

ABSTRACT
Hantavirus infections are characterized by vascular hyperpermeability and neutrophilia. However, the pathogenesis of this disease is poorly understood. Here, we demonstrate for the first time that pulmonary vascular permeability is increased by Hantaan virus infection and results in the development of pulmonary edema in C.B-17 severe combined immunodeficiency (SCID) mice lacking functional T cells and B cells. Increases in neutrophils in the lung and blood were observed when pulmonary edema began to be observed in the infected SCID mice. The occurrence of pulmonary edema was inhibited by neutrophil depletion. Moreover, the pulmonary vascular permeability was also significantly suppressed by neutrophil depletion in the infected mice. Taken together, the results suggest that neutrophils play an important role in pulmonary vascular hyperpermeability and the occurrence of pulmonary edema after hantavirus infection in SCID mice.
IMPORTANCE Although hantavirus infections are characterized by the occurrence of pulmonary edema, the pathogenic mechanism remains largely unknown. In this study, we demonstrated for the first time in vivo that hantavirus infection increases pulmonary vascular permeability and results in the development of pulmonary edema in SCID mice. This novel mouse model for human hantavirus infection will be a valuable tool and will contribute to elucidation of the pathogenetic mechanisms. Although the involvement of neutrophils in the pathogenesis of hantavirus infection has largely been ignored, the results of this study using the mouse model suggest that neutrophils are involved in the vascular hyperpermeability and development of pulmonary edema in hantavirus infection. Further study of the mechanisms could lead to the development of specific treatment for hantavirus infection.
INTRODUCTION
Hantaviruses are enveloped and negative-sense RNA viruses with a tripartite genome comprising large (L), medium (M), and small (S) segments. These genomic segments encode the RNA-dependent RNA polymerase, the glycoprotein precursor, and the nucleocapsid protein (N), respectively (1). The genus Hantavirus, family Bunyaviridae, comprises rodent-borne viruses that cause two diseases in humans, hemorrhagic fever with renal syndrome (HFRS) in the Old World and hantavirus pulmonary syndrome (HPS) in the New World (2, 3). HFRS is characterized by fever, hemorrhage, renal impairment, and thrombocytopenia (4–7). The prototype hantavirus, Hantaan virus (HTNV), causes a severe form, while Seoul virus has been reported to cause a more moderate form of HFRS, mainly in Asia. In Europe, HFRS is caused by Dobrava virus and Puumala virus (PUUV) (8). On the other hand, HPS is characterized by fever and noncardiogenic pulmonary edema (9). Sin Nombre virus (SNV), New York virus, Black Creek Canal virus, Andes virus (ANDV), and Laguna Negra virus regularly cause HPS in the Americas (10). However, respiratory impairment and noncardiogenic pulmonary edema were observed in HFRS patients infected with HTNV, Dobrava virus, and PUUV (11–18). In addition, pulmonary edema was noted in ca. 10 to 60% of fatal HFRS cases (12, 13, 15). On the other hand, renal impairment was observed in HPS cases (19–21). Thus, renal involvement and pulmonary edema are not specific to HFRS or HPS but are common among hantavirus diseases. Since both symptoms are caused by capillary leakage, hyperpermeability after hantavirus infection in endothelial cells, which are the predominant target cells for hantaviruses, is considered to be a plausible mechanism of the pathogenesis of HFRS and HPS. Since infected endothelial cells showed no cytopathic effect, immune-mediated pathogenesis has been suggested (22–26). In particular, T-cell mediated immunopathology has been suggested by the detection of increased immunoblasts and activated T cells in acute-phase HPS and HFRS patients (24, 26–29) and elevated levels of T cell cytokines in HFRS patients (30–34).
On the other hand, neutrophilia has been documented in patients with hantavirus infection (26, 35–37). However, neutrophilia is far less common in viral infections than in bacterial infections (38, 39). Neutrophils are thought to play a key role in the progression of acute respiratory distress syndrome, which clinically resembles HPS (40, 41). Furthermore, it has been shown that neutrophils produce various vascular permeability factors (42–46). Therefore, there is a possibility that neutrophils play a key role in vascular hyperpermeability and occurrence of pulmonary edema in hantavirus infection in humans.
Animal models have been developed to uncover the pathogenicity of hantaviruses. Hooper and coworkers demonstrated that immunocompetent adult Syrian hamsters develop a lethal vascular leak syndrome resembling clinical human HPS after ANDV infection (47, 48). However, infection with SNV, which is responsible for most human deaths in North America (47), and infection with Old World hantaviruses (i.e., HTNV, Seoul virus, and PUUV), which is responsible for HFRS, did not cause any disease symptoms in immunocompetent Syrian hamsters (49, 50). Various attempts have been made to develop an animal model using mice. However, most immunocompetent laboratory strains of mice are resistant to hantavirus infections (51). Seto et al. reported that HTNV strain AA57 caused pulmonary disease symptoms such as accumulation of pleural effusion and perivascular edema in 2-week-old ICR mice, but pulmonary edema was not observed (52).
We report here that HTNV causes severe pulmonary edema, closely resembling that in human cases of hantavirus infection, in infected adult C.B-17/Icr-scid/scidJcl (SCID) mice, indicating that T cells and B cells are not responsible for the occurrence of pulmonary edema. The role of neutrophils in the induction of hyperpermeability and pulmonary edema was also examined by using SCID mice.
MATERIALS AND METHODS
Virus.
Hantaan virus strain 76-118 cl-1 (53) was grown in Vero E6 cells (ATCC C1008, CRL1586) maintained in minimum essential medium with Eagle salts (MEM; Invitrogen, Carlsbad, CA) that was supplemented with 2 mM l-glutamine (Invitrogen), 5% fetal calf serum (FCS), 0.5% nonessential amino acids, 5% insulin-transferrin-selenium (Invitrogen), and 1% penicillin-streptomycin (Invitrogen). The culture supernatant was used in this experiment.
Animals.
Six- to eight-week-old female C.B-17/Icr-scid/scidJcl (SCID) mice (Nihon Clea, Tokyo, Japan) were used in the present study and were maintained at the Laboratory of Animal Experiments, School of Medicine, Hokkaido University. All animal experiments were approved by the Animal Studies Ethical Committee at Hokkaido University. All of the mice were treated according to the laboratory animal control guidelines of the Hokkaido University Institutional Animal Care and Use Committee. Experiments involving virus infections were performed in a BSL-3 facility. Mice exhibiting weight loss in excess of 25% of initial body weight were euthanized except for mice in a pilot experiment.
Infection and treatment of mice.
HTNV (4,000 focus-forming units [FFU] in MEM) was inoculated by intraperitoneal injection into adult mice. After viral challenge, the mice were observed, and body weights were measured at 0 (preinfection) and 7, 14, 21, 28, 33, 35, and 42 days postinoculation (dpi). At these time points, some of the animals were euthanized, and sera were collected and stored at −30°C. The lungs, kidneys, spleens, livers, hearts, and brains were collected and stored at −80°C for RNA extraction. The organs of some mice were fixed in 10% phosphate-buffered formalin for histological analysis. Blood samples from the remaining mice were collected at the same time course into microhematocrit capillary tubes (Fisherbrand, Pittsburgh, PA) for hematology.
