

Abstract
Robinow syndrome is a skeletal dysplasia with both autosomal dominant and autosomal recessive inheritance patterns. It is characterized by short stature, limb shortening, genital hypoplasia and craniofacial abnormalities. The etiology of dominant Robinow syndrome is unknown, however the phenotypically more severe autosomal recessive form of Robinow syndrome has been associated with mutations in the orphan tyrosine kinase receptor, ROR2, which has recently been identified as a putative WNT5A receptor. Here we show that two different missense mutations in WNT5A, which result in amino acid substitutions of highly conserved cysteines, are associated with autosomal dominant Robinow syndrome. One mutation has been found in all living affected members of the original family described by Meinhard Robinow and another in a second unrelated patient. These missense mutations result in decreased WNT5A activity in functional assays of zebrafish and Xenopus development. This work suggests that a WNT5A/ROR2 signal transduction pathway is important in human craniofacial and skeletal development, and that proper formation and growth of these structures is sensitive to variations in WNT5A function.
Introduction
The Wnt family of signaling proteins in humans has 19 identified members, with the major subgroup of Wnts (WNT1, WNT3A, WNT8) signaling through activation of β-catenin dependent transcription (Nusse, 2005). This canonical, or β-catenin dependent Wnt signaling pathway functions primarily to activate cell proliferation and cell fate change during development. A non-canonical, β-catenin independent signal transduction pathway that controls cell polarity or movement has also been identified (Heisenberg et al., 2000). Wnt5a has been shown to signal in a non-canonical fashion, modulating cellular movements independent of β-catenin (Heisenberg et al., 2000; Slusarski et al., 1997). Non-canonical Wnt signaling is necessary for the directional cell migration of pancreatic islet cell progenitors during pancreas formation in zebrafish and mice (Kim et al., 2005). In addition, non-canonical Wnt5a signaling regulates directional cell migration necessary for secondary palate fusion during mouse development (He et al., 2008). Non-canonical Wnt signaling is an area of active research in developmental biology, and it may involve multiple downstream pathways. Evidence exists for a Wnt5a signaling cascade involving the intracellular activation of calcium/calmodulin-dependent protein kinase and protein kinase C via the Frizzled2 transmembrane receptor, resulting in Ca2+ fluxes (Kohn and Moon, 2005). Other studies suggest that Wnt5a can signal through the orphan tyrosine kinase receptor, Ror2, but there may be multiple downstream mediators of this ligand-receptor complex (Mikels and Nusse, 2006; Oishi et al., 2003; Schambony and Wedlich, 2007). In the work presented here, we demonstrate that mutations in WNT5A are associated with human phenotypes similar to those found with loss-of-function ROR2 mutations (Afzal et al., 2000; van Bokhoven et al., 2000), suggesting a role for this newly identified pathway in human development and disease.
In 1969, Meinhard Robinow and colleagues described a human syndrome characterized by short stature, mesomelic limb shortening, hypertelorism, mandibular hypoplasia, irregular dental alignment and hypoplastic external genitalia (Robinow et al., 1969). Based on the initial pedigree, Robinow syndrome was recognized as an autosomal dominant inherited syndrome [MIM 180700] with high penetrance. Over 100 patients with Robinow syndrome have since been identified in families with both autosomal dominant and autosomal recessive inheritance patterns (Patton and Afzal, 2002).
The autosomal recessive form of Robinow syndrome [MIM 268310], which is characterized by more severe skeletal, vertebral and craniofacial abnormalities (Mazzeu et al., 2007b; Patton and Afzal, 2002) is often caused by loss-of-function mutations in the gene encoding the tyrosine kinase-like orphan receptor 2, ROR2 (van Bokhoven et al., 2000),(Afzal et al., 2000). Recently, Ror2 has been identified as a putative receptor for Wnt5a (Mikels and Nusse, 2006; Schambony and Wedlich, 2007). Wnt5a and Ror2 are expressed in adjacent and partially overlapping domains during mouse embryogenesis, and Wnt5a can directly bind to the extracellular cysteine-rich domain of Ror2 (Nomi et al., 2001; Oishi et al., 2003; Schleiffarth et al., 2007). Wnt5a null mice and Ror2 null mice exhibit phenotypes grossly similar to those found in Robinow syndrome patients, including shortening of the anterior-posterior axis, facial dysmorphism, genital hypoplasia and cardiac defects (DeChiara et al., 2000; Oishi et al., 2003; Schleiffarth et al., 2007; Yamaguchi et al., 1999). Wnt5a null mice have a more pronounced phenotype compared to Ror2 null mice (Oishi et al., 2003), but these differences can be explained by functional compensation by the related gene Ror1 in Ror2 nulls (Nomi et al., 2001). Ror1/Ror2 double mutant mice exhibit a Robinow syndrome-like phenotype that more closely resembles Wnt5a null mice (Nomi et al., 2001; Oishi et al., 2003; Yamaguchi et al., 1999). Wnt5a null mice exhibit a phenotype that is more severe than human dominant Robinow syndrome patients with perinatal lethality, 100% penetrance of cardiac defects, and presence of rib fusions (Schleiffarth et al., 2007; Yamaguchi et al., 1999). Heterozygous mutations in ROR2 that cause brachydactyly type B [MIM 113000] do not result in simple loss-of-function like those ROR2 mutations described in recessive Robinow syndrome. Rather, the ROR2 mutations reported in patients with brachydactyly type B are thought to result in dominant-negative mutations (Oldridge et al., 2000; Schwabe et al., 2000; Stricker et al., 2006). These studies suggest that mutations in other genes in a ROR2 signal transduction pathway may be associated with dominant Robinow syndrome. We tested the hypothesis that mutations in WNT5A are associated with autosomal dominant Robinow syndrome, thereby identifying an important role for this signaling pathway in human craniofacial and skeletal development.
Results
Wnt5a null mice and Ror2 null mice exhibit phenotypes similar to those found in Robinow syndrome patients, including shortening of the anterior-posterior axis, facial dysmorphism, genital hypoplasia and cardiac defects (Afzal et al., 2000; Mazzeu et al., 2007b; Oishi et al., 2003; Robinow et al., 1969; Schwabe et al., 2004; van Bokhoven et al., 2000). Figure 1 illustrates the phenotypic characteristics of this well described mouse model and patients with a clinical diagnosis of dominant Robinow syndrome (Mazzeu et al., 2007b; Oishi et al., 2003; Robinow et al., 1969; Yamaguchi et al., 1999). These observations led us to screen for WNT5A mutations in patients with dominant Robinow syndrome.
Figure 1. Wnt5a−/− mice have anatomical defects phenotypically similar to patients with autosomal dominant Robinow syndrome.
