

Abstract
Schizophrenia and bipolar disorder are highly heritable psychiatric disorders. Associated genetic and gene expression changes have been identified, but many have not been replicated and have unknown functions. We identified groups of genes whose expressions varied together, i.e. co-expression modules, then tested them for association with schizophrenia. Using Weighted Gene Co-expression Network Analysis, we show that two modules were differentially expressed in patients versus controls. One, up-regulated in cerebral cortex, was enriched with neuron differentiation and neuron development genes, as well as disease GWAS genetic signals; the second, altered in cerebral cortex and cerebellum, was enriched with genes involved in neuron protection functions. The findings were preserved in five expression data sets, including sets from three brain regions, from a different microarray platform, and from bipolar disorder patients. From those observations, we propose neuron differentiation and development pathways may be involved in etiologies of both schizophrenia and bipolar disorder, and neuron protection function participates in pathological process of the diseases.
Keywords: Gene Expression, Schizophrenia, WGCNA, Neuron differentiation, Neuron protection
Introduction
Schizophrenia (SCZ) and bipolar disorder (BD) are psychiatric disorders with world-wide lifetime prevalences of around 1%1-4. These disorders have been shown to be highly heritable by monozygotic and dizygotic twin studies and by adoption studies: the heritability estimates for SCZ range from 70 to 85% and, for BD, from 60 to 85%5. Over the past two decades, genetic studies of thousands of samples have identified hundreds of candidate genes6-8. Most of these findings have not been supported by genome-wide studies. Moreover, functional genomic roles have not yet been determined for most of the associated genes.
Gene expression can bridge the gap between genetic variation and disease susceptibility as an intermediate phenotype that is regulated by a combination of genetic and epigenetic factors. Gene expression transcription profiling is widely used and has been thoroughly validated by the MicroArray Quality Control (MAQC) project9,10. Expression microarray studies test thousands of gene transcripts for differential expression simultaneously. When all these genes are tested for association with disease, it creates a multiple testing problem: to minimize false positives, the multiple test correction sets the significance threshold so high that true positives might be missed as well. Additionally, testing individual genes for disease association also ignores the interaction between genes. So far, hundreds of gene expression microarray studies of psychiatric diseases have been reported, including studies of SCZ, BD and autism. However, the significant gene changes detected by one study are seldom replicated in another, let alone across three or more11.
Gene expression network analyses are an alternative approach for analyzing expression data that reduce the sample space tested (that is, the number of hypotheses to be tested). Gene expression networks are constructed from expression data from thousands of genes, and describe the interactions among groups of transcripts. They can be used to observe systematic alterations in expression associated with complex diseases such as psychiatric disorders. Horvath and colleagues have developed an algorithm for creating networks, called Weighted Gene Co-expression Network Analysis (WGCNA), which is widely used12,13. This method identifies groups of genes within a network whose expressions are highly correlated. These groups, called modules, can then be compared between cases and controls, among different tissues, species, or other phenotypes or clinical traits14-17.
Oldham et al. were the first to apply this method to expression in brain. They compared the gene expression across different brain regions and demonstrated that the modules reflect the underlying cell-type composition of the regions18.
The method was first applied to a psychiatric disease by Torkamani et al., who detected SCZ-associated gene co-expression modules in one microarray data analysis, and found that aging affected gene expression in normal controls, but not in SCZ patients19. They made the interesting distinction between constructing networks from case and control data separately and constructing a network from a combination of case and control data. Constructing modules from case and control networks separately allows comparison of basic module structure between the two groups. If modules are not substantially preserved in cases, a conclusion can be drawn that fundamental relationships among genes have been disrupted. If modules are substantially preserved in cases, the case and control data can be combined for network construction, and the detected modules assessed for differential expression between cases and controls. Torkamani found that modules were substantially preserved in cases, and identified five SCZ-associated modules; Gene Ontogeny (GO) enrichment analysis showed those five modules were associated with oxidative phosphorylation, angiogenesis, neuron differentiation, chromatin and nucleosome assembly, and inositol phosphate metabolism, separately.
Voineagu et al. applied WGCNA to another psychiatric disease, autism. In addition to a GO enrichment analysis, they found one of the differentially expressed modules was enriched in disease genome-wide association study (GWAS) signals. They interpreted this as evidence that the module's member genes were causally associated with autism. They also found another module with altered expression but not enriched in GWAS signals and took it as an indication of a non-genetic etiology20.
Note that all three of these network studies were based on analysis of a single microarray data set; their findings have not yet been replicated in other data sets at the network level.
To demonstrate the robustness and reproducibility of WGCNA findings, we used multiple microarray data sets to study co-expression networks in brains of psychiatric patients. We constructed gene expression networks, and then identified gene co-expression modules within the networks. The modules were tested for association with SCZ. We assessed whether the disease-associated modules were detected in independent data sets, including sets from several different brain collections and different brain regions. Reproducible, or preserved, modules were then evaluated for case-control differences in the independent data sets.
