

Abstract
Motivation: MicroRNAs (miRNAs) are small non-coding RNAs that are extensively involved in gene expression regulation. One major roadblock in functional miRNA studies is the reliable prediction of genes targeted by miRNAs, as rules defining miRNA target recognition have not been well-established to date. Availability of high-throughput experimental data from a recent CLASH (cross linking, ligation and sequencing of hybrids) study has presented an unprecedented opportunity to characterize miRNA target recognition patterns, which may provide guidance for improved miRNA target prediction.
Results: The CLASH data were analysed to identify distinctive sequence features that characterize canonical and non-canonical miRNA target types. Most miRNA targets were of non-canonical type, i.e. without involving perfect pairing to canonical miRNA seed region. Different miRNAs have distinct targeting patterns, and this miRNA-to-miRNA variability was associated with seed sequence composition. Specifically, seed-based canonical target recognition was dependent on the GC content of the miRNA seed. For miRNAs with low GC content of the seed region, non-canonical targeting was the dominant mechanism for target recognition. In contrast to canonical targeting, non-canonical targeting did not lead to significant target downregulation at either the RNA or protein level.
Contact: xwang@radonc.wustl.edu
1 INTRODUCTION
MicroRNAs (miRNAs) are a family of small non-coding RNAs that play important regulatory roles in many physiological and disease processes (Ambros, 2004; Miska, 2005). About 2000 human miRNAs have been discovered to date (Kozomara and Griffiths-Jones, 2011), and collectively these miRNAs regulate the expression of thousands of genes at both post-transcriptional and translational levels (Baek et al., 2008; Selbach et al., 2008). Thus, identification of gene targets is critical for functional characterization of miRNAs.
Currently, most researchers rely on computational tools to initially identify target candidates for further experimental validation. Significant progress has been made in recent years for the identification of sequence features relevant to target prediction, such as the seed sequence at the 5′ end of the miRNA as well as the sequence context surrounding the seed binding site of the target transcript (Bartel, 2009). However, despite the progress, current bioinformatics target prediction tools still have suboptimal performance with both high false-positive and false-negative rates (Saito and Saetrom, 2010). One major obstacle in computational target prediction is the lack of guidance from experimental observations. To address this issue, multiple high-throughput studies have been performed in recent years to systematically identify miRNA targets at the transcriptome level. For example, one major experimental strategy is to identify target transcripts associated with functional RNA-induced silencing complex (RISC; Chi et al., 2009; Hafner et al., 2010). These methods were able to identify a large number of miRNA target transcripts that were bound to RISC by cross-linking immunoprecipitation followed by high-throughput RNA sequencing (CLIP-seq). In addition, proteomics and ribosome studies have been performed to systematically characterize target expression at the translational level (Baek et al., 2008; Guo et al., 2010; Hendrickson et al., 2009; Selbach et al., 2008).
More recently, CLASH (cross linking, ligation and sequencing of hybrids), an improved CLIP-seq method, has been developed to directly identify miRNA-target pairs by ligation and sequencing of miRNA-target RNA duplexes (Helwak et al., 2013).This new method can simultaneously identify both the miRNAs and their cognate binding sites in the target transcripts, allowing direct characterization of sequence features relevant to miRNA target recognition. With CLASH, thousands of miRNA-target pairs were identified (Helwak et al., 2013). Interestingly, the majority of the target sites contained no canonical seed pairing site, suggesting that other sequence features of the miRNAs played a dominant role in target recognition. In our study, we systematically analysed the sequence composition of the miRNAs and paired target sites from the CLASH data, and identified sequence features that distinguished canonical from non-canonical miRNA targets.
2 METHODS
2.1 Data retrieval
The CLASH dataset was available as Supplementary Data in (Helwak et al., 2013). This dataset was downloaded from the journal’s website and analysed in our study. In this published study, HEK293 cells overexpressing human AGO1 were crosslinked by ultraviolet irradiation. The crosslinked RNA-induced silencing complex (RISC), containing both miRNAs and cognate mRNA target transcripts, were immunoprecipitated using an antibody that specifically binds to the tagged hAGO1 in the RISC. RNA–RNA ligation was then performed for the isolated RISC complexes to covalently ligate the miRNAs and cognate mRNA target transcripts. Ligated RNA products were finally used as a template for RNA-seq library construction, and the identity of both miRNAs and their paired mRNA target transcripts were determined by sequencing chimeric miRNA-target RNA. Altogether, 18 514 miRNA-target pairs were identified in this way.