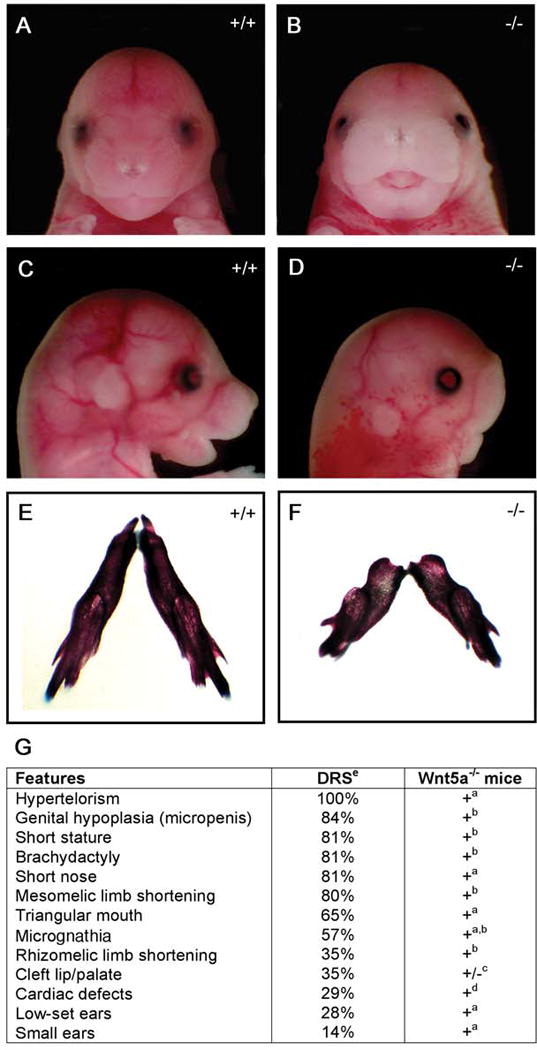
Wnt5a−/− mouse embryos at E16.5 (B) have widely spaced eyes (hypertelorism) and a triangular mouth compared to controls (A). Lateral views (C, D) reveal flattening of the facial profile, micrognathia, and small, low set ears in the Wnt5a−/− embryos (D) compared to control littermates (C). Skeletal preparations at birth show mandibular hypoplasia in Wnt5a mutants (F) compared to controls (E). G) Percentage of reported findings in dominant Robinow syndrome patients compared to phenotypes in Wnt5a−/− mice. aData presented in this figure, b (Yamaguchi et al., 1999) c(Yang et al., 2003) d(Schleiffarth et al., 2007). ePercentages of dominant Robinow syndrome (DRS) patients with each abnormality was adapted from ((Mazzeu et al., 2007b).
Our initial screening for WNT5A mutations in patients with dominant Robinow syndrome focused on the family in which the syndrome was initially described (Robinow et al., 1969). In that report, the skeletal, genital and craniofacial abnormalities of four living affected family members were described in detail. We obtained DNA from the original four patients initially described by Dr. Robinow and colleagues, and from all additional living family members both affected and unaffected (Fig. 2C). Family members not previously reported (Generation V), who are offspring of the affected female sibling of the proband described in the original report (Robinow et al., 1969), manifested mesomelic limb shortening, short stature and characteristic facial features. The affected male family members have no children.
Figure 2. Index Family – Robinow syndrome.
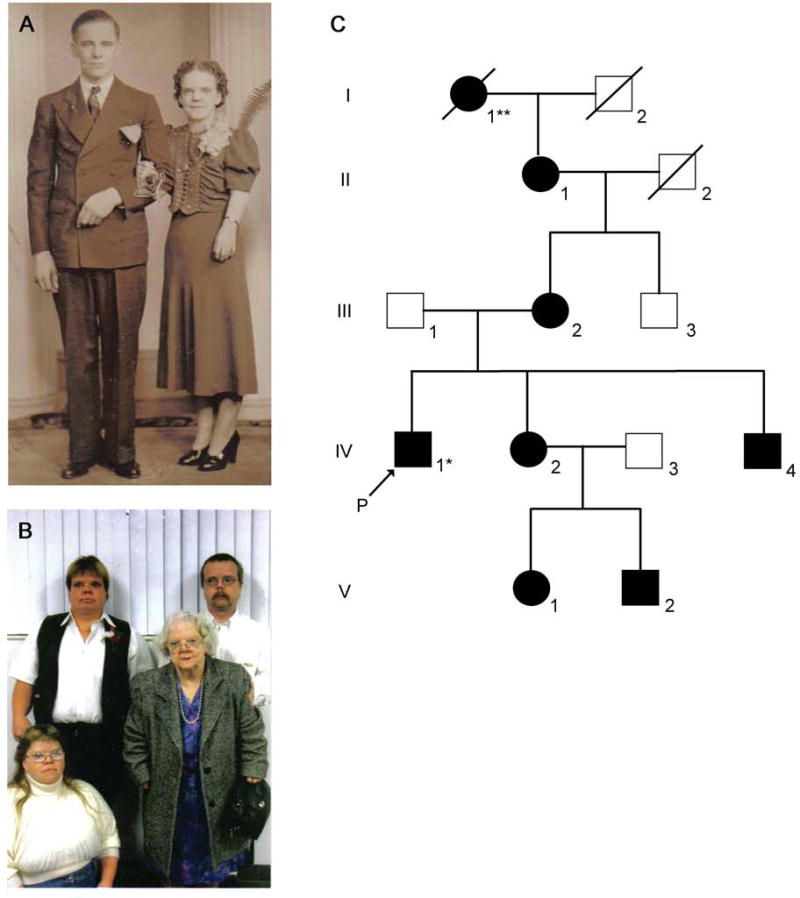
A) Photograph of an unaffected male (II-2) and his wife (II-1) with dominant Robinow syndrome. Features evident in the affected female include short stature, hypertelorism, and mesomelic limb shortening. B) Proband (Robinow et al., 1969), siblings and grandmother, who is the female pictured in panel A. Photograph in panel B from left to right, top row: male IV-4, male IV-1 (*proband); bottom row: female IV-2, and female II-1. Robinow syndrome features (described above) are evident in all four individuals. Additional images of all four individuals have been reported previously (Robinow et al., 1969). C) Pedigree of the index family with dominant Robinow syndrome originally described in (Robinow et al., 1969). An additional generation (V) is shown in this pedigree. ** Original pedigree reported male I-2 and not female I-1 had dominant Robinow syndrome (Robinow et al., 1969). We confirmed female I-1, and not male I-2, had dominant Robinow syndrome by consulting the family.
PCR amplification (Tables 1) and sequencing of the coding regions of WNT5A in this family revealed heterozygous missense mutations in two sequential base pairs in exon 4 of WNT5A. This mutation of WNT5A, 544-545CT→TC (Fig. 3D) results in an amino acid substitution, C182R, at a highly conserved cysteine residue (Fig. 3B). This mutation is present in all seven affected family members and not present in the only living unaffected family member, (III-3) (Fig. 2C and data not shown). This mutation is in a highly conserved region of the WNT5A protein (Figure 3D), and was not identified in 196 unaffected control samples by ARMS PCR or sequencing, or by review of human single nucleotide polymorphism (SNP) databases (http://hapmap.org/, GeneCards™ genome-http://www.stanford.edu/cgi-bin/genecards/index.shtml, and UCSC Genome Browser http://www.genome.ucsc.edu/), which showed no human SNPs at WNT5A 544-545. No other mutations in the coding regions of WNT5A were identified in this family (Fig. 2C).
Table 1.
