–52).
Plasma IL-18 levels also were increased in patients with AMI compared with aged matched controls and correlated with increased atrial natriuretic peptide (ANP) levels, suggesting that IL-18 may play a role in inducing ANP (53). Immunohistochemistry revealed increased IL-18 and IL-18Rα expression in endothelial cells, macrophages and cardiomyocytes and decreased IL-18BP in the myocardium of patients with end-stage HF undergoing heart transplantation (54). Parallel to increased levels of IL-18, an increase in IL-18BP levels is seen in many disease processes. The naturally occurring IL-18BP in the circulation neutralizes the circulating IL-18 so that the “free” IL-18 levels are much lower than the total IL-18 levels, and the “free” IL-18 levels seem to better correlate with disease activity than the total IL-18 levels (55).
INTERLEUKIN-18 IN IN VIVO AND IN VITRO MODELS
Cardiomyocyte Hypertrophy
A cardiac muscle cell line, HL-1, treated with IL-18 became hypertrophic and increased expression of ANP, as shown by ANP promoter-driven luciferase activity (56). This activation was dependent on the activation of phosphatidylinositol 3 kinase (PI3K), Akt and the transcription factor GATA4 (56) (see Figure 1). ANP normally is expressed only early in development and its expression in adults is a sign of myocardial hypertrophy (57). Daily administration of IL-18 to healthy mice induces ANP expression in the heart, myocardial hypertrophy, and contractile dysfunction (58,59). IL-18 knockout (KO) mice subjected to pressure overload developed less hypertrophy, which is a key feature of the pathology, but also had worse contractile function (60). IL-18 KO mice, as well as mice treated with an IL-18 blocking antibody or with rh-IL-18BP showed a significantly blunted hypertrophic response to isoproterenol (IPO); the effects on cardiac function were not reported (61). These results suggest that IL-18 is involved in the hypertrophic response in overload cardiomyopathy. Whether pharmacological blockade of IL-18 would have a deleterious effect by inhibiting the necessary hypertrophic response, or a beneficial effect by limiting pathologic myocardial hypertrophy and diastolic dysfunction, remains unclear.
Myocardial Ischemia and Infarction
In a mouse model of AMI, increased IL-18 in the serum and pro-IL-18 in smooth muscle and endothelial cells was found in both the infarcted and noninfarcted regions of the heart (62). A similar increase was observed in the serum and in the post-infarcted myocardium of mice subjected to experimental ischemia followed by reperfusion (63). The rapid rise in pro-IL-18 mRNA and IL-18 protein was followed by a delayed increase in IL-18BP (62). The use of an IL-18-neutralizing antibody, given to the mouse 1 h before ischemia, reduced infarct size (63). Furthermore, injection of mesenchymal stem cells derived from mice overexpressing IL-18BP into the coronary artery or myocardium of rats before ischemia resulted in increased left ventricular developed pressure (LVDP), improved ejection fraction and decreased infarct size (64). These results show that IL-18 is increased after myocardial infarction and that blockade of IL-18 may be beneficial. The mechanism by which IL-18 blockade reduces infarct size has not been fully characterized (63). Further studies are needed to see if blocking IL-18 confers a sustained benefit to the heart in terms of more favorable cardiac remodeling and preserved function.
Acute Myocarditis
A study conducted using biopsy and autopsy specimens collected from patients with acute myocarditis highlighted the presence of the inflammasome in the heart tissue of patients but not of control subjects (65). The analysis also revealed that the presence of the inflammasome in the heart correlated with the severity of HF symptoms and predicted lack of functional recovery at follow up (65). This suggests that the inflammasome (and related cytokines including IL-1β and IL-18) may play an important role in the tissue injury response to myocarditis. A recent study evaluated the levels of IL-18, IL-18Rα, IL-18Rβ and IL-18BP in the heart of rats with autoimmune myocarditis (61). All four proteins were strongly increased in myeloid cells and mildly increased in cardiomyocytes. Hydrodynamic-based IL-18BP gene delivery ameliorated left ventricle (LV) remodeling, preserved LV function, reduced myocardial leukocyte infiltrates and myocardial hypertrophy and reduced the expression of brain natriuretic peptide (BNP) and several proinflammatory cytokines (61).
