

Abstract
A high-throughput screening campaign was conducted to interrogate a 380,000+ small-molecule library for novel D2 dopamine receptor modulators using a calcium mobilization assay. Active agonist compounds from the primary screen were examined for orthogonal D2 dopamine receptor signaling activities including cAMP modulation and β-arrestin recruitment. Although the majority of the subsequently confirmed hits activated all signaling pathways tested, several compounds showed a diminished ability to stimulate β-arrestin recruitment. One such compound (MLS1547; 5-chloro-7-[(4-pyridin-2-ylpiperazin-1-yl)methyl]quinolin-8-ol) is a highly efficacious agonist at D2 receptor–mediated G protein–linked signaling, but does not recruit β-arrestin as demonstrated using two different assays. This compound does, however, antagonize dopamine-stimulated β-arrestin recruitment to the D2 receptor. In an effort to investigate the chemical scaffold of MLS1547 further, we characterized a set of 24 analogs of MLS1547 with respect to their ability to inhibit cAMP accumulation or stimulate β-arrestin recruitment. A number of the analogs were similar to MLS1547 in that they displayed agonist activity for inhibiting cAMP accumulation, but did not stimulate β-arrestin recruitment (i.e., they were highly biased). In contrast, other analogs displayed various degrees of G protein signaling bias. These results provided the basis to use pharmacophore modeling and molecular docking analyses to build a preliminary structure-activity relationship of the functionally selective properties of this series of compounds. In summary, we have identified and characterized a novel G protein–biased agonist of the D2 dopamine receptor and identified structural features that may contribute to its biased signaling properties.
Introduction
Dopamine receptors mediate the many actions of dopamine in the brain and periphery. In mammals, five distinct dopamine receptors have been characterized, which are divided into two subfamilies based on their structure, pharmacology, and signaling properties (Sibley et al., 1992). D1-like receptors (D1R and D5R) are Gαs/olf coupled, whereas D2-like receptors (D2R, D3R. and D4R) are Gαi/o coupled (Sibley et al., 1992). Dopamine receptors have also been shown to signal through recruitment and activation of the scaffolding protein β-arrestin (Beaulieu et al., 2007a). Among the dopamine receptors, the D2R is one of the most validated drug targets in neurology and psychiatry. However, most drugs targeting the D2R are problematic, either being less efficacious than desired or possessing adverse side effects due to the activation or blockade of multiple parallel signaling pathways. Despite recent advances, it remains unclear which signaling arms of the D2R are involved in the therapeutic effects of various agents used to treat neuropsychiatric disease states associated with the D2R.
One of the best characterized signaling pathways of the D2R is Gi/Go-mediated inhibition of adenylate cyclase, which reduces intracellular cAMP levels and thereby attenuates phosphorylation of the 32-kDa dopamine and cAMP-regulated phosphoprotein (DARPP-32) (Bateup et al., 2008) by protein kinase A. DARPP-32 is a protein phosphatase that acts as an integrator of cell signaling of many neurotransmitters, including dopamine (Svenningsson et al., 2004). Reduction of DARPP-32 phosphorylation inhibits its activity and associated downstream signaling pathways (Svenningsson et al., 2004; Bateup et al., 2008). Notably, administration of antipsychotic drugs, such as haloperidol or clozapine, has been shown to increase the level of DARPP-32 phosphorylation (Pozzi et al., 2003). A more recently characterized signaling pathway for the D2R is activation of glycogen synthase kinase 3 β (GSK3β), which is G protein–independent and occurs through agonist recruitment of β-arrestin-2 to the receptor. This leads to the formation of a β-arrestin–dependent complex of protein kinase B (Akt) and protein phosphatase 2A, which results in dephosphorylation of Akt and subsequent activation of GSK3β (Beaulieu et al., 2007b, 2008). Interestingly, Caron and colleagues have argued that inhibition of this pathway in D2R-expressing neurons is correlated with antipsychotic properties (Masri et al., 2008; Urs et al., 2012), whereas Roth and colleagues have suggested that stimulation of the D2R–β-arrestin pathway may actually enhance antipsychotic efficacy (Allen et al., 2011).
A promising approach to dissecting the importance of these signaling pathways, and resolving these associated controversies, is to study them using ligands that exhibit functionally selective or biased signaling properties (Urban et al., 2007; Whalen et al., 2011). Many G protein–coupled receptors (GPCRs) are able to transduce signals through more than one intracellular pathway. Although in most cases the endogenous transmitter will activate all signaling pathways, synthetic agonists may preferentially activate one signaling pathway over another, or even activate one while inhibiting another (Kenakin, 2007a,b, 2008; Mailman, 2007; Urban et al., 2007). Although the mechanisms underlying functional selectivity are not known, a leading hypothesis is that GPCRs can adopt multiple functionally “active” conformational states that are either stabilized or induced by these selective ligands (Kobilka and Deupi, 2007; Wess et al., 2008). Although relatively few biased ligands have been described for the D2R (Kilts et al., 2002; Mottola et al., 2002; Gay et al., 2004; Lane et al., 2007, 2008), existing examples strongly support the concept of the D2R being able to adopt multiple signaling-biased confirmations. Recently, a structural basis for functional selectivity of several GPCRs has been proposed (Liu et al., 2012; Dror et al., 2013; Kruse et al., 2013; Wacker et al., 2013; Abdul-Ridha et al., 2014) suggesting that rational design of functionally selective compounds may be possible.
Recently, Jin and colleagues developed and characterized a series of analogs of the atypical antipsychotic aripiprazole that are partial agonists of D2R-mediated β-arrestin recruitment, yet fail to stimulate Gi-linked cAMP inhibition (Allen et al., 2011; Chen et al., 2012). In contrast, no biased ligands have been described with the opposing pharmacology for the D2R, that is, stimulation of G protein signaling pathways without activation of β-arrestin recruitment. We now report the discovery of a novel, highly efficacious G protein–biased agonist for the D2R that also antagonizes β-arrestin recruitment to the receptor. Identification of such functionally selective ligands may provide the requisite pharmacological probes with which to dissect these two signaling pathways and elucidate their function in vivo. Functionally selective agonists may also result in improved therapies for certain neuropsychiatric disorders, such as Parkinson’s disease, in which D2R stimulation is desired, and schizophrenia, where inhibition of D2R signaling is the goal.
Materials and Methods
Calcium Mobilization Assay.