Since it is widely accepted that SCZ and BD have genetic factors in common21,22, we tested whether the gene modules perturbed in SCZ were similarly perturbed in BD. We also tested whether the disease-associated modules were enriched with genetic variants that had been previously associated with disease by GWASs23,24.
Subjects and Methods
Samples and quality control
Cerebellum (CB) and parietal cortex (PCX) brain tissues were obtained from Stanley Medical Research Institute (SMRI)25,. They came from the SMRI's Neuropathology Consortium and Array collections, and included 50 SCZ samples, 50 BD samples, and 50 unaffected control samples. Expression data for these samples came from the NCBI Gene Expression Omnibus (GEO) database (GSE35978). One of the two prefrontal cortex (PFC) brain expression data sets came from Dobrin's group using SMRI samples (PFC-SMRI)25, while the second came from the Victorian Brain Bank Network (PFC-VBBN)26 and was obtained from at GEO (GSE21138, sample information and preparation in Supplementary File).
ComBat, a batch effects adjustment program that we have previously shown to be the best available, was used to remove batch effects from the data sets (see Supplementary File for detailed preprocessing steps)27,28.
Gene network construction and module detection
We used weighted gene co-expression network analysis (WGCNA)13 to identify modules of co-expressed genes within gene expression networks. To construct the network, the absolute values of Pearson correlation coefficients were calculated for all possible gene pairs. Values were entered into a matrix, and the data were transformed so that the matrix followed an approximate scale-free topology (see Supplementary File for detailed information). A dynamic tree cut algorithm was used to detect network modules29. WGCNA and the dynamic tree cut algorithm were implemented in R12,29.
We ran singular value decomposition (SVD) on each module's expression matrix and used the resulting module eigengene, which is equivalent to the first principal component 30, to represent the overall expression profiles of the modules.
Module preservation statistics
We utilized the module preservation statistic Zsummary, to assess the module preservation from different expression data sets33 (see Supplementary File for formal definition). Unlike the cross-tabulation test, Zsummary not only takes into account the overlap in module membership, but also the density and connectivity patterns of modules. In addition, for our study, network-based preservation statistics only require that module membership be identified in the original data sets, reducing the variation coming from various parameters setting to identify new modules in validation data sets.
We converted the probe-level measurements into gene-level measurements to make data from different platforms comparable. The probe within a gene that had the highest coefficient of variation was used to represent that gene. Overall, 8497 genes were retained in our preservation calculation.
Differential expression test for modules
We used multiple linear regressions on the modules’ eigengenes to remove the effects of sex, age, pH and PMI from the SMRI samples and VBBN samples. The residual eigengene values were then used to test the disease association using Pearson's correlation test. We used false discovery rate (FDR) for multiple testing correction49.
Module-based GWAS signal enrichment test
GAIN-SCZ was the genome-wide association study of the GAIN SCZ data set, which comprised 4591 cases and controls (1217 European-American cases, 1442 European-American controls, 953 African-American cases, 979 African-American controls). The data was downloaded from dbGaP (http://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs000021.v3.p2). GAIN-BD was a genome-wide association study of the GAIN BD data set, which comprised 3261 cases and controls (1079 European-American cases, 1081 European-American controls, 415 African-American cases, 686 African-American controls). The data was downloaded from dbGaP (http://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs000017.v3.p1). Whole genome genotyping of GAIN data was done with the Affymetrix Genome-Wide Human SNP Array 6.0.
The TGen-BD GWAS data came from the genome-wide association study of the Translational Genomics Research Institute's (TGen) BD data (http://www.tgen.org/), which comprised 1,190 BD cases and 401 controls. Sample genotyping was conducted using the Affymetrix GeneChip Mapping 5.0K Array. We performed imputation using MaCH v1.0 to increase the density of interrogated SNPs31, with HapMap data as reference. Overall, 2,593,107 SNPs in GAIN-SCZ, 3,281,319 SNPs in GAIN-BD, and 2,542,706 SNPs in TGen-BD were included after imputation.
Imputed GWAS data was used to test whether the two disease-associated modules were enriched in SCZ/BD association signals32. We used a previously-reported procedure to run the enrichment test32. First, the max −log(P-value) of a SNP located between 20kb upstream and downstream of a gene was assigned to represent the gene, then the gene set's enrichment scores (ES) were calculated based the gene's rank. SNP level permutation was applied to generate the distribution of the ES and then the distribution was normalized. For multiple gene sets, FDR was calculated by joining all the distributions of ESs, each for one gene set, generated by permutation. As the difference of gene length distribution between genes in one module was significant (p=5.47e-27), we also applied a permutation procedure to verify whether there was a gene length bias in the genetic signals enrichment test43, 50, and there was no bias (see Supplementary Methods section 8).