Microarray data from the photoactivatable-ribonucleoside (PAR)-CLIP study (Hafner et al., 2010) were used to evaluate the impact of miRNAs on target RNA expression regulation. In this microarray study, 25 miRNAs were concurrently suppressed in HEK293 cells by a pool of antisense oligonucleotide inhibitors, and the impact on target expression regulation at the transcriptional level was determined by Affymetrix Human U133Plus2 chips. Raw microarray data were downloaded from the NCBI GEO database (Barrett et al., 2013), and array signals were normalized using the robust multiarray averaging method from the Bioconductor package (http://www.bioconductor.org). Signals from transcripts of the same gene were combined and averaged using Gene mapping index files downloaded from the NCBI ftp site (Maglott et al., 2011). The log2-transformed fold change of each gene after miRNA depletion was calculated by comparing with the negative control arrays (mock transfections).
Proteomic data from (Selbach et al., 2008) were retrieved from the online Supplementary File at the journal’s website, and used to evaluate the impact of miRNA on target protein expression. In the Selbach study, five miRNAs (let-7b, miR-1, miR-16, miR-30a and miR-155) were individually overexpressed in HeLa cells and changes in protein synthesis were determined with mass spectrometry-based proteomic analysis. Protein expression changes were represented by log2-transformed fold change as compared with the negative control (mock transfections).
2.2 Data analysis
Thermodynamic stability of miRNA-target RNA duplex, as represented by the ΔG value, was calculated with RNAfold (Hofacker, 2003). The stability of perfect base pairing between miRNA seed/non-seed sequence and the target binding site was calculated with the nearest neighbor method and thermodynamic parameters for RNA–RNA base pairing (Xia et al., 1998). Canonical seed is defined as any 6-mer sequence within positions 2–8 or 7-mer sequence within positions 1–8 of the miRNA; extended seed is defined as any 6-mer sequence within miRNA positions 4–10; and non-seed sequence is any sequence downstream of the extended seed region. Sequences of the miRNAs and their cognate target sites from the CLASH dataset were compared to identify base pairing patterns, including perfect pairing, imperfect pairing with a GU mismatch and imperfect pairing with a non-GU mismatch. As negative control, the target transcript from each miRNA-target pair was replaced by a randomly selected transcript from the CLASH dataset (shuffled control), and stochastic pairing pattern between the miRNA and random transcript was determined. The enrichment ratio of each base-pairing pattern between the miRNAs and paired target sites was calculated by comparing with the negative control. Statistical difference between various targeting patterns and the negative control for thermodynamic stability or gene expression change was calculated with Student’s t-test.
3 RESULTS
3.1 Canonical seed pairing was absent in most miRNA target sites
Using the CLASH method, 18 514 pairs of miRNAs and cognate target binding sites were identified (Helwak et al., 2013). Here, we reanalysed this dataset to identify sequence features that are significantly associated with miRNA targets. One well-characterized feature is the perfect pairing between canonical miRNA seed (positions 2–8) and the target binding site. The 3′ portion of the miRNA was generally considered to play an auxiliary role, with perfect seed pairing as the dominant determinant (Bartel, 2009). To reassess the relative importance of both seed and non-seed sequences in target recognition, all possible 6-mers from the miRNA sequences were compared with the corresponding paired target sites. Specifically, for each miRNA-target pair, the entire miRNA sequence was scanned to identify all position-specific 6-mers that were matched to the target sequence. As negative control, each miRNA sequence was compared with a randomly assigned transcript from the CLASH dataset (shuffled control). Overall, 6-mers from positions 3–8 were most enriched, which fall within the canonical seed region (Fig. 1). The other 6-mer position from the canonical seed region (nucleotides 2–7) was ranked as the third most enriched along the miRNA sequences. Thus, our analysis was consistent with previous findings that perfect canonical seed pairing is an important parameter for miRNA target recognition.
Fig. 1.