WNT5A Primer Sequences
| Exons | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| 1 | CGCTTCAGCTCCGGTTCACT | CGACGCTGGAGTTCCAGCTT |
| 2 | CGGATGCTTCCGTTGTTT | CCAGCATTAAATATTGCCGC |
| 3 | GGGACTGCACAATACCTCCG | GAGCTGGAAGGCATCCTCCT |
| 4 | CTCCCTACCCTCTACCCCAA | AGGACGGAGCTACAGGGAGA |
| 5 | CTCCATGAATGTGTGTATTG | GCTGCGTGCTGGGTGGCA |
Figure 3. Dominant Robinow syndrome is associated with C83S or C182R WNT5A mutations.
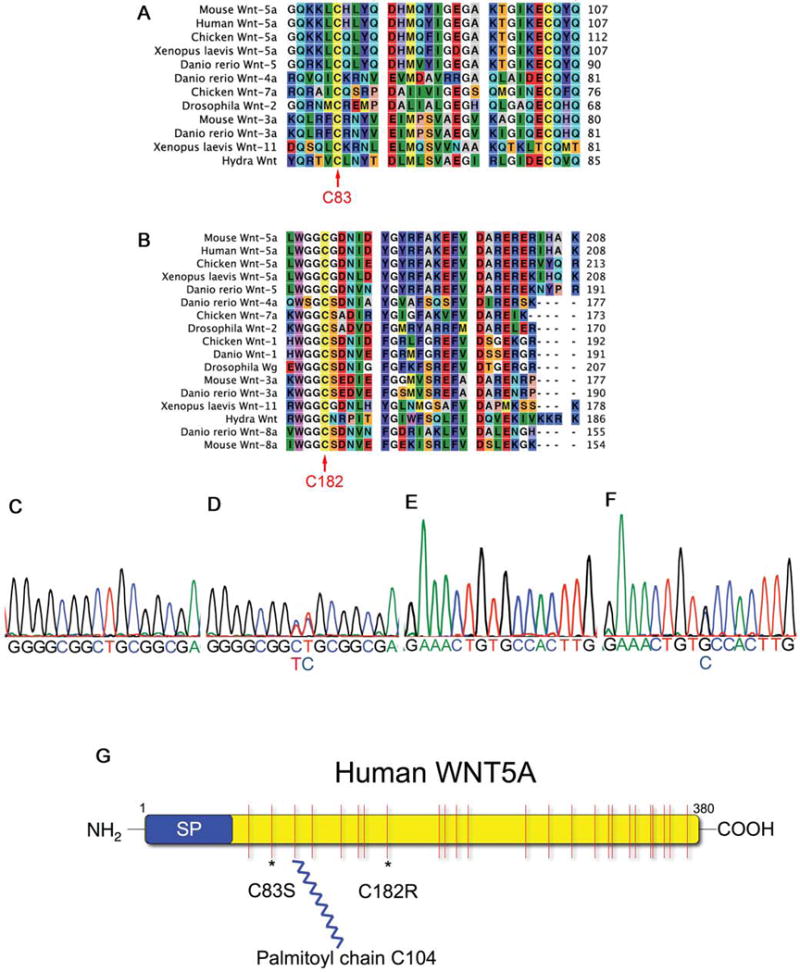
A, B) Sequence alignments show the cysteines (C83 and C182, red arrows) are conserved from human to hydra. C) Sequence data of a WNT5A exon 4 PCR product showing the wild type sequence of base pairs 537–552. D) Exon 4 PCR sequence data of a patient IV-1 showing a missense heterozygous mutation 544-545CT→TC (C182R). E) Control PCR sequence data of WNT5A bases 240–255. F) Sequence of an individual with autosomal dominant Robinow syndrome with a missense heterozygous mutation 248G→C (C83S). G) Schematic of the human WNT5A protein with a signal peptide (SP), 24 cysteines shown in red with the C83S and C182R mutation sites * and the palmitoylation site on C104 (Kurayoshi et al., 2007).
Linkage analysis for this family (Fig. 2) was performed concurrent with WNT5A exon sequencing by using a total of 382 fluorescently labeled short tandem repeat (STR) polymorphic markers (ABI PRISM Linkage Mapping Set MD10.A). All of the affected family members (Fig. 2) showed linkage with a LOD score of 1.78 at chromosome 3 in a region containing the human WNT5A locus. A LOD score of 1.78 is the maximum predicted score achievable with the number of meioses (individuals) available for this pedigree. Importantly, linkage to other candidate genes implicated in WNT5A signaling, including ROR2 and the WNT5A receptor FRIZZLED2, was excluded by this analysis.
We identified a second heterozygous missense WNT5A mutation 248G→C in an unrelated adult patient with a sporadic dominant Robinow syndrome phenotype using the same methods. This mutation predicts an amino acid substitution (C83S)(Fig. 3F) affecting a second highly conserved cysteine residue (Fig. 3A). This patient’s phenotype included marked hypertelorism, short nose, short stature, and mesomelic shortening of the limbs. The parents were unaffected clinically, however no parental DNA is available for testing. Sequence data from 173 control DNA samples did not show the 248G→C missense mutation in exon 3, nor was it found in the SNP databases as described above. Coding regions of WNT5A were sequenced in 23 additional unrelated patients with clinical diagnosis of dominant Robinow syndrome, and no WNT5A mutations resulting in amino acid substitutions were identified. This suggests that mutations in the coding regions of WNT5A cause only a subset of this syndrome as defined by current clinical criteria (Mazzeu et al., 2007b).
Mutations of cysteines in Drosophila wingless (wg), and mammalian Wnts negatively affect Wnt function in multiple assay systems (Mason et al., 1992; McMahon and Moon, 1989; van den Heuvel et al., 1993; Willert et al., 2003). We hypothesize that mutating any of the 24 conserved cysteines might inhibit WNT5A function by altering protein folding, secretion, palmitoylation, or receptor binding. PolyPhen (Polymorphism Phenotyping) protein structure modeling (http://genetics.bwh.harvard.edu/pph/) supports our prediction that the WNT5A mutations identified in our study affect WNT5A function. Based on this modeling, the calculated PSIC software score difference of 3.151 and 4.513 for the WNT5AC83S and WNT5AC182R mutations, respectively, is characterized as “probably damaging”. Therefore, both the WNT5AC83S and WNT5AC182R mutations are predicted to, with a high level of confidence, affect protein function.
The functional relevance of the WNT5A mutations C182R and C83S was tested in vivo by overexpression of mRNA in zebrafish embryos. We have previously shown that wnt5 signaling is required for normal pancreatic islet cell coalescence in zebrafish and mice, and that this process requires a gradient of signal along the anterior-posterior axis in zebrafish (Kim et al., 2005). Disruption of this signaling gradient by mRNA overexpression of human WNT5A results in a failure of insulin-expressing cell coalescence into a pancreatic islet (Fig. 4D, F) when compared to control embryos (Fig. 4B, E). Overexpression of WNT5AC182R mRNA (Fig. 4G, H) and WNT5AC83S mRNA (not shown) are both less effective at disruption of islet cell coalescence than overexpression of wild type WNT5A mRNA in this assay of Wnt signaling function (WNT5AC182R vs control, p = 0.00005; WNT5AC83S vs control, p = 0.002) (Fig 4L). This data suggests that both the C182R and C83S mutations create phenotypes due to expression of less active (hypomorphic) forms of WNT5A.
Figure 4. Cysteine mutations in WNT5A reduce the ability of this protein to activate non-canonical Wnt signaling in zebrafish embryos.