Flp-In T-Rex 293 cells were stably transfected with human D2SR and Gqi5 protein using the Flp-In T-Rex expression system (Life Technologies, Grand Island, NY). The cell line was constructed by cotransfecting SFD2s/FRT/TO and pOG44, followed by hygromycin B selection. Gqi5/pIRESpuro3 (Clontech, Mountain View, CA) was then transfected into the D2R stable cell line, followed by selection with puromycin. D2R expression is controlled by tetracycline induction, whereas Gqi5 is continuously expressed.
D2R-stimulated calcium mobilization was measured using methods similar to those previously published by our laboratory (Chun et al., 2013). Cells were induced with 1 μM tetracycline added directly to the culture media and plated in 384- or 1536-well, optical, clear-bottom, black-walled plates (Greiner Bio-One, Monroe, NC). Twenty microliters per well (20,000 cells/well) were added to 384-well plates, and 3 μl/well (4000 cells/well) to a 1536-well plate. The next day, cells were incubated for 60 minutes at room temperature in the dark with Fluo-8 NW calcium dye in the presence of an extracellular signal quencher (Screen Quest Fluo-8 NW Calcium Assay Kit; AAT Bioquest, Inc., Sunnyvale, CA), as recommended by the manufacturer. The plates were then treated with various concentrations of agonist in the presence of 0.2 mM sodium metabisulfite. Plates were read kinetically in real time (every 0.6 second) for 2 minutes after agonist addition. Compound additions were done in unison using a 384-tip onboard robotic pipette (384-well assays) or an onboard 1536-pintool (1536-well assays) while continuously reading at an excitation wavelength of 480 nm and an emission wavelength of 540 nm on a functional drug screening system (FDSS) μCell (384-well assays) or an FDSS 7000 (1536-well assays) (Hamamatsu, Bridgewater, NJ). Data were recorded and quantified as maximum minus minimum (max−min) relative florescence units within the assay window using FDSS software.
cAMP Inhibition Assay.
D2R-mediated inhibition of forskolin-stimulated cAMP production was assayed using the DiscoveRx HitHunter assay kit (DiscoveRx Inc., Fremont, CA). CHO-K1 cells stably expressing the human D2R long isoform (DiscoveRx) were seeded in Cell Plating Media 2 (DiscoveRx Inc.) at a density of 5000 cells/well in 384-well black, clear-bottom plates. After 16–24 hours of incubation at 37°C, 5% CO2, and 90% humidity, the medium was removed and replaced with 5 μl/well phosphate-buffered saline (PBS). Cells were treated with 2.5 μl of various concentrations of compound diluted in PBS in the presence of an ∼EC80 concentration of forskolin (100 μM) and 0.2 mM sodium metabisulfite and incubated for 60 minutes at 37°C, 5% CO2, and 90% humidity. DiscoveRx HitHunter reagents were then added, and cells were incubated in the dark at room temperature, according to manufacturer recommendations. Luminescence was measured on a Hamamatsu FDSS μCell (Hamamatsu) for 8.5 seconds. Data were collected as relative luminescence units (RLUs), and values were normalized to a percentage of the maximum forskolin-stimulated cAMP signal. Data fit to a single site model and the Hill coefficients of the concentration response curves did not significantly differ from unity.
β-Arrestin Recruitment Assay.
The ability of the agonist-activated receptor to recruit β-arrestin-2 was determined using the DiscoveRx PathHunter technology (DiscoveRx) that involves enzyme complementation of fusion-tagged receptor along with an arrestin recruitment modulating sequence and β-arrestin-2 proteins. Control experiments determined that this PathHunter receptor construct will couple to G protein–mediated signaling with similar efficacy as an unmodified construct (data not shown). Assays were conducted, with minor modifications, as previously published by our laboratory (Banala et al., 2011; Bergman et al., 2013). In brief, CHO-K1 cells expressing D2R long isoform (DiscoveRx) were seeded in Cell Plating Media 2 (DiscoveRx) at a density of 2625 cells/well in 384-well black, clear-bottom plates. Following 24 hours of incubation, the cells were treated with multiple concentrations of compound in PBS containing 1% dimethylsulfoxide and incubated at 37°C for 90 minutes. DiscoveRx reagent was then added to cells according to the manufacturer’s recommendations followed by a 30–60-minute incubation at room temperature. Luminescence was measured on a Hamamatsu FDSS μCell reader. Data were collected as RLUs and subsequently normalized to a percentage of the control luminescence seen with a maximum concentration of dopamine, with 0% being RLUs seen in the absence of any compound. The Hill coefficients of the concentration response curves did not significantly differ from unity.
β-Arrestin Bioluminescence Resonance Energy Transfer Assay.
To directly assess induction of D2R–β-arrestin-2 interaction, we used a bioluminescence resonance energy transfer (BRET) assay that utilizes a cell line stably transfected with an Rluc-8 fusion-tagged D2R (short isoform) under a tetracycline-inducible promoter, as well as mVenus fusion-tagged β-arrestin-2 (Hamdan et al., 2005; Klewe et al., 2008). The cell line was constructed using Flp-In T-Rex 293 cells (Invitrogen, Life Technologies) transfected with pIRESpuro3/mVenus/β-arrestin-2, where the mVenus tag is on the N terminus of the human β-arrestin-2. Clones were then analyzed for expression of the construct following selection with 2 μg/ml puromycin. The cell line with the highest level of expression was then transfected with pcDNA5/FRT/TO-SFD2LRluc-8 and POG44 followed by hygromycin selection and subsequent functional screening to select the final stable line. Addition of the Rluc-8 substrate coelenterazine h results in an emission at 485 nm. However, when in close proximity to mVenus, resonance energy transfer leads to a shift in the emission spectrum from 485 to 510–540 nm, thereby quantifying the interaction between the receptor and the β-arrestin-2 protein. Cells were induced for 24 hours by addition of 1 μM tetracycline directly to the culture media, resulting in membrane receptor expression of approximately 5.8 pmol/mg protein. Cells were then removed from the plates using Earle’s balanced salt solution without calcium (EBSS−), pelleted by centrifugation, resuspended (200,000 cells/ml) in Dulbecco’s phosphate-buffered saline (Mediatech, Manassas, VA) plus 0.05 g/500 ml sucrose, and seeded into 96-well, solid-bottom white assay plates (20,000 cells/well) (Greiner Bio-One). Cells were allowed to sit for 45 minutes at room temperature and were then treated with 5 μM coelenterazine h (Nanolight Technology, Pinetop, AZ), incubated for 5 minutes, and then stimulated with agonist using an onboard robotics 8-channel pipet head in a Flexstation III (Molecular Devices, Sunnyvale, CA). Original data were collected 5 minutes after agonist addition as a ratio of 525/485-nm emission. Data are expressed as normalized to the percentage of the maximum dopamine-induced ratio.