Contractile Dysfunction
Similar to other inflammatory cytokines like IL-1β and tumor necrosis factor-α (TNF-α), IL-18 has cardiodepressant effects (58,66). However, given that IL-18 also induces the production of these same proinflammatory cytokines, it has been difficult to isolate the individual contributions of each cytokine (67–69). Reciprocally, IL-1, TNF, IL-6, IL-10 and other cytokines induce production of IL-18 (70).
As noted previously, daily administration of IL-18 in healthy mice decreased LV contractility and relaxation (58). Blocking IL-18 with IL-18BPa improved contractile function when added to the perfusate in an ischemia-reperfusion model using human atrial myocardium (71). Neutralization of IL-18 with an anti-mouse IL-18 antibody prevented LPS-induced myocardial dysfunction, reduced myocardial neutrophil infiltration by 55% and attenuated ICAM-1 and VCAM-1 myocardial expression by 50 percent and 37 percent, respectively (72). In vitro IL-18 increased peak and diastolic calcium transients but reduced shortening of isolated cardiomyocytes (58,62). The increase in calcium may be a compensatory mechanism to overcome the cardiodepressant effects of IL-18, but may also lead to decreased responsiveness of the myofilaments.
IL-18 also activates human neutrophils and primes them for free radical production through activation of p38MAPK in vitro and increases neutrophil accumulation in the peritoneal cavity of mice (43,66,73). Neutralizing IL-18 after LPS injection had no effect on TNF-α production, however, TNF-α KO mice had reduced levels of IL-18, suggesting that TNF-α-induced cardiodepressant effects may be mediated by the induction of IL-18 (72). A separate study using daily administration of IL-18 to induce myocardial dysfunction, however, saw no increase in the production of TNF-α or ICAM-1 (58).
In vivo IL-18 induces contractile dysfunction induced by IL-1β or by plasma collected from patients with HF through p38 MAPK (74). Blockade of IL-18 with IL-18BPa, a neutralizing antibody, or with genetic manipulation, prevents the development of systolic dysfunction induced by IL-1β but does not alter IL-6 levels (74). These findings suggest that the blockade of IL-18 improves the systolic function by preventing the activation of p38 MAPK without interfering with other effects of IL-1, such as the activation of COX-2.
β-Adrenergic Receptor Signaling
There is a close interplay between β-adrenergic signaling and HF progression. Hyperactivity of β-adrenergic receptors results in HF, and patients with high circulating catecholamines have less functional (that is, desensitized) β-adrenergic receptors (β-ARs) (75–77). This effect can be reproduced in animal models by chronic exposure to high doses of β-adrenergic agonists, such as isoproterenol (IPO; Figure 3) (78,79). Simulation of β-adrenergic receptors with IPO increases β2-AR and NF-κB signaling, resulting in increased IL-18 mRNA in mouse myocardial tissue and serum as early as 2 h and continuing for up to 72 h, with protein concentrations peaking at 3 h (79). Direct administration of IL-18 induces further β-AR dysfunction as measured by reduced responsiveness to IPO (51). IPO also increases production of IL-18BP from adult mouse cardiomyocytes for up to 24 h (78).
Figure 3.
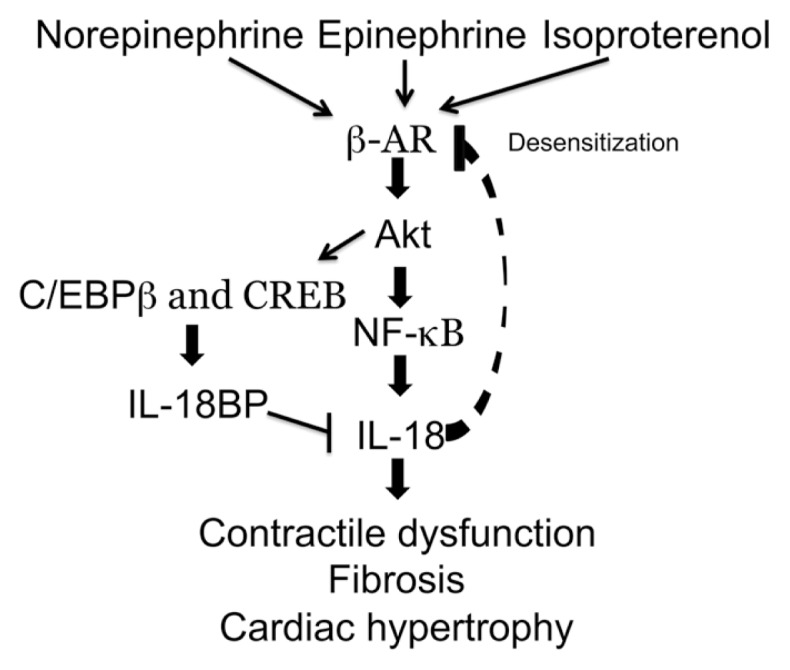
The role of IL-18 in β-adrenergic receptor (β-AR) signaling. Decreased response to isoproterenol in mice treated daily with IL-18 (58) indicates a desensitization of β-ARs, however the mechanism is unclear.