Histopathology and IHC.
Tissues were fixed in 10% phosphate-buffered formalin at 4°C within 14 days and routinely embedded in paraffin, sectioned (2.0 μm), and stained with hematoxylin and eosin. To investigate the extent of pulmonary edema, the left lungs were sectioned at constant positions, and the number of alveoli with exudate was counted in more than 300 alveoli and in more than 25 high power fields (see Fig. 4F). For immunohistochemistry (IHC), the antigens were retrieved by hydrolytic autoclaving for 10 min at 121°C in 10 mM sodium citrate-sodium chloride buffer (pH 6.0). Endogenous peroxidase activity was blocked by incubation with 1% hydrogen peroxide in methanol for 30 min. The first antibody added was monoclonal antibody (MAb) clone E5/G6 (1 μg/ml; incubated overnight at 4°C) that recognizes a linear epitope common to hantavirus N (54). The Vector M.O.M. immunodetection kit (Vector Laboratories, Burlingame, CA) was used to visualize HTNV antigens in the sections.
FIG 4.

Pulmonary lesions in the lungs of HTNV-infected SCID mice on day 35 dpi. (A and B) HTNV antigens were detected using an E5/G6 MAb against the N of hantavirus (B, inset). Original magnifications: ×25 (A and B), ×100 (C and D), and ×250 (insets). Cellular proliferation was observed in the lung (A). Adhesion of neutrophils and degenerated and multilayered endothelial cells were seen in a middle-sized vein (A, inset). Br, bronchi; Al, alveoli; V, vein. Many virus antigen-positive cells were present in the alveolar area (B). Alveolar cells were positive for virus antigens (B, inset). (C and D) Pulmonary effusion, macrophages (arrows), and neutrophils (arrowheads) were observed in the alveoli. Inflammatory cells were increased in the alveolar wall. (E) Ratio of alveoli with exudate in each individual. There were four mice per group except for the 35 dpi group (n = 7). (F) Method for calculating the ratio of alveoli with exudate. To investigate the extent of pulmonary edema, the left lungs were sectioned at constant positions, and the number of alveoli with exudate was counted in more than 300 alveoli and in more than 25 high-power fields. Exudate is seen in asterisk-labeled alveoli in the lower panels. Since there is little exudate in a normal lung, an alveolus was considered to be an alveolus with exudate if the alveolus was filled with exudate of more than one-tenth of the area.
RNA extraction and real-time PCR.
Total RNA was extracted from each tissue and serum of infected mice by using ISOGEN (Nippon Gene, Japan) and a QIAamp viral RNA minikit (Qiagen, Hilden, Germany) in accordance with the manufacturers' protocols. The RNA was then reverse transcribed by using a first-strand cDNA synthesis kit (GE Healthcare, United Kingdom). Real-time PCRs were carried out on a LightCycler 480 (Roche Diagnostics, Vienna, Austria) using TaqMan Universal master mix II (Applied Biosystems, Carlsbad, CA). The following primers and probe were used: real-time primer HTNV F (5′-TTATTGTGCTCTTCATGGTTGC-3′), real-time primer HTNV R (5′-CATCCCCTAAGTGGAAGTTGTC-3′), and probe 75 (5′-GGAGGCTG-3′) (all from Roche Diagnostics).
Recovery of lung leukocytes from mice.
Animals were euthanized at 28 dpi, and the trachea was cannulated by using a 22-gauge catheter. Bronchoalveolar lavage (BAL) was performed twice with 0.8 ml of ice-cold phosphate-buffered saline (PBS; pH 7.4) each time. After collection of BAL fluid, animals were perfused with 3 ml of ice-cold PBS via the heart. The lungs were excised, minced, and enzymatically digested for 1 h in 1 ml of RPMI 1640 medium supplemented with 10% FCS, 1% penicillin-streptomycin, 50 μM 2-mercaptoethanol, 1 mg of type VIII collagenase (Sigma-Aldrich)/ml, and 30 μg of DNase I (Sigma-Aldrich)/ml. The undigested fragment was further dispersed by double passage through 0.80- and 0.45-μm-pore-size nylon mesh filters. The total cells were pelleted, and any contaminating erythrocytes were eliminated from the cell pellet by adding 0.83% NH4Cl in 10 mM Tris buffer (pH 7.4). After spinning, the pellet was resuspended in 10 ml of complete RPMI 1640 medium (10% FCS, 1% penicillin-streptomycin, 50 μM 2-mercaptoethanol). Cell suspensions were filtered through a 0.45-μm-pore-size nylon mesh filter and pelleted for flow cytometry.
White blood cell counts.
The total numbers of white blood cells were counted after staining with Turk's solution (Kanto Chemical Co., Tokyo, Japan) on a hemocytometer.
Flow cytometry.
All flow cytometry data were collected on a FACSCalibur (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (TreeStar, Inc., San Carlos, CA). For the analysis of cell surface markers, the cells were resuspended in fluorescence-activated cell sorting (FACS) buffer (1% bovine serum albumin and 0.1% NaN3 in PBS). The cells were stained at 4°C for 30 min, and the following MAbs were used: phycoerythrin (PE)-conjugated anti-mouse Gr-1 (clone RB6-8C5; eBioscience, San Diego, CA), fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD11b (clone M1/70; BioLegend, San Diego, CA), PE-Cy5 anti-mouse CD3ε (clone 145-2C11; BioLegend), PE–anti-mouse NK1.1 (clone PK136, BD Biosciences), PE–anti-mouse CD11c (clone HL3, BD Biosciences), and FITC–anti-mouse I-A/I-E (clone M5/114.15.2; BioLegend). The cells were pretreated with anti-mouse CD16/CD32 (clone 93; BioLegend). After washing, the cells were fixed in 2% paraformaldehyde and then resuspended in FACS buffer for analysis.
In vivo depletion of neutrophils, macrophages, and NKT cells.