Radioligand Binding Assays.
Radioligand competition binding assays were conducted with slight modifications as previously described by our laboratory (Chun et al., 2013). Human embryonic kidney 293 cells stably transfected with human D1R, D2R, D3R, D4R, or D5R (Codex Biosolutions, Inc., Gaithersburg, MD) were dissociated from plates using EBSS−, and intact cells were collected by centrifugation at 900g for 10 minutes. Cells were resuspended and lysed using 5 mM Tris-HCl and 5 mM MgCl2 at pH 7.4 at 4°C. Cell lysate was pelleted by centrifugation at 30,000g for 30 minutes and resuspended in EBSS with calcium at pH 7.4. Cell lysates (100 μl, containing ∼8 μg of protein for D2-like receptor assays or ∼10 μg of protein for D1-like receptor assays) were incubated for 90 minutes at room temperature with the indicated concentrations of MLS1547 (5-chloro-7-[(4-pyridin-2-ylpiperazin-1-yl)methyl]quinolin-8-ol) and either 0.5 nM [3H]SCH23390 (8-chloro-3-methyl-5-phenyl-1,2,4,5-tetrahydro-3-benzazepin-7-ol) (D1R and D5R) or 0.5 nM [3H]methylspiperone (D2R, D3R, and D4R) in a final reaction volume of 250 μl. Nonspecific binding was determined in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from free by filtration through a PerkinElmer Unifilter-96 GF/C 96-well microplate using the PerkinElmer Unifilter-96 Harvester (PerkinElmer, Waltham, MA), washing three times with 1 ml/well ice-cold assay buffer. After drying, 50 μl of liquid scintillation cocktail (MicroScint PS; PerkinElmer) was added to each well, and plates were sealed and analyzed on a PerkinElmer Topcount NXT.
Pharmacophore Modeling.
All modeling was performed using tools in the Schrödinger suite (Schrödinger Inc., New York, NY). Although the main goal of pharmacophore modeling is to explain the molecular features that are associated with active compounds, such capability can benefit from the inclusion of “inactive” compounds in the model-building procedure. In our case, to differentiate G protein–biased from G protein–nonbiased agonists in such a model, we defined a so-called “biased activity” measure that is similar to the “bias” factor in Kenakin et al. (2012), but without taking the exponential form:
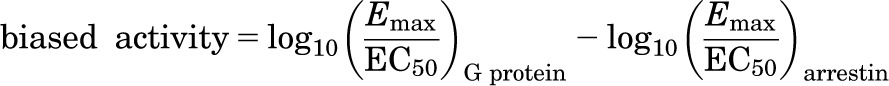 |
For G protein–biased agonists that did not exhibit measurable β-arrestin stimulation, we used the log10 (Emax/EC50) values from the cAMP assay to quantitate the biased activity of these ligands.
Based on the biased activity calculations described earlier, 10 compounds displaying either the strongest or weakest G protein bias were selected: NCGC9125, NCGC9126, NCGC6387, NCGC9141, and MLS1547 for the biased set, and NCGC9134, NCGC9132, NCGC5872, NCGC9131, and NCGC6388 for the nonbiased set (see Results and Discussion). For each compound, the three-dimensional structure was built using the program Maestro (version 9.5; Schrödinger LLC), and a single protonation state was chosen, with the 4 position of the piperazine bearing the positive charge in all cases. Multiple conformers were generated using the ConfGen program (version 2.5; Schrödinger LLC). At least 10 conformers per ligand were generated, which required sampling with the “intermediate” search strategy of the program.
The goal of a pharmacophore search is to identify the largest set of pharmacophore features with specific three-dimentional relationships, i.e., interfeature distances that are common among all active compounds. The pharmacophore model was built using the program Phase (version 3.6; Schrödinger LLC). The default pharmacophore features include positive (P), negative (N), hydrogen-bond acceptor (A), hydrogen-bond donor (D), aromatic (R), and hydrophobic (H) types. For the compounds studied here, the following functional groups were assigned to the H feature using a procedure that has been described in Greene (1994): isopropyl, aromatic halogens, aromatic CH3, and methoxy-CH3. The location of a given hydrophobic site is a weighted average of the positions of the nonhydrogen atoms in the associated fragment.
In addition, to allow ambiguous alignment of nonpolar features, aromatic groups in the ligands were assigned to both the R and H feature types. When we assigned these features to the selected ligands, the largest number of features in a pharmacophore hypothesis that produced matches for all five biased compounds was found to be 4. These 4-point pharmacophores were of the HHPR, AHHP, HPRR, and AHPR variants (the order of the features is arbitrary). For each of these variants, a number of three-dimensional hypotheses were enumerated and clustered based on all occurring interfeature distances. From this initial set of hypotheses, the one that best matched the actives was then determined by a more rigorous custom scoring function (“survival score”), which consisted of 1) the alignment score based on the root-mean-square-deviation of feature positions, 2) the vector score of aligned features with a directional character (aromatic, donor, and acceptor), and 3) the pairwise volume overlap of all the ligands aligned to the pharmacophore. This default scoring of all possible variants revealed that the HHPR and HRPR hypotheses were best able to explain the data. To eliminate the hypotheses that also matched the nonbiased compounds, the five most nonbiased compounds were used to rescore the hypotheses (“inactive score”). The finally selected pharmacophore was the HRPR variant (see Results and Discussion) with the best survival-inactive score.
Construction of a Novel Active D2R Model.