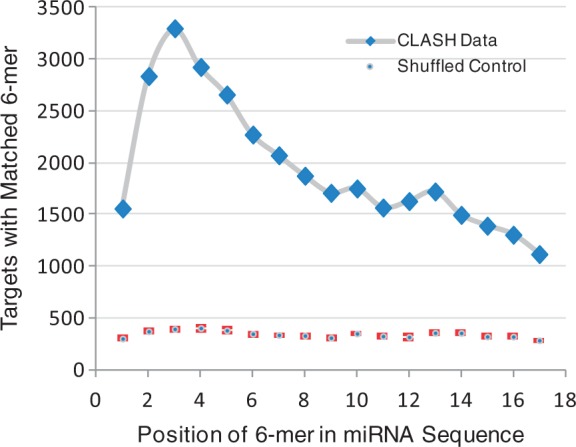
Occurrence of target sites matching to miRNA 6-mer words. For each miRNA-target pair, the 6-mer word from each position of the miRNA sequence was compared with the target sequence to identify any perfect base pairing. The numbers of miRNA-target pairs with 6-mer match at the same miRNA positions were summarized and presented in the graph. As negative control, the cognate target sequence in each miRNA-target pair was replaced with a randomly assigned non-target transcript sequence for 6-mer word analysis (shuffled control). The negative control simulation run was repeated 10 times and the average number of matched 6-mers at each position was presented (±SD)
Interestingly, 6-mers from all positions of the miRNA sequence were paired to the target sites at significantly higher rates as compared with the negative control (Fig. 1). In particular, 6-mers from two positions, nucleotides 4–9 and 5–10 were the second and fourth most enriched, respectively, although they fall outside the canonical seed region. Thus, nucleotides 4–10 were considered as the “extended” seed region in our analysis as 6-mers from this region partly overlap with canonical seeds, matching to the target sites at similarly high rates to canonical seeds. Further, 6-mers from all other positions in the miRNA sequence were also significantly enriched, although to a lesser extent as compared with canonical or extended 6-mer seeds (Fig. 1). As shown in Table 1, perfect matches to canonical 6-mer seeds were identified in 22.4% of all target sites, which was 6.8-fold enriched as compared with the shuffled control. Similarly, 6-mers from the extended seed region were matched to 20.9% of all target sites, which was 6.3-fold enriched. Seed pairing was even more significant when 7-mer seeds were considered, with 11.0- and 11.5-fold in enrichment for canonical and extended seed regions, respectively. When 6-mers from both canonical and extended seed regions were considered collectively, seed matches were found in 32.9% of all target sites. Thus, the majority of the target sites did not have perfect pairing to any miRNA seed even when both canonical and extended seeds were considered.
Table 1.
Characteristics of seed-pairing sites in miRNA target transcripts
| Seed type | Targets with seed-pairing site (%) | Shuffled control (%) | Enrichment ratio |
|---|---|---|---|
| 6-mer perfect match | |||
| Canonical region (nucleotides 2–8) | 22.4 | 3.3 | 6.8 |
| Extended region (nucleotides 4–10) | 20.9 | 3.3 | 6.3 |
| 6-mer with one G-U mismatch | |||
| Canonical region (nucleotides 2–8) | 13.5 | 6.0 | 2.3 |
| Extended region (nucleotides 4–10) | 16.0 | 6.6 | 2.4 |
| 7-mer perfect match | |||
| Canonical region (nucleotides 1–8) | 12.1 | 1.1 | 11.0 |
| Extended region (nucleotides 3–10) | 12.6 | 1.1 | 11.5 |
| 7-mer with an internal non-GU mismatch | |||
| Canonical region (nucleotides 1-8) | 8.9 | 6.4 | 1.4 |
| Extended region (nucleotides 3–10) | 8.5 | 6.5 | 1.3 |
Similar to perfect base pairing, G-U wobble pairs can also provide thermostability in the RNA duplex. However, a G-U mismatch in the seed region may potentially interfere with target binding based on structural analysis (Bartel, 2009). The impact of G-U mismatch on seed-target binding is still a debatable topic, as some studies suggested that the presence of a G-U mismatch would mostly abolish the miRNA targeting ability, while some other studies identified functional target sites with G-U mismatch to the miRNA seed (Brodersen and Voinnet, 2009; Vella et al., 2004). This issue was revisited using the CLASH data to analyse any 6-mer seed with a single G-U mismatch to the target site. As shown in Table 1, there were significantly more 6-mers from both canonical and extended seed regions, with 2.3- and 2.4-fold in enrichment, respectively, for target pairing with a G-U mismatch as compared with the shuffled control. Thus, our analysis indicated that imperfect seed pairing with a G-U mismatch to the target site may still be biologically relevant. However, the enrichment ratios were much lower than those of perfect seed match, suggesting significantly less important role of G-U mismatched seed pairing in miRNA target recognition. Furthermore, the impact of non-GU mismatches was also examined. A 7-mer with an internal non-GU mismatch was only slightly enriched in the target sites (1.3- and 1.4-fold in enrichment, Table 1), indicating that a non-GU internal mismatch in seed pairing would mostly abolish miRNA target recognition. Combined together, 51.1% of all target sites were paired perfectly to canonical/extended seeds, or imperfectly with a G-U mismatch.