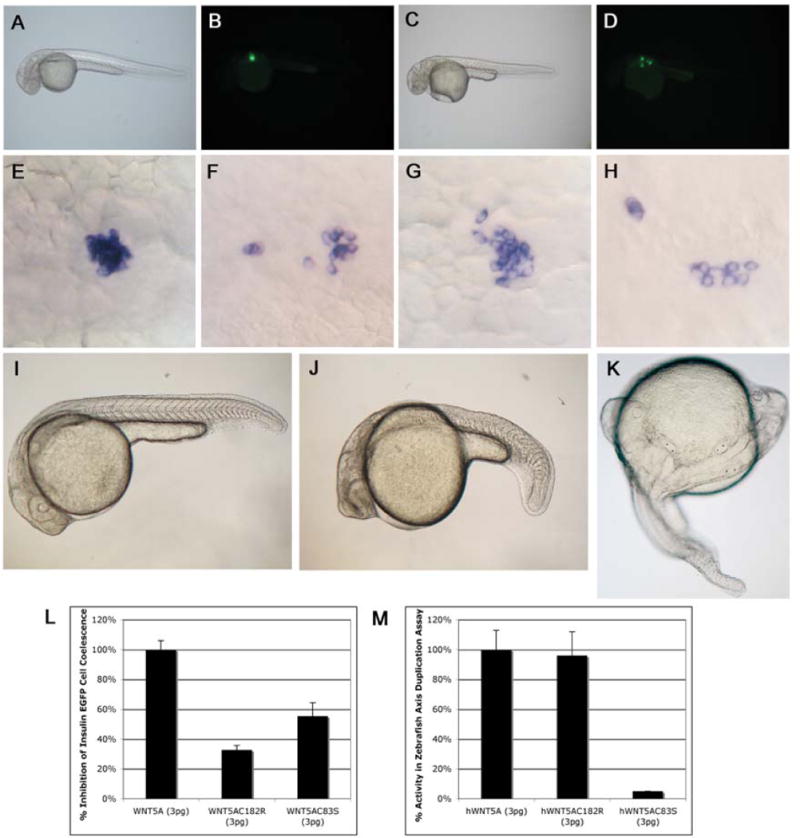
Uninjected brightfield (A) and fluorescent images of insulin-eGFP transgenic zebrafish embryos (B) at 30hpf showing insulin-positive cells have coalesced to form an islet. Insulin-eGFP positive cells in embryos injected with WNT5A mRNA (C, D) do not coalesce properly, providing an in vivo assay for non-canonical Wnt5a activity. In situ hybridization showing insulin-expressing cells coalescing to form the zebrafish islet at 30hpf (E). Injections with WNT5A mRNA (F), WNT5AC182R mRNA (G, H) or WNT5AC83R mRNA (not shown) show scattered insulin expressing cells at 30 hpf, but the efficacy of this effect is reduced in WNT5AC182R (32%±3% n=93 P=0.00005) and WNT5AC83R (55%±9% n=152 P=0.002) mRNA injected embryos compared to WNT5A mRNA injected embryos (100%±6% n=260) (L). I) Uninjected zebrafish embryo at 26hpf. J) Zebrafish embryo at 26hpf injected with dnWNT5A mRNA (25pg) showing a bent tail phenotype similar to pipetail (Wnt5) mutants. Injections of WNT5A mRNA (K) or WNT5AC182R mRNA (not shown) into zebrafish embryos at the one-cell stage results in duplications of the embryonic axis, providing an overexpression-based in vivo activity assay for Wnt signaling competency by the WNT5A sequence variants. Normalizing the axis duplicating activity of WNT5A mRNA injected embryos to 100%±13% n=196, we noted indistinguishable axis duplicating activity 96%±16% n=65 in WNT5AC182 mRNA injected embryos (M). Injections of 3pg of WNT5AC83S mRNA resulted in a significantly different (P=0.001), low percentage of axis duplication activity 3%±0% n=102 compared to the full duplicating activity of WNT5A mRNA at the same dose (3pg) (M). In contrast, dnWNT5A injections did not duplicate the embryonic axis (J).
WNT5A can precociously activate canonical, β-catenin dependent Wnt signaling during early axis formation in frogs (He et al., 1997). Although this scientific scenario is an artificial test for WNT5A because WNT5A does not perform this function in vivo, it is nevertheless a test of biochemical potential for activation of canonical Wnt signaling. We tested the ability of both wild type WNT5A and WNT5AC182R mRNAs to induce duplicate axis formation upon injection into zebrafish embryos, and we noted a similar incidence of duplicated embryonic axes when assayed at 24hpf (Fig. 4). Interestingly, and in sharp contrast to the similarly reduced signaling ability in a non-cell fate Wnt pathway, injection of WNT5AC83S mRNA exhibited a significantly lower incidence of duplicated embryonic axes compared to wild type injected embryos (p = 0.001) (Fig. 4M), suggesting a significant change in the biochemical potential for canonical Wnt signaling in this WNT5A sequence variant. As we do not yet understand the rules whereby a cell distinguishes between the canonical and non-canonical signaling potential of Wnt proteins, this observation suggests a loss of a single cysteine residue can serve as a major delineating structural feature.
To specifically test the hypothesis that these human variants generate antimorphic proteins, we compared the injections of these human variants with injections of mRNA encoding a dominant-negative WNT5A protein form. Injections with a C-terminal truncation, dominant negative mutant, (dn) WNT5A (Sen et al., 2001), did not cause axis duplication, but instead produced phenotypes similar to the Wnt5 loss of function embryos (pipetail) (Hammerschmidt et al., 1996) (Fig. 4J). This data suggests that the C83S and C182R mutations are not acting as dominant-negative inhibitors of Wnt signaling, but they are instead acting as partial loss of function or hypomorphic alleles of WNT5A.
Additional studies were performed to test the function of these mutations in altering convergent-extension movements during Xenopus gastrulation (Moon et al., 1993) and to place them in a novel ROR2-mediated noncanonical Wnt pathway (Schambony and Wedlich, 2007). In 1993, Moon et al showed that Xwnt5A blocks activin-mediated elongation of blastula stage animal caps without altering mesoderm formation (Moon et al., 1993). In order to determine the effect of the WNT5AC182R and WNT5AC83S mutations on cell movement in Xenopus embryos, we compared control animal caps isolated at prebastula stages to control animal caps exposed to activin protein, and to animal caps harvested from embryos that had been injected at the two cell stage with either moderate dose (30pg) or high dose (200 pg) HWNT5A, or WNT5AC182R and WNT5AC83S mutant constructs (Fig. 5) and then exposed to activin at blastula stages. Non-activin treated uninjected (Fig 5A) and embryos injected with 200 pg WNT5AC182R mRNA (Fig. 5C) are also shown. These experiments demonstrated reduced ability of the mutant forms of WNT5A to block activin-mediated cell movement during gastrulation at moderate doses (Fig. 5 D,E,F), however at higher doses all three forms were effective at blocking activin-mediated cell movement and animal cap elongation (Fig. 5 G,H,I). This observation confirms that while both the WNT5AC182R and WNT5AC83S mutations have reduced activity in this assay system, some functional ability to activate WNT5A signaling is retained and supports our hypothesis that these mutations represent an alteration of WNT5A function due to a hypomorphic allele, and not a dominant negative mechanism of action.