Our active D2R model was based on an active model of the D3R, for which an inactive crystal structure is available (Chien et al., 2010). The active D3R model was created by applying a set of spatial constraints obtained by comparing the inactive and active states of the β2-adrenergic receptor. In brief, the inactive and active structures of the β2-adrenergic receptor [PDB ID 2RH1 (Cherezov et al., 2007) and PDB ID 3SN6 (Rasmussen et al., 2011), respectively] were aligned and, by subtracting the coordinates of the Cα atoms of the inactive from the active form, a set of delta coordinates were created. These deltas were added to the coordinates of the aligned D3R structure to generate a set of spatial constraints. Using these constraints, the inactive model of the D3R was transformed into an active form by using a hybrid minimization–Monte Carlo scheme, implemented in the program Prime (version 3.3; Schrödinger LLC). The active D3R model was then used as a template in building the D2R homology model using Prime. Docking of compounds into the active-state model of the D2R was achieved using the program Glide (version 6.0; Schrödinger LLC), using the SP scoring function. To determine the binding mode of the congeneric series, a single reference compound (MLS1547) was first docked in the orthosteric binding site (OBS) revealed by the bound eticlopride in the D3R structure, which is formed by residues from transmembranes (TMs) 3, 5, 6, and 7. We found the pyridine moiety of MLS1547 preferred to point toward TM2. Interestingly, for two well studied D2R antagonists, spiperone and azaperone, that share a common 4-fluophenyl-4-butanone moiety, as proposed/validated previously (Boeckler et al., 2005), if we assume that such a moiety is bound in the OBS, then the pyridine moiety of azaperone points toward TM2, similar to MLS1547 (data not shown). We also docked MLS1547 in another D2R model in an active conformation based on a D3R active model (Newman et al., 2012) and equilibrated using molecular dynamics simulations. Encouragingly, results from both D2R models were consistent. All other compounds were then docked using core constraints on the core substructure shared by all compounds to ensure the core in the OBS adopted a similar binding mode.
Results and Discussion
In an effort to discover G protein–biased agonists of the D2R, we screened the ∼380,000-compound small-molecule library in the Molecular Libraries Probe Production Center Network at the National Center for Advancing Translational Sciences. The primary screen used a stably transfected cell line expressing the human D2R and a chimeric Gqi5 protein enabling robust calcium mobilization upon activation of the D2R. This screen identified 2288 compounds with significant D2-Gqi5 agonist activity, defined as compounds that, when screened at 40 μM, elicited a response larger than 3-fold over the standard deviation of mean signal from control wells with no compound. Agonists were also screened for calcium mobilization in a parental, nontransfected cell line, and any compound showing activity was eliminated. The active compounds were then subjected to verification by generating full concentration-response curves for each compound. Subsequently, the hit compounds that exhibited full dose response–activity relationships were evaluated in two orthogonal assays—one was to evaluate their ability to inhibit cAMP accumulation, a primary G protein signaling mechanism for the D2R (Bateup et al., 2008), and the other was to test their ability to recruit β-arrestin, an important signaling response independent of G protein activation (Beaulieu et al., 2007a, 2008). As would be expected, our results revealed that the vast majority of the hit compounds were equally active in both the G protein– and β-arrestin–mediated signaling assays (e.g., see Supplemental Fig. 1). However, a small subset of hits (about two dozen) showed greatly diminished efficacy and/or potency in the β-arrestin recruitment assay (see Supplemental Material for screening details). One of these compounds, MLS000051547 (MLS1547; Fig. 1A), was selected for further characterization as it appeared to have high efficacy in the G protein–mediated signaling assays, but no measurable activity in the β-arrestin recruitment assay.
Fig. 1.

MLS1547 stimulates D2R G protein–mediated signaling. (A) Structure of MLS000051547 (MLS1547). (B) Human embryonic kidney 293 cells stably expressing D2R and Gqi5 were assayed for MLS1547 stimulation of calcium accumulation, as described in Materials and Methods. Cells were stimulated with the indicated concentrations of dopamine (DA) or MLS1547 (1547). EC50 and Emax values were obtained for dopamine (2.5 ± 0.3 nM and 101.7% ± 0.1%, respectively) and MLS1547 (0.37 ± 0.2 μM and 89.3% ± 4.3%, respectively) (mean ± S.E.M., n = 3). (C) CHO cells stably expressing the D2R were assayed for MLS1547 inhibition of forskolin-stimulated cAMP, as described in Materials and Methods. Cells were stimulated with the indicated concentration of dopamine or MLS1547 (1547). EC50 and Emax values were obtained for dopamine (0.06 ± 0.02 μM and 100.4% ± 1.6%, respectively) and MLS1547 (0.26 ± 0.07 μM and 97.1% ± 3.7%, respectively) (mean ± S.E.M., n = 5). (D) CHO cells stably expressing D2R were assayed for sulpiride reversal of MLS1547 inhibition of forskolin-stimulated cAMP accumulation. Cells were stimulated with an EC80 concentration of MLS1547 (1547) in the presence of increasing concentrations of the D2R antagonist sulpiride. The IC50 value obtained for sulpiride was 22.0 ± 2.8 nM (mean ± S.E.M., n = 4). Data are representative of three to five independent experiments run in triplicate and plotted as a percentage of the maximum response observed with dopamine (B and C), or as a percentage of the response seen with an EC80 concentration of MLS1547 (D), as indicated.
Indeed, Fig. 1B shows that MLS1547 behaved as a highly efficacious agonist in the D2-Gqi5 calcium mobilization assay, with an EC50 value of 0.37 μM and an Emax of ∼90%. To ensure that this activity is physiologic, and not restricted to the chimeric G protein nature of the Gqi5 assay, we examined the ability of MLS1547 to inhibit forskolin-stimulated cAMP accumulation in cells stably expressing the D2R. Figure 1C shows that, similar to dopamine, MLS1547, completely inhibited the stimulation of cAMP by forskolin, with an EC50 of 0.26 μM suggestive of it having high efficacy at D2R-mediated G protein–linked signaling. We further found that the inhibition of cAMP accumulation by MLS1547 was completely blocked by cotreatment with the D2R antagonist sulpiride, as shown in Fig. 1D. The IC50 of 22 nM for sulpiride’s inhibition of MLS1547’s action is similar to sulpiride’s potency for blocking dopamine’s response in this cAMP assay (data not shown). Taken together, these data indicate that MLS1547 is a highly efficacious agonist at the D2R for stimulating G protein–mediated signaling.