We further analysed the relevance of non-seed sequence of miRNA in target recognition. Among all seed-pairing target sites (9465 in total), 43.5% of them were also paired to non-seed region of miRNA, which was a significant enrichment over shuffled control (Table 2). Furthermore, as to non-canonical target sites without any seed match, 40.9% were paired to the non-seed region of the miRNA. Thus, pairing to non-seed region of the miRNA was a significant feature for both canonical and non-canonical targets.
Table 2.
Presence of non-seed 6-mer match in miRNA target sites
| Target type | CLASH data | Shuffled control | Enrichment ratio |
|---|---|---|---|
| Targets with seed match | 9465 | 3027 | |
| Targets also with non-seed 6-mer match | 4121 | 541 | |
| Percentage of targets with non-seed 6-mer match | 43.5% | 17.9% | 2.4 |
| Targets with no seed match | 9049 | 15 537 | |
| Targets with non-seed 6-mer match | 3705 | 1529 | |
| Percentage of targets with non-seed 6-mer match | 40.9% | 9.8% | 4.2 |
3.2 GC content of miRNA seed was strongly correlated to target recognition patterns
All 18 514 target sites from the CLASH dataset were analysed to identify sequence features that characterized perfect seed pairing. First, canonical 6-mer seeds, involving either positions 2–7 or 3–8, were characterized. Interestingly, there was a very strong correlation between GC content and perfect pairing of 6-mer seeds within the canonical region, with correlation coefficients (r) of 0.91 and 0.96 for nucleotides 2–7 and 3–8, respectively (Fig. 2A and B). Few targets were paired to any canonical seed when the GC content of the seed was low. On the other hand, a much higher percentage of seed-pairing targets were found for miRNAs with high GC content of the seed. GC content is commonly used to represent thermodynamic stability of RNA duplex. Thus, the thermostability of seed-target binding was also evaluated by calculating the free energy (ΔG) of seed-target RNA duplex with RNAfold (Hofacker, 2003). As expected, the ΔG of seed-target duplex was strongly correlated with the percentage of targets with seed pairing (r = −0.90 and −0.95 for nucleotides 2–7 and 3–8, respectively; Fig. 2C and D). Thus, seed-based target recognition was dependent on the thermostability of seed pairing to the target site. For miRNAs with weak canonical seed-pairing stability, non-canonical targeting was the dominant mechanism for target recognition.
Fig. 2.