Results
Gene modules in parietal cortex
We first analyzed the parietal cortex (PCX) brain gene expression in 50 SCZ patients and 50 normal controls from the Stanley Medical Research Institute (SMRI) using the Affymetrix Human Gene 1.0 ST Array25. After a series of sample and array-level quality control measures (see Supplementary methods for details), we retained 45 SCZ patients and 46 normal controls with measures of 19,884 transcripts.
Networks were constructed in two different ways: first, we constructed one network from control data and one network from case data, then identified modules in the control network and assessed their preservation in the case network. Second, we constructed a network from the combined case and control data, and identified modules within it.
Structure of co-expression modules in schizophrenia cases and controls
Case and control sample gene networks were constructed separately to detect whether there was any gene co-regulation disruption or creation in cases relative to controls. We assessed module preservation using a permutation-based preservation statistic, Zsummary, developed by Langfelder et al., which assesses whether the connectivity level and pattern of a module in one data set is preserved in another33. The authors suggest the following significance thresholds: Zsummary < 2 implies no evidence for module preservation, 2 < Zsummary < 10 implies weak to moderate evidence, and Zsummary > 10 implies strong evidence for module preservation. In our study, all modules detected in the control data had Zsummary greater than 10 in the case data (Fig. 1), suggesting well-preserved membership and connectivity of the control modules in the SCZ cases. This is consistent with two previous gene network-based psychosis studies20,34, where the case modules had no obvious perturbations relative to control modules.
Figure 1. Gene network modules from PCX from schizophrenia patients and normal controls are well preserved.
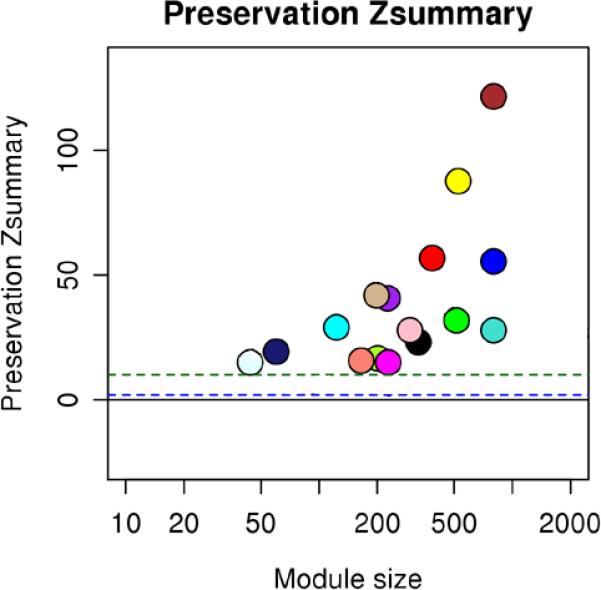
Zsummary is the summary preservation statistics, using the control modules as reference modules. Y-axis represents preservation statistics for the corresponding module in the case data sets, and x-axis is the gene numbers in each module. The dashed blue and green lines indicate the thresholds Z=2 and Z=10, respectively. Zsummary < 2 implies no evidence for module preservation, 2 < Zsummary < 10 implies weak to moderate evidence, and Zsummary > 10 implies strong evidence for module preservation.
To test the reliability of the module construction results, we compared the modules identified from control samples to published gene expression networks. Cross-tabulation showed that our control modules have similar module membership to modules previously reported by Oldham et al. 18(Supplementary Fig. 1). Slight differences, including some modules in our controls collapsing into one module in the Oldham data, might have been due to the differences in samples and platforms, data-filtering steps and/or parameters used for network construction and module detection.
Differential expression of modules in schizophrenia patients
Since there was no significant difference in module structure between cases and controls, modules identified in a gene network constructed from both case and control data were analyzed for differential expression in SCZ patients. Twenty-four modules were detected (Supplementary Fig. 2). We used an eigengene to summarize each module's expression profile30 (see Methods for formal eigengene definition). After multiple linear regressions on the eigengenes to remove the effects of sex, pH and post-mortem interval (PMI), three covariates that can affect measures of gene expression11, the residual module eigengenes for each individual were tested for disease association. The eigengenes of two PCX modules, referred to as M1A and M3A, were significantly associated with SCZ after multiple testing correction (Fig. 2A). M1A comprised 490 genes, while M3A comprised 106. NOTCH2 and MT1X, respectively, were the hub genes of M1A and M3A, meaning that, of all the genes in their modules, these two genes had the strongest correlation with the module's eigengene, as well as being highly connected to the other genes in their modules (Supplementary Tables 5 and 6).
Figure 2. Module and hub genes’ association test results.

Eigengene-based test detected 24 modules in PCX (A), listed on the x-axis. The y-axis indicates the -log10 of association p value. The red line represents the p=0.05 threshold. M3A and M1A modules were significant after multiple test correction, with FDR q value<0.05 (green bars with red stars on the top). NOTCH2 in M1A (B) and MT1X in M3A (C), with disease in replicate data sets. Green bars represent the significance of association in SCZ and control networks, while red bars represent the significance of association in BD and control networks. The purple line marks the significance threshold of p=0.05.