Apoptosis
IL-18 induces apoptosis in many models but appears to be cell-type specific. When treated with IL-18, human cardiac microvascular endothelial cells (HCMEC), which act as an important barrier between the lumen of the vessel and the tissue surrounding it, decreased production of the antiapoptotic protein Bcl-2 and increased production of the proapoptotic proteins Fas and Fas ligand (Fas-L) via activation of NF-κB (80). Fas-L binds the Fas receptor, which recruits the Fas-associated death domain (FADD), activates caspase-8 and starts a proapoptotic cascade. IL-18 also has been shown to induce Fas-L-mediated cytotoxicity in murine Th1 cells and NK cells independently of IFN-γ or TNF-α production (81,82). After treatment with IL-18, HCMECs also showed an increase in the activation of the proapoptotic protein BH3, interacting domain death agonist (BID) into the truncated form, tBID, which induces the release of cytochrome c from mitochondria and activation of caspase-9/-3 dependent apoptosis (80). Further studies revealed that IL-18 also prevented prosurvival signals through increased transcription of the tumor suppressor phosphatase and tensin homologue deleted on chromosome 10 (PTEN) by p38MAPK and subsequent NF-κB activation, resulting in increased apoptosis of HCMECs (83) (see Figure 1). However, IL-18 does not appear to induce apoptosis in neutrophils (73). The effect of IL-18 on cardiomyocyte apoptosis is not established.
Extracellular Matrix Remodeling
An eight-fold increase in interstitial collagen content, indicative of increased fibrosis and subsequent wall stiffness, was seen in cardiac tissue of mice after 7 d of treatment with IL-18 (59). Fibrosis is induced through an increase in osteopontin (OPN), an extracellular matrix protein involved in the regulation of collagen levels in the heart (84). OPN has been implicated in a number of adverse cardiac remodeling events and is found in the serum of patients with advanced HF (85). Induction of pressure overload in mice resulted in increased IL-18 and OPN gene and protein expression and increased fibrosis, while IL-18 alone was sufficient to induce OPN both in vivo and in vitro in murine cardiac fibroblasts (84). The addition of a neutralizing antibody to the IL-18R blocked the increase in OPN, and silencing of the transcription factor IRF1 decreased IL-18 expression and subsequently impaired OPN gene expression in cardiac fibroblasts (Figure 1) (84). IL-18 also induces fibronectin expression in human cardiac fibroblasts, which is another way by which IL-18 could exert its profibrotic effects (86). Rat cardiac fibroblasts exposed to IL-18 had increased expression of collagen type I and III, periostin and matrix metalloproteinase 2 (MMP-2) and altered migratory properties (87).
Interleukin-18 Blockade
Although still in the initial phases, trials investigating the safety and efficacy of the recombinant human IL-18BP (r-hIL-18BP) and of a blocking IL-18 antibody have been designed over the past years (88–90). The r-hIL-18BP can be given as a short acting subcutaneous injection and must be given every 2 d to maintain blood levels (89). In healthy volunteers and patients with moderate-to-severe rheumatoid arthritis (RA) or plaque psoriasis, r-hIL-18BP showed favorable safety profiles with adverse events mild to moderate in severity and most commonly including injection site reactions (89). When given every 2 d, steady state levels of r-hIL-18BP were achieved within 4 to 6 d with the half-life of the molecule between 33.7 and 40.1 h (89). The efficacy of an antibody blocking IL-18 in type 2 diabetes is under investigation in an ongoing clinical trial (91). The primary advantage of an IL-18 neutralizing/blocking antibody would be a longer elimination half-life that may allow monthly or quarterly administration.