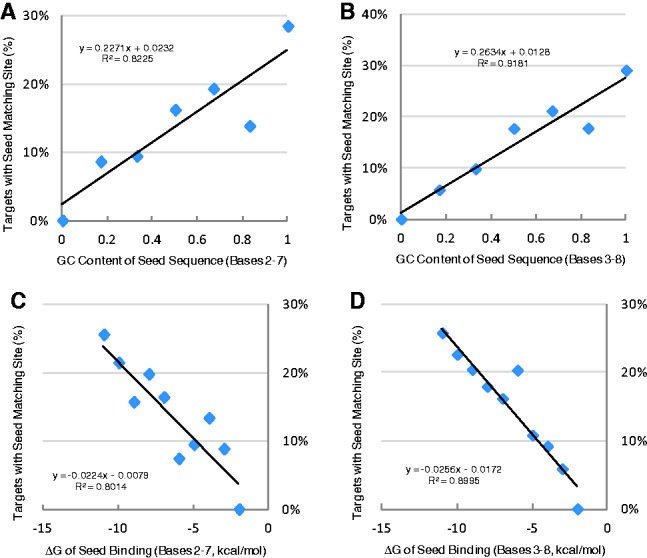
GC content and thermostability of canonical seeds that were paired to the target sites. (A and B) Correlation between GC content of canonical seeds (positions 2–7 or 3–8) and percentage of seed-pairing target sites. (C and D) Correlation between thermostability of seed binding, as measured by free energy (ΔG) and percentage of seed-pairing target sites
Besides targets involving canonical seed pairing, we also analysed non-canonical targets including pairing to extended seeds, imperfect pairing with a G-U mismatch or pairing to non-seed sequences. Similar to canonical seed pairing, 6-mer matches to the extended seed region (positions 4–10) were strongly correlated to sequence GC content (r = 0.99, Fig. 3A), which reflected the stability of seed-target duplex as represented by ΔG (r = −0.98, Fig. 3B). Combined together, the GC content of both canonical and extended seeds within the full seed region was a strong predictor of perfect seed pairing to the target site. In addition to perfect seed pairing, imperfect seed pairing with a G-U mismatch was also analysed. As shown in Figure 3C, low GC content (≤50%) of the seed was also a significant predictor of poor seed pairing (r > 0.99). Interestingly, no such correlation was observed when the GC content was higher (>50%). Pairing between non-seed 6-mers in the miRNA sequence and the target sites was also strongly dependent on the GC content of the 6-mers, in a similar way to seed pairing of the target site (r = 0.98, Fig. 3D). Thus, binding stability of the paired nucleotides was a major determinant of miRNA targeting patterns, whether involving seed or non-seed sequences.
Fig. 3.
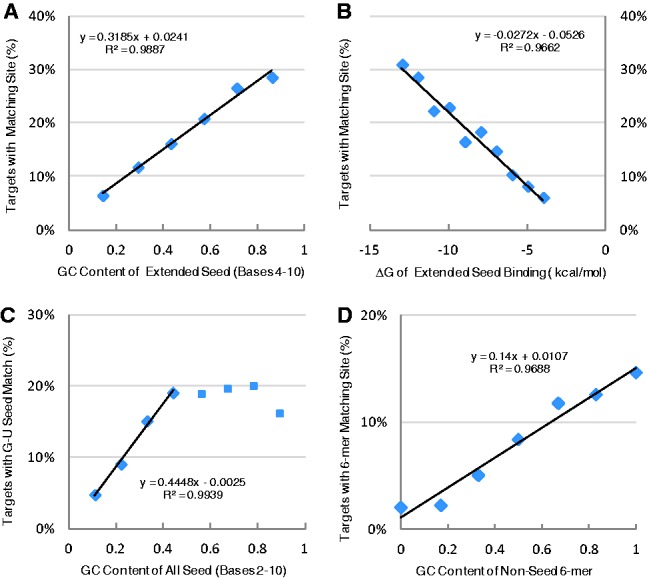
GC content and thermostability of non-canonical seeds that were paired to the target sites. (A) Correlation between GC content of non-canonical extended seeds (any 6-mer within positions 4–10) and percentage of perfect seed-pairing target sites. (B) Correlation between thermostability of extended seed binding, as measured by free energy (ΔG) and percentage of seed-pairing target sites. (C) Correlation between any 6-mer seed and percentage of imperfect seed-pairing target sites containing a G-U mismatch. (D) Correlation between non-seed 6-mers in miRNA sequence and percentage of targets with 6-mer matching sites
3.3 Canonical and non-canonical targeting had similar thermostability but distinct impacts on target expression
Overall thermostability of the miRNA-target RNA duplex was assessed for both canonical and non-canonical targeting patterns. The following non-canonical target types were included in the analysis: targets pairing to extended seed region, pairing to non-seed region and with no pairing anywhere in the miRNA sequence. These non-canonical targets were compared with canonical targets as well as randomly assigned non-target transcripts from the CLASH dataset (shuffled control). Specifically, overall distribution of thermostability for each type of target binding, as represented by ΔG, was determined with RNAfold (Hofacker, 2003). As shown in Figure 4, binding of miRNAs to their cognate targets was significantly more stable than binding to randomly matched transcripts (P < 10−300 with Student’s t-test for all target types). The mean binding free energies were −24.6, −24.8, −24.4 and −20.9 kcal/mol for targets involving canonical seed pairing, extended seed pairing, non-seed 6-mer pairing and no pairing anywhere in the miRNA sequence, respectively. There was no difference among the first three target types, all of which had significantly lower free energy than the last type. In contrast, binding between miRNAs and randomly matched transcripts were much more unstable, with mean ΔG of −17.1 kcal/mol.