The ability of MLS1547 to stimulate recruitment of β-arrestin to the D2R was evaluated using the DiscoveRx β-arrestin PathHunter assay, which relies on the complementation and activation of β-galactosidase when β-arrestin-2 is recruited to the receptor. Notably, β-arrestin-2 is the β-arrestin protein functionally coupled to the D2R in vivo (Skinbjerg et al., 2009). Whereas incubation with dopamine resulted in robust recruitment of β-arrestin-2 in a dose-dependent manner, MLS1547 failed to exhibit activity in this assay (Fig. 2A), despite it being a highly efficacious agonist of G protein–mediated signaling. However, other possible explanations for these discrepant results could include interference of the compound with the enzyme-linked signaling, resulting in a suppression of signal. To address these alternative explanations, we used a BRET assay that directly measures the physical interactions between the β-arrestin-2 protein and the D2R. Furthermore, this also serves as a control for a different cell background, as these assays were conducted in human embryonic kidney 293 cells. As seen in Fig. 2B, however, MLS1547 failed to stimulate any observable recruitment of β-arrestin-2 to the D2R in this assay, despite robust recruitment by dopamine. Taken together, these findings indicate that, although MLS1547 is a highly efficacious agonist at G protein–mediated signaling, it is unable to stimulate measurable β-arrestin recruitment, thus establishing it as an extremely G protein–biased agonist at the D2R.
Fig. 2.

MLS1547 acts as an antagonist for dopamine (DA)-stimulated β-arrestin recruitment to the D2R. (A) DiscoveRx PathHunter cells were assayed for agonist-induced recruitment of β-arrestin-2 to the D2R as described in Materials and Methods. Cells were stimulated with the indicated concentrations of dopamine or MLS1547 (1547), and EC50 and Emax values were obtained for dopamine (0.09 ± 0.03 μM and 99.0% ± 0.6%, respectively; mean ± S.E.M., n = 3). MLS1547 failed to stimulate measurable β-arrestin-2 recruitment. (B) Human embryonic kidney 293 cells were stably transfected with Rluc-8–fused D2R and mVenus-fused β-arrestin-2, stimulated with various concentrations of dopamine or MLS1547 (1547) as indicated, and examined for BRET as described in Materials and Methods. EC50 and Emax values obtained for dopamine were 0.05 ± 0.01 μM and 99.8% ± 1.9%, respectively (mean ± S.E.M., n = 3). MLS1547 failed to stimulate any measurable D2R–β-arrestin-2 interactions. Data are representative of three to five independent experiments run in triplicate and plotted as a percentage of maximum response observed with dopamine as indicated. (C) DiscoveRx PathHunter cells were stimulated with an EC80 concentration of dopamine (1 μM), then assayed for the ability of MLS1547 to antagonize this response. Data are expressed as the percentage of the maximum response observed with 1 μM dopamine and represent the mean ± S.E.M. values of three individual experiments performed in triplicate. The IC50 value for MLS1547 was calculated to be 9.9 ± 0.9 μM. (D) The same cells described for the BRET assay in B were stimulated with an EC80 of dopamine (1 μM) and assayed for the ability of MLS1547 to antagonize this response. Data are expressed as mean values of six independent experiments run in quadruplicate. The IC50 value for MLS1547 was 3.8 ± 1.8 μM (mean ± S.E.M., n = 6).
Since MLS1547 activates G protein–linked pathways, yet does not stimulate β-arrestin recruitment, MLS1547 would be expected to antagonize the dopamine-induced β-arrestin response by blocking dopamine binding to the receptor. As seen in Fig. 2C, MLS1547 fully antagonized dopamine-mediated β-arrestin recruitment to the D2R in the DiscoveRx assay, with an IC50 of 9.9 μM. Similar results were obtained when MLS1547 was examined for antagonist activity in the D2R β-arrestin BRET assay, demonstrating an IC50 of 3.8 μM (Fig. 2D). In summary, although MLS1547 is an agonist of D2R-stimulated G protein–mediated signaling, it acts as an antagonist of the β-arrestin–mediated signaling pathway.
To determine the affinity of MLS1547 for the D2R, we used standard radioligand binding competition analyses. MLS1547 was found to completely displace [3H]methylspiperone binding to the D2R, with a calculated Ki value of 1.2 ± 0.2 μM (Fig. 3). Displacement studies were also conducted on the other dopamine receptor subtypes (Supplemental Fig. 2), resulting in mean ± S.E.M. Ki values of 2.3 ± 0.2 μM (D3R) and 0.32 ± 0.04 μM (D4R) (n = 3), suggesting a less than 10-fold difference in affinity between the members of the D2-like family. When the D1-like receptors were examined by measuring the ability of MLS1547 to displace [3H]SCH23390 binding to D1 and D5 receptors, the extrapolated Ki values for both receptors were >50 μM (Supplemental Fig. 2).
Fig. 3.

Radioligand binding competition assay using MLS1547 and the D2R. Membranes from human embryonic kidney 293 cells stably transfected with the human D2R were harvested for radioligand competition binding assays as described in Materials and Methods. Membranes were incubated with the indicated concentrations of MLS1547 and 0.5 nM [3H]methylspiperone. The data are representative of four independent experiments and expressed as a percentage of the binding seen in the absence of any competing ligand. The Ki for MLS1547 was calculated to be 1.2 ± 0.2 μM (mean ± S.E.M., n = 4).
A series of MLS1547 analogs were either obtained from commercial sources or, in a few cases, synthesized to investigate the structure-activity relationship (SAR) of pathway selectivity for this set of compounds (for purity and manufacturer, see Supplemental Table 1). These analogs were assayed in the D2R cAMP assay as well as in the DiscoveRx β-arrestin recruitment assay (Tables 1 and 2). Although none of the analogs were more potent than MLS1547, a subset of the analogs was also extremely G protein–biased such that they lacked β-arrestin recruitment activity (Table 1). However, another subset of compounds exhibited agonist activity in both the cAMP and β-arrestin assays with variable degrees of G protein signaling bias (Table 2). These findings were used to formulate a preliminary SAR of G protein bias in this series of compounds.
TABLE 1.