CONCLUSION AND FUTURE DIRECTIONS
Over the last 20 years, IL-18 has proven to be much more than simply an IFN-γ inducing factor. An increase in IL-18 activity has been correlated with a number of human pathologies including AMI, HF, and pressure-overload. Considering the role of IL-18 in mediating acute cardiac effects such as contractile dysfunction and β-AR desensitization as well as chronic cardiac changes like hypertrophy and fibrosis, it is reasonable to hypothesize that IL-18 blockade may represent a valuable strategy in both acute and chronic cardiac diseases (Figure 4). Genetic deletion or neutralization of IL-18 reduces myocyte hypertrophy in models of pressure overload or LPS-induced myocardial dysfunction (60,72). Preventing IL-18 activity by pretreatment with a neutralizing antibody, IL-18BPa, NLRP3 inhibitors or caspase-1 inhibitors improves contractile function and decreases infarct size after ischemia-reperfusion injury, providing the basis for additional studies of these molecules in AMI (63,71,90). Inhibition of IL-18 may, however, impede the physiologic hypertrophic response following pressure overload, and lead to maladaptive remodeling (53). Preclinical studies of IL-18 targeted treatments with IL-18BP or IL-18Ab given during ischemia or at the time of reperfusion are lacking. If such studies were able to confirm a protective effect of IL-18 that is maintained over time and not associated with an impairment in infarct healing in preclinical AMI modes, then pilot clinical trials in ST-segment elevation AMI (STEMI) and non-STEMI may be warranted, as has occurred with IL-1 blockers (92–95). A comparison between IL-18 and IL-1 blockers in AMI would provide further understanding of whether the two cytokines have overlapping or synergistic effects or whether IL-18 mediates IL-1 effects (as suggested in [74]). The acute cardiodepressant effect of IL-18 suggests that IL-18 blockade may represent a valuable treatment for acute decompensated or chronic symptomatic HF. The effects of IL-18 appear to affect both systolic and diastolic function (51,52). Preliminary data with IL-1 blockade show that IL-1 activity may be a modifiable factor in patients with HF (1,3,94,96,97) and the mechanistic links between IL-18 and IL-1 suggest that IL-18 blockade may have similar beneficial effects (1,74). Therefore IL-18 blockade may be used in those patients with elevated IL-1 to improve LV function without altering other IL-1 mediated effects.
Figure 4.

Schematic of IL-18 activation and signaling with potential targets for pharmacologic intervention.
Heart failure with preserved ejection fraction (HFPEF) is a disease characterized by impaired filling of the LV due to increased fibrosis and cardiomyocyte hypertrophy (98). Inflammatory cytokines contribute to the pathophysiology of HFPEF (96,99), thus IL-18 is a potential target for HFPEF due to its prohypertrophic and profibrotic effects (56–60,84,86,87). The chronic prohypertrophic and profibrotic effects of IL-18 in animal studies suggest that a longer duration of IL-18 blockade may prevent, or even reverse, the development of pathologic cardiac hypertrophy; however it also may prevent development of physiologic hypertrophy (53).
Targeted intervention of IL-18 might be advantageous considering the differences in signaling with IL-1 on COX-2 induction (39). The effects of IL-18 blockade on the host defense against opportunistic infections in patients will, however, need to be assessed (100–102). The decision to proceed with one or more pilot studies in this area will likely depend on the safety and pharmacokinetic profiles of the agents. Short-acting agents (for example, r-hIL-18BP) may be advantageous for conditions in which a rapid on/rapid off effect is desired and a short-term treatment is envisioned; whereas long-acting agents (for example, an IL-18 neutralizing antibody) will likely serve well for conditions in which longer duration of treatment is necessary. Many antiinflammatory strategies found effective in the animal models of heart disease have, however, failed to show beneficial effects in clinical trials, and therefore caution should be used when extrapolating preclinical results to clinical use (3).