Fig. 4.
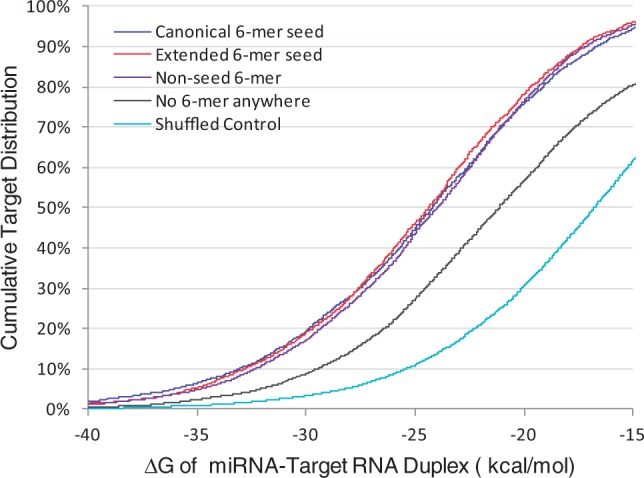
Thermostability of miRNA-target duplex for individual targeting patterns. A cumulative distribution of targets in relation to ΔG was presented for each target type. The following target groups were considered: targets matching to at least one canonical 6-mer seed, but not to any extended seed; targets not matching to any canonical seed, but to at least one 6-mer extended seed; target not matching to any type of seed, but to at least one 6-mer in the non-seed region; targets with no 6-mer match anywhere in the miRNA sequence. As negative control, the cognate target sequence was replaced by a randomly assigned transcript sequence for miRNA pairing analysis (shuffled control)
It was shown previously that binding of miRNA to the target generally resulted in suppressed gene expression at both the transcriptional and translational levels (Baek et al., 2008; Selbach et al., 2008). However, most previous studies were focused on canonical miRNA targeting involving perfect seed pairing, and the impact of miRNA binding to non-canonical targets has not been well characterized. In our analysis, the CLASH dataset was combined with a public microarray dataset, which studied the impact of concurrent depletion of 25 miRNAs on mRNA levels in HEK 293 cells (Hafner et al., 2010). As negative control of the analysis, each target transcript in the miRNA-target pairs from the CLASH dataset was replaced by a randomly assigned non-target transcript with similar expression level to the target as determined by microarrays. Specifically, the expression difference between a target transcript and its random control was <0.5 (log2-transformed expression value). Consistent with the Hafner study, canonical targets with perfect seed pairing sites were significantly upregulated by miRNA depletion as compared with the negative control (mean fold change of 0.044 and P < 10−17 by Student’s t-test, Fig. 5A). In contrast, all three types of non-canonical targets were upregulated only moderately as compared with the negative control. The mean fold changes for individual non-canonical target types were in the range of 0.014 to 0.021 and P-values in the range of 0.003 to 0.0002. Thus, miRNA binding to target transcripts led to distinctively different outcomes for gene expression regulation at the mRNA level, which were dependent on the type of miRNA targets.
Fig. 5.