Replication of findings in different data sets
Replication of module structure
We tested co-expression module preservation across different microarray data sets. We again used the module preservation statistic, Zsummary, to compare modules. Data sets included prefrontal cortex (PFC) tissues and cerebellum (CB) tissues, also from the Stanley Medical Research Institute (SMRI) but performed by a different research group25, and another PFC data set, where samples came from the Victorian Brain Bank Network (VBBN)26.
As measured by the Zsummary statistic, six of the 18 modules identified in the SMRI PCX data were strongly preserved in the SMRI PFC data set, including M1A. Six modules were moderately preserved, including M3A, and four modules were not preserved (Supplementary Fig. 3). Similarly, M1A was strongly preserved and M3A moderately preserved in the SMRI CB and VBBN PFC data sets (Table 1; Supplementary Fig. 4 and 5).
Table 1.
Preservation statistic of SMRI-PCX-SZ M1A (top) and M3A (bottom) on VBBN-PFC-SZ, SMRI-PFC-SZ, SMRI-CB-SZ, SMRI-PFC-BD, SMRI-CB-BD data sets.
| Test Modules | Brain banks | Platform | Diseases | Brain regions | Zsummary * |
|---|---|---|---|---|---|
| M1A | VBBN | Affy U133 | Schizophrenia | PFC | 17 |
| SMRI | Affy U133 | Schizophrenia | PFC | 21 | |
| SMRI | Affy HG 1.0 | Schizophrenia | CB | 29 | |
| SMRI | Affy HG 1.0 | Bipolar disorder | PCX | 29 | |
| SMRI | Affy HG 1.0 | Bipolar disorder | CB | 27 | |
| M3A | VBBN | Affy U133 | Schizophrenia | PFC | 3 |
| SMRI | Affy U133 | Schizophrenia | PFC | 6 | |
| SMRI | Affy HG 1.0 | Schizophrenia | CB | 8 | |
| SMRI | Affy HG 1.0 | Bipolar disorder | PCX | 7 | |
| SMRI | Affy HG 1.0 | Bipolar disorder | CB | 9 |
VBBN, victorian brain bank network; SMRI, Stanley medical research institute; PFC, prefrontal cortex; CB, cerebellum; PCX, parietal cortex.
Zsummary < 2 implies no evidence for module preservation, 2 < Zsummary < 10 implies weak to moderate evidence, and Zsummary > 10 implies strong evidence for module preservation.
Replication of disease association
We tested the modules’ hub genes for disease association, since hub genes by definition are highly correlated with their modules’ eigengenes35. After controlling for the effects of covariates, NOTCH2, the hub gene of M1A, was significantly associated with SCZ in VBBN and SMRI PFC data (p=0.049 and 0.022, respectively), but not in SMRI CB data (p=0.583). MT1X, the hub gene of M3A, was consistently upregulated in SCZ in VBBN PFC, SMRI PFC and SMRI CB data (p=3.5E-03, 0.026 and 1.3E-04, respectively) (Fig. 2B, 2C). These results suggest that M1A gene network perturbation between SCZ and controls occurs in cerebral cortex but not in cerebellum, while M3A perturbation happens across all brain regions.
Modules shared between schizophrenia and bipolar disorder
We were interested in whether the M1A and M3A modules associated with SCZ were also associated with BD. One BD data set with two regions studied was tested, PCX and CB from SMRI. The control samples here overlapped with the control samples used to construct the SCZ case and control network and modules. We first confirmed that the M1A and M3A in PCX were well-preserved in the BD case-control data sets (Table 1; Supplementary Fig. 6 and 7), then we tested whether NOTCH2 and MT1X gene expression were associated with BD. NOTCH2 showed an association trend in PCX (p=0.054) but not in CB (p=0.117), while MT1X showed significant association in both regions (p=0.015 in PCX and 1.5e-3 in CB) (Fig. 2B, 2C), indicating M1A in PCX and M3A in two brain regions were altered in BD, and were shared by two diseases.
Characterization of two disease-associated modules
NOTCH2, a member of the Notch gene family, had the highest intramodular connectivity in the SMRI PCX M1A module. The Notch signaling pathway plays a key role in cell to cell communication, and was reported to play multiple roles in central nervous system development36-38. Functional enrichment analyses were performed with The Database for Annotation, Visualization and Integrated Discovery (DAVID) (Fig. 3, Supplementary Table 7)39. Consistent with the hub gene's function, M1A was highly significantly enriched with gene ontology terms related to neuron differentiation and neuron development genes (p=7.50e-08 and 3.63e-06, FDR q=1.27e-04 and 6.14e-03, respectively).
Figure 3. Global gene module characterization-M1A.