ACKNOWLEDGMENT
A Abbate and BW Van Tassell are supported by research grants from the American Heart Association and the National Institutes of Health. E Mezzaroma and S Toldo are supported by American Heart Association postdoctoral grants.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Van Tassell BW, et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PloS One. 2012;7:e33438. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go AS, et al. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Amer Coll Cardiol. 2014;63:1593–603. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Gullestad L, et al. Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 2012;122:23–35. doi: 10.1159/000338166. [DOI] [PubMed] [Google Scholar]
- 5.Kalogeropoulos AP, Georgiopoulou VV, Butler J. From risk factors to structural heart disease: the role of inflammation. Heart Fail Clin. 2012;8:113–23. doi: 10.1016/j.hfc.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Dinarello CA, Pomerantz BJ. Proinflammatory cytokines in heart disease. Blood Purif. 2001;19:314–21. doi: 10.1159/000046960. [DOI] [PubMed] [Google Scholar]
- 7.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–98. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 8.Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–53. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura K, Okamura H, Wada M, Nagata K, Tamura T. Endotoxin-induced serum factor that stimulates gamma interferon production. Infec Immun. 1989;57:590–5. doi: 10.1128/iai.57.2.590-595.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–8. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okamura H, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 12.Ahn HJ, et al. A mechanism underlying synergy between IL-12 and IFN-gamma-inducing factor in enhanced production of IFN-gamma. J Immunol. 1997;159:2125–2131. [PubMed] [Google Scholar]
- 13.Bazan JF, Timans JC, Kastelein RA. A newly defined interleukin-1? Nature. 1996;379:591. doi: 10.1038/379591a0. [DOI] [PubMed] [Google Scholar]
- 14.Artlett CM. The role of the NLRP3 inflammasome in fibrosis. Open Rheumatol J. 2012;6:80–6. doi: 10.2174/1874312901206010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mezzaroma E, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Nat Acad Sci U S A. 2011;108:19725–30. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu Y. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997;275:206–9. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 17.Ghayur T, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–23. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 18.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Nat Acad Sci U S A. 1999;96:2256–61. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–61. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 20.Toldo S, et al. Interleukin-1beta immunoneutralization improves cardiac remodeling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol. 2013;98:734–45. doi: 10.1113/expphysiol.2012.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawaguchi M, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi M. NLRP3 Inflammasome as a novel player in myocardial infarction. Int Heart J. 2014;55:101–5. doi: 10.1536/ihj.13-388. [DOI] [PubMed] [Google Scholar]
- 23.Saxena A, et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–48. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugawara S, et al. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol. 2001;167:6568–75. doi: 10.4049/jimmunol.167.11.6568. [DOI] [PubMed] [Google Scholar]
- 25.Joosten LA, et al. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009;60:3651–62. doi: 10.1002/art.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Omoto Y, et al. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J Immunol. 2006;177:8315–9. doi: 10.4049/jimmunol.177.12.8315. [DOI] [PubMed] [Google Scholar]
- 27.Omoto Y, et al. Granzyme B is a novel interleukin-18 converting enzyme. J Dermatol Sci. 2010;59:129–35. doi: 10.1016/j.jdermsci.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–32. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson SE, et al. Expression and alternative processing of IL-18 in human neutrophils. Eur J Immunol. 2006;36:722–31. doi: 10.1002/eji.200535402. [DOI] [PubMed] [Google Scholar]
- 30.Bellora F, et al. M-CSF induces the expression of a membrane-bound form of IL-18 in a subset of human monocytes differentiating in vitro toward macrophages. Eur J Immunol. 2012;42:1618–26. doi: 10.1002/eji.201142173. [DOI] [PubMed] [Google Scholar]