Analogs of MLS1547 active in cAMP assays yet lacking β-arrestin recruitment activity
The compounds were assayed in agonist mode using the DiscoveRx cAMP and β-arrestin assays as described in Materials and Methods.
| Structure | Compound ID | D2R cAMP Response |
D2R β-Arrestin Recruitment |
|||
|---|---|---|---|---|---|---|
| Emax (% Control ± S.E.M.) | EC50 (μM ± S.E.M.) | Log (Emax/EC50) | Emax (% Control ± S.E.M.) | EC50 (μM ± S.E.M.) | ||
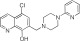 |
MLS000051547 | 97.1 ± 3.7 | 0.26 ± 0.07 | 2.57 | Inactive | NA |
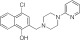 |
NCGC00319124 | 87.3 ± 14.7 | 5.5 ± 0.9 | 1.20 | Inactive | NA |
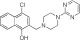 |
NCGC00319127 | 93.9 ± 4.6 | 4.7 ± 1.7 | 1.30 | Inactive | NA |
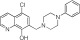 |
NCGC00319125 | 66.8 ± 3.9 | 2.8 ± 1.2 | 1.38 | Inactive | NA |
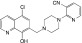 |
NCGC00346387 | 94.8 ± 2.1 | 1.4 ± 0.8 | 1.83 | Inactive | NA |
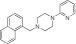 |
NCGC00319141 | 89.5 ± 5.2 | 0.7 ± 0.7 | 2.11 | Inactive | NA |
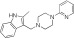 |
MLS000860449 | 84.9 ± 2.7 | 4.5 ± 3.0 | 1.28 | Inactive | NA |
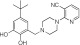 |
NCGC0319129 | 74.8 ± 23.8 | 9.5 ± 5.6 | 0.90 | Inactive | NA |
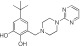 |
NCGC00319126 | 62.8 ± 7.7 | 1.1 ± 0.6 | 1.76 | Inactive | NA |
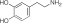 |
Dopamine | 100 | 0.06 ± 0.02 | 3.22 | 100 | 0.09 ± 0.03 |
NA, not applicable.
TABLE 2.
Analogs of MLS1547 active in both cAMP and β-arrestin assays
The compounds were assayed in agonist mode using the DiscoveRx cAMP and β-arrestin assays as described in Materials and Methods.
| Structure | Compound ID | D2R cAMP Response |
D2R β-Arrestin Recruitment |
Bias Activity | ||||
|---|---|---|---|---|---|---|---|---|
| Emax (% Control ± S.E.M.) | EC50 (μM ± S.E.M.) | Log (Emax/EC50) | Emax (% Control ± S.E.M.) | EC50 (μM ± S.E.M.) | Log (Emax/EC50) | |||
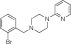 |
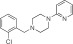
NCGC00319139 | 95.5 ± 2.0 | 0.03 ± 0.01 | 3.50 | 89.3 ± 9.4 | 2.0 ± 0.5 | 1.65 | 1.85 |
 |
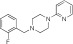
NCGC00319137 | 84.1 ± 4.5 | 0.02 ± 0.001 | 3.62 | 84.7 ± 2.5 | 1.6 ± 0.3 | 1.72 | 1.90 |
 |
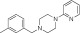
NCGC00319136 | 93.8 ± 1.4 | 0.1 ± 0.04 | 2.97 | 76.4 ± 2.9 | 0.7 ± 0.07 | 2.04 | 0.93 |
 |
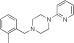
NCGC00092785 | 77.0 ± 10.9 | 0.04 ± 0.01 | 3.28 | 73.8 ± 1.2 | 0.4 ± 0.02 | 2.27 | 1.02 |
 |
NCGC00319134 | 86.1 ± 14.4 | 0.1 ± 0.04 | 2.94 | 74.7 ± 1.8 | 0.3 ± 0.03 | 2.40 | 0.54 |
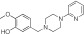 |
NCGC00319131 | 89.6 ± 0.5 | 0.2 ± 0.1 | 2.65 | 66.0 ± 0.2 | 0.8 ± 0.01 | 1.92 | 0.73 |
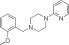 |
NCGC00319132 | 78.9 ± 10 | 0.1 ± 0.01 | 2.90 | 59.2 ± 4.2 | 0.3 ± 0.04 | 2.30 | 0.60 |
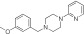 |
NCGC00319133 | 86.5 ± 14.7 | 0.01 ± 0.001 | 3.94 | 87.6 ± 2.0 | 0.2 ± 0.03 | 2.64 | 1.30 |
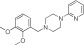 |
NCGC00319130 | 71.0 ± 17.1 | 0.1 ± 0.02 | 2.85 | 66.0 ± 0.2 | 2.1 ± 0.4 | 1.50 | 1.35 |
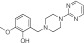 |
NCGC00319135 | 83.3 ± 14.4 | 1.4 ± 0.1 | 1.77 | 68.9 ± 4.9 | 13.2 ± 6.6 | 0.72 | 1.06 |
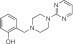 |
NCGC00319140 | 92.5 ± 5.5 | 2.0 ± 0.2 | 1.67 | 66.2 ± 18.6 | 20.5 ± 9.6 | 0.51 | 1.16 |
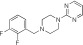 |
NCGC00319128 | 97.2 ± 0.7 | 0.4 ± 0.1 | 2.39 | 70.0 ± 5.9 | 3.4 ± 0.5 | 1.31 | 1.07 |
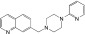 |
NCGC00346388 | 90.9 ± 1.9 | 0.2 ± 0.1 | 2.66 | 56.8 ± 7.1 | 1.0 ± 0.6 | 1.75 | 0.90 |
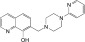 |
NCGC00345872 | 97.1 ± 0.9 | 0.3 ± 0.1 | 2.51 | 58.0 ± 8.4 | 0.9 ± 0.3 | 1.81 | 0.70 |
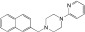 |
NCGC00345873 | 93.1 ± 1.6 | 0.1 ± 0.07 | 2.97 | 63.3 ± 8.9 | 0.9 ± 0.6 | 1.85 | 1.12 |
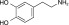 |
Dopamine | 100 | 0.06 ± 0.02 | 3.22 | 100 | 0.09 ± 0.03 | 3.05 | 0.18 |
To do this, a pharmacophore model was constructed to distinguish between the completely G protein–biased (Table 1) and less/nonbiased D2R agonists (Table 2). Specifically, we selected 10 compounds, displaying either the strongest or weakest G protein bias, and then assigned default pharmacophore features [positive (P), acceptor (A), donor, aromatic (R), and hydrophobic (H)] to these ligands (see Materials and Methods). We found that a 4-point HRPR hypothesis was best able to explain the experimental data (Fig. 4, A and B). Specifically, the alignment of the H feature in these compounds is the key difference between the completely biased (Table 1) and less/nonbiased (Table 2) agonists of this scaffold. As shown by two representative biased/nonbiased pairs in Fig. 4, the completely biased compounds align optimally to the H feature, whereas the less/nonbiased compounds do not (Fig. 4, A and B).
Fig. 4.