Figure 5. WNT5AC182R and WNT5AC182S have reduced ability to block activin-mediated cell movements during Xenopus gastrulation.

Control (uninjected) blastula stage animal caps round up in normal culture conditions (A) and elongate when cultured in activin protein (B). Embryos injected with 200 pg of WNT5AC182R mRNA into each blastomere at a two cell stage form normal appearing animal caps under control conditions (C). Moderate dose (30 pg) WNT5A mRNA (D) and High dose (200 pg WNT5A mRNA) (G) block activin-induced animal cap elongation. In contrast, moderate dose (30 pg) WNT5AC182 mRNA (E) and WNT5AC83S mRNA (F) do not inhibit activin-induced elongation fully. In contrast, higher doses (200 pg) of WNT5AC182R mRNA (H) and WNT5AC83S mRNA (I) injected at a two cell stage do inhibit activin-induced animal cap elongation.
Recent studies have demonstrated Wnt5A/ROR2 mediated activation of noncanonical Wnt signaling pathways and of Xenopus paraxial protocadherin (XPAPC)(Schambony and Wedlich, 2007). We used a simple assay of XPAPC activation as evidenced by induction of mRNA expression to determine if the mutations described in patients with Robinow syndrome acted through the WNT5a/ROR2 signaling pathway. We found that in this assay, the WNT5AC83S mutation had a significantly reduced ability to induce XPAPC expression when compared to wild type HWNT5A (Fig. 6) and that the WNT5AC182R mutation trended to reduced ability to alter XPAPC expression when compared to wild type mRNA, without reaching statistical significance (data not shown).
Figure 6. XPAPC induction after wild type and mutant WNT5A overexpression as assessed by qPCR.
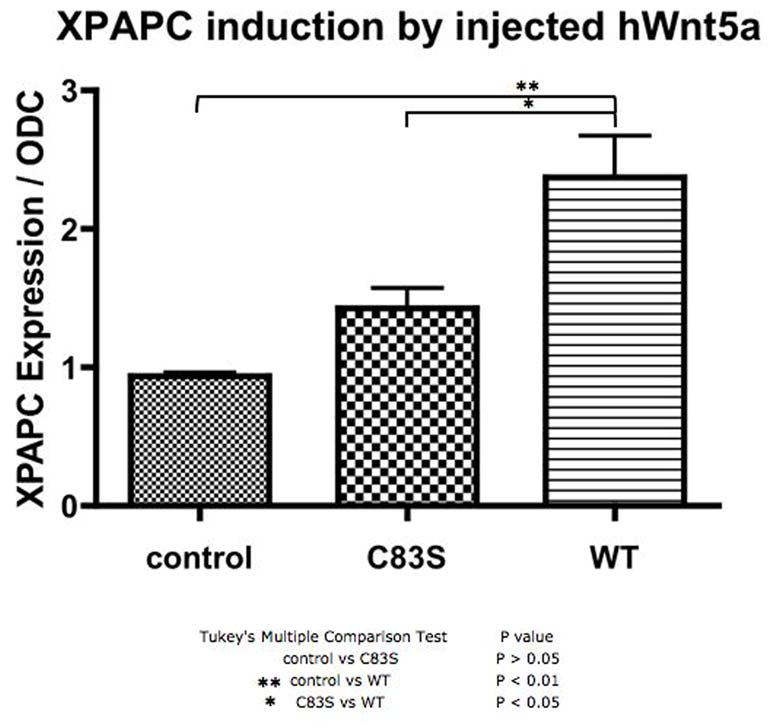
Control embryos at stage 10.5, and embryos injected with the WNT5AC83S mRNA or wild type HWNT5A mRNA at the four cell stage were harvested at 10.5 and were assayed for XPAPC expression normalized to ODC expression. There is a greater than two fold increase in XPAPC expression after injection with wild type HWNT5A. There is significantly less induction of XPAPC expression by the WNT5AC83S mutant than HWNT5A.
Discussion
The overlapping phenotypes displayed by the dominant and recessive forms of Robinow syndrome implicate WNT5A as a ligand for the tyrosine kinase receptor ROR2. Ror2 function in non-canonical Wnt signaling is newly described during Xenopus gastrulation (Schambony and Wedlich, 2007), as no prior experimentation has demonstrated a role for the highly conserved kinase domain of Ror2 in any known Wnt-dependent process (Kim and Forrester, 2003; Mikels and Nusse, 2006; Oishi et al., 2003). The array of mutations in ROR2 associated with recessive Robinow syndrome demonstrates a role for the conserved kinase-like domain of ROR2 in normal human development (Schwabe et al., 2000). This work, describing WNT5A mutations in dominant Robinow syndrome, supports a non-canonical signaling model in which a Wnt ligand signals via a tyrosine kinase receptor, and implicates the WNT5A/ROR2 pathway in human craniofacial, skeletal and genital development.
In this work, we describe WNT5A coding sequence variations in the original family in which Robinow syndrome was described and a second unrelated patient. Robinow syndrome characteristics include mesomelic shortening of the limbs, a flat facial profile, prominent forehead, and hypertelorism (Robinow et al., 1969). Recently, Wnt signaling has been shown to be a major factor controlling differences in facial development between chicken and mice (Brugmann et al., 2007). Comparisons of the dominant and recessive forms of Robinow syndrome suggest that during normal development the concentration of ligand governs the degree of limb outgrowth and continued development of facial structures. Humans with heterozygous ROR2 loss-of-function mutations are unaffected carriers (Afzal et al., 2000; van Bokhoven et al., 2000). Heterozygous Ror2 null mice are also phenotypically normal (DeChiara et al., 2000; Schwabe et al., 2004; Takeuchi et al., 2000). Loss-of-function mutations in both copies of ROR2 are necessary for the more severe phenotypic manifestation of recessive Robinow syndrome (Afzal et al., 2000; van Bokhoven et al., 2000). One normal functioning copy and one copy of WNT5A with reduced function are associated with dominant Robinow syndrome. Despite this phenotype arising from one partially functioning WNT5A allele in humans, external examination of heterozygous mice in late gestation revealed none of the characteristic craniofacial, skeletal or growth abnormalities associated with the homozygous Wnt5a−/− mutation. There is certainly precedent for a disease-causing heterozygous mutation in humans to be grossly undetectable in mice, and for mouse phenotype to vary with genetic background, as demonstrated in DiGeorge Syndrome (22q11 deletion/TBX1 mutation)(Jerome and Papaioannou, 2001) and holoprosencephaly (SHH mutation)(Belloni et al., 1996; Roessler et al., 1996).
The molecular abnormalities described here in dominant Robinow syndrome suggest a model by which the WNT5A/ROR2 signal transduction pathway is regulated by the local concentration of the ligand and the receptor concentration is not rate limiting. In this report we describe attenuation of WNT5A function associated with defects in limb outgrowth and abnormal craniofacial morphogenesis. This suggests that modest activity changes in WNT5A could be a mechanism for some normal variation of limb and facial development in humans.