To deplete neutrophils, mice were treated with protein G-purified anti-Gr-1 MAb (RB6-8C5). RB6-8C5 hybridoma was provided by the Cell Resource Center for Biomedical Research Institute of Development, Aging, and Cancer, Tohoku University, and the MAb was purified from culture supernatant or ascitic fluid of mice by using a HiTrap protein G HP column (GE Healthcare). Each mouse was given 250 μg of anti-Gr-1 MAb or control IgG from rat serum (Sigma-Aldrich) by intramuscular injection every day from 19 to 33 dpi. Macrophage and monocyte depletion with liposomal clodronate was performed as previously described (55). Briefly, 10 mg of clodronate (Sigma-Aldrich)/ml in PBS was mixed with a phosphatidylcholine (Nakarai, Kyoto, Japan)-cholesterol (Nakarai) (3:1 molar ratio) solution in chloroform and incubated for 2 h at 37°C. Vesicles were formed by vacuum desiccation overnight, suspended in 5 ml of PBS, and purified by dialysis. Each mouse was given 200 μl of purified clodronate liposome PBS or control PBS by intraperitoneal injection at 18, 21, 25, and 29 dpi. To deplete NKT cells, mice were treated with anti-NK1.1 MAb (PK136), which depletes both NKT and NK cells. Each mouse was given 100 μg of anti-NK1.1 MAb or control IgG from mouse serum (Sigma-Aldrich) by intramuscular injection at 20, 25, and 30 dpi (56). Effective neutrophil depletion (>90%) at 33 dpi was confirmed by morphological analysis and flow cytometry of peripheral blood (data not shown). The extent of macrophage depletion (75 to 90%) was determined by flow cytometry of splenocytes (data not shown). Likewise, the extent of NKT cell depletion (75 to 90%) was determined by flow cytometry of peripheral blood (data not shown).
Evaluation of vascular permeability.
Vascular permeability was analyzed by the Evans blue extravasation method (57). Mice were inoculated intravenously with 200 μl of a 2% solution of Evans blue dye dissolved in normal saline (0.85% sodium chloride). After 1 h, the mice were anesthetized, and the lungs were perfused with 5 ml of normal saline through the left cardiac ventricle. The homogenized lung tissue was incubated with 1.5 ml of formamide for 16 h at 55°C and centrifuged at 4°C for 15 min at 6,000 × g. The amount of Evans blue dye in the supernatant was measured spectrophotometrically at 650 nm.
Statistical analysis.
Data were analyzed using the nonparametric Mann-Whitney U test and statistical significance was accepted when the P value was <0.05.
RESULTS
Body weight change after infection with HTNV.
Infected mice showed gradual, progressive weight loss and slowing of activity and they exhibited a rough coat after 21 dpi (Fig. 1). Statistically significant differences in mean body weight were observed between groups of HTNV-infected mice and uninfected mice from 21 dpi (P < 0.01). Three of the eleven infected mice had weight losses of >20% at 35 dpi. The HTNV-infected mice displayed terminal symptoms at 42 dpi with shivering and cessation of feeding and drinking and of spontaneous movement. Four of the five infected mice had weight losses of >25% at 42 dpi.
FIG 1.

Percent body weight change in SCID mice following HTNV infection. Mice were inoculated intraperitoneally with Hantaan virus strain 76-118 cl-1 (HTNV+) or not treated (control). Values are means ± the standard errors (SE); there were five to eleven mice per group. Asterisks denote a significant difference from the control mice (**, P < 0.01 [Mann-Whitney U test]).
Viral loads in the organs of mice following inoculation with HTNV.
The viral genome was detected in the lungs, kidneys, heart, spleen, liver, brain, and serum from 7 dpi (Fig. 2). The viral loads were high in all organs examined at 7 dpi. The genomes in all organs except for the brain and serum were increased to a plateau level at 14 dpi. On the other hand, those of the brain and serum were gradually increased.
FIG 2.

Mean viral genome load in SCID mice following HTNV infection. The viral load in each organ was measured by quantitative real-time PCR. Values are means + the SE. The number of mice tested is shown below the names of organs or serum.
Histopathological examination and degree of pulmonary edema.
To examine the distribution of the virus, IHC was carried out with an E5/G6 MAb generated against the N of hantavirus. In accordance with the viral genome load, viral antigen was first observed in the spleen from 7 dpi (Table 1). In the spleen, viral antigen-positive cells were seen around white pulp at 14 dpi (data not shown). After 14 dpi, viral antigens were observed in multiple organs (Table 1). In the lungs, virus antigen-positive cells were detected, and the numbers of virus antigen-positive cells sequentially increased in the alveoli at 14, 21, 28, and 35 dpi (Fig. 3). For some reason, the number of antigen-positive cells at 42 dpi was slightly less than that at 35 dpi. In the kidney, heart, liver, and brain, virus antigen-positive cells were seen in small vessels at 14 dpi (data not shown). As shown in Fig. 5, the virus had spread from blood vessels to surrounding parenchymal cells, such as cardiac myocytes, hepatocytes, and neuronal cells, at 35 dpi. However, there was no antigen in epithelial cells of the bronchus or in renal tubule cells even at 35 dpi (Fig. 4B and 5D).
TABLE 1.
IHC results using specific hantavirus nucleocapsid protein monoclonal antibody E5/G6
| Time postinoculation (dpi) | IHC result for various organsa |
|||||
|---|---|---|---|---|---|---|
| Lung | Kidney | Heart | Spleen | Liver | Brain | |
| 0 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 |
| 7 | 0/4 | 0/4 | 0/4 | 2/2 | 0/2 | 0/4 |
| 14 | 4/4 | 4/4 | 3/4 | 2/2 | 4/4 | 3/4 |
| 21 | 4/4 | 4/4 | 4/4 | 1/1 | 2/2 | 4/4 |
| 28 | 4/4 | 4/4 | 4/4 | 2/2 | 4/4 | 4/4 |
| 35 | 4/4 | 4/4 | 4/4 | 2/2 | 4/4 | 4/4 |
| 42 | 4/4 | 4/4 | 4/4 | 2/2 | 4/4 | 4/4 |
Each entry indicates the number of positive organs/the number organs tested.
FIG 3.

Histopathological changes in the lung from HTNV-infected SCID mice at various times. In the lung, virus antigen-positive cells were detected and sequentially increased in the alveoli at 14, 21, 28, and 35 dpi. Low magnification, ×25; high magnification ×100. Br, bronchi; Al, alveoli; V, vein.
FIG 5.

Histopathology in HTNV-infected SCID mice. Tissues were obtained from HTNV-infected SCID mice on day 35 dpi. Hematoxylin and eosin staining was performed (A to C, G to I, and M to N). HTNV antigens were detected using an E5/G6 MAb against the N of hantavirus (D to F, J to L, and P to Q). Original magnification, ×100. Virus antigen-positive cells were seen in the kidney (A and D, renal cortex; B and E, renal medulla), heart (C and F), spleen (G and J), liver (H and K), and brain (I and L, cerebral cortex; M and P, cerebellar cortex; N and Q, choroid plexus) without inflammatory reactions. Hemosiderin was stained blue by Berlin blue stain (J).