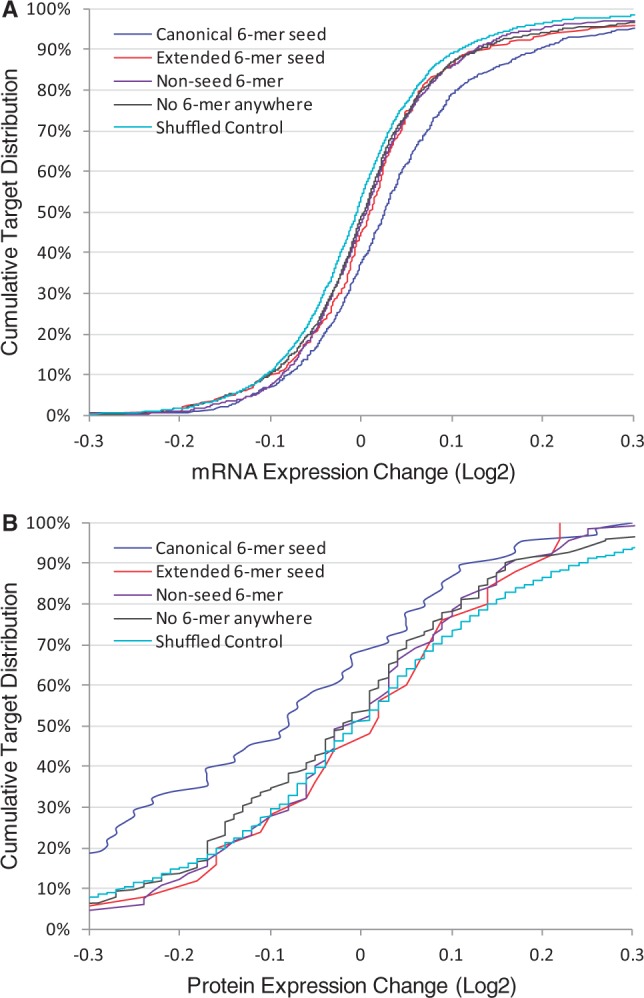
Impact on target gene expression was dependent on miRNA targeting patterns. The following target groups were considered: targets matching to at least one canonical 6-mer seed, but not to any extended seed; targets not matching to any canonical seed, but to at least one 6-mer extended seed; target not matching to any type of seed, but to at least one 6-mer in the non-seed region; targets with no 6-mer match anywhere in the miRNA sequence. A cumulative distribution of targets in relation to gene expression changes was presented for each target type. (A) Target mRNA expression changes resulting from simultaneous depletion of 25 miRNAs (Hafner et al., 2010). As negative control, the cognate target sequence was replaced by a randomly assigned transcript sequence for miRNA pairing analysis (shuffled control). (B) Target protein expression changes resulting from individually overexpressing five miRNAs (Selbach et al., 2008). All 330 miRNA-target pairs present in both the proteomic and CLASH datasets were included in the analysis of different miRNA targeting patterns. As negative control, the cognate target protein was replaced by a randomly assigned protein from the proteomic dataset (shuffled control)
Furthermore, the impact on target protein expression by different miRNAs was evaluated with proteomic data. Selbach et al. performed a proteomic study to identify the global impact of miRNA overexpression on protein synthesis (Selbach et al., 2008). In the Selbach study, five miRNAs were individually transfected into HeLa cells and changes in protein synthesis due to miRNA overexpression were determined by proteomic studies. In our analysis, different miRNA targeting types were evaluated with this proteomic dataset. Specifically, all miRNA-target pairs from the CLASH dataset were cross-checked with the proteomic dataset, and 330 common miRNA-target pairs presented in both datasets were identified. From these identified pairs, different miRNA targeting patterns were correlated to changes in target protein expression due to overexpression of individual miRNAs. As shown in Figure 5B, canonical targets with perfect 6-mer seed match were significantly suppressed by miRNA overexpression (mean fold change of −0.114 and P = 0.0004 by comparing with the negative control by Student’s t-test). In contrast, none of the other miRNA targeting patterns were significantly associated with suppression of target protein expression (mean fold change ranging from −0.027 to 0.001 and P-values ranging from 0.36 to 0.87 by Student’s t-test).
4 DISCUSSION
Multiple experimental strategies have been established to identify both miRNAs and target transcripts that are bound to the RISC complex (Chi et al., 2009; Hafner et al., 2010). However, one major challenge of these strategies is that RNA sequences were identified after pooling miRNAs and target transcripts from all RICS complexes together, which represents a many-to-many relationship for miRNAs and their targets. Thus, information about specific miRNA-target pairs in each RISC complex was unknown. Although multiple computational algorithms were proposed recently for prediction of miRNA-target pairs from CLIP data (Khorshid et al., 2013; Liu et al., 2013; Reczko et al., 2012), the most straightforward and reliable way would be to directly identify miRNA-target pairs experimentally. Using the CLASH method, Helwak et al. (2013) identified thousands of specific miRNA-target transcript pairs that were bound to the same RISC complexes. Their work presented an unprecedented opportunity to study miRNA target recognition patterns, especially those involving non-canonical target sites.