A number of reasons may explain why WNT5A mutations were detected in only a subset of patients. First, since we used direct sequencing of the WNT5A coding regions, our analysis would not detect mutations in the promoter or other regulatory elements, or other large deletions or duplications. Subsequent to our sequence analysis an alternatively spliced form of exon 1 has been identified and confirmation and future sequencing of this exon would be appropriate in patients with clinically diagnosed dominant Robinow syndrome (Katoh, 2009). Second, it is likely that dominant Robinow syndrome is genetically heterogeneous, and mutations in other components of the WNT5A/ROR2 signaling or regulatory pathways may also be responsible for dominant Robinow syndrome. Recently, Mazzeu, et al have identified abnormalities of Chromosome 1 associated with limb, skeletal, genital and craniofacial characteristics commonly found in Robinow syndrome (Mazzeu et al., 2007a). Potential genetic heterogeneity is confounded by difficulties in accurate clinical diagnosis of craniofacial and skeletal syndromes. Recessive Robinow syndrome has unique characteristic rib and vertebral anomalies and pronounced limb and craniofacial abnormalities that improve clinical diagnostic accuracy and are well correlated with ROR2 mutations (Mazzeu et al., 2007a). In contrast, the phenotype of dominant Robinow syndrome is reported to be more subtle, with less severe skeletal and craniofacial changes when compared to the recessive phenotype. Identification of a functional mutation in WNT5A in the initially described family with dominant Robinow syndrome sets a phenotypic standard with molecular correlation that will help us begin to sort out genotype/phenotype correlations and identify molecular etiologies in Robinow syndrome. The knowledge developed through the characterization of ROR2 mutations in patients with recessive Robinow syndrome has been limited by the paucity of information about the role of this molecule in development. The identification of WNT5A mutations in dominant Robinow syndrome patients places this WNT5A/ROR2 signaling pathway in an important role in craniofacial and skeletal development and disease.
Experimental Procedures
Human Studies
Approval for this study was obtained from the Institutional Review Boards of the participating institutions (University of Minnesota IRB Code #0603M83366 and Radboud University Nijmegen Medical Center). Patients with Robinow syndrome were identified based on clinical phenotype (Mazzeu et al., 2007b) and pedigree consistent with autosomal dominant transmission by one of our investigators (SB, JFM, HGB). All living members of the original family described by Dr. Robinow were enrolled. The patient with the 248G→C mutation is a female with a typical phenotypic presentation of the autosomal dominant Robinow syndrome phenotype. Informed consent was obtained from the patient, parent, or guardian in all cases, and 5 milliliters of blood were obtained by venipuncture. DNA was isolated with the VersaGene™ Genomic DNA Purification Kit (Gentra Systems, Inc., Minneapolis, MN) using a standard protocol for DNA extraction from whole blood. Control DNA for complete WNT5A coding sequencing was obtained from phenotypically normal unrelated individuals who consented to participate in this study and was isolated from whole blood as above.
Wnt5a Genomic PCR
100 nanograms of DNA were used in PCR reactions performed with intronic WNT5A primer pairs (Supplementary Table 1) spanning across each individual exon for all patient samples and controls. Negative control reactions for each primer set were run without DNA template to assay for DNA contamination.
PCR products were separated on a 1.5% agarose gel and amplicons were gel extracted using the S.N.A.P. Gel Purification Kit (Invitrogen, Carlsbad, CA). 30ng of purified DNA was then sequenced by our on site facility. A DNA ABI PRISM 3730xl DNA Analyzer was used in sequencing reactions of PCR products using dye-labeled terminator chemistry. Sequencing was conducted in both 5′ and 3′ directions using the same primers used for the PCR reactions (Table 1). Sequence variants were confirmed by re-sequencing in both direction three times, and compared to verified sequence of an additional one hundred seventy three controls for exon 3 obtained from phenotypically normal individuals (JLL and HGB). One hundred ninety six controls from normal banked DNA were also analyzed for the mutation identified in exon 4 by ARMS-PCR (179 controls)(Perrey et al., 1999) or sequencing (17 controls). Primers used for ARMS-PCR recognized wild type WNT5A exon 4:
(5′-GGACTGGCTCTGGGGCGGCT-3′, 5′-GAGGAGAGGACGGAGCTACA-3′) and the C182R mutation in WNT5A exon 4:
(5′-GGACTGGCTCTGGGGCGGTC, 5′-GAGGAGAGGACGGAGCTACA-3′).
40 ng of genomic DNA was used in ARMS-PCR analysis. PCR parameters were 94°C for 30 seconds, 62°C for 60 seconds, 72°C for 60 seconds, loop thirty times, 72°C for 10 minutes.
Linkage analysis was performed using a total of 382 fluorescently labeled short tandem repeat (STR) polymorphic markers (ABI PRISM Linkage Mapping Set MD10.A) with easyLINKAGE Plus pairwise and multipoint comparison. Data was analyzed using GeneHunter v2lr5 software, with parametric analysis for a dominant model of inheritance. We assumed complete penetrance, and a frequency of the disease allele of 1:10,000.
Genotyping of mice
Embryos used in this study were obtained by intercrossing Wnt5a+/− mice (Yamaguchi et al., 1999). Wild type littermates were used as controls. Genomic DNA was PCR amplified as described (Kim et al., 2005). The study has been approved by the University of Minnesota Animal Care and Use Committee.
Skeletal preparation and histology
The skeletons of wild-type and Wnt5a−/− embryos were stained with alizarin red and alcian blue as described (Hogan et al., 1986).
mRNA overexpression
WNT5A, WNT5AC83S, WNT5AC182R, and dnWNT5A were PCR engineered from cDNA clone (catalog # MHS1010-9204179, Open Biosystems, Huntsville AL) incorporating a 5′ BglII site, a Kozak translation site (GCCACC), start codon, coding sequence, stop codon and a 3′ SpeI restriction site. These constructs were ligated into pT3TS (Hyatt and Ekker, 1999) using BglII and SpeI engineered restriction sites. To make the dnWNT5A, a stop codon was inserted after the SPDYC motif as described (Sen et al., 2001). The WNT5A mutant constructs were generated by PCR using overlapping primers containing the mutated bases (base pairs 544-545, CT/TC) or (base pair 248, G/C) to incorporate the mutations into full-length constructs of WNT5AC182R and WNT5AC83S respectively. All clones where sequence-verified from both directions with primers flanking the mutation site (not shown). All four clones (pT3TS WNT5A, pT3TS WNT5AC182R, pT3TS WNT5AC83S, and pT3TS dnWNT5A) were linearized with XbaI and 5′ (7-methyl guanosine) capped mRNA was transcribed with T3 RNA polymerase (T3 Message Machine, Ambion, Austin, TX).
Zebrafish maintenance and injections
Wild-type Danio rerio (Segrest Farms, Gibsonton, FL) and insulin-eGFP (Huang et al., 2001) embryos were raised at 30°C and spawning was performed as previously described (Kimmel et al., 1995). WNT5A, WNT5AC83S, WNT5AC182R, and dnWNT5A mRNAs were injected into one-cell embryos. Axis duplication was scored at 24hpf in wild type embryos. Insulin-eGFP embryos were injected with mRNAs and scored for islet formation defects at 30hpf.
Whole mount in situ hybridization
pCRIITOPO Danio rerio insulin clone containing base pairs 38-369 NCBI accession # NM_131056 was linearized with NotI and DIG labeled riboprobe was synthesized with T7 RNA polymerase (Roche, Mannheim, Germany). Whole mount in situ hybridization was performed as described (Jowett, 1999).