Marked histopathological change was observed in the lung. In accordance with the viral loads, cells in the lung sequentially proliferated and the rate of cellular proliferation was significantly higher at 35 dpi than at 0 dpi (Fig. 3 and Fig. 4A to D). Adhesion of neutrophils and degenerated and multilayered endothelial cells were seen in a middle-sized vein (Fig. 4A, inset). Pulmonary effusion, macrophages, and neutrophils were observed in the alveoli (Fig. 4C and D). Inflammatory cells were increased in the alveolar wall (Fig. 4C and D). No inflammatory cell infiltration was seen in those tissues except for the lung. The degree of pulmonary edema was assessed by calculation of the ratio of alveoli with exudate (Fig. 4E). Pulmonary edema transiently appeared from 28 dpi and peaked at 35 dpi. Correspondingly, the relative lung weight also increased with an almost 2-fold increase at 28 dpi and peaked with an increase of almost two and a half times at 35 dpi (data not shown).
White blood cell and neutrophil counts.
Since the present study focused on the role of neutrophils in hantavirus infection, white blood cell and neutrophil counts in peripheral blood were determined. White blood cell count in HTNV-infected SCID mice showed a significant increase from 7 dpi and was twice that in uninfected mice at 28 dpi (Fig. 6A). Neutrophil count significantly increased from 21 dpi and was three times higher than that in uninfected mice at 28 dpi (Fig. 6B). The numbers of white blood cells and neutrophils were decreased at 33 dpi compared to those in mice at 28 dpi.
FIG 6.
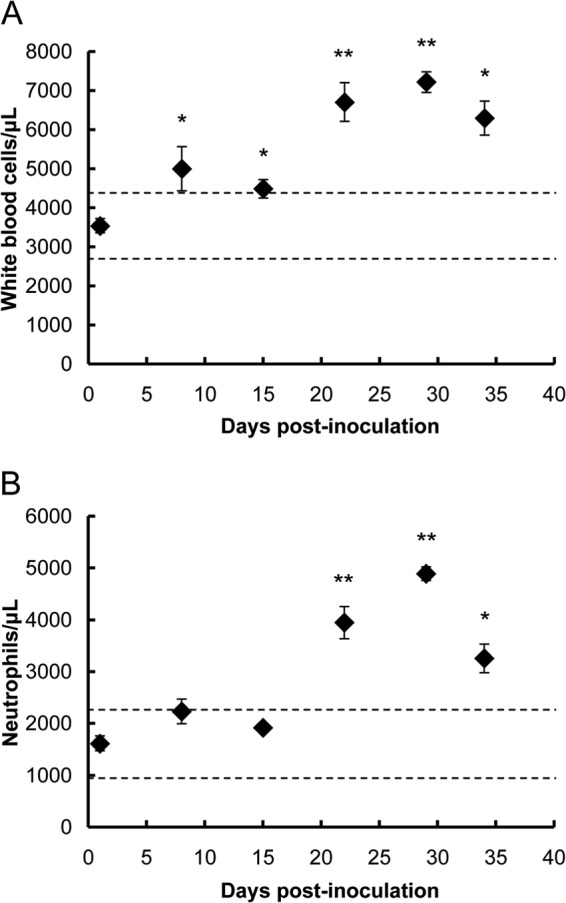
Total white blood cell counts (A) and neutrophil counts (B) in SCID mice following HTNV infection. Values are means ± the SE; there were four to six mice per group. Asterisks denote a significant difference from the control (i.e., uninfected) (*, P < 0.05; **, P < 0.01 [Mann-Whitney U test]). The dashed line represents average ± 2 standard deviations of control mice (n = 5).
Recruitment of inflammatory cells to the lung.
Since intra-alveolar edema began to be observed at 28 dpi, the change in proportion of leukocytes in the lung was investigated (Fig. 7). The percentages of CD11b+ Gr-1hi (neutrophils), CD11b+ Gr-1int (macrophages), and CD3+ NK1.1+ (NKT cells) in lung tissues from infected mice were significantly higher than those in lung tissues from uninfected mice (Fig. 7B).
FIG 7.

Representative FACS plots (A) and percent positive expression (B) for lung homogenate stained with MAbs to Gr-1, CD11b, CD3, NK1.1, CD11c, and MHC class II at 28 dpi with HTNV. Values are means + the SE; there were four or five mice per group. Asterisks denote a significant difference from control (uninfected) mice (*, P < 0.05 [Mann-Whitney U test]).
Influence of depletion on occurrence of pulmonary edema.
Since the percentages of neutrophils, macrophages, and NKT cells were increased in lung tissues from infected mice, each subset was depleted to investigate whether these cells are involved in the occurrence of pulmonary edema. Gross observation of left lungs from HTNV-infected mice with rat IgG at 33 dpi demonstrated that the lungs were highly edematous (Fig. 8A, inset). Correspondingly, these mice had clear histopathologic changes in their lungs (Fig. 8A). On the other hand, HTNV-infected mice with anti-Gr-1 MAb showed no pulmonary edema (Fig. 8A). Moreover, the degree of pulmonary edema was significantly reduced by neutrophil depletion in HTNV-infected mice (Fig. 8B). In HTNV-infected SCID mice, the gross pulmonary edema and elevated degree of pulmonary edema were unchanged even with macrophage depletion and NKT cell depletion (Fig. 8C and D).
FIG 8.

Effect of neutrophil depletion on pulmonary edema. (A) Histopathological findings in lungs collected from HTNV-infected and uninfected SCID mice in the presence of anti-Gr-1 MAb or rat IgG at 33 dpi. Br, bronchi; Al, alveoli; V, vein. Upper and middle panels show hematoxylin and eosin staining; lower panels show immunohistochemistry using an E5/G6 MAb against the N of hantavirus. Original magnification, ×25 (upper panels) and ×100 (middle and lower panels). Gross observation of left lungs from mice in neutrophil depletion experiments (A, upper panels, inset). HTNV-infected mice with anti-Gr-1 MAb showed no pulmonary edema. (B) Ratio of alveoli with exudate in each individual in neutrophil depletion experiments using anti-Gr-1 MAb. There were three to five mice per group. (C) Ratio of alveoli with exudate and representative gross observation of left lungs from mice in macrophage depletion experiments using clodronate liposome. There were three mice per group. (D) Ratio of alveoli with exudate and representative gross observation of left lungs from mice in NKT cell depletion experiments using anti-NK1.1 MAb. There were three or four mice per group. Asterisks denote a significant difference between HTNV-infected mice with anti-Gr-1 MAb and with rat IgG (*, P < 0.05 [Mann-Whitney U test]).
Pulmonary vascular permeability.
To investigate whether the pulmonary edema in the mice is caused by HTNV-induced hyperpermeability, vascular permeability in the lung was examined by measurement of the extravasation of Evans blue dye. Pulmonary vascular permeability was significantly higher in the infected mice than in uninfected mice at 33 dpi (Fig. 9). The vascular permeability at 28 dpi was slightly enhanced compared to that in control mice, but the difference was not significant. The elevated pulmonary vascular permeability conformed with the degree of pulmonary edema. Furthermore, to investigate whether neutrophils increase pulmonary vascular permeability, pulmonary vascular permeability was examined in mice with neutrophil depletion. It was found that the level of hyperpermeability was significantly reduced by neutrophil depletion at 33 dpi (Fig. 9).