- 31.Dinarello CA. Membrane interleukin-18 revisits membrane IL-1alpha in T-helper type 1 responses. Eur J Immunol. 2012;42:1385–7. doi: 10.1002/eji.201242635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akita K, et al. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J Biol Chem. 1997;272:26595–603. doi: 10.1074/jbc.272.42.26595. [DOI] [PubMed] [Google Scholar]
- 33.Torigoe K, et al. Purification and characterization of the human interleukin-18 receptor. J Biol Chem. 1997;272:25737–42. doi: 10.1074/jbc.272.41.25737. [DOI] [PubMed] [Google Scholar]
- 34.Born TL, Thomassen E, Bird TA, Sims JE. Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J Biol Chem. 1998;273:29445–50. doi: 10.1074/jbc.273.45.29445. [DOI] [PubMed] [Google Scholar]
- 35.Parnet P, Garka KE, Bonnert TP, Dower SK, Sims JE. IL-1Rrp is a novel receptor-like molecule similar to the type I interleukin-1 receptor and its homologues T1/ST2 and IL-1R AcP. J Biol Chem. 1996;271:3967–70. doi: 10.1074/jbc.271.8.3967. [DOI] [PubMed] [Google Scholar]
- 36.Boraschi D, et al. IL-37: a new anti-inflammatory cytokine of the IL-1 family. Eur Cytokine Net. 2011;22:127–47. doi: 10.1684/ecn.2011.0288. [DOI] [PubMed] [Google Scholar]
- 37.Nold MF, et al. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11:1014–22. doi: 10.1038/ni.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riva F, et al. TIR8/SIGIRR is an interleukin-1 receptor/toll like receptor family member with regulatory functions in inflammation and immunity. Front Immunol. 2012;3:322. doi: 10.3389/fimmu.2012.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JK, et al. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A. 2004;101:8815–20. doi: 10.1073/pnas.0402800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adachi O, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 41.Kanakaraj P, et al. Defective interleukin (IL)-18-mediated natural killer and T helper cell type 1 responses in IL-1 receptor-associated kinase (IRAK)-deficient mice. J Exp Med. 1999;189:1129–38. doi: 10.1084/jem.189.7.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kojima H, et al. Interleukin-18 activates the IRAK-TRAF6 pathway in mouse EL-4 cells. Biochem Biophys Res Comm. 1998;244:183–6. doi: 10.1006/bbrc.1998.8236. [DOI] [PubMed] [Google Scholar]
- 43.Wyman TH, et al. Physiological levels of interleukin-18 stimulate multiple neutrophil functions through p38 MAP kinase activation. J Leukoc Biol. 2002;72:401–9. [PubMed] [Google Scholar]
- 44.Novick D, et al. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–36. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 45.Kim SH, et al. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Nat Acad Sci U S A. 2000;97:1190–5. doi: 10.1073/pnas.97.3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mallat Z, et al. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598–603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- 47.Hong SN, et al. Atherosclerotic biomarkers and aortic atherosclerosis by cardiovascular magnetic resonance imaging in the Framingham Heart Study. J. Am. Heart Assoc. 2013;2:e000307. doi: 10.1161/JAHA.113.000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blankenberg S. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106:24–30. doi: 10.1161/01.cir.0000020546.30940.92. [DOI] [PubMed] [Google Scholar]
- 49.Jefferis BJ, et al. Interleukin 18 and coronary heart disease: prospective study and systematic review. Atherosclerosis. 2011;217:227–33. doi: 10.1016/j.atherosclerosis.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartford M, et al. Interleukin-18 as a predictor of future events in patients with acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2010;30:2039–46. doi: 10.1161/ATVBAHA.109.202697. [DOI] [PubMed] [Google Scholar]
- 51.Blankenberg S, et al. Interleukin-18 and the risk of coronary heart disease in European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME) Circulation. 2003;108:2453–9. doi: 10.1161/01.CIR.0000099509.76044.A2. [DOI] [PubMed] [Google Scholar]
- 52.Kaptoge S, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Europ Heart J. 2013;35:578–89. doi: 10.1093/eurheartj/eht367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seta Y, et al. Interleukin 18 in acute myocardial infarction. Heart. 2000;84:668. doi: 10.1136/heart.84.6.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mallat Z, et al. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. FASEB J. 2004;18:1752–4. doi: 10.1096/fj.04-2426fje. [DOI] [PubMed] [Google Scholar]
- 55.Gangemi S, et al. Increased circulating Interleukin-18 levels in centenarians with no signs of vascular disease: another paradox of longevity? Exp Gerontol. 2003;38:669–72. doi: 10.1016/s0531-5565(03)00061-5. [DOI] [PubMed] [Google Scholar]