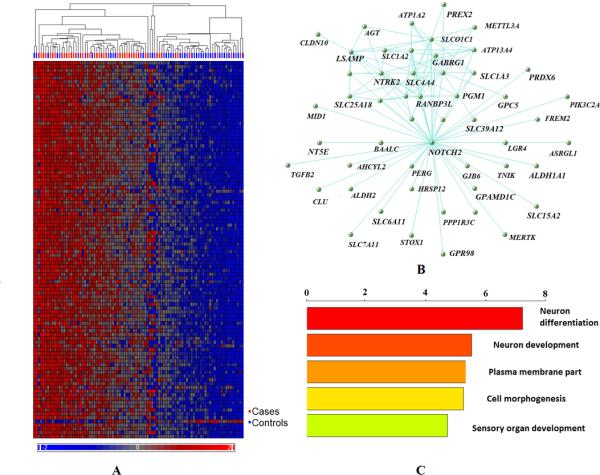
A. Heatmap of genes in M1A. Samples are listed in columns and genes in rows. Samples bar under the hierarchal clustering tree were marked as red indicating cases and as blue indicating controls. Normalized expression values ranged between -2 and 2, as shown in the color legend under the heatmap. B. Highly connected genes in M1A network. C. Top five enriched gene ontology categories of highly corrected genes in the M1A module.
We also found that 46 genes in M1A overlapped with a synaptic gene group associated with SCZ identified by Lips et al.40. These overlapping genes are interesting because ten of them have previously been reported to be associated with either SCZ or BD, which may suggest a relationship between synaptic dysfunction and these two psychiatric diseases. These genes include ABLIM1, APOE, AQP4, SLC1A2, SLC1A3, SLC4A4, GABRG1, NTRK2, ADD3 and MAOA.
Functional enrichment analysis showed M3A to be highly enriched in gene ontology categories related to metallothioneins (MT) and metal binding site functions (p=2.41e-05, FDR q=0.031) (Fig. 4, Supplementary Table 8). MT genes respond to several stimuli, including metals, oxidative agents, inflammation and stress; they influence cognition, protect against neurotoxicity and play a role in the astrocytic response to CNS injuries41. The expressions of our MT gene family members, including the hub gene MT1X, and MT1E, MT1F and MT2A have previously been reported to be differentially expressed in SCZ42.
Figure 4. Global gene module characterization-M3A.
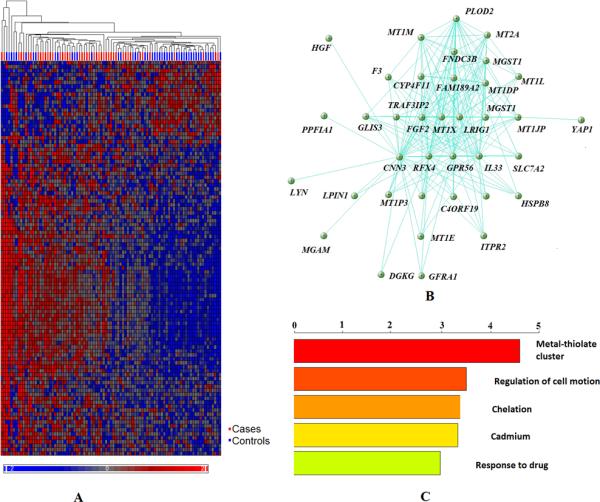
A. Heatmap of genes in M3A. Samples listed in column and genes in rows. Samples bar under the hierarchal clustering tree were marked as red indicating cases and as blue indicating controls. Normalized expression values ranged between -2 and 2, as shown in the color legend under the heatmap. B. Highly connected genes in M3A core network. C. Top five gene ontology categories enriched in the M3A module.
Enrichment of genetic association signals
To determine whether the expression alterations to modules M1A and M3A had genetic bases, we tested the modules for enrichment with SCZ and BD genetic association signals. M1A showed significant enrichment of signals from both SCZ and BD GWASs (GAIN, SCZ, p<0.001, FDR q=0.009; GAIN-BD, p<0.001, FDR q=0.0015; TGen, BD, p<0.001, FDR q=0.002) (Supplementary Fig. 8, 9 and 10) (see Supplementary materials for further descriptions of GWAS data sets). Those significances were retained after controlling for gene length bias43. M3A was not enriched in signals from either disease. We also used other GWAS data, from non-psychiatric disease type 2 diabetes (T2D), as a negative control for our enrichment analysis. Neither M1A nor M3A were enriched with signals from a T2D GWAS.
Discussion
We looked for case-control differences at the level of gene co-expression regulation, rather than the level of individual genes. We found strong evidence of such differences in one expression data set, and then demonstrated robust gene network preservation and disease association across five additional different data sets. These data sets came from different brain banks and different brain regions, and were produced on different expression microarray platforms; they contained data from patients with SCZ and BD, as well as normal controls. To the best of our knowledge, this is the first time gene expression alterations associated with disease have been replicated across so many gene expression data sets. This reproducibility suggests that our results are reliable and that the WGCNA network-based method increased the statistical power of our expression study.