FIG 9.
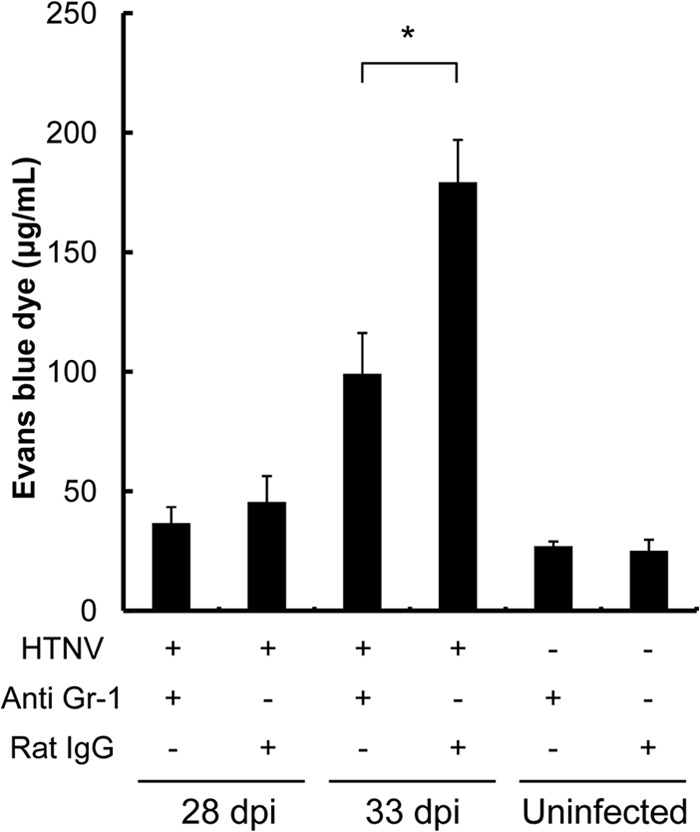
Pulmonary vascular permeability in HTNV-infected mice. To assess changes in pulmonary vascular permeability, the Evans blue dye accumulation in lung tissue at 28 and 33 dpi was measured. Moreover, to investigate the involvement of neutrophils in pulmonary vascular permeability, the permeability was examined in mice with neutrophil depletion (anti-Gr-1 +) at 28 and 33 dpi. There were four mice per group except for the control group with Rat IgG (n = 3). Asterisks denote a significant difference between HTNV-infected mice with anti-Gr-1 MAb and with Rat IgG (*, P < 0.05 [Mann-Whitney U test]).
DISCUSSION
We demonstrated in this study for the first time that pulmonary vascular hyperpermeability is caused by HTNV infection and results in the development of pulmonary edema in SCID mice. Increases in neutrophils in the lung and blood were observed at 28 dpi, when pulmonary edema began to be observed. Increased neutrophils in blood have also been observed by hantavirus infection in humans and animal models (26, 35–37, 48), but their role in the pathogenesis has remained unclear. It has been reported that activated neutrophils induce a respiratory burst and degranulation of neutrophil granules (58). In the present study, the adhesion of neutrophils was observed in the vessels of infected mice, suggesting that neutrophils in the lung are activated. In the HTNV-infected mice, the extent of pulmonary edema peaked at 35 dpi and, for some reason, was reduced at 42 dpi even though the virus titers were high. The number of neutrophils in blood peaked at 28 dpi and was decreased at 33 dpi, suggesting that the number of neutrophils correlates with the extent of pulmonary edema 1 week earlier. Furthermore, the occurrence of pulmonary edema was inhibited by neutrophil depletion. Pulmonary vascular permeability was also significantly suppressed by neutrophil depletion in infected mice. Taken together, the results indicate that neutrophils play an important role in the induction of pulmonary vascular hyperpermeability and occurrence of pulmonary edema after hantavirus infection in the mice.
It has been shown that neutrophils produce various vascular permeability factors including vascular endothelial growth factor (VEGF), matrix metalloproteinases, heparin-binding protein, tumor necrosis factor alpha, interleukin-1β, gamma interferon, and nitric oxide (42–46). VEGF has been noted recently as a vascular permeability factor in hantavirus infection. Shrivastava-Ranjan et al. demonstrated that in vitro ANDV infection alone induces VEGF secretion from endothelial cells, which occurs simultaneously with internalization of vascular endothelial cadherin composing adherens junctions (59). Moreover, elevated VEGF levels were observed in serum, pulmonary edema fluid and peripheral blood mononuclear cells from patients with acute HPS (59, 60). Hypoxia itself also promotes the secretion of VEGF from endothelial cells (61–63).
There are different opinions regarding the role of T cells in the pathogenesis. T cells have thus far been regarded as playing a key role in the immunopathology of hantavirus infection based on circumstantial evidence (24, 26, 64–70). For instance, studies of fatal HPS cases have revealed mononuclear cell infiltrates in pulmonary tissue consisting largely of CD4+ and CD8+ T cells (24, 26). In addition, very large proportions of SNV-specific CD8+ T cells were circulating in HPS patients during the acute phase, and the proportion of SNV-specific T cells was significantly higher in patients with severe HPS than in patients with moderate disease (65, 66). Our previous studies showed that adoptive transfer of immune splenocytes (predominantly lymphocytes: CD4+ T cells, ca. 25%; CD8+ T cells, 14%; and B cells, 50%) into SCID mice that had been infected with HTNV resulted in an increase of serum blood urea nitrogen level in the mice (71). Furthermore, adoptive transfer of immune whole splenocytes and immune T cells alone induced pulmonary edema in recipient mice earlier than that in untransferred HTNV-infected mice (T. Koma, K. Yoshimatsu, and J. Arikawa, unpublished data).
On the other hand, depletion of T cells in ANDV-infected Syrian hamsters has no effect on the onset, symptoms, or severity of lethal pulmonary edema, suggesting that T cell responses may not be determinants of the pathogenesis in the hamster model (72, 73). A recent study demonstrated that SNV caused a disease such as pulmonary edema in immunosuppressed Syrian hamsters (74). In that study, it seemed that the pathological change was correlated not with the number of blood neutrophils but with virus titer. In the present SCID mice study, the appearance of pulmonary vascular hyperpermeability and pulmonary edema were markedly reduced by neutrophil depletion. However, the elevated permeability was not completely normalized (approximately half inhibition) by neutrophil depletion in infected SCID mice. Therefore, it is thought that in addition to neutrophils, T cell-mediated and other mechanisms are involved in vascular hyperpermeability and the pathogenesis of hantavirus infection.
ACKNOWLEDGMENTS
RB6-8C5 hybridoma was kindly provided by the Cell Resource Center for Biomedical Research Institute of Development, Aging, and Cancer, Tohoku University. We are grateful to Miho Nodagashira and Shinya Tanaka of the Graduate School of Medicine, Hokkaido University, for their expert technical advice. We also acknowledge Stewart Chisholm of the Stewart English School for revising the grammar in the final manuscript.