- 56.Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005;280:4553–67. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- 57.Gardner D. Natriuretic peptides: markers or modulators of cardiac hypertrophy? Trends Endocrinol Metabol. 2003;14:411–6. doi: 10.1016/s1043-2760(03)00113-9. [DOI] [PubMed] [Google Scholar]
- 58.Woldbaek PR, et al. Daily administration of interleukin-18 causes myocardial dysfunction in healthy mice. Am J Physiol Heart Circ Physiol. 2005;289:H708–14. doi: 10.1152/ajpheart.01179.2004. [DOI] [PubMed] [Google Scholar]
- 59.Platis A, et al. The effect of daily administration of IL-18 on cardiac structure and function. Perfusion. 2008;23:237–42. doi: 10.1177/0267659108101511. [DOI] [PubMed] [Google Scholar]
- 60.Colston JT, et al. Interleukin-18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun. 2007;354:552–8. doi: 10.1016/j.bbrc.2007.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang H, et al. Effect of hydrodynamics-based delivery of IL-18BP fusion gene on rat experimental autoimmune myocarditis. Clin Exp Med. 2013 2013 Oct 12; doi: 10.1007/s10238-013-0260-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 62.Woldbaek PR, et al. Increased cardiac IL-18 mRNA, pro-IL-18 and plasma IL-18 after myocardial infarction in the mouse; a potential role in cardiac dysfunction. Cardiovasc Res. 2003;59:122–31. doi: 10.1016/s0008-6363(03)00339-0. [DOI] [PubMed] [Google Scholar]
- 63.Venkatachalam K, et al. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem. 2009;284:7853–65. doi: 10.1074/jbc.M808824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang M, et al. IL-18 binding protein-expressing mesenchymal stem cells improve myocardial protection after ischemia or infarction. Proc Nat Acad Sci U S A. 2009;106:17499–504. doi: 10.1073/pnas.0908924106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toldo S, et al. Formation of the inflammasome in acute myocarditis. Int. J. Cardiol. 2014;171:e119–21. doi: 10.1016/j.ijcard.2013.12.137. [DOI] [PubMed] [Google Scholar]
- 66.Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev. 2010;15:543–62. doi: 10.1007/s10741-010-9168-4. [DOI] [PubMed] [Google Scholar]
- 67.Puren AJ, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J Clin Invest. 1998;101:711–21. doi: 10.1172/JCI1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morel JC, Park CC, Kumar P, Koch AE. Interleukin-18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFkappaB activation. Lab Invest. 2001;81:1371–83. doi: 10.1038/labinvest.3780351. [DOI] [PubMed] [Google Scholar]
- 69.Netea MG, Kullberg BJ, Verschueren I, Van Der Meer JW. Interleukin-18 induces production of proinflammatory cytokines in mice: no intermediate role for the cytokines of the tumor necrosis factor family and interleukin-1beta. Eur J Immunol. 2000;30:3057–60. doi: 10.1002/1521-4141(200010)30:10<3057::AID-IMMU3057>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 70.Dinarello CA. IL-18: A TH1-inducing, pro inflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol. 1999;103:11–24. doi: 10.1016/s0091-6749(99)70518-x. [DOI] [PubMed] [Google Scholar]
- 71.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci. 2001;98:2871–6. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raeburn CD, et al. Neutralization of IL-18 attenuates lipopolysaccharide-induced myocardial dysfunction. Am J Physiol Heart Circ Phys. 2002;283:H650–7. doi: 10.1152/ajpheart.00043.2002. [DOI] [PubMed] [Google Scholar]
- 73.Leung BP, et al. A role for IL-18 in neutrophil activation. J Immunol. 2001;167:2879–86. doi: 10.4049/jimmunol.167.5.2879. [DOI] [PubMed] [Google Scholar]
- 74.Toldo S, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025–31. doi: 10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fowler MB, Laser JA, Hopkins GL, Minobe W, Bristow MR. Assessment of the beta-adrenergic receptor pathway in the intact failing human heart: progressive receptor down-regulation and subsensitivity to agonist response. Circulation. 1986;74:1290–302. doi: 10.1161/01.cir.74.6.1290. [DOI] [PubMed] [Google Scholar]
- 76.Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac beta-adrenergic receptors, and heart failure. Circulation. 2000;101:1634–7. doi: 10.1161/01.cir.101.14.1634. [DOI] [PubMed] [Google Scholar]
- 77.Naga Prasad SV, Nienaber J, Rockman HA. Beta-adrenergic axis and heart disease. Trends Gen. 2001;17:S44–49. doi: 10.1016/s0168-9525(01)02487-8. [DOI] [PubMed] [Google Scholar]
- 78.Murray DR, et al. Beta2 adrenergic activation induces the expression of IL-18 binding protein, a potent inhibitor of isoproterenol induced cardiomyocyte hypertrophy in vitro and myocardial hypertrophy in vivo. J Mol Cell Cardiol. 2012;52:206–18. doi: 10.1016/j.yjmcc.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chandrasekar B, et al. Beta-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Comm. 2004;319:304–11. doi: 10.1016/j.bbrc.2004.04.185. [DOI] [PubMed] [Google Scholar]