Xenopus Embryos and Injections
Xenopus embryos were obtained by in vitro fertilization after induction of ovulation in female frogs using human chorionic gonadotropin (Sigma). Animal cap experiments were performed as described in (Moon et al., 1993). Briefly, the animal poles of both blastomeres were injected with 30 or 200 pg of HWNT5A, WNT5AC83S mRNA or WNT5AC182R mRNA at the two cell stage. The upper one fourth of the embryo was harvested using an eyebrow knife at blastula stage (stage 8) and cultured in 1/3 strength modified Ringer’s solution and 5 ng/ml activin protein (R & D Systems, Minneapolis, MN). Embryos were assayed for elongation when control embryos were at stage 40. For XPAPC induction, the two dorsal blastomeres were injected with 200 pg of mRNA at the four cell stage and embryos rapidly frozen at stage 10.5 (early gastrulation) for RNA isolation, cDNA synthesis and quantitative PCR. (Methods were adapted from (Schambony and Wedlich, 2007).
XPAPC Induction by Quantitative PCR
XPAPC induction in injected Xenopus embryos was assessed by qPCR. Total RNA was isolated from embryos using TRIzol reagent (Invitrogen Life Technologies). cDNA was synthesized and amplifications were performed in triplicate with 1x RT2 Real-Time SYBR green mix (SuperArray) on a Stratagene Mx3000P Real-Time PCR system with the following parameters: 45 cycles, 95 °C for 30 s, 55°C for 60s, and 72°C for 60 s. qPCR data were normalized using ornithine decarboxylase (ODC) as a control. Primer sequences (Schambony and Wedlich, 2007) for XPAPC were: 5′- cccagtcggtctcttcttctttg 3′ (forward), 5′-ttgctgatgctgctcttggttag 3′ (reverse), and ODC 5′- gccattgtgaagactctctccattc3′ (forward), 5′-ttcgggtgattccttgccac 3′ (reverse). Data are presented as the mean ± s.e. using GraphPad Prizm4.
Statistics
The transgenic insulin-eGFP experimental values are the individual scores from each embryo. The transgenic insulin-eGFP positive cell coalescence experimental mean was then calculated for each experiment. The mean value of the means of each individual experiment was calculated [(sum of individual experiment means)/number of individual experiment means=mean of the means]. The mean variance of the means was also calculated. The % of embryos injected with wild type WNT5A mRNA in pancreatic islet coelesence and axis duplication assays was normalized to 100% activity and the % activity of WNT5AC182R and WNT5AC83S mRNA injected embryos was compared to wild type WNT5A mRNA injected activity.
We used Student’s t-test to compare inhibition of insulin-eGFP cell coalescence in embryos injected with WNT5A mRNA compared to embryos injected with WNT5AC83S, or WNT5AC182R mRNA. p values <0.05 were considered significant.
Acknowledgments
We would like to dedicate this manuscript to the memory of Dr. Ian N. Jongewaard.
We are indebted to our patients and their families for their participation in and encouragement of this study. We thank The Robinow Syndrome Foundation for assistance with this research effort and Drs. Harry Orr, Saulius Sumanas, Hyon Kim, and William Oetting for their scientific comments. This research was supported by Minnesota Medical Foundation Research Grants to AP and JRS; Howard Hughes Fellowship to JRS; a gift from the Sit Investment Associates Foundation to JLL; and FAPESP grant support for JFM. Drs. Anthony Person and Mara Robu were supported by the MinnCResT Training Program T32-DE07288-07 (NIDCR). C.J.B. was supported by the Musculoskeletal Training Grant NIH-NIAMS, T32 AR050938.
Footnotes
Web Resources
Accession numbers and URLs for data presented herein are as follows:
Genebank, http://www.ncbi.nlm.nih.gov/Genbank/ (for human WNT5A [NM003392], mouse Wnt5a [NM009524], human ROR2 [NM004560], mouse Ror2 [NM013846], and Danio rerio insulin [NM131056]).
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
HapMap, http://hapmap.org/,
GeneCards™ genome, http://www.stanford.edu/cgi-bin/genecards/index.shtml,
UCSC Genome Browser http://www.genome.ucsc.edu/
Author Contributions
Study design by SCE, SB, JLL, LAS. Human subjects approval obtained by JLL and HGB. Sample retrieval and phenotyping done by JH, SB, JFM, JLL, and HGB. ADP, CMS, ANN, SH were involved in PCR reactions and sequencing of PCR products. Generation of constructs pT3TSWNT5A, pT3TSWNT5AC182R, pT3TSWNT5AC83S, and pT3TSdnWNT5A by ADP. Zebrafish experiments done by ADP and MER. Xenopus Experiments and qPCR were done by ANN,CMS,CJB and JLL. Mouse data and Fig. 1 by JRS, AP, and MER. Linkage analysis done by HGB and HVB. Manuscript written by ADP, SCE, JLL, HGB, HVB, LAS and JFM. All authors have agreed to all content in the manuscript, including the data presented.
Competing Interest Statement
The authors declare that they have no competing financial interests.