This study was supported in part by the Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists program (23-5017). This study was also supported in part by a grant from the Global COE program (Establishment of International Collaboration Centers for Zoonosis Control) and by Grants-in-Aid for Research on Emerging and Re-Emerging Infectious Diseases from the Ministry of Health, Labor, and Welfare, including H22-emerging-ippan-006.
Footnotes
Published ahead of print 9 April 2014
REFERENCES
- 1.Plyusnin A, Vapalahti O, Vaheri A. 1996. Hantaviruses: genome structure, expression and evolution. J. Gen. Virol. 77:2677–2687. 10.1099/0022-1317-77-11-2677 [DOI] [PubMed] [Google Scholar]
- 2.Nichol ST, Spiropoulou CF, Morzunov S, Rollin PE, Ksiazek TG, Feldmann H, Sanchez A, Childs J, Zaki S, Peters CJ. 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262:914–917. 10.1126/science.8235615 [DOI] [PubMed] [Google Scholar]
- 3.Lee HW, Lee PW, Johnson KM. 1978. Isolation of the etiologic agent of Korean Hemorrhagic fever. J. Infect. Dis. 137:298–308. 10.1093/infdis/137.3.298 [DOI] [PubMed] [Google Scholar]
- 4.Lee HW, van der Groen G. 1989. Hemorrhagic fever with renal syndrome. Prog. Med. Virol. 36:62–102 [PubMed] [Google Scholar]
- 5.Lee HW. 1982. Korean hemorrhagic fever. Prog. Med. Virol. 28:96–113 [PubMed] [Google Scholar]
- 6.Vapalahti O, Mustonen J, Lundkvist A, Henttonen H, Plyusnin A, Vaheri A. 2003. Hantavirus infections in Europe. Lancet Infect. Dis. 3:653–661. 10.1016/S1473-3099(03)00774-6 [DOI] [PubMed] [Google Scholar]
- 7.Muranyi W, Bahr U, Zeier M, van der Woude FJ. 2005. Hantavirus infection. J. Am. Soc. Nephrol. 16:3669–3679. 10.1681/ASN.2005050561 [DOI] [PubMed] [Google Scholar]
- 8.Plyusnin A, Kruger DH, Lundkvist A. 2001. Hantavirus infections in Europe. Adv. Virus Res. 57:105–136. 10.1016/S0065-3527(01)57002-5 [DOI] [PubMed] [Google Scholar]
- 9.Enria DA, Briggiler AM, Pini N, Levis S. 2001. Clinical manifestations of New World hantaviruses. Curr. Top. Microbiol. Immunol. 256:117–134 [DOI] [PubMed] [Google Scholar]
- 10.Jonsson CB, Figueiredo LT, Vapalahti O. 2010. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 23:412–441. 10.1128/CMR.00062-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsergouli K, Papa A. 2013. Vascular endothelial growth factor levels in Dobrava/Belgrade virus infections. Viruses 5:3109–3118. 10.3390/v5123109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D. 1965. Clinical analysis of 111 fatal cases of epidemic hemorrhagic fever. Am. J. Med. 39:218–220. 10.1016/0002-9343(65)90044-6 [DOI] [PubMed] [Google Scholar]
- 13.Steer A. 1955. Pathology of hemorrhagic fever: a comparison of the findings; 1951 and 1952. Am. J. Pathol. 31:201–221 [PMC free article] [PubMed] [Google Scholar]
- 14.Clement J, Colson P, McKenna P. 1994. Hantavirus pulmonary syndrome in New England and Europe. N. Engl. J. Med. 331:545–548. 10.1056/NEJM199408253310813 [DOI] [PubMed] [Google Scholar]
- 15.Lukes RJ. 1954. The pathology of thirty-nine fatal cases of epidemic hemorrhagic fever. Am. J. Med. 16:639–650. 10.1016/0002-9343(54)90270-3 [DOI] [PubMed] [Google Scholar]
- 16.Rasmuson J, Andersson C, Norrman E, Haney M, Evander M, Ahlm C. 2011. Time to revise the paradigm of hantavirus syndromes? Hantavirus pulmonary syndrome caused by European hantavirus. Eur. J. Clin. Microbiol. Infect. Dis. 30:685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen AT, Penalba C, Bernadac P, Jaafar S, Kessler M, Canton P, Hoen B. 2001. Respiratory manifestations of hemorrhagic fever with renal syndrome: retrospective study of 129 cases in Champagne-Ardenne and Lorraine. Presse Med. 30:55–58 (In French.) [PubMed] [Google Scholar]
- 18.Seitsonen E, Hynninen M, Kolho E, Kallio-Kokko H, Pettila V. 2006. Corticosteroids combined with continuous veno-venous hemodiafiltration for treatment of hantavirus pulmonary syndrome caused by Puumala virus infection. Eur. J. Clin. Microbiol. Infect. Dis. 25:261–266. 10.1007/s10096-006-0117-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hjelle B, Jenison SA, Goade DE, Green WB, Feddersen RM, Scott AA. 1995. Hantaviruses: clinical, microbiologic, and epidemiologic aspects. Crit. Rev. Clin. Lab. Sci. 32:469–508. 10.3109/10408369509082592 [DOI] [PubMed] [Google Scholar]
- 20.Hjelle B, Goade D, Torrez-Martinez N, Lang-Williams M, Kim J, Harris RL, Rawlings JA. 1996. Hantavirus pulmonary syndrome, renal insufficiency, and myositis associated with infection by Bayou hantavirus. Clin. Infect. Dis. 23:495–500. 10.1093/clinids/23.3.495 [DOI] [PubMed] [Google Scholar]
- 21.Pergam SA, Schmidt DW, Nofchissey RA, Hunt WC, Harford AH, Goade DE. 2009. Potential renal sequelae in survivors of hantavirus cardiopulmonary syndrome. Am. J. Trop. Med. Hyg. 80:279–285 [PMC free article] [PubMed] [Google Scholar]
- 22.Lahdevirta J. 1982. Clinical features of HFRS in Scandinavia as compared with East Asia. Scand. J. Infect. Dis. Suppl. 36:93–95 [PubMed] [Google Scholar]
- 23.Pensiero MN, Sharefkin JB, Dieffenbach CW, Hay J. 1992. Hantaan virus infection of human endothelial cells. J. Virol. 66:5929–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nolte KB, Feddersen RM, Foucar K, Zaki SR, Koster FT, Madar D, Merlin TL, McFeeley PJ, Umland ET, Zumwalt RE. 1995. Hantavirus pulmonary syndrome in the United States: a pathological description of a disease caused by a new agent. Hum. Pathol. 26:110–120. 10.1016/0046-8177(95)90123-X [DOI] [PubMed] [Google Scholar]
- 25.Yanagihara R, Silverman DJ. 1990. Experimental infection of human vascular endothelial cells by pathogenic and nonpathogenic hantaviruses. Arch. Virol. 111:281–286. 10.1007/BF01311063 [DOI] [PubMed] [Google Scholar]