M1A was enriched with neuron development and neuron differentiation genes, among others. Using module preservation statistics, we determined that the M1A module was highly preserved in the five other data sets, and was consistently up-regulated in SCZ and BD cerebral cortex relative to controls, but not in cerebellum. This result is consistent with Torkamani et al's finding that modules enriched in neuron differentiation are differentially expressed between SCZ patients and normal controls19. A test for enrichment with GWAS signals suggested that M1A expression changes may have a genetic basis. Based on those observations, we speculate that the etiology of psychiatric diseases could be related to developmental dysfunction in the cerebral cortex, extending the neurodevelopmental hypothesis of SCZ44, which has recently received further support from imaging studies45 and deep sequencing analysis46.
M3A was enriched with metallothionein genes. It was moderately conserved in other data sets and consistently showed differential expression in both cerebral cortex and cerebellum between SCZ and BD patients, relative to controls. M3A was not significantly enriched in GWAS signals. Expression changes in these MT genes in SCZ and BD has been previously reported42,47, but, so far, no positive genetic associations of these genes have been reported. Considering that the major physiological function of MT genes is binding of heavy metals to protect the central nervous system (CNS) against neurotoxicity or other injuries41, the change in the expression of M3A may be a downstream effect of disease or may be related to environmental insult. Alternatively, loci regulating M3A or its member genes may exist, but not have been detected by GWAS.
One advantage of module-based association studies is that they substantially reduce the multiple testing correction burden inherent in genome-wide association studies51 and microarray studies, particularly in brain expression studies where sample size is always limited by tissue availability. Microarray studies test tens of thousands of genes at one time, which require that p-values be very small to reach genome-wide significance. However, WGCNA produced tens of modules of highly correlated genes to be tested for association. In our case, we found M1A to be significant in most of our data sets, but when we ran an individual gene-based analysis, the hub gene of M1A, NOTCH2, was nominally significant but did not survive the multiple testing correction (data not shown). WGCNA may also reveal the function of an otherwise poorly characterized gene, if that gene is a member of a module highly enriched in a particular function. In addition, by comparing results from different data sets, we demonstrated that findings at the network/pathway level are consistent.
Our results suggested that the observed similarities between SCZ and BD are manifested at the transcriptional level. Shared genetic elements have previously been demonstrated21,22,48, and some expression studies reported overlapping aberrant individual genes at the transcriptional level47. Our study explored the shared pathology defect at a higher level, revealing a larger scale gene pathway-based perturbation. Further functional work can focus on those genes, especially the hub genes, which may reveal the molecular bases of the symptoms shared between these two diseases.
In summary, our study revealed that 1) SCZ and BD shared altered expression of neuron differentiation and neuron development genes (M1A) and metallothionein genes (M3A), which may indicate common etiology or pathology; 2) M1A's up-regulation in cerebral cortex could be regulated by genetic variants already found to be associated with SCZ and BD risk; 3) M3A expression change was found in both cerebral cortex and cerebellum, and was not detectably regulated by genetic variants.
Supplementary Material
Acknowledgement
This work was funded by 5R01MH080425 (to C.L.), and supported by Eklund Family. We thank Elizabeth Thomas and Ali Torkamani for sharing Victorian Brain Bank Network (VBBN) brain expression data with us. We thank investigators of Wellcome Trust Case Control Consortium (WTCCC) for sharing non-psychiatric disease type 2 diabetes (T2D) GWAS data. We thank the investigators of Bipolar Genome Study (BiGS) for generating those bipolar GAIN-BD and Tgen-BD GWAS data, thank investigators of GAIN for generating SZ GWAS data. We thank Seth E. Dobrin, Maree Webster and other collaborators at the Stanley Medical Research Institute for providing the Stanley Online Genomics Database and brain samples. We also thank Lorenzo Pesce, Alex Rodriguez and collaborators at Computation Institute, University of Chicago for supplying their supercomputing devices and services.
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
Supplementary information is available at Molecular Psychiatry's website.
Contributor Information
Chao Chen, Psychiatry Department, the University of Illinois at Chicago, Chicago, IL, US 60607.
Lijun Cheng, Psychiatry Department, the University of Illinois at Chicago, Chicago, IL, US 60607.
Kay Grennan, Department of Psychiatry and Behavioral Neuroscience, The University of Chicago, Chicago, IL, US 60637.
Fabio Pibiri, Department of Pediatrics, the University of Illinois at Chicago, Chicago, IL, US 60607.
Chunling Zhang, Psychiatry Department, the University of Illinois at Chicago, Chicago, IL, US 60607.
Judith A. Badner, Department of Psychiatry and Behavioral Neuroscience, The University of Chicago, Chicago, IL, US 60637.
Elliot S. Gershon, Department of Psychiatry and Behavioral Neuroscience, The University of Chicago, Chicago, IL, US 60637
Chunyu Liu, Psychiatry Department, the University of Illinois at Chicago, Chicago, IL, US 60607.