Pharmacophore model for G protein–biased and nonbiased agonist interactions with the D2R. (A and B) HRPR pharmacophore with aligned biased compounds MLS1547 and NCGC9141. Biased compounds align well to all four features: two aromatic (beige), one hydrophobic (green), and a positive (blue) feature. (Middle) Compared with the biased compound MLS1547, the nonbiased compound NCGC5872 (left) lacks the −Cl group and cannot align both the hydrophobic and aromatic features, whereas the different attachment points of the naphthalene ring in compounds NCGC9141 and NCGC5873 cause the former, but not the latter, to align well to the 4-point pharmacophore. In this case, one of the aromatic rings in NCGC9141 aligns the hydrophobic feature. (C and D) Docked poses of biased (cyan) and unbiased (gray) compounds in an active model of the D2R. Note the more extensive interaction of nonpolar features in the biased compounds with a hydrophobic pocket formed by residues in EL2 and TM5.
In parallel, we docked these compounds into a novel active-state model of the D2R using MLS1547 as a reference compound. For the MLS1547 compound, we observed two plausible orientations with the pyridine moiety pointing toward either TM5 in the OBS or toward TMs 2 and 7, away from the OBS (data not shown). Based on the SAR derived from Table 1, it appears that the N in the pyridine ring likely makes an H bond with a receptor residue, as there is a ∼10-fold increase in the potency of MLS1547 compared with that of NCGC319125. When the pyridine points toward TM5, however, a corresponding H-bond donor cannot be identified in the OBS. Thus, it is more likely that MLS1547 adopts the alternate orientation with the pyridine pointing toward TMs 2 and 7, in which the N on the pyridine ring may interact with Thr369 in TM7 to form an H bond. These docking results were confirmed using a model with the D2R in an active conformation based on another D3R active model (Newman et al., 2012) and equilibrated using molecular dynamics simulations (data not shown). Encouragingly, results from both models were similar.
Using the MLS1547 pose with the pyridine pointing toward TMs 2 and 7, we used a core-restrained protocol to dock the other compounds into the active D2R model to compare the binding modes of the fully G protein–biased versus less/nonbiased agonists. The resulting poses show that the completely biased compounds from Table 1 have a significantly higher tendency to interact with the extracellular portion of TM5. Specifically, the chloro (−Cl) group of MLS1547 interacts with a hydrophobic pocket formed by Ile184EL2, Phe1895.38, and Val1905.41 (Fig. 4) (PDB ID D2, MLS1547; Supplemental Data). Interestingly, this is consistent with our finding from the pharmacophore modeling that the fully G protein–biased, but not the less/nonbiased compounds, can be well aligned to the H feature of the aforementioned HRPR pharmacophore (PDB ID NCGC9141, NCGC5872, NCGC5873; Supplemental Data). Although recent publications suggest a possible role for aromatic −Cl groups in an H-bond formation, it is not atypical to consider −Cl as a hydrophobic group as well. Indeed, there are multiple examples in the literature where a methyl/−Cl swap resulted in roughly the same potency, and was better tolerated than the unsubstituted hydrogen (Baum et al., 2009).
Thus, it is tempting to speculate that the structural basis for the different efficacies of these two groups of agonists critically depends on whether a compound can interact with the hydrophobic pocket near the extracellular portion of TM5 mentioned earlier. Interestingly, interactions with the extracellular portion of TM5 have previously been proposed as the structural basis for the β-arrestin signaling bias of the serotonergic agonist ergotamine at the 5-hydroxytryptamine (5HT)2B receptor, whereas ergotamine is notably nonbiased at the related 5HT1B receptor (Wacker et al., 2013). Thus, comparing crystal structures of ergotamine bound to both receptors, it was found that the extracellular portion of TM5 is tilted significantly more toward the OBS in the 5HT2B receptor, compared with the 5HT1B receptor (Wacker et al., 2013). This suggests a mechanism whereby agonists that prevent such a tilting would bias toward G protein activation, compared with the β-arrestin pathway. Future experiments to test this SAR prediction for the D2R and other GPCRs will be required. It is interesting to note, however, that in the D2R, mutations of four residues at the cytoplasmic end of TM5 disrupt β-arrestin recruitment much more than they impact cAMP signaling (Lan et al., 2009), suggesting that differential propagation of signals through TM5 may play an important role in determining signaling bias.
In summary, MLS1547 is the first example of a G protein–biased agonist of the D2R. Although it robustly activates G protein–mediated signaling, the compound does not promote β-arrestin recruitment to the receptor. Rather, through occupancy of the receptor, MLS1547 functions as an antagonist of dopamine-induced β-arrestin recruitment to the D2R. Administration of compounds with this pharmacological profile to animals would be expected to stimulate G protein–based signaling of the D2R while simultaneously inhibiting signaling through the β-arrestin/Akt/GSK3β pathway. Such compounds may also engender less β-arrestin–mediated receptor desensitization or internalization, thereby further amplifying the G protein signaling arm. Importantly, MLS1547, despite displaying no measurable β-arrestin recruitment in our assays, appears to be a highly efficacious agonist at G protein–mediated signaling for the D2R. Recently, β-arrestin–biased agonists have been developed for the D2R that exhibit varying degrees of agonist efficacy (Allen et al., 2011; Chen et al., 2012). Functionally selective probes for both of the major signaling arms of the D2R should now help to dissect their roles in normal physiology and behavior as well as elucidate their involvement in the therapeutic effects of various pharmaceutical agents.
Supplementary Material
Acknowledgments
The authors thank Paul Shinn and Danielle VanLeer for compound management support.
Abbreviations
- A
acceptor
- Akt
protein kinase B
- BRET
bioluminescence resonance energy transfer
- −Cl
chloro
- DARPP-32
32-kDa dopamine and cAMP-regulated phosphoprotein
- D1R
D1 dopamine receptor
- D2R
D2 dopamine receptor
- D3R
D3 dopamine receptor
- D4R
D4 dopamine receptor
- D5R
D5 dopamine receptor
- EBSS
Earle’s balanced salt solution
- FDSS
functional drug screening system
- GPCR
G protein–coupled receptor
- GSK3β
glycogen synthase kinase 3 β
- Η
hydrophobic
- 5HT
5-hydroxytryptamine
- MLS1547
5-chloro-7-[(4-pyridin-2-ylpiperazin-1-yl)methyl]quinolin-8-ol
- OBS
orthosteric binding site
- P
positive
- PBS
phosphate-buffered saline
- R
aromatic
- RLU
relative luminescence unit
- SAR
structure-activity relationship
- TM
transmembranes
Authorship Contributions
Participated in research design: Free, Chun, Xiao, Dulcey, Titus, Ferrer, Javitch, Shi, Southall, Marugan, Sibley.