- 26.Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, Feddersen RM, Zumwalt RE, Miller GL, Khan AS, Rollin PE, Ksiazek TG, Nichol ST, Mahy BWJ, Peters CJ. 1995. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–579 [PMC free article] [PubMed] [Google Scholar]
- 27.Koster F, Foucar K, Hjelle B, Scott A, Chong YY, Larson R, McCabe M. 2001. Rapid presumptive diagnosis of hantavirus cardiopulmonary syndrome by peripheral blood smear review. Am. J. Clin. Pathol. 116:665–672. 10.1309/CNWF-DC72-QYMR-M8DA [DOI] [PubMed] [Google Scholar]
- 28.Huang C, Jin B, Wang M, Li E, Sun C. 1994. Hemorrhagic fever with renal syndrome: relationship between pathogenesis and cellular immunity. J. Infect. Dis. 169:868–870. 10.1093/infdis/169.4.868 [DOI] [PubMed] [Google Scholar]
- 29.Markotic A, Dasic G, Gagro A, Sabioncello A, Rabatic S, Kuzman I, Zgorelec R, Smoljan I, Beus I, Zupanc TA, Dekaris D. 1999. Role of peripheral blood mononuclear cell (PBMC) phenotype changes in the pathogenesis of haemorrhagic fever with renal syndrome (HFRS). Clin. Exp. Immunol. 115:329–334. 10.1046/j.1365-2249.1999.00790.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Temonen M, Mustonen J, Helin H, Pasternack A, Vaheri A, Holthofer H. 1996. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: an immunohistochemical study. Clin. Immunol. Immunopathol. 78:47–55. 10.1006/clin.1996.0007 [DOI] [PubMed] [Google Scholar]
- 31.Krakauer T, Leduc JW, Krakauer H. 1995. Serum levels of tumor necrosis factor-alpha, interleukin-1, and interleukin-6 in hemorrhagic fever with renal syndrome. Viral Immunol. 8:75–79. 10.1089/vim.1995.8.75 [DOI] [PubMed] [Google Scholar]
- 32.Krakauer T, Leduc JW, Morrill JC, Anderson AO, Krakauer H. 1994. Serum levels of alpha and gamma interferons in hemorrhagic fever with renal syndrome. Viral Immunol. 7:97–101. 10.1089/vim.1994.7.97 [DOI] [PubMed] [Google Scholar]
- 33.Linderholm M, Ahlm C, Settergren B, Waage A, Tarnvik A. 1996. Elevated plasma levels of tumor necrosis factor (TNF)-alpha, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 173:38–43. 10.1093/infdis/173.1.38 [DOI] [PubMed] [Google Scholar]
- 34.Makela S, Mustonen J, Ala-Houhala I, Hurme M, Koivisto AM, Vaheri A, Pasternack A. 2004. Urinary excretion of interleukin-6 correlates with proteinuria in acute Puumala hantavirus-induced nephritis. Am. J. Kidney Dis. 43:809–816. 10.1053/j.ajkd.2003.12.044 [DOI] [PubMed] [Google Scholar]
- 35.Pal E, Strle F, Avsic-Zupanc T. 2005. Hemorrhagic fever with renal syndrome in the Pomurje region of Slovenia: an 18-year survey. Wien Klin. Wochenschr. 117:398–405. 10.1007/s00508-005-0359-2 [DOI] [PubMed] [Google Scholar]
- 36.Hjelle B, Jenison S, Mertz G, Koster F, Foucar K. 1994. Emergence of hantaviral disease in the southwestern United States. West. J. Med. 161:467–473 [PMC free article] [PubMed] [Google Scholar]
- 37.Mir MA. 2010. Hantaviruses. Clin. Lab. Med. 30:67–91. 10.1016/j.cll.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lilius EM, Nuutila J. 2012. Bacterial infections, DNA virus infections, and RNA virus infections manifest differently in neutrophil receptor expression. Sci. World J. 2012:527347. 10.1100/2012/527347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dale DC. 2010. Neutropenia and neutrophilia, p 939–950 In Kaushansky K, Beutler E, Seligsohn U, Lichtman MA, Kipps TJ, Prchal J. (ed), Williams hematology, 8th ed. McGraw-Hill Book Co, Inc, New York, NY [Google Scholar]
- 40.Ksiazek TG, Peters CJ, Rollin PE, Zaki S, Nichol S, Spiropoulou C, Morzunov S, Feldmann H, Sanchez A, Khan AS, Mahy BWJ, Wachsmuth K, Butler JC. 1995. Identification of a new North American hantavirus that causes acute pulmonary insufficiency. Am. J. Trop. Med. Hyg. 52:117–123 [DOI] [PubMed] [Google Scholar]
- 41.Abraham E. 2003. Neutrophils and acute lung injury. Crit. Care Med. 31:S195–S199. 10.1097/01.CCM.0000057843.47705.E8 [DOI] [PubMed] [Google Scholar]
- 42.Taichman NS, Young S, Cruchley AT, Taylor P, Paleolog E. 1997. Human neutrophils secrete vascular endothelial growth factor. J. Leukoc. Biol. 62:397–400 [DOI] [PubMed] [Google Scholar]
- 43.Mantovani A, Cassatella MA, Costantini C, Jaillon S. 2011. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11:519–531. 10.1038/nri3024 [DOI] [PubMed] [Google Scholar]
- 44.Gueders MM, Foidart JM, Noel A, Cataldo DD. 2006. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur. J. Pharmacol. 533:133–144. 10.1016/j.ejphar.2005.12.082 [DOI] [PubMed] [Google Scholar]
- 45.Tapper H, Karlsson A, Morgelin M, Flodgaard H, Herwald H. 2002. Secretion of heparin-binding protein from human neutrophils is determined by its localization in azurophilic granules and secretory vesicles. Blood 99:1785–1793. 10.1182/blood.V99.5.1785 [DOI] [PubMed] [Google Scholar]
- 46.DiStasi MR, Ley K. 2009. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 30:547–556. 10.1016/j.it.2009.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hooper JW, Larsen T, Custer DM, Schmaljohn CS. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289:6–14. 10.1006/viro.2001.1133 [DOI] [PubMed] [Google Scholar]
- 48.Wahl-Jensen V, Chapman J, Asher L, Fisher R, Zimmerman M, Larsen T, Hooper JW. 2007. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 81:7449–7462. 10.1128/JVI.00238-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chu YK, Jennings GB, Schmaljohn CS. 1995. A vaccinia virus-vectored Hantaan virus vaccine protects hamsters from challenge with Hantaan and Seoul viruses but not Puumala virus. J. Virol. 69:6417–6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmaljohn CS, Chu YK, Schmaljohn AL, Dalrymple JM. 1990. Antigenic subunits of Hantaan virus expressed by baculovirus and vaccinia virus recombinants. J. Virol. 64:3162–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Araki K, Yoshimatsu K, Lee BH, Okumura M, Kariwa H, Takashima I, Arikawa J. 2004. Age-dependent hantavirus-specific CD8+ T-cell responses in mice infected with Hantaan virus. Arch. Virol. 149:1373–1382. 10.1007/s00705-003-0285-4 [DOI] [PubMed] [Google Scholar]