References
- 1.Jablensky A. The 100-year epidemiology of schizophrenia. Schizophrenia Research. 1997 Dec 19;28(2-3):111–125. doi: 10.1016/s0920-9964(97)85354-6. [DOI] [PubMed] [Google Scholar]
- 2.McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Archives of General Psychiatry. 2003 May;60(5):497–502. doi: 10.1001/archpsyc.60.5.497. [DOI] [PubMed] [Google Scholar]
- 3.Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RMA, Petukhova M, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the national comorbidity survey replication. Archives of General Psychiatry. 2007 May;64(5):543–552. doi: 10.1001/archpsyc.64.5.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGrath J, Saha S, Chant D, Welham J. Schizophrenia: A Concise Overview of Incidence, Prevalence, and Mortality. Epidemiologic Reviews. 2008 Nov 1;30(1):67–76. doi: 10.1093/epirev/mxn001. [DOI] [PubMed] [Google Scholar]
- 5.Burmeister M, McInnis MG, Zollner S. Psychiatric genetics: progress amid controversy. Nature Reviews Genetics. 2008 Jul;9(7):527–540. doi: 10.1038/nrg2381. [DOI] [PubMed] [Google Scholar]
- 6.Sanders J, Gill M. Unravelling the genome: a review of molecular genetic research in schizophrenia. Irish Journal of Medical Science. 2007 Mar;176(1):5–9. doi: 10.1007/s11845-007-0004-3. [DOI] [PubMed] [Google Scholar]
- 7.Hattori E, Liu CY, Badner JA, Bonner TI, Christian SL, Maheshwari M, et al. Polymorphisms at the G72/G30 gene locus, on 13q33, are associated with bipolar disorder in two independent pedigree series. American Journal of Human Genetics. 2003 May;72(5):1131–1140. doi: 10.1086/374822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Donovan MC, Williams HJ, Owen MJ. New findings from genetic association studies of schizophrenia. Journal of Human Genetics. 2009 Jan;54(1):9–14. doi: 10.1038/jhg.2008.7. [DOI] [PubMed] [Google Scholar]
- 9.Shi LM, Campbell G, Jones WD, Campagne F, Wen ZN, Walker SJ, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nature Biotechnology. 2010 Oct;:S5–S16. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi LM, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature Biotechnology. 2006 Sep;24(9):1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sequeira PA, Martin MV, Vawter MP. The first decade and beyond of transcriptional profiling in schizophrenia. Neurobiol Dis. 2011 Jan;45(1):23–36. doi: 10.1016/j.nbd.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath S, Langfelder P. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008 Dec 29;9(559) doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 14.Cai CC, Langfelder P, Fuller TF, Oldham MC, Luo R, van den Berg LH, et al. Is human blood a good surrogate for brain tissue in transcriptional studies? BMC Genomics. 2010 Oct 20;:11. doi: 10.1186/1471-2164-11-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mammalian Genome. 2007 Jul;18(6-7):463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu XM, Li MF, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011 Oct 27;478(7370):483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene co-expression networks in human and chimpanzee brains. Proceedings of the National Academy of Sciences of the United States of America. 2006 Nov 21;103(47):17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nature Neuroscience. 2008 Nov;11(11):1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torkamani A, Dean B, Schork NJ, Thomas EA. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Research. 2010 Apr;20(4):403–412. doi: 10.1101/gr.101956.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geschwind DH, Voineagu I, Wang XC, Johnston P, Lowe JK, Tian Y, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011 Jun 16;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. Genome-wide association study identifies five new schizophrenia loci. Nature Genetics. 2011 Oct;43(10):969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009 Aug 6;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi JX, Levinson DF, Duan JB, Sanders AR, Zheng YL, Pe'er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009 Aug 6;460(7256):753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goes FS, Zandi PP, Miao K, McMahon FJ, Steele J, Willour VL. Mood-incongruent psychotic features in bipolar disorder: Familial aggregation and suggestive linkage to 2p11-q14 and 13q21-33. American Journal of Psychiatry. 2007 Feb;164(2):236–247. doi: 10.1176/ajp.2007.164.2.236. [DOI] [PubMed] [Google Scholar]
- 25.Torrey EF, Webster M, Knable M, Johnston N, Yolken RH. The Stanley Foundation brain collection and Neuropathology Consortium. Schizophrenia Research. 2000 Aug 3;44(2):151–155. doi: 10.1016/S0920-9964(99)00192-9. [DOI] [PubMed] [Google Scholar]