Conducted experiments: Free, Chun, Moritz, Miller, Doyle, Conroy, Padron, Meade, Xiao, Dulcey, Titus, Bryant-Genevier, Barnaeva.
Contributed new reagents or analytic tools: Xiao, Dulcey, Han, Duan, Javitch, Marugan.
Performed data analysis: Free, Chun, Moritz, Miller, Doyle, Conroy, Padron, Meade, Xiao, Hu, Dulcey, Titus, Bryant-Genevier, Ferrer, Beuming, Shi, Southall, Marugan.
Wrote or contributed to the writing of the manuscript: Free, Xiao, Ferrer, Javitch, Beuming, Shi, Southall, Sibley.
Footnotes
This work was supported, in part, by the Intramural Research Program of the National Institutes of Health [National Institute of Neurological Disorders and Stroke].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Abdul-Ridha A, López L, Keov P, Thal DM, Mistry SN, Sexton PM, Lane JR, Canals M, Christopoulos A. (2014) Molecular determinants of allosteric modulation at the M1 muscarinic acetylcholine receptor. J Biol Chem 289:6067–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, et al. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci USA 108:18488–18493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. (2011) N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem 54:3581–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Svenningsson P, Kuroiwa M, Gong S, Nishi A, Heintz N, Greengard P. (2008) Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat Neurosci 11:932–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B, Mohamed M, Zayed M, Gerlach C, Heine A, Hangauer D, Klebe G. (2009) More than a simple lipophilic contact: a detailed thermodynamic analysis of nonbasic residues in the s1 pocket of thrombin. J Mol Biol 390:56–69 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. (2007a) The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci 28:166–172 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Wetsel WC, Lefkowitz RJ, Gainetdinov RR, Caron MG. (2008) A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132:125–136 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, Borrelli E, Caron MG. (2007b) Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci 27:881–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P. (2013) Modification of cocaine self-administration by buspirone (buspar®): potential involvement of D3 and D4 dopamine receptors. Int J Neuropsychopharmacol 16:445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckler F, Lanig H, Gmeiner P. (2005) Modeling the similarity and divergence of dopamine D2-like receptors and identification of validated ligand-receptor complexes. J Med Chem 48:694–709 [DOI] [PubMed] [Google Scholar]
- Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. (2012) Structure-functional selectivity relationship studies of β-arrestin-biased dopamine D₂ receptor agonists. J Med Chem 55:7141–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, et al. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. (2013) D1-D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol 84:190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, et al. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503:295–299 [DOI] [PubMed] [Google Scholar]
- Gay EA, Urban JD, Nichols DE, Oxford GS, Mailman RB. (2004) Functional selectivity of D2 receptor ligands in a Chinese hamster ovary hD2L cell line: evidence for induction of ligand-specific receptor states. Mol Pharmacol 66:97–105 [DOI] [PubMed] [Google Scholar]
- Greene J, Kahn S, Savoj H, Sprague P, Teig S. (1994) Chemical function queries for 3D database search. J Chem Inf Comput Sci 34:1297–1308 [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. (2005) High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen 10:463–475 [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2007a) Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci 28:407–415 [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2007b) Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol 72:1393–1401 [DOI] [PubMed] [Google Scholar]
- Kenakin TP. (2008) Pharmacological onomastics: what’s in a name? Br J Pharmacol 153:432–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 3:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilts JD, Connery HS, Arrington EG, Lewis MM, Lawler CP, Oxford GS, O’Malley KL, Todd RD, Blake BL, Nichols DE, et al. (2002) Functional selectivity of dopamine receptor agonists. II. Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. J Pharmacol Exp Ther 301:1179–1189 [DOI] [PubMed] [Google Scholar]
- Klewe IV, Nielsen SM, Tarpø L, Urizar E, Dipace C, Javitch JA, Gether U, Egebjerg J, Christensen KV. (2008) Recruitment of beta-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology 54:1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. (2007) Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 28:397–406 [DOI] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504:101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H, Liu Y, Bell MI, Gurevich VV, Neve KA. (2009) A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Mol Pharmacol 75:113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, Powney B, Wise A, Rees S, Milligan G. (2007) Protean agonism at the dopamine D2 receptor: (S)-3-(3-hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1,Gi2, and Gi3. Mol Pharmacol 71:1349–1359 [DOI] [PubMed] [Google Scholar]
- Lane JR, Powney B, Wise A, Rees S, Milligan G. (2008) G protein coupling and ligand selectivity of the D2L and D3 dopamine receptors. J Pharmacol Exp Ther 325:319–330 [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335:1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman RB. (2007) GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci 28:390–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. (2008) Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA 105:13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR, Booth RG, Hyslop DK, Piercey M, Wightman RM, et al. (2002) Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J Pharmacol Exp Ther 301:1166–1178 [DOI] [PubMed] [Google Scholar]
- Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, et al. (2012) Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem 55:6689–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi L, Håkansson K, Usiello A, Borgkvist A, Lindskog M, Greengard P, Fisone G. (2003) Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J Neurochem 86:451–459 [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley DR, Monsma FJ, Jr, McVittie LD, Gerfen CR, Burch RM, Mahan LC. (1992) Molecular neurobiology of dopamine receptor subtypes. Neurochem Int 20 (Suppl):17S–22S [DOI] [PubMed] [Google Scholar]
- Skinbjerg M, Ariano MA, Thorsell A, Heilig M, Halldin C, Innis RB, Sibley DR. (2009) Arrestin3 mediates D(2) dopamine receptor internalization. Synapse 63:621–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. (2004) DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol 44:269–296 [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13 [DOI] [PubMed] [Google Scholar]
- Urs NM, Snyder JC, Jacobsen JP, Peterson SM, Caron MG. (2012) Deletion of GSK3β in D2R-expressing neurons reveals distinct roles for β-arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci USA 109:20732–20737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013) Structural features for functional selectivity at serotonin receptors. Science 340:615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Han SJ, Kim SK, Jacobson KA, Li JH. (2008) Conformational changes involved in G-protein-coupled-receptor activation. Trends Pharmacol Sci 29:616–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 17:126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.