References
- Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tuysuz B, Murday VA, Patton MA, Wilkie AO, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet. 2000;25:419–422. doi: 10.1038/78107. [DOI] [PubMed] [Google Scholar]
- Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Goodnough LH, Gregorieff A, Leucht P, Ten Berge D, Fuerer C, Clevers H, Nusse R, Helms JA. Wnt signaling mediates regional specification in the vertebrate face. Development. 2007;134:3283–3295. doi: 10.1242/dev.005132. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Kimble RB, Poueymirou WT, Rojas J, Masiakowski P, Valenzuela DM, Yancopoulos GD. Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nat Genet. 2000;24:271–274. doi: 10.1038/73488. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Pelegri F, Mullins MC, Kane DA, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Haffter P, Heisenberg CP, et al. Mutations affecting morphogenesis during gastrulation and tail formation in the zebrafish, Danio rerio. Development. 1996;123:143–151. doi: 10.1242/dev.123.1.143. [DOI] [PubMed] [Google Scholar]
- He F, Xiong W, Yu X, Espinoza-Lewis R, Liu C, Gu S, Nishita M, Suzuki K, Yamada G, Minami Y, et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 2008;135:3871–3879. doi: 10.1242/dev.025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Saint-Jeannet JP, Wang Y, Nathans J, Dawid I, Varmus H. A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science. 1997;275:1652–1654. doi: 10.1126/science.275.5306.1652. [DOI] [PubMed] [Google Scholar]
- Heisenberg CP, Tada M, Rauch GJ, Saude L, Concha ML, Geisler R, Stemple DL, Smith JC, Wilson SW. Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature. 2000;405:76–81. doi: 10.1038/35011068. [DOI] [PubMed] [Google Scholar]
- Hogan B, Costantini F, Lacy E. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory; 1986. [Google Scholar]
- Huang H, Vogel SS, Liu N, Melton DA, Lin S. Analysis of pancreatic development in living transgenic zebrafish embryos. Mol Cell Endocrinol. 2001;177:117–124. doi: 10.1016/s0303-7207(01)00408-7. [DOI] [PubMed] [Google Scholar]
- Hyatt TM, Ekker SC. Vectors and techniques for ectopic gene expression in zebrafish. Methods Cell Biol. 1999;59:117–126. doi: 10.1016/s0091-679x(08)61823-3. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Jowett T. Analysis of protein and gene expression. Methods Cell Biol. 1999;59:63–85. doi: 10.1016/s0091-679x(08)61821-x. [DOI] [PubMed] [Google Scholar]
- Katoh M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med. 2009;23:763–769. doi: 10.3892/ijmm_00000190. [DOI] [PubMed] [Google Scholar]
- Kim C, Forrester WC. Functional analysis of the domains of the C elegans Ror receptor tyrosine kinase CAM-1. Dev Biol. 2003;264:376–390. doi: 10.1016/j.ydbio.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Schleiffarth JR, Jessurun J, Sumanas S, Petryk A, Lin S, Ekker SC. Wnt5 signaling in vertebrate pancreas development. BMC Biol. 2005;3:23. doi: 10.1186/1741-7007-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Kurayoshi M, Yamamoto H, Izumi S, Kikuchi A. Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signalling. Biochem J. 2007;402:515–523. doi: 10.1042/BJ20061476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JO, Kitajewski J, Varmus HE. Mutational analysis of mouse Wnt-1 identifies two temperature-sensitive alleles and attributes of Wnt-1 protein essential for transformation of a mammary cell line. Mol Biol Cell. 1992;3:521–533. doi: 10.1091/mbc.3.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzeu JF, Krepischi-Santos AC, Rosenberg C, Szuhai K, Knijnenburg J, Weiss JM, Kerkis I, Mustacchi Z, Colin G, Mombach R, et al. Chromosome abnormalities in two patients with features of autosomal dominant Robinow syndrome. Am J Med Genet A. 2007a;143A:1790–1795. doi: 10.1002/ajmg.a.31661. [DOI] [PubMed] [Google Scholar]
- Mazzeu JF, Pardono E, Vianna-Morgante AM, Richieri-Costa A, Ae Kim C, Brunoni D, Martelli L, de Andrade CE, Colin G, Otto PA. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am J Med Genet A. 2007b;143:320–325. doi: 10.1002/ajmg.a.31592. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Moon RT. Ectopic expression of the proto-oncogene int-1 in Xenopus embryos leads to duplication of the embryonic axis. Cell. 1989;58:1075–1084. doi: 10.1016/0092-8674(89)90506-0. [DOI] [PubMed] [Google Scholar]
- Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006;4:e115. doi: 10.1371/journal.pbio.0040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT, Campbell RM, Christian JL, McGrew LL, Shih J, Fraser S. Xwnt-5A: a maternal Wnt that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development. 1993;119:97–111. doi: 10.1242/dev.119.1.97. [DOI] [PubMed] [Google Scholar]
- Nomi M, Oishi I, Kani S, Suzuki H, Matsuda T, Yoda A, Kitamura M, Itoh K, Takeuchi S, Takeda K, et al. Loss of mRor1 enhances the heart and skeletal abnormalities in mRor2-deficient mice: redundant and pleiotropic functions of mRor1 and mRor2 receptor tyrosine kinases. Mol Cell Biol. 2001;21:8329–8335. doi: 10.1128/MCB.21.24.8329-8335.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- Oldridge M, Fortuna AM, Maringa M, Propping P, Mansour S, Pollitt C, DeChiara TM, Kimble RB, Valenzuela DM, Yancopoulos GD, et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat Genet. 2000;24:275–278. doi: 10.1038/73495. [DOI] [PubMed] [Google Scholar]
- Patton MA, Afzal AR. Robinow syndrome. J Med Genet. 2002;39:305–310. doi: 10.1136/jmg.39.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrey C, Turner SJ, Pravica V, Howell WM, Hutchinson IV. ARMS-PCR methodologies to determine IL-10, TNF-alpha, TNF-beta and TGF-beta 1 gene polymorphisms. Transpl Immunol. 1999;7:127–128. doi: 10.1016/s0966-3274(99)80030-6. [DOI] [PubMed] [Google Scholar]
- Robinow M, Silverman FN, Smith HD. A newly recognized dwarfing syndrome. Am J Dis Child. 1969;117:645–651. doi: 10.1001/archpedi.1969.02100030647005. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Schambony A, Wedlich D. Wnt-5A/Ror2 regulate expression of XPAPC through an alternative noncanonical signaling pathway. Dev Cell. 2007;12:779–792. doi: 10.1016/j.devcel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Schleiffarth JR, Person AD, Martinsen BJ, Sukovich DJ, Neumann A, Baker CV, Lohr JL, Cornfield DN, Ekker SC, Petryk A. Wnt5a is required for cardiac outflow tract septation in mice. Pediatr Res. 2007;61:386–391. doi: 10.1203/pdr.0b013e3180323810. [DOI] [PubMed] [Google Scholar]
- Schwabe GC, Tinschert S, Buschow C, Meinecke P, Wolff G, Gillessen-Kaesbach G, Oldridge M, Wilkie AO, Komec R, Mundlos S. Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am J Hum Genet. 2000;67:822–831. doi: 10.1086/303084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe GC, Trepczik B, Suring K, Brieske N, Tucker AS, Sharpe PT, Minami Y, Mundlos S. Ror2 knockout mouse as a model for the developmental pathology of autosomal recessive Robinow syndrome. Dev Dyn. 2004;229:400–410. doi: 10.1002/dvdy.10466. [DOI] [PubMed] [Google Scholar]
- Sen M, Chamorro M, Reifert J, Corr M, Carson DA. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001;44:772–781. doi: 10.1002/1529-0131(200104)44:4<772::AID-ANR133>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Slusarski DC, Yang-Snyder J, Busa WB, Moon RT. Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev Biol. 1997;182:114–120. doi: 10.1006/dbio.1996.8463. [DOI] [PubMed] [Google Scholar]
- Stricker S, Verhey van Wijk N, Witte F, Brieske N, Seidel K, Mundlos S. Cloning and expression pattern of chicken Ror2 and functional characterization of truncating mutations in Brachydactyly type B and Robinow syndrome. Dev Dyn. 2006;235:3456–3465. doi: 10.1002/dvdy.20993. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Takeda K, Oishi I, Nomi M, Ikeya M, Itoh K, Tamura S, Ueda T, Hatta T, Otani H, et al. Mouse Ror2 receptor tyrosine kinase is required for the heart development and limb formation. Genes Cells. 2000;5:71–78. doi: 10.1046/j.1365-2443.2000.00300.x. [DOI] [PubMed] [Google Scholar]
- van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, Skovby F, Kerr B, Percin EF, Akarsu N, et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat Genet. 2000;25:423–426. doi: 10.1038/78113. [DOI] [PubMed] [Google Scholar]
- van den Heuvel M, Harryman-Samos C, Klingensmith J, Perrimon N, Nusse R. Mutations in the segment polarity genes wingless and porcupine impair secretion of the wingless protein. Embo J. 1993;12:5293–5302. doi: 10.1002/j.1460-2075.1993.tb06225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- Yang Y, Topol L, Lee H, Wu J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development. 2003;130:1003–1015. doi: 10.1242/dev.00324. [DOI] [PubMed] [Google Scholar]