- 26.Narayan S, Tang B, Head SR, Gilmartin TJ, Sutcliffe JG, Dean B, et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Research. 2008 Nov 6;1239:235–248. doi: 10.1016/j.brainres.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Grennan K, Badner J, Zhang DD, Gershon E, Jin L, et al. Removing Batch Effects in Analysis of Expression Microarray Data: An Evaluation of Six Batch Adjustment Methods. PLoS One. 2011 Feb 28;6(2) doi: 10.1371/journal.pone.0017238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007 Jan;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 29.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008 Mar 1;24(5):719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 30.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Systems Biology. 2007 Nov 21;:1. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: Using Sequence and Genotype Data to Estimate Haplotypes and Unobserved Genotypes. Genetic Epidemiology. 2010 Dec;34(8):816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Zhang KL, Cui SJ, Chang SH, Zhang LY. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Research. 2010 Jul;38:W90–W95. doi: 10.1093/nar/gkq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langfelder P, Luo R, Oldham MC, Horvath S. Is My Network Module Preserved and Reproducible? PLoS Computational Biology. 2011 Jan;7(1) doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas EA, Torkamani A, Dean B, Schork NJ. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Research. 2010 Apr;20(4):403–412. doi: 10.1101/gr.101956.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saris CGJ, Horvath S, van Vught PWJ, van Es MA, Blauw HM, Fuller TF, et al. Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genomics. 2009 Aug 27;:10. doi: 10.1186/1471-2164-10-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexson TO, Hitoshi S, Coles BL, Bernstein A, van der Kooy D. Notch signaling is required to maintain all neural stem cell populations - Irrespective of spatial or temporal niche. Developmental Neuroscience. 2006;28(1-2):34–48. doi: 10.1159/000090751. [DOI] [PubMed] [Google Scholar]
- 37.Breunig JJ, Silbereis J, Vaccarino FM, Sestan N, Rakic P. Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proceedings of the National Academy of Sciences of the United States of America. 2007 Dec 18;104(51):20558–20563. doi: 10.1073/pnas.0710156104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lathia JD, Mattson MP, Cheng A. Notch: from neural development to neurological disorders. Journal of Neurochemistry. 2008 Dec;107(6):1471–1481. doi: 10.1111/j.1471-4159.2008.05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 40.Lips ES, Cornelisse LN, Toonen RF, Min JL, Hultman CM, Consortium tIS, et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West AK, Hidalgo J, Eddins D, Levin ED, Aschner M. Metallothionein in the central nervous system: Roles in protection, regeneration and cognition. Neurotoxicology. 2008 May;29(3):489–503. doi: 10.1016/j.neuro.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi KH, Elashoff M, Higgs BW, Song J, Kim S, Sabunciyan S, et al. Putative psychosis genes in the prefrontal cortex: combined analysis of gene expression microarrays. BMC Psychiatry. 2008 Nov 7;:8. doi: 10.1186/1471-244X-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peilin Jia Jian Tian, Zhao Zhongming. Assessing gene length biases in gene set analysis of Genome-Wide Association Studies. International Journal of Computational Biology and Drug Design. 2010;3(4):297–310. doi: 10.1504/IJCBDD.2010.038394. [DOI] [PubMed] [Google Scholar]
- 44.Weinberger DR. Implications of Normal Brain-Development for the Pathogenesis of Schizophrenia. Archives of General Psychiatry. 1987 Jul;44(7):660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 45.Pantelis C, Yucel M, Wood SJ, Velakoulis D, Sun DQ, Berger G, et al. Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophrenia Bulletin. 2005 Jul;31(3):672–696. doi: 10.1093/schbul/sbi034. [DOI] [PubMed] [Google Scholar]
- 46.Myers RA, Casals F, Gauthier J, Hamdan FF, Keebler J, Boyko AR, et al. A Population Genetic Approach to Mapping Neurological Disorder Genes Using Deep Resequencing. PLoS Genetics. 2011 Feb;7(2) doi: 10.1371/journal.pgen.1001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shao L, Vawter MP. Shared gene expression alterations in schizophrenia and bipolar disorder. Biological Psychiatry. 2008 Jul 15;64(2):89–97. doi: 10.1016/j.biopsych.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Craddock N O'Donovan MC, Owen MJ. Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophrenia Bulletin. 2006 Jan;32(1):9–16. doi: 10.1093/schbul/sbj033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storey JD, Tibshirani R. Statistical significance for genome-wide studies. Proc Natl Acad Sci USA. 2003 Apr 1;100(7):3889–94. [Google Scholar]
- 50.Jia P, Wang L, Fanous AH, Chen X, Kendler KS, International Schizophrenia Consortium et al. A bias-reducing pathway enrichment analysis of genome-wide association data confirmed association of the MHC region with schizophrenia. J Med Genet. 2012 Feb;49(2):96–10. doi: 10.1136/jmedgenet-2011-100397. [DOI] [PubMed] [Google Scholar]
- 51.Jia P, Wang L, Fanous AH, Pato CN, Edwards TL, The Internatio nal Schizophrenia Consortium et al. Network-Assisted Investigation of Combined Causal Signals from Genome-Wide Association Studies in Schizophrenia. PLoS Computational Biology. 2012;8(7):e1002587. doi: 10.1371/journal.pcbi.1002587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.