- 52.Seto T, Nagata N, Yoshikawa K, Ichii O, Sanada T, Saasa N, Ozaki Y, Kon Y, Yoshii K, Takashima I, Kariwa H. 2011. Infection of Hantaan virus strain AA57 leading to pulmonary disease in laboratory mice. Virus Res. 163:284–290. 10.1016/j.virusres.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 53.Tamura M, Asada H, Kondo K, Tanishita O, Kurata T, Yamanishi K. 1989. Pathogenesis of Hantaan virus in mice. J. Gen. Virol. 70(Pt 11):2897–2906. 10.1099/0022-1317-70-11-2897 [DOI] [PubMed] [Google Scholar]
- 54.Yoshimatsu K, Arikawa J, Tamura M, Yoshida R, Lundkvist A, Niklasson B, Kariwa H, Azuma I. 1996. Characterization of the nucleocapsid protein of Hantaan virus strain 76-118 using monoclonal antibodies. J. Gen. Virol. 77(Pt 4):695–704. 10.1099/0022-1317-77-4-695 [DOI] [PubMed] [Google Scholar]
- 55.Nijnik A, Madera L, Ma S, Waldbrook M, Elliott MR, Easton DM, Mayer ML, Mullaly SC, Kindrachuk J, Jenssen H, Hancock RE. 2010. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. J. Immunol. 184:2539–2550. 10.4049/jimmunol.0901813 [DOI] [PubMed] [Google Scholar]
- 56.Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI. 2000. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 191:661–668. 10.1084/jem.191.4.661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaturvedi UC, Dhawan R, Khanna M, Mathur A. 1991. Breakdown of the blood-brain barrier during dengue virus infection of mice. J. Gen. Virol. 72(Pt 4):859–866 [DOI] [PubMed] [Google Scholar]
- 58.Lacy P, Eitzen G. 2008. Control of granule exocytosis in neutrophils. Front. Biosci. 13:5559–5570. 10.2741/3099 [DOI] [PubMed] [Google Scholar]
- 59.Shrivastava-Ranjan P, Rollin PE, Spiropoulou CF. 2010. Andes virus disrupts the endothelial cell barrier by induction of vascular endothelial growth factor and downregulation of VE-cadherin. J. Virol. 84:11227–11234. 10.1128/JVI.01405-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gavrilovskaya I, Gorbunova E, Koster F, Mackow E. 2012. Elevated VEGF levels in pulmonary edema fluid and PBMCs from patients with acute hantavirus pulmonary syndrome. Adv. Virol. 2012:674360. 10.1155/2012/674360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. 1995. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 375:577–581. 10.1038/375577a0 [DOI] [PubMed] [Google Scholar]
- 62.Pham I, Uchida T, Planes C, Ware LB, Kaner R, Matthay MA, Clerici C. 2002. Hypoxia upregulates VEGF expression in alveolar epithelial cells in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L1133–L1142 [DOI] [PubMed] [Google Scholar]
- 63.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. 2004. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 6:485–495. 10.1016/j.ccr.2004.09.026 [DOI] [PubMed] [Google Scholar]
- 64.Mori M, Rothman AL, Kurane I, Montoya JM, Nolte KB, Norman JE, Waite DC, Koster FT, Ennis FA. 1999. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 179:295–302. 10.1086/314597 [DOI] [PubMed] [Google Scholar]
- 65.Kilpatrick ED, Terajima M, Koster FT, Catalina MD, Cruz J, Ennis FA. 2004. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 172:3297–3304 [DOI] [PubMed] [Google Scholar]
- 66.Ennis FA, Cruz J, Spiropoulou CF, Waite D, Peters CJ, Nichol ST, Kariwa H, Koster FT. 1997. Hantavirus pulmonary syndrome: CD8+ and CD4+ cytotoxic T lymphocytes to epitopes on Sin Nombre virus nucleocapsid protein isolated during acute illness. Virology 238:380–390. 10.1006/viro.1997.8827 [DOI] [PubMed] [Google Scholar]
- 67.Borges AA, Campos GM, Moreli ML, Souza RL, Aquino VH, Saggioro FP, Figueiredo LT. 2006. Hantavirus cardiopulmonary syndrome: immune response and pathogenesis. Microbes Infect. 8:2324–2330. 10.1016/j.micinf.2006.04.019 [DOI] [PubMed] [Google Scholar]
- 68.Tuuminen T, Kekalainen E, Makela S, Ala-Houhala I, Ennis FA, Hedman K, Mustonen J, Vaheri A, Arstila TP. 2007. Human CD8+ T cell memory generation in Puumala hantavirus infection occurs after the acute phase and is associated with boosting of EBV-specific CD8+ memory T cells. J. Immunol. 179:1988–1995 [DOI] [PubMed] [Google Scholar]
- 69.Terajima M, Hayasaka D, Maeda K, Ennis FA. 2007. Immunopathogenesis of hantavirus pulmonary syndrome and hemorrhagic fever with renal syndrome: do CD8+ T cells trigger capillary leakage in viral hemorrhagic fevers? Immunol. Lett. 113:117–120. 10.1016/j.imlet.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rasmuson J, Pourazar J, Linderholm M, Sandstrom T, Blomberg A, Ahlm C. 2011. Presence of activated airway T lymphocytes in human Puumala hantavirus disease. Chest 140:715–722. 10.1378/chest.10-2791 [DOI] [PubMed] [Google Scholar]
- 71.Yoshimatsu K, Arikawa J, Ohbora S, Itakura C. 1997. Hantavirus infection in SCID mice. J. Vet. Med. Sci. 59:863–868. 10.1292/jvms.59.863 [DOI] [PubMed] [Google Scholar]
- 72.Hammerbeck CD, Hooper JW. 2011. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. J. Virol. 85:9929–9944. 10.1128/JVI.05356-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prescott J, Safronetz D, Haddock E, Robertson S, Scott D, Feldmann H. 2013. The adaptive immune response does not influence hantavirus disease or persistence in the Syrian hamster. Immunology 140:168–178. 10.1111/imm.12116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brocato RL, Hammerbeck CD, Bell TM, Wells JB, Queen LA, Hooper JW. 2014. A lethal disease model for hantavirus pulmonary syndrome in immunosuppressed Syrian hamsters infected with Sin Nombre virus. J. Virol. 88:811–819. 10.1128/JVI.02906-13 [DOI] [PMC free article] [PubMed] [Google Scholar]