Most previous miRNA target analyses were focused on target sites pairing to canonical miRNA seed region. The CLASH data indicate that many targets are not paired to any canonical miRNA seed. In fact, canonical miRNA targets represented only 22% of all targets in the CLASH dataset (Table 1). However, it was not clear why canonical or non-canonical targets were preferentially associated with certain miRNAs. A major novelty of this work is the identification of sequence determinants for miRNA-to-miRNA variability in target recognition patterns. Specifically, sequence features related to miRNA seed composition were identified that distinguish canonical miRNA targeting from non-canonical targeting. Our previous work on a limited number of miRNAs (Nelson et al., 2011) suggested that there is significant miRNA-to-miRNA variability associated with target recognition patterns. Interestingly, reanalysis of the CLASH data containing a large number of miRNAs revealed that composition of the miRNA seed sequence was strongly correlated to targeting patterns. For miRNAs with weak seed binding stability, they recognized target transcripts mostly through non-canonical mechanisms without involving canonical seed pairing. Thus, the current work suggested that variability in seed sequence composition resulted in variable miRNA-dependent targeting patterns, and this seed variability issue should be taken into consideration when predicting targets for individual miRNAs. To this end, we are currently working on incorporating the seed composition feature into an improved target prediction algorithm for more accurate prediction of suppression of target gene expression.
Overall, non-canonical targeting was the norm rather than exception when all miRNAs were considered collectively. However, the functional consequences of miRNA targeting were dependent on target recognition patterns. Canonical targeting, involving perfect pairing to the canonical seed region, generally led to significant downregulation of target transcripts. In contrast, only marginal transcriptional downregulation was observed for non-canonical types of targets based on microarray analysis (Fig. 5A). Similarly, canonical targeting, but not any other targeting type, led to significant suppression of target protein expression based on proteomic analysis (Fig. 5B). Thus, our target expression analysis agrees with previous observations that miRNAs typically suppress target expression at both the transcriptional and translational levels (Baek et al., 2008; Guo et al., 2010; Hendrickson et al., 2009; Selbach et al., 2008). Consistent with our analysis, previous experimental studies demonstrated that miRNAs with weak seed binding stability had low proficiency on target downregulation (Didiano and Hobert, 2006; Garcia et al., 2011), which can be explained by our prediction that most targets of these miRNAs were of non-canonical type whose expression was not effectively impacted by miRNA binding.
The target-type dependency for gene expression regulation raised an interesting question as to what constitutes an effective miRNA target. Non-canonical targets were bound by miRNA and this binding could be functionally important in ways other than direct target downregulation. For example, non-canonical targets may serve as competing endogenous RNAs (ceRNAs) to sequester miRNAs from regulating canonical targets (Salmena et al., 2011). If this is the case, non-canonical targets could behave more like a reservoir of ‘miRNA regulators’, rather than targets of miRNA. Thus, it is likely that concurrent interactions between miRNAs and canonical/non-canonical targets are important to maintain the steady state of gene expression regulation networks mediated by miRNAs.
Funding: National Institutes of Health (grant R01GM089784 to X.W.).
Conflict of Interest: none declared.
REFERENCES
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Baek D, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat. Rev. Mol. Cell Biol. 2009;10:141–148. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- Chi SW, et al. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didiano D, Hobert O. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat. Struct. Mol. Biol. 2006;13:849–851. doi: 10.1038/nsmb1138. [DOI] [PubMed] [Google Scholar]
- Garcia DM, et al. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, et al. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, et al. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, et al. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009;7:e1000238. doi: 10.1371/journal.pbio.1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorshid M, et al. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods. 2013;10:253–255. doi: 10.1038/nmeth.2341. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, et al. CLIP-based prediction of mammalian microRNA binding sites. Nucleic Acids Res. 2013;41:e138. doi: 10.1093/nar/gkt435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, et al. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–D57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005;15:563–568. doi: 10.1016/j.gde.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Nelson PT, et al. Specific sequence determinants of miR-15/107 microRNA gene group targets. Nucleic Acids Res. 2011;39:8163–8172. doi: 10.1093/nar/gkr532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczko M, et al. Functional microRNA targets in protein coding sequences. Bioinformatics. 2012;28:771–776. doi: 10.1093/bioinformatics/bts043. [DOI] [PubMed] [Google Scholar]
- Saito T, Saetrom P. MicroRNAs - targeting and target prediction. N Biotechnol. 2010:243–249. doi: 10.1016/j.nbt.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Salmena L, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Vella MC, et al. Architecture of a validated microRNA::target interaction. Chem. Biol. 2004;11:1619–1623. doi: 10.1016/j.chembiol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Xia T, et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]