- 80.Chandrasekar B, et al. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004;279:20221–33. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 81.Dao T, Ohashi K, Kayano T, Kurimoto M, Okamura H. Interferon-gamma-inducing factor, a novel cytokine, enhances Fas ligand-mediated cytotoxicity of murine T helper 1 cells. Cell Immunol. 1996;173:230–5. doi: 10.1006/cimm.1996.0272. [DOI] [PubMed] [Google Scholar]
- 82.Tsutsui H, et al. IFN-gamma-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J Immunol. 1996;157:3967–73. [PubMed] [Google Scholar]
- 83.Chandrasekar B, Valente AJ, Freeman GL, Mahimainathan L, Mummidi S. Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-kappaB-dependent PTEN activation. Biochem Biophys Res Comm. 2006;339:956–63. doi: 10.1016/j.bbrc.2005.11.100. [DOI] [PubMed] [Google Scholar]
- 84.Yu Q, et al. IL-18 induction of osteopontin mediates cardiac fibrosis and diastolic dysfunction in mice. Am J Physiol Heart Circ Physiol. 2009;297:H76–85. doi: 10.1152/ajpheart.01285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stawowy P, et al. Increased myocardial expression of osteopontin in patients with advanced heart failure. Eur J Heart Fail. 2002;4:139–46. doi: 10.1016/s1388-9842(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 86.Reddy VS, et al. Interleukin-18 stimulates fibronectin expression in primary human cardiac fibroblasts via PI3K-Akt-dependent NF-kappaB activation. J Cell Physiol. 2008;215:697–707. doi: 10.1002/jcp.21348. [DOI] [PubMed] [Google Scholar]
- 87.Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine. 2011;53:19–28. doi: 10.1016/j.cyto.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.First Time in Human Study of Intravenous Interleukin-18 Antibody (A18110040) [Internet] Bethesda (MD): US National Institutes of Health, U.S. National Library of Medicine; [updated 2012 Dec 19; cited 2014 May 2]. Available from: http://clinicaltrials.gov/ct2/show/NCT01035645. NLM identifier, NCT01035645. [Google Scholar]
- 89.Tak PP, Bacchi M, Bertolino M. Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur J Drug Metab Pharmacokinet. 2006;31:109–16. doi: 10.1007/BF03191127. [DOI] [PubMed] [Google Scholar]
- 90.Marchetti C, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury following ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2013;63:316–22. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Investigate the Efficacy and Safety of GSK1070806 in Obese Subjects With T2DM [Internet] Bethesda (MD): US National Institutes of Health, U.S. National Library of Medicine; [updated 2014 Apr 14; cited 2014 May 2]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01648153?term=GSK1070806&rank=1. NLM identifier, NCT01648153. [Google Scholar]
- 92.Crossman DC, et al. Investigation of the effect of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-HEART Study) Trials. 2008;9:8. doi: 10.1186/1745-6215-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abbate A, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–7.e1. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 94.Abbate A, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013;111:1394–400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.IL-1 Blockade in Acute Myocardial Infarction (VCU-ART3) [Internet] Bethesda (MD): US National Institutes of Health, U.S. National Library of Medicine; [updated 2013 Sep 20; cited 2014 May 2]. Available from: http://clinicaltrials.gov/ct2/show/NCT01950299?term=nct01950299&rank=1. NLM identifier, NCT01950299. [Google Scholar]
- 96.Van Tassell BW, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study) Am J Cardiol. 2014;113:321–7. doi: 10.1016/j.amjcard.2013.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. 2013;128:1910–23. doi: 10.1161/CIRCULATIONAHA.113.003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–9. doi: 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Westermann D, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 100.Stuyt RJ, et al. Role of interleukin-18 in host defense against disseminated Candida albicans infection. Infect Immun. 2002;70:3284–6. doi: 10.1128/IAI.70.6.3284-3286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huang X, McClellan SA, Barrett RP, Hazlett LD. IL-18 contributes to host resistance against infection with Pseudomonas aeruginosa through induction of IFN-gamma production. J Immunol. 2002;168:5756–63. doi: 10.4049/jimmunol.168.11.5756. [DOI] [PubMed] [Google Scholar]
- 102.Liu B, et al. Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J Gen Virol. 2004;85:423–8. doi: 10.1099/vir.0.19596-0. [DOI] [PubMed] [Google